
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the photocatalysis evolution of heteroatom-doped Ag4M2 nanoclusters†
Xinglian Cheng‡
a,
Xin Sui‡b,
Jiayu Xua,
Xu Liu *a,
Mingyang Chen*bc and
Yan Zhua
*a,
Mingyang Chen*bc and
Yan Zhua
aSchool of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: xuliu@nju.edu.cn
bCenter for Green Innovation, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: mychen@ustb.edu.cn
cBeijing Computational Science Research Center, Beijing 100193, China
First published on 4th October 2021
Abstract
Atomically precise metal nanoclusters doped with one or more heteroatom of other metals have exhibited extraordinary catalytic properties. Here we report a series of thiolate-protected Ag4M2 (M is dopant Ni, Pd and Pt) nanoclusters that adopt a similar structural framework like a distorted hexahedron, in which four Ag atoms are located at the midpoints of four side edges and two metal heteroatoms reside on the centres of the top and the bottom planes. The opposite orders of the catalytic performances of the three catalysts for the photocatalytic degradation of the methyl orange and rhodamine B dyes are found, which is attributed to two different types of inter-molecular recombination mechanisms. In both photocatalytic systems, both the catalyst and the dye are visible-light active, and the inter-molecular recombination of the photo-excited hole in the catalyst and the photo-excited electron in the dye leads to charge separation across the system comprising the catalyst and the dye. The study represents an important step towards developing the precise tailoring of the composition and structure to control the physicochemical properties of metal nanoclusters.
Introduction
Atomically precise metal nanoclusters recently have drawn research attention, owing to their precise crystal structures and unique physicochemical properties.1–6 Such metal nanoclusters have found potential applications in optics, electronics, catalysis, etc.7–11 Tailoring the size, structure, and composition of the metal nanoclusters can elaborately tune the optical, electronic and catalytic properties, which can provide opportunities to not only expand their practical applications but also gain fundamental understanding of the intrinsic aspects of the nanoclusters.12–15 For example, it has been reported that the editing on the surface structure of Au23(SR)16 can modulate the product selectivity for the click reaction of phenylacetylene and benzyl azide.14 The excited-state carrier lifetimes can vary with crystal structure of gold nanoclusters where the body-centered cubic Au38 has a much longer carrier lifetime than the face-centered cubic Au36 and hexagonal close-packed Au30.15Doping other metals into the parent nanoclusters is of paramount importance to attain atomically precise bimetal nanoclusters and to use them as well-defined materials for various applications, in particular, catalytic applications.16–20 The monoplatinum-doped PtAu24(SR)18 nanocluster as a heterogeneous catalyst exhibited higher activity than Au25(SR)18 for the styrene oxidation.16 Au47Cd2(TBBT)31 from Au44(TBBT)28 (TBBT = 4-tert-butylbenzenethiol) doped with Cd atoms into the surface as an electrocatalyst gave rise to very high faradaic efficiencies for CO2 electroreduction.17 The gold nanocluster doped with Ru atoms as a photocatalyst showed an excellent photocatalytic performance for N2 reduction.18 Therefore, the doped metal nanoclusters have emerged as a new class of promising catalysts for widely catalysis fields.
Here we study the catalytic performances of a series of Ag4M2 nanoclusters doped with Ni, Pd and Pt atoms, in which a new Ag4Pt2 nanocluster is synthesized with structurally similar to previously reported Ag4Ni2 and Ag4Pd2.21,22 The distinguishable catalysis evolution behaviours of the three Ag4M2 nanoclusters can be found in the photocatalytic reaction and the corresponding mechanism is revealed.
Results and discussion
The three Ag4M2(SR)8 (M = Ni, Pd, Pt; SR = SPhMe2; short for Ag4M2) nanoclusters adopt a similar structural pattern. For each Ag4M2, as shown in Fig. 1a–c, four Ag atoms are located at the midpoints of four side edges of the hexahedron framework and two metal heteroatoms reside on the centres of the top and the bottom planes. The S atoms of the 2,4-dimethylbenzenethiol each binding to one Ag atom and one Ni/Pd/Pt atom form a rectangular cuboid. Viewed from the plane which consists of a Ni/Pd/Pt atom and four S atoms, each four thiol ligands are arranged as clockwise. Based on the steric hindrance, the two ligands bound to the same Ag are staggered rather than eclipsed. The average angle of S–Ag–S in Ag4M2(SR)8 (M = Ni, Pd, Pt) is 171.25°, 171.42° and 171.12°, respectively, indicating that the two S atoms are not fully eclipsed. For the Ag–S bond length, Ag4Pt2(SR)8 has the longest Ag–S bond average length of 2.390 Å, followed by 2.387 Å for Ag4Ni2(SR)8 and then 2.370 Å for Ag4Pd2(SR)8. All of the three clusters' S–M–S (M = Ni, Pd, Pt) average angle is found to be 90°, which means the four S atoms and the M atom are in a plane. Ag4Pd2(SR)8 and Ag4Pt2(SR)8 have almost the same M–S bond average length, e.g., 2.342 Å and 2.343 Å. The Ni–S bond length of Ag4Ni2(SR)8 is the shortest, that is, 2.235 Å. Ag–Ag bond average distances in the three clusters are presented in a descending sequence: 3.109 Å (Ag4Pt2(SR)8) > 3.065 Å (Ag4Pd2(SR)8) > 2.987 Å (Ag4Ni2(SR)8). Notably, the dopant Ni, Pd, Pt atoms in the three clusters are unshielded by the organic ligands, which is beneficial for catalytic performances.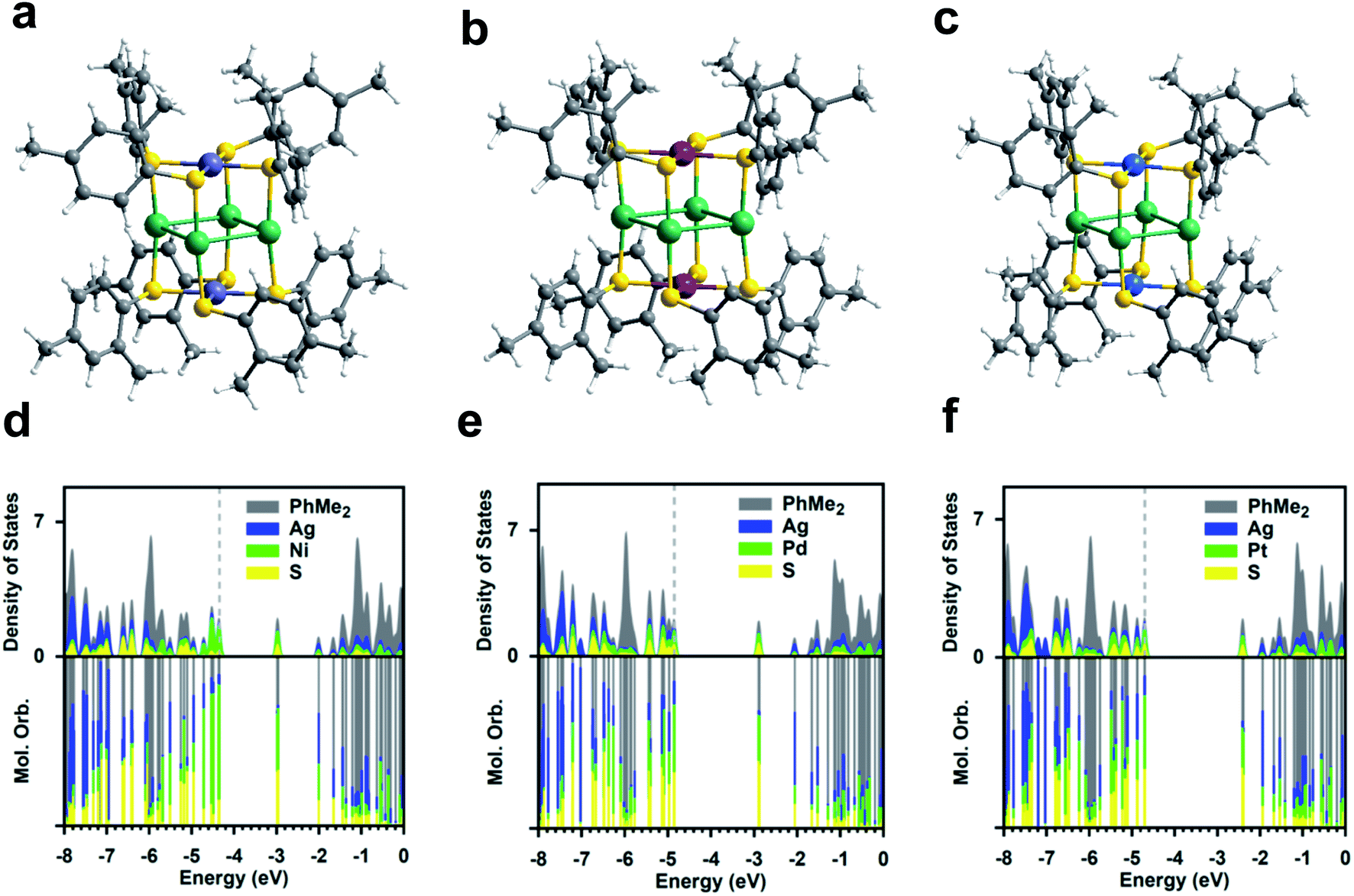 | ||
| Fig. 1 Crystal structures of (a) Ag4Ni2(SPhMe2)8, (b) Ag4Pd2(SPhMe2)8, and (c) Ag4Pt2(SPhMe2)8. PDOS for Ag4M2(SPhMe2)8 at the DFT PBE/Aug-cc-pVDZ level, M = (d) Ni, (e) Pd, and (f) Pt. Color labels: purple = Ni; red = Pd; blue = Pt; green = Ag; yellow = S; gray = C; white = H. | ||
The partial densities of states (PDOSs) for the three clusters are given in Fig. 1d–f. It reveals that both the HOMO and LUMO are dominated by the doubly-degenerate atomic bonding and antibonding M–S orbitals for Ag4Pd2 and Ag4Pt2; for Ag4Ni2, the HOMO is dominated by the localized Ni d orbital possibly due to the small ionic radius for Ni and the LUMO is dominated by the antibonding Ni–S orbital. It is aware that the HOMO–LUMO photoexcitation for the three catalysts, despite being visible-light active, does not lead to spatial charge separation and the resultant exciton may undergo ultrafast recombination before it could be utilized due to the nature of the d–d* excitation. For all three catalysts, the LUMO + 2 is dominated by the unoccupied Ag orbital, which lies <3 eV above the HOMO.
The heteroatoms doping induces the alterations in the electronic structures of the three clusters, which is indicated by their UV-vis optical spectra. As shown in Fig. 2a, the Ag4Ni2 cluster shows two broad absorption peaks at 381 and 445 nm, respectively. The Ag4Pd2 cluster has two prominent absorption peaks at 321 and 408 nm, respectively, which are blue-shift compared to Ag4Ni2. For Ag4Pt2, one sharp absorption peak at 319 nm and two shoulder peaks at 281 and 415 nm can be observed, which is further blue-shift from Ag4Pd2. Plotting UV-vis absorption spectra on the photon energy scale and extrapolating the absorbance to zero can give rise to optical energy gaps (Fig. 2b). The corresponding highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) gaps of Ag4Ni2, Ag4Pd2 and Ag4Pt2 are measured as 1.60, 2.24 and 2.61 eV, respectively.
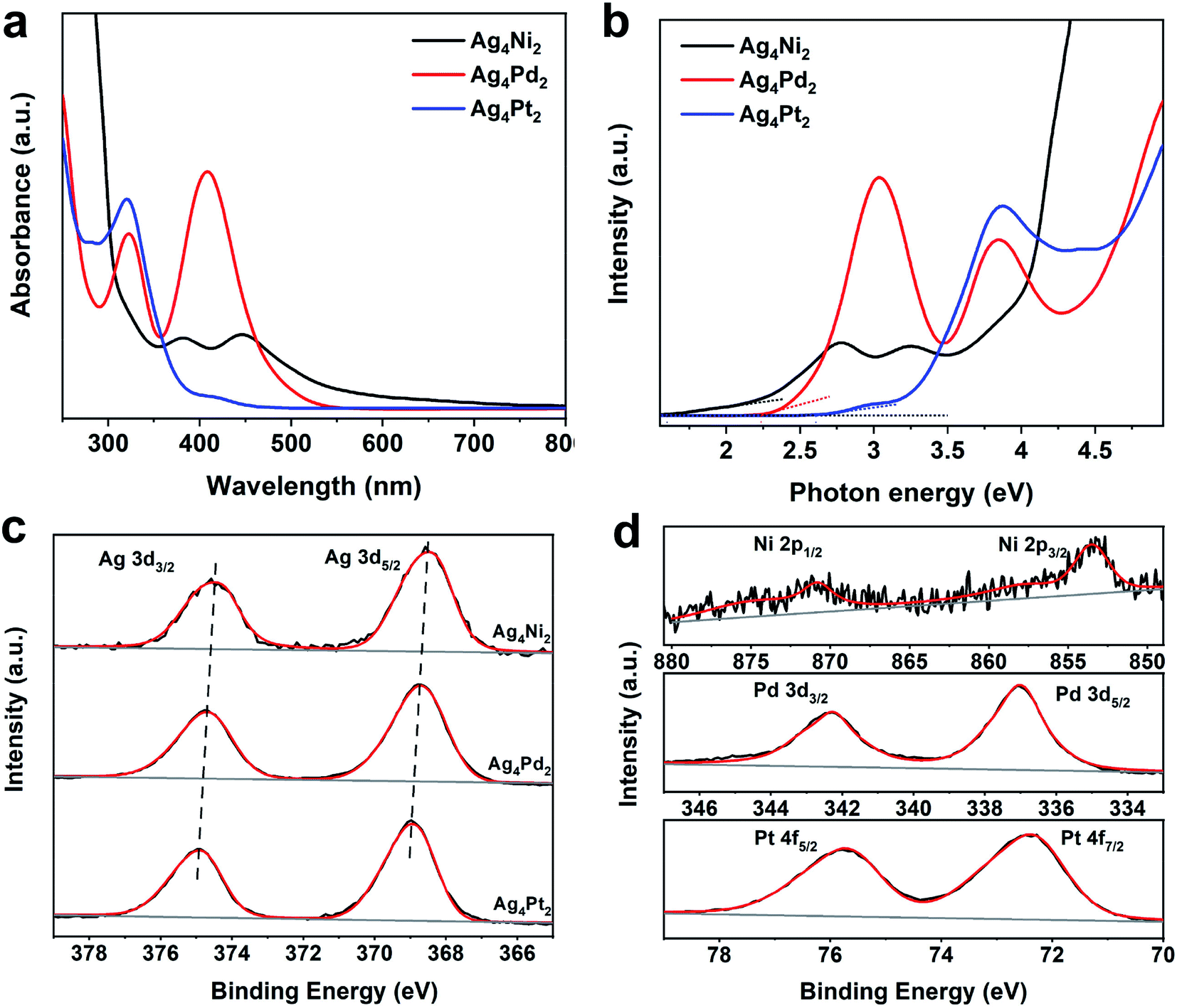 | ||
| Fig. 2 (a) UV-vis absorption spectra of Ag4Ni2, Ag4Pd2 and Ag4Pt2. (b) UV-vis absorption spectra plotted on the photon energy scale. (c) Ag 3d XPS profiles of Ag4Ni2, Ag4Pd2 and Ag4Pt2. (d) Ni 2p, Pd 3d and Pt 4f XPS profiles of Ag4M2 (M = Ni, Pd, Pt). | ||
Furthermore, X-ray photoelectron spectroscopy (XPS) is used to measure the metal charge states of the Ag4Ni2, Ag4Pd2 and Ag4Pt2 clusters. The binding energies of Ag 3d5/2 and Ag 3d3/2 in Ag4Ni2 are located at 368.5 and 374.5 eV (Fig. 2c), which are close to the Ag1+ according to the previous report.23 The binding energies of Ag 3d in Ag4Pd2 and Ag4Pt2 are respectively positively shifted by 0.2 and 0.4 eV (the corresponding binding energies are listed in Table S1†), further revealing that the heteroatoms doping could change the electronic properties in parent clusters. Fig. 2d shows that the XPS analysis of Ni, Pd and Pt in the three clusters illustrates the charge states of the three dopants are likely +2.21–23
The atomic-level structures and electronic properties of the three Ag4M2 clusters can have a substantial influence on the catalytic properties. Thus, the three clusters are acted as ideal model catalysts to evaluate the distinct catalysis at an atomic-level. Photocatalysis of the three Ag4M2 nanoclusters was explored by using them supported on TiO2 as photocatalysts for photocatalytic degradation of the methyl orange and rhodamine B (Fig. 3). The three Ag4M2/TiO2 catalysts exhibited more effective photocatalytic activity than pure TiO2. During the photocatalytic degradation of the methyl orange, the Ag4Pd2 catalyst gave rise to the best photocatalytic performance among the three cluster catalysts. Within 18 min, the methyl orange can be completely degraded on the Ag4Pd2 catalyst under illumination, while the degradation of the methyl orange on the Ag4Pt2 catalyst need 42 min with the visible light, and the photodegradation process on the Ag4Ni2 catalyst spent 54 min. From Fig. 3a, the plots of the methyl orange degradation on the catalysts versus reaction time further imply the photocatalysis evolution behaviors of the three clusters: Ag4Pd2 > Ag4Pt2 > Ag4Ni2. On the contrary, the order of activity in the photocatalytic degradation of the rhodamine B is Ag4Ni2 > Ag4Pt2 > Ag4Pd2 (Fig. 3b). The three clusters were robust during the catalytic processes (Fig. S1†). Deduced from the electrochemical impedance spectroscopy (EIS) of the three Ag4M2/TiO2 catalysts and pure TiO2 (Fig. S2†), which is measured for the interfacial charge-transfer,24 the semicircular diameters of the three cluster samples were smaller than that of pure TiO2. The smaller impedance of the three cluster catalysts revealed the faster interfacial-charge transfer between the clusters and TiO2. It can account in part for higher photocatalytic activity of the three cluster catalysts than pure TiO2 for the two photocatalytic degradation reactions.
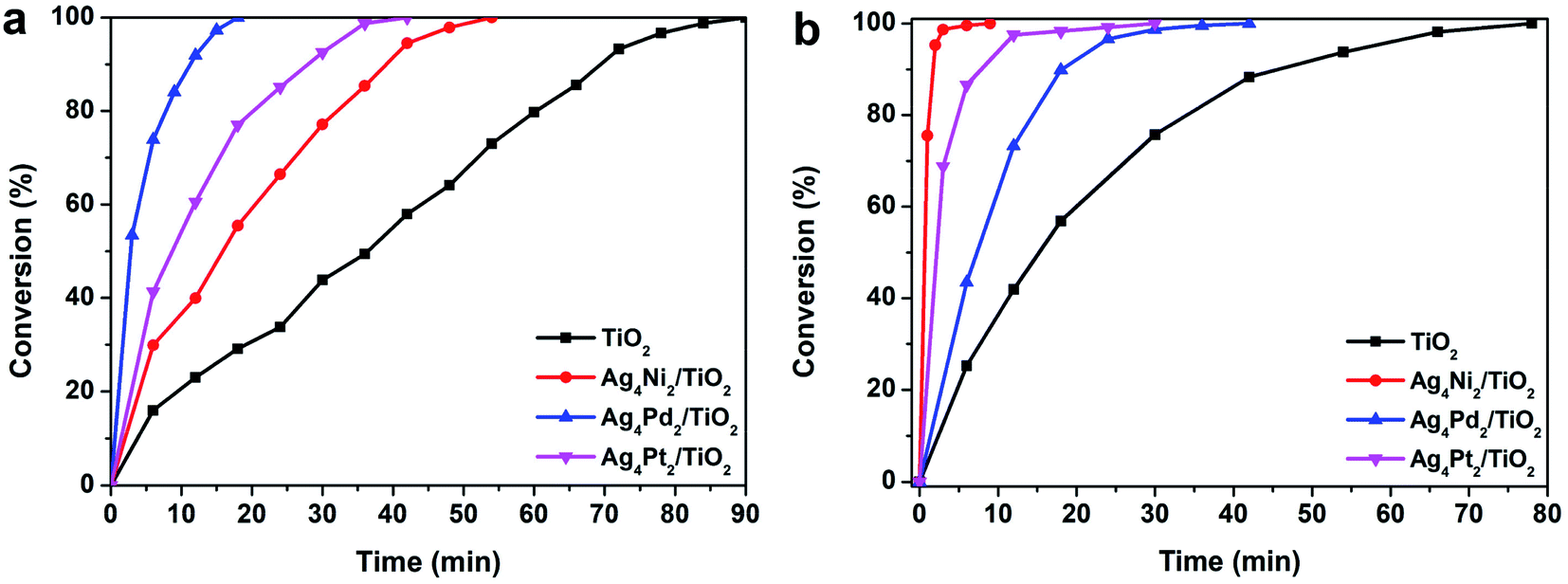 | ||
| Fig. 3 Photocatalytic degradation of (a) methyl orange and (b) rhodamine B. Reaction conditions: 50 mg Ag4M2/TiO2, 20 mL H2O and 0.1 mL (1 g L−1) methyl orange or rhodamine B, under visible light. | ||
To understand the origins for the different photocatalytic performance of the three Ag4M2 cluster catalysts for the catalytic reduction of methyl orange and rhodamine B, the electronic structures of the Ag4M2(SPhMe2)8, M = Ni, Pd and Pt, were analyzed with the full atomistic structural model. The predicted HOMO–LUMO gaps are predicted to be 1.37, 1.95 and 2.28 eV for Ag4Ni2, Ag4Pd2, and Ag4Pt2 at the DFT PBE/aug-cc-pVDZ(-pp) level (Fig. 4), indicating that all of the three clusters are visible light active. The predicted HOMO–LUMO gaps are ∼15% smaller than the experimentally measured gaps for the three catalysts, but the predicted HOMO–LUMO gap ratio of Ag4Ni2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag4Pd2:Ag4Pt2 = 1:1.42:1.66 is in excellent agreement with the experimental ratio of 1:1.40:1.63.
:Ag4Pd2:Ag4Pt2 = 1:1.42:1.66 is in excellent agreement with the experimental ratio of 1:1.40:1.63.
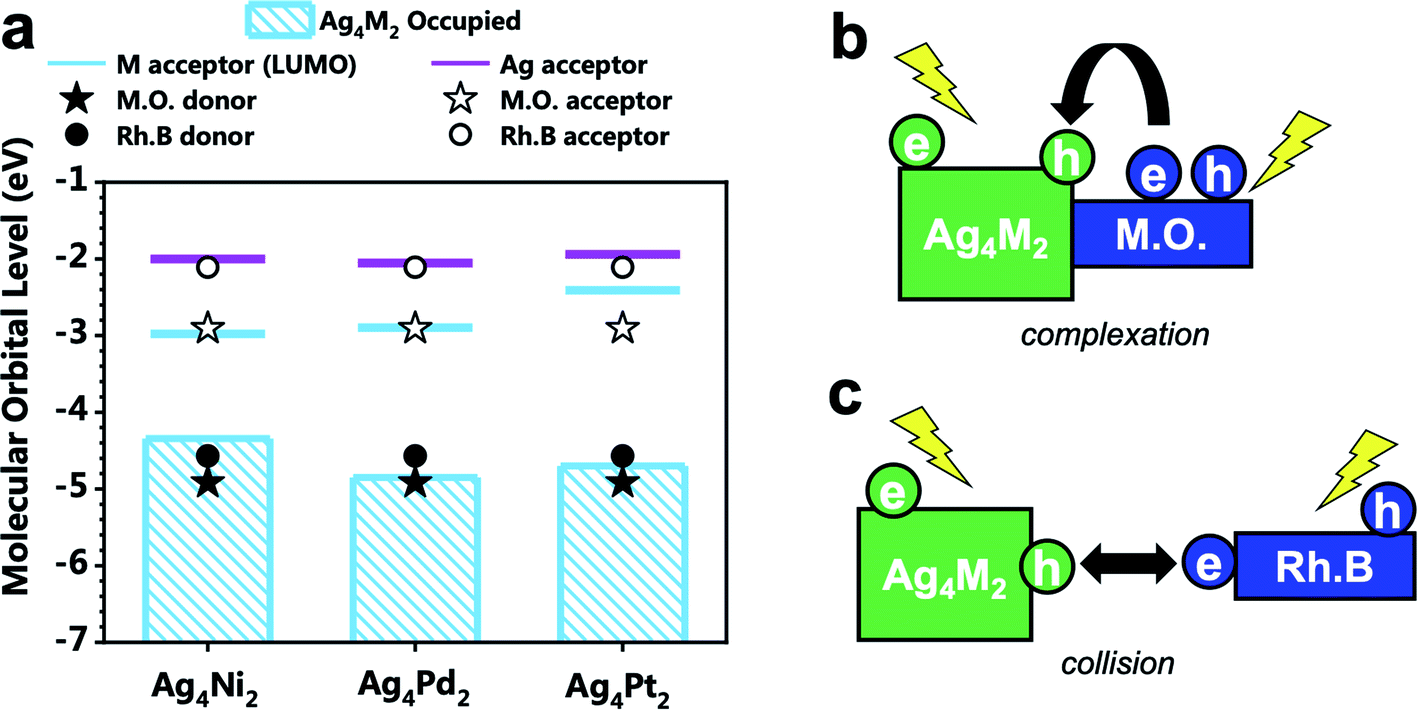 | ||
| Fig. 4 (a) Energy level alignment of the electron donors and acceptors of Ag4M2(SPhMe2)8 (M = Ni, Pd, and Pt), methyl orange (denoted by M.O.), and rhodamine B (denoted by Rh.B). (b) Scheme for the intermolecular recombination via complexation mechanism for the photodegradation of methyl orange catalyzed by Ag4M2. (c) Scheme for the intermolecular recombination via collision mechanism for the photodegradation of rhodamine B catalyzed by Ag4M2. | ||
The molecular orbital energy levels and PDOSs for methyl orange and rhodamine B are given in Fig. S3.† Both the methyl orange and rhodamine B dyes are found to be visible-light active, with HOMO–LUMO gaps of 2.01 and 2.47 eV respectively. Under visible light, the exciton of methyl orange mainly comes from the excitation from the bonding NN (azo) orbital to the anti-bonding NPh orbital, and the exciton of rhodamine B involves the NEt2 donor and the COOH acceptor.
The alignment of the electron donor and acceptor levels for the isolated Ag4M2 catalysts and dyes are shown in Fig. 4a. The Ag donors for the three catalysts are found to be at essentially the same energy level, but the energy levels of the M acceptors of the three catalysts differ. The energy level of the Ni donor is significantly lower than those of the Pt and Pd donors, which can be in part ascribed to the lower atomic ionization potential for Ni (7.64 eV) than those for Pt (8.3 eV), and Pd (8.96 eV).25–27 The energy gap between the M-donor state and Ag-acceptor state is 2.34, 2.80, and 2.75 eV for Ag4Ni2, Ag4Pd2, and Ag4Pt2, respectively. The donor levels of methyl orange and rhodamine B are found to be near the M-donor levels of Ag4Ni2, suggesting that the photocatalytic degradation mechanism should not involve hole injection from the excited Ag4M2 to the ground state dyes.
Next, the complexation of the two dyes with the Ag4M2 catalysts are investigated computationally. Methyl orange can form complex with Ag4M2 at the M–S or Ag sites with comparable exergonicities at 298 K. At the M–S site, the counterion (Na+) of methyl orange is found at ∼2.8 Å from S and an O of SO3 is at ∼4.0 Å from M (Fig. S4†); the complexation exergonicity is 5.3 kcal mol−1 for Ag4Pt2, 3.7 kcal mol−1 for Ag4Pd2, and 7.8 kcal mol−1 for Ag4Ni2. At the Ag site, SO3 of methyl orange forms non-bond O–Ag interaction with a distance of ∼3.2 Å (Fig. S5†), for which the complexation exergonicity is 5.3 kcal mol−1 for Ag4Pt2, 2.4 kcal mol−1 for Ag4Pd2, and 8.7 kcal mol−1 for Ag4Ni2. It is also found that the M site of the catalyst repels the organic moiety of methyl orange and thus does not form complex with direct interaction between methyl orange and M. The PDOS for the complex of methyl orange and Ag4M2 indicates that the complexation exhibits little influence on the orbital energy level alignment. Rhodamine B does not form complex with Ag4M2 spontaneously. The formation of weak complex between rhodamine B and Ag4M2 (Fig. S6†) has an endergonicity of ∼10 kcal mol−1 at 298 K, and the complex contains no short-range dye–metal interaction.
Experiments showed that the orders of the three Ag4M2 catalysts for their performances in the photocatalytic degradation of methyl orange and rhodamine B are opposite. Highly likely, the two photodegradation systems have different mechanisms. Previous studies have shown that the dominant mechanism of methyl orange is N-de-ethylation,28 whereas the degradation of rhodamine B may involve the cleavage of the azo moiety.29 Based on such information and our DFT predictions, two variants of intermolecular recombination mechanisms are proposed for the two photodegradation systems. The photocatalytic degradation of methyl orange is facilitated by intermolecular recombination via complexation (Fig. 4b and 5a). Complexation is first formed between methyl orange and the catalyst under dark condition. Under incident radiation, electron is excited from the M donor state into the Ag acceptor state in Ag4M2 and is excited from NN to NPh in methyl orange. The photoexcited electron of methyl orange and the hole of Ag4M2 migrate to recombine, leading to a substantial charge separation with an excessive electron on Ag of Ag4M2 and an excessive hole on NN, which might induce the further degradation of methyl orange at the azo moiety. Such intermolecular recombination is facilitated by the complexation between methyl orange and the catalyst, with the electron and hole traveling across the junction via resonance. The recombination is driven by the energy gap between the NPh acceptor state of methyl orange and the M donor state of Ag4M2. The above intermolecular donor–acceptor gap exhibits an ascendant order for the complexes of methyl orange with the three catalysts, with Ag4Ni2 (1.43 eV) < Ag4Pt2 (1.79) < Ag4Pd2 (1.94), which is consistent with the order of the measured catalytic performance.
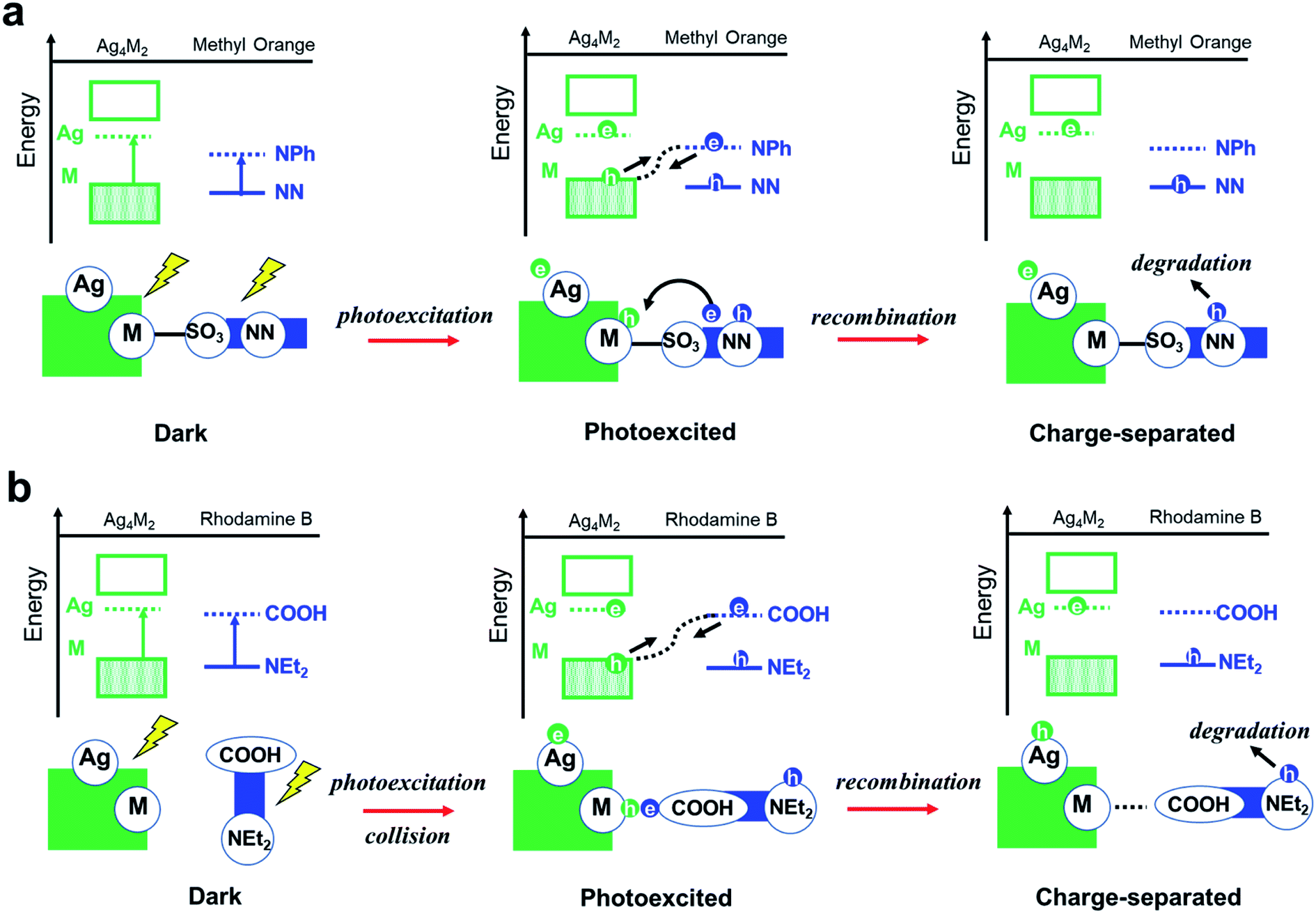 | ||
| Fig. 5 Proposed reaction pathways for photocatalytic degradation of (a) methyl orange and (b) rhodamine B over the Ag4M2(SPhMe2)8 (M = Ni, Pd, and Pt) catalyst. | ||
The photocatalytic degradation of rhodamine B is facilitated by intermolecular recombination via collision (Fig. 4c and 5b). No complexation is formed between rhodamine B and the catalyst under dark condition. The isolated dye and catalyst are then photoexcited. Electron is excited from the M donor state into the Ag acceptor state in Ag4M2 and is excited from NEt2 to COOH in rhodamine B. The excitation therefore induces polarization of both the dye and the catalyst, which promotes the collision between these two. When the COOH of rhodamine B collides into the M or M–S site of the catalysts, the photoexcited electron of rhodamine B and the hole of Ag4M2 recombine, which leaves an excessive electron on Ag of Ag4M2 and an excessive hole on NEt2. The further degradation of rhodamine B is likely to occur at the NEt2. The collision-induced intermolecular recombination between rhodamine B and Ag4M2 is limited by the concentration of photoexcited catalyst and dye species. In our experiments, the concentration of the dye species is essentially invariant between the reactions catalyzed by the three catalysts, while the concentration of the photoexcited catalyst species is related to the light-harvesting capability of the catalysts. According to the Shockley–Queisser limit, when the gap between the donor and acceptor states is greater than 1.4 eV, the smaller gap leads better visible-light harvesting capability. With the gap between the M donor and Ag acceptor in the ascendant order with Ag4Ni2 (2.34 eV) < Ag4Pt2 (2.75) < Ag4Pd2 (2.80), we expect the catalytic performance of the three catalysts in catalyzing the photodegradation of rhodamine B: Ag4Ni2 > Ag4Pt2 > Ag4Pd2, which is in line with the experimental results.
There are several reasons for the photocatalytic degradation of methyl orange and rhodamine B exhibiting different mechanisms. First, rhodamine B is more sterically bulky than methyl orange, which leads to their different complexation energetics and behaviors with the catalyst protected by bulky thiolates. Second, the major degradation site of methyl orange (NN) is at the middle of the dye molecule and is at the terminal region of rhodamine B (NEt2). Consequently, collision of the degradable moiety of methyl orange with the catalyst may not be possible. Overall, the donor and acceptor alignments, as well as the structural effects of the dye (such as steric effect and positions of the chromophores), determine the choices of the intermolecular recombination mechanisms.
Conclusions
In summary, we synthesize a series of the structurally similar Ag4M2 (M = Ni, Pd, Pt) nanoclusters and study their electronic properties and photocatalysis. We demonstrate that M and Ag form an electron donor–acceptor pair that plays essential roles in the photocatalytic activity. The difference in catalytic performances of the three catalysts on the photocatalytic reaction may be mainly ascribed to the alignment of the molecular orbitals near the HOMO–LUMO gap, rather than their geometric structures. We hypothesize that the catalytic activities of the three catalysts in the degradation of methyl orange and rhodamine B are due to both the catalysts and the dyes being visible-light active while having proper donor and acceptor alignment and due to the inter-molecular recombination yields cross-fragment charge separation. The opposite orders of the catalytic performances of the three catalysts for the photocatalytic degradation of the methyl orange and rhodamine B dyes are attributed to two different types inter-molecular recombination mechanisms, the complexation-induced recombination for methyl orange and the collision-induced recombination for rhodamine B. Such a difference is because of the different thermodynamics for the complexation between the catalysts and the two dyes. The work is expected to open up an avenue for fundamental understanding of the heteroatoms doped into nanoclusters effects on photocatalysis at the atomic-precision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge supports from Scientific and Technological Innovation Foundation of Shunde Graduate School, USTB (BK19BE024). This computational work is supported by the Tianhe2-JK computing time award at the Beijing Computational Science Research Center (CSRC).References
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1585 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- H. Shen, G. Deng, S. Kaappa, T. Tan, Y. Z. Han, S. Malola, S. C. Lin, B. K. Teo, H. Häkkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 CrossRef CAS.
- S. Tian, Y. Z. Li, M. B. Li, J. Yuan, J. Yang, Z. Wu and R. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS.
- X. Wan, J. Wang, Z. Nan and Q. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS.
- S. Knoppe, R. Azoulay, A. Dass and T. Bürgi, J. Am. Chem. Soc., 2012, 134, 20302–20305 CrossRef CAS PubMed.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- T. Ohta, M. Shibuta, H. Tsunoyama, Y. Negishi, T. Eguchi and A. Nakajima, J. Phys. Chem. C, 2013, 117, 3674–3679 CrossRef CAS.
- Q. Li, T. Luo, M. G. Taylor, S. Wand, X. Zhu, Y. Song, G. Mpourmpakis, N. L. Rosi and R. Jin, Sci. Adv., 2017, 3, e1603193 CrossRef PubMed.
- M. B. Li, S. K. Tian, Z. Wu and R. Jin, Chem. Mater., 2016, 28, 1022–1025 CrossRef.
- S. Li, H. Chen, X. Liu, H. Liu, J. Ma and Y. Zhu, Chem. Sci., 2020, 11, 8000–8004 RSC.
- M. Zhou, T. Higaki, G. Hu, M. Y. Sfeir, Y. Chen, D. Jiang and R. Jin, Science, 2019, 364, 279–282 CrossRef.
- H. Qian, D. E. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 132, 3097–3101 CrossRef.
- Y. Sun, W. Pei, M. Xie, S. Xu, S. Zhou, J. Zhao, K. Xiao and Y. Zhu, Chem. Sci., 2020, 11, 2440–2447 RSC.
- S. Takano, H. Hirai, S. Muramatsu and T. Tsukuda, J. Am. Chem. Soc., 2018, 140, 8380–8383 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D. E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef.
- G. Sun, X. Kang, S. Jin, X. Li, D. Hu, S. Wang and M. Zhu, Acta Phys.-Chim. Sin., 2018, 34, 799–804 CAS.
- X. Liu, J. Yuan, J. Chen, J. Yang and Z. Wu, Part. Part. Syst. Charact., 2019, 36, 1900003 CrossRef.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Surf. Interface Anal., 1979, 3, 1981 Search PubMed.
- J. Xia, J. Di, H. Li, H. Xu, H. Li and S. Guo, Appl. Catal., B, 2016, 181, 260–269 CrossRef.
- T. Kessler, K. Brück, C. Baktash, J. R. Beene, C. Geppert, C. C. Havener, H. F. Krause, Y. Liu, D. R. Schultz, D. W. Stracener, C. R. Vane and K. Wendt, J. Phys. B: At., Mol. Opt. Phys., 2007, 40, 4413–4432 CrossRef.
- T. Kobayashi, C. R. Locke and K. Midorikawa, J. Appl. Phys., 2017, 56, 010302 CrossRef.
- Z. J. Jakubek and B. Simard, J. Phys. B: At., Mol. Opt. Phys., 2000, 33, 1827–1841 CrossRef CAS.
- A. A. Al-Kahtani, J. Biomater. Nanobiotechnol., 2017, 8, 66–82 CrossRef CAS.
- W. Liu, C. Liu, L. Liu, Y. You, J. Jiang, Z. Zhou and Z. Dong, Ecotoxicol. Environ. Saf., 2017, 141, 9–16 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2020873. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06388j |
| ‡ X. C. and X. S. have equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |