DOI:
10.1039/D1RA02787E
(Paper)
RSC Adv., 2021,
11, 22951-22959
Catechol oxidation promoted by bridging phenoxo moieties in a bis(μ-phenoxo)-bridged dicopper(II) complex†
Received
10th April 2021
, Accepted 16th June 2021
First published on 29th June 2021
Abstract
A dinuclear copper(II) complex [Cu2(papy)2(CH3OH)2] has been synthesized by reaction of one equiv. of Cu(OAc)2·2H2O with one equiv. of the tetradentate tripodal ligand H2papy [N-(2-hydroxybenzyl)-N-(2-picolyl)glycine] and has been characterized by various spectroscopic techniques and its solid state structure has been confirmed by X-ray crystal structure analysis. The single-crystal structure of the complex reveals that the two copper centers are hexa-coordinated and bridged by two O-atoms of the phenoxo moieties. The variable temperature magnetic susceptibility measurement of the complex reveals weak ferromagnetic interactions among the Cu(II) ions with a J value of 1.1 cm−1. The catecholase activity of the complex has been investigated spectrophotometrically using 3,5-di-tert-butyl catechol as a model substrate in methanol solvent under aerobic conditions. The Michaelis–Menten kinetic treatment has been applied using different excess substrate concentrations. The parameters obtained from the catecholase activity by the complex are KM 2.97 × 10−4 M, Vmax 2 × 10−4 M s−1, and kcat 7.2 × 103 h−1. A reaction mechanism has been proposed based on experimental findings and theoretical calculations. The catechol substrate binds to dicopper(II) centers and subsequently two electrons are transferred to the metal centers from the substrate. The bridging phenoxo moieties participate as a Brønsted base by accepting protons from catechol during the catalytic cycle and thereby facilitating the catechol oxidation process.
1 Introduction
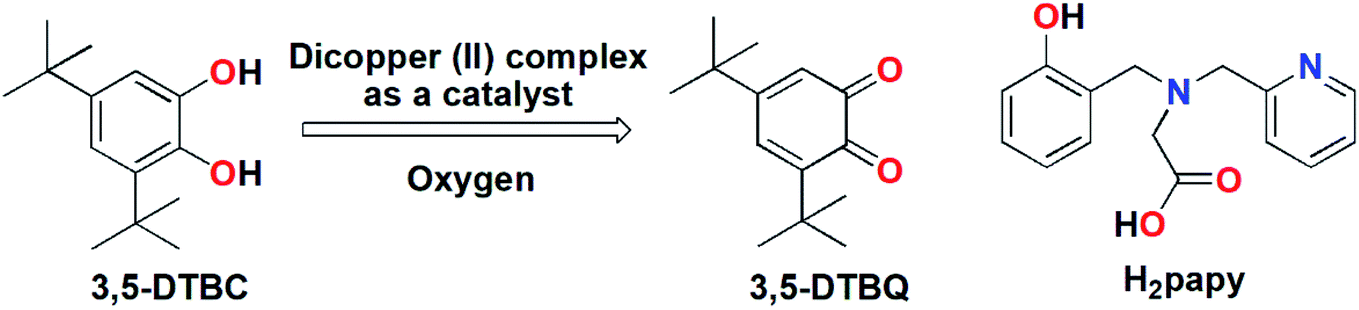
Metalloenzymes that contain dinuclear copper centers at their active sites are involved in diverse and biologically important reactions such as dioxygen activation and transport, electron transfer, the two-electron reduction of N2O, hydrolysis and many more.1–11 Catechol oxidase is one of the well-known representatives of type 3 copper enzymes responsible for catalyzing the oxidation of a wide range of ortho-diphenols to ortho-quinones.12–16 The active site of this enzyme contains a dicopper core and in the reduced state the Cu(I) ions is coordinated to three N-atoms of the histidine residues while in the oxidized state a hydroxo group is bridged between the two Cu(II) ions leading to penta-coordination environment around each metal ion.13–16 Synthetically prepared dinuclear copper(II) complexes that can mimic the structural or/and the functional aspect of catechol oxidase are well reported in literature.17–37 In most cases 3,5-di-tert-butyl catechol is used as a model substrate for catechol activity owing to its low reduction potential and the presence of bulky tert-butyl groups prevents further side reaction such as ring opening.38–40 From the structural–functional correlation it has been found that the catecholase activity exhibited by model dicopper complexes is dependent on several parameters including metal–metal distance, electronic and steric properties of coordinated ligands and the nature of bridging ligands. Bridging ligands (exogenous/endogenous) such as hydroxide,17,23 alkoxide,41 phenoxide,22,37 imidazolate,42,43 pyrazolate,44,45 and carboxylate19,25 accelerate the catecholase activity because they can be displaced readily by the incoming catechol substrate and can also facilitate the deprotonation of catechol substrate by abstracting protons. In this context studies of ligand systems containing phenoxyl donor group(s) is of great interest as phenoxyl group has dramatic influence on the catecholase reactivity.30,37 Vittal and co-workers have reported several bis(μ-phenoxo)-bridged dicopper(II) complexes with reduced Schiff base ligands from N-(2-hydroxybenzyl)-amino acids in which the Cu ions were penta-coordinated and they exhibited diverse catecholase reactivity (kcat in the range 133–4612 h−1) that was dependent on the length of the methylene side-arm of the reduced Schiff base ligands.37,46–50 We have previously worked with non-innocent ligand systems and have observed that reducible moieties (azo) present in such systems can facilitate to attain rare low-valent metal oxidation state51 and promote ortho-alkylation reactions.52 Therefore, we envision that bridging phenoxyl moieties, being oxidizable in nature, may have positive influence on the catechol oxidation and needs to be addressed. For this purpose, we have employed the tripodal tetradentate ligand H2papy [N-(2-hydroxybenzyl)-N-(2-picolyl)glycine]53 (cf. Scheme 1) having a phenoxyl donor moiety. Herein we report the synthesis and detailed structural characterizations of a bis(μ-phenoxo)-bridged dicopper(II) complex, its magnetic properties and catechol oxidation kinetics and elucidate the catechol oxidation mechanism by means of a combined experimental and theoretical approaches.
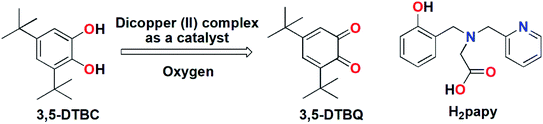 |
| | Scheme 1 (Left) Biomimetic oxidation of 3,5-DTBC by dicopper(II) complex; (Right) H2papy ligand. | |
2 Results and discussion
2.1 Synthesis and characterization of metal complex
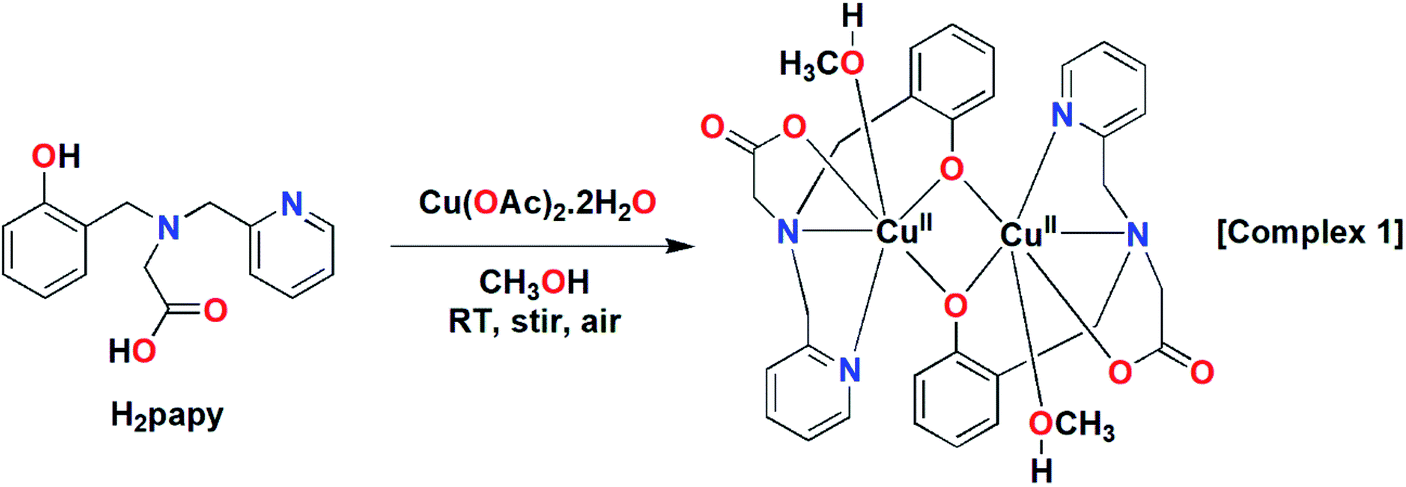
The tetradentate ligand H2papy was prepared following the literature procedure.53 Reaction of one equivalent of the ligand with one equivalent of Cu(OAc)2·2H2O in CH3OH under stirring at room temperature resulted formation of a green compound (cf. Scheme 2). The compound was isolated and characterized as a dinuclear Cu(II) complex, [Cu2(papy)2(CH3OH)2] (1) by various spectroscopic techniques.
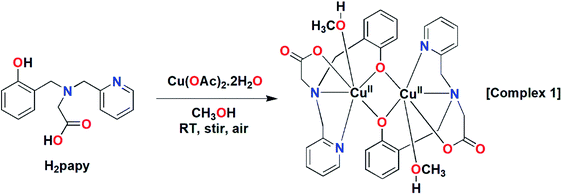 |
| | Scheme 2 The synthesis of dicopper(II) complex, 1. | |
The ESI-MS of complex 1 showed prominent mass peaks at m/z 667.0657, 689.0203 and 707.0227 corresponding to the molecular formation of [Cu2(papy)2 + H]+, [Cu2(papy)2 + Na]+ and [Cu2(papy)2(H2O) + Na]+ respectively (cf. Fig. S1, ESI†). The FTIR spectrum of the complex (cf. Fig. S2, ESI†) showed a broad band around 3417 cm−1 that can be assigned to O–H stretches of methanol and residual water. A couple of prominent bands in the range 1652–1280 cm−1 may be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN, CC and C–O/phenolate stretching modes. The notable feature is the shift of stretching modes of carboxylate group from free ligand to the dicopper(II) complex. For the free ligand, H2papy, the antisymmetric stretching frequency appears at 1634 cm−1 and the symmetric one at 1386 cm−1.54 In the complex, 1, the antisymmetric stretching frequency is shifted to 1645 cm−1 and the symmetric stretching frequency shifted to 1382 cm−1. The separation between these two stretches (Δ) is 263 cm−1. This separation (Δν) is often employed to infer the type of carboxylate coordination with the metal-center.55 A value of Δν > 200 is considered to be an indication of monodentate coordination to a metal, while Δν < 200 indicates bridging or chelating coordination modes. The Δν value of 263 cm−1 in complex 1 is in agreement with monodentate coordination of the ligand carboxylate moiety to a Cu(II) ion. The UV/vis spectrum of 1 showed broad peaks at λmax values of 667 and 402 nm (cf. Fig. S3, ESI†). The lower energy band corresponds to the d–d transition in a d9-ion (Cu2+) in octahedral environment, while the band at higher energy region is due to the ligand-to-metal charge transfer (LMCT).
O, CN, CC and C–O/phenolate stretching modes. The notable feature is the shift of stretching modes of carboxylate group from free ligand to the dicopper(II) complex. For the free ligand, H2papy, the antisymmetric stretching frequency appears at 1634 cm−1 and the symmetric one at 1386 cm−1.54 In the complex, 1, the antisymmetric stretching frequency is shifted to 1645 cm−1 and the symmetric stretching frequency shifted to 1382 cm−1. The separation between these two stretches (Δ) is 263 cm−1. This separation (Δν) is often employed to infer the type of carboxylate coordination with the metal-center.55 A value of Δν > 200 is considered to be an indication of monodentate coordination to a metal, while Δν < 200 indicates bridging or chelating coordination modes. The Δν value of 263 cm−1 in complex 1 is in agreement with monodentate coordination of the ligand carboxylate moiety to a Cu(II) ion. The UV/vis spectrum of 1 showed broad peaks at λmax values of 667 and 402 nm (cf. Fig. S3, ESI†). The lower energy band corresponds to the d–d transition in a d9-ion (Cu2+) in octahedral environment, while the band at higher energy region is due to the ligand-to-metal charge transfer (LMCT).
2.2 Crystal and molecular structure of complex 1
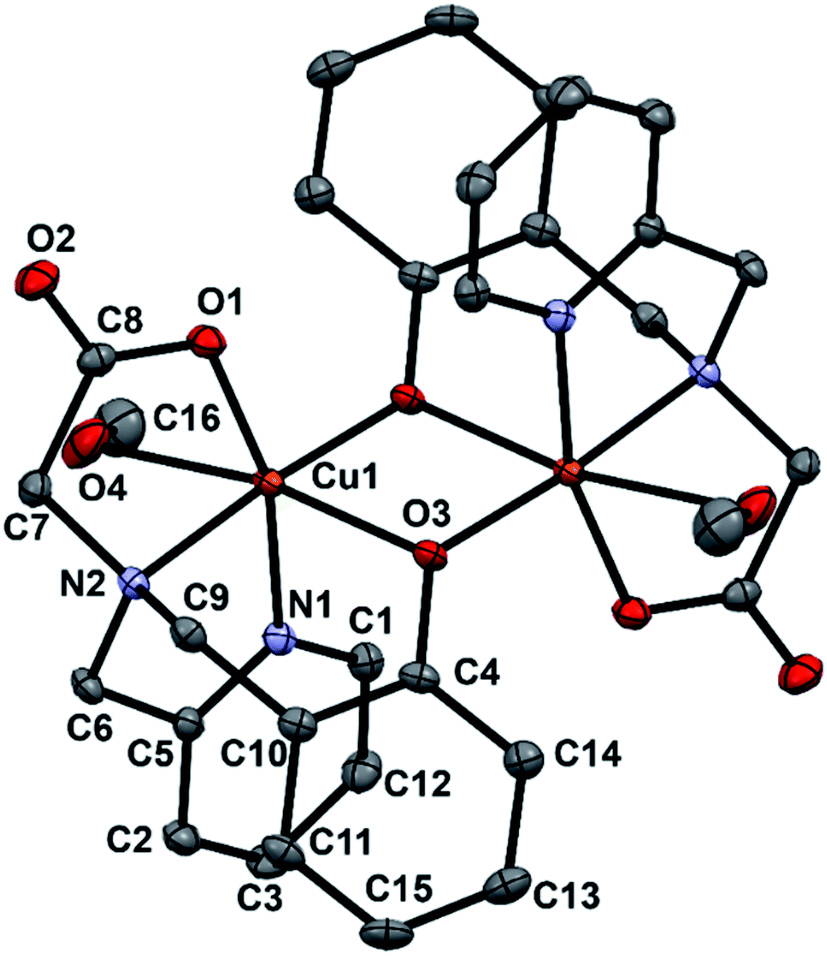
Single crystals of complex 1 were grown by slow evaporation of methanol solution containing pure complex. X-ray studies reveals that complex 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A view of 1 is represented in Fig. 1 and selected bond lengths and angles are listed in Table 1. Complex 1 has a centrosymmetric structure with a binuclear Cu2(μ-O)2 symmetrical rectangular core. The asymmetric unit of 1 contains one-half of the dinuclear complex molecule in general positions. An inversion symmetry operation generates the full molecule, giving a coordination compound of formula [Cu2(papy)2(CH3OH)2]. Each Cu(II) ion resides in a distorted octahedral environment being coordinated to pyridine N-atom, amine N-atom, carboxylate O-atom and two bridging O-atoms of phenoxyl groups while the sixth coordination site is occupied by a solvent molecule (methanol). The two bridged phenyl rings are out of the Cu2(μ-O)2 rectangular plane (out-of-plane shift (τ) of the phenyl group is 42.17°) and are trans to each other. The Cu–Ophenolate distances are 1.9307(11) and 2.3542(11) Å, the Cu–Ophenolate–Cu and Ophenolate–Cu–Ophenolate angles are 96.46(4) and 83.54(4)° respectively. The Cu⋯Cu separation in 1 is 3.208 Å and falls in the ranges as observed for similar complexes having bis(μ-phenoxo)-bridged Cu2(μ-O)2 core.46–51,56,57 It is to be mentioned that one of the prerequisite conditions for a dicopper(II) complex to function as a catecholase enzyme model is that the Cu⋯Cu distance should fall in the range of 2.9–3.2 Å.58
. A view of 1 is represented in Fig. 1 and selected bond lengths and angles are listed in Table 1. Complex 1 has a centrosymmetric structure with a binuclear Cu2(μ-O)2 symmetrical rectangular core. The asymmetric unit of 1 contains one-half of the dinuclear complex molecule in general positions. An inversion symmetry operation generates the full molecule, giving a coordination compound of formula [Cu2(papy)2(CH3OH)2]. Each Cu(II) ion resides in a distorted octahedral environment being coordinated to pyridine N-atom, amine N-atom, carboxylate O-atom and two bridging O-atoms of phenoxyl groups while the sixth coordination site is occupied by a solvent molecule (methanol). The two bridged phenyl rings are out of the Cu2(μ-O)2 rectangular plane (out-of-plane shift (τ) of the phenyl group is 42.17°) and are trans to each other. The Cu–Ophenolate distances are 1.9307(11) and 2.3542(11) Å, the Cu–Ophenolate–Cu and Ophenolate–Cu–Ophenolate angles are 96.46(4) and 83.54(4)° respectively. The Cu⋯Cu separation in 1 is 3.208 Å and falls in the ranges as observed for similar complexes having bis(μ-phenoxo)-bridged Cu2(μ-O)2 core.46–51,56,57 It is to be mentioned that one of the prerequisite conditions for a dicopper(II) complex to function as a catecholase enzyme model is that the Cu⋯Cu distance should fall in the range of 2.9–3.2 Å.58
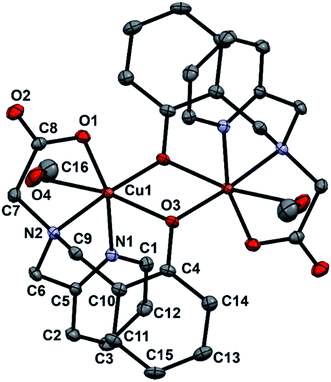 |
| | Fig. 1 An ORTEP representation of the molecular structure of complex 1 with atom numbering scheme. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. | |
Table 1 Selected bond lengths [Å] and angles [°] for 1a
| Symmetry transformations used to generate equivalent atoms: #1 −x + 1, −y + 1, −z. |
| Cu(1)–O(3) |
1.9307(11) |
| Cu(1)–O(1) |
1.9600(11) |
| Cu(1)–N(1) |
1.9970(13) |
| Cu(1)–N(2) |
2.0335(13) |
| Cu(1)–O(3)#1 |
2.3542(11) |
| Cu(1)–O(3)–Cu(1)#1 |
96.46(4) |
| O(3)–Cu(1)–O(1) |
99.82(5) |
| O(3)–Cu(1)–N(1) |
94.05(5) |
| O(1)–Cu(1)–N(1) |
163.38(5) |
| O(3)–Cu(1)–N(2) |
175.00(5) |
| O(1)–Cu(1)–N(2) |
83.43(5) |
| N(1)–Cu(1)–N(2) |
83.38(5) |
| O(3)–Cu(1)–O(3)#1 |
83.54(4) |
| O(1)–Cu(1)–O(3)#1 |
98.52(4) |
| N(1)–Cu(1)–O(3)#1 |
92.06(5) |
| N(2)–Cu(1)–O(3)#1 |
92.24(4) |
2.3 Magnetism and magnetostructural correlation
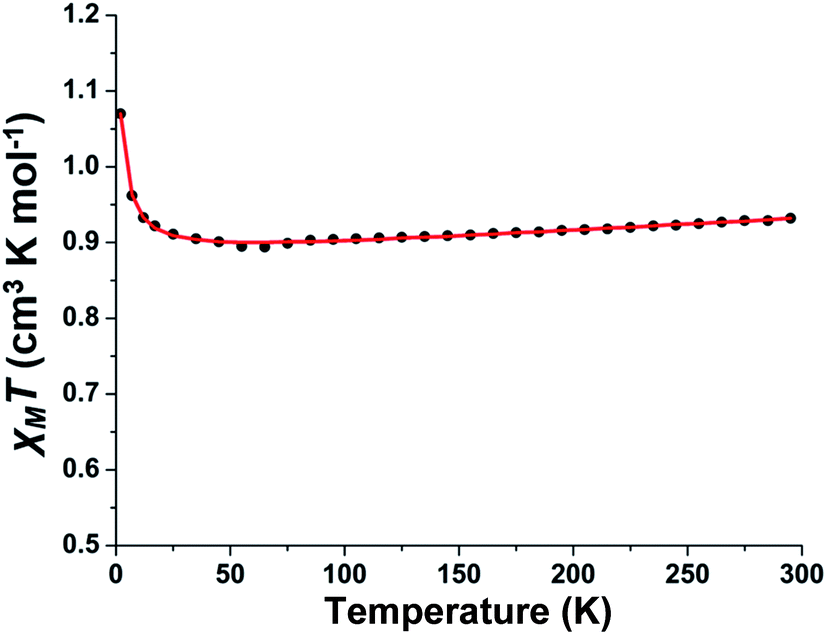
The variable-temperature magnetic susceptibility measurement study for polycrystalline sample of complex 1 was carried out in the temperature range 2–295 K. The plot of χMT versus T for the phenoxo-bridged binuclear Cu(II) complex is shown in Fig. 2. The value of 0.93 cm3 K mol−1 of χMT at 295 K is in agreement with two S = ½ spin and a g parameter deviating from 2.00 as usually found for Cu(II) ions (vide infra). The χMT remained unchanged down to 35 K and below this temperature, it sharply increased to 0.107 cm3 K mol−1 for 2 K indicating the occurrence of ferromagnetic interactions between the two Cu(II) centers. The experimental data have been analyzed using the equation for dinuclear Cu(II) complexes with the corresponding Hamiltonian in the form H = −JS1S2 (ref. 59) (the exchange coupling parameter J is negative for antiferromagnetic interactions and positive for ferromagnetic interactions) and fitted with Bleaney–Bowers equation.60 The best fit parameters obtained for 1 are g = 2.165 and J = +1.1 cm−1 (TIP (fixed) = 1.7 × 10−4 cm3 mol−1) confirming the existence of a very weak ferromagnetic interactions between the Cu(II) ions.
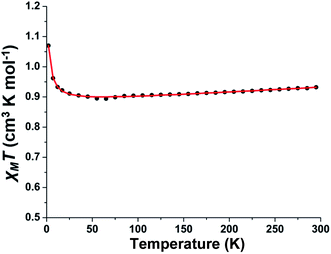 |
| | Fig. 2 Experimental (black dots) and simulated (red line) χMT versus T curve for complex 1. | |
It is well-reviewed in literature that bis(μ-phenoxo)-bridged dinuclear copper(II) complexes show magnetic properties strongly dependent on several structural factors.61 For example, the Cu–O–Cu bridging angle, the out-of-plane shift of the phenyl rings, the Cu–O bond length, the planarity of the Cu2O2 core, and the conformation and rotation of the phenyl groups can influence the magnetic properties of this type of complexes. In complex 1, the Cu–O–Cu bridging angle is relatively large (96.46(4)°); however it falls into the class of limited bis(μ-phenoxo)-bridged dicopper(II) complexes exhibiting ferromagnetic interactions.56,62 To rationalize the magnetic behaviour of 1 other structural parameters have to be considered. The comparatively large average Cu–Ophenolate bond lengths (2.2849 Å) in conjunction with large out-of-plane shift (τ = 42.17°) and the anti conformation of the phenyl rings in 1 may be responsible for the observed weak ferromagnetic coupling. It is to be mentioned that this behaviour is similar to that reported by Ruiz et al. for bis(μ-hydroxo)-bridged dicopper(II) complexes63,64 where a large out-of-plane displacement of R (R = H, phenyl) favours ferromagnetic interactions.
2.4 Electrochemistry
The electrochemical behavior of 1 has been investigated using Pt working electrode and Ag/AgCl reference electrode in methanol solvent at a scan rate of 100 mV s−1. The cyclic voltammogram of 1 showed two irreversible reductive responses at Epc values of −0.41 and −0.74 V vs. Ag/AgCl, respectively and two irreversible oxidative responses at Epa values of 0.32 and 0.73 V vs. Ag/AgCl, respectively (cf. Fig. S4, ESI†). The reductive responses are tentatively assigned to CuIICuII → CuIICuI and CuIICuI → CuICuI redox processes. Negative reduction potentials have been observed for dicopper(II) complexes containing μ-OH, μ-alkoxo and/or μ-phenoxo bridges and O-donor ligands leading to accumulation of electron density on the metal centers.22,65,66 The peak separation for cathodic responses [i.e. ΔEpc = Epc1 − Epc2] is 0.33 V and lies in the range as observed for dicopper(II) complexes with endogenous bis-phenoxo bridges exhibiting electrochemical reduction responses via two one electron transfer steps with a separation of ca. 0.35–0.80 V.34,67
2.5 Catecholase activity study
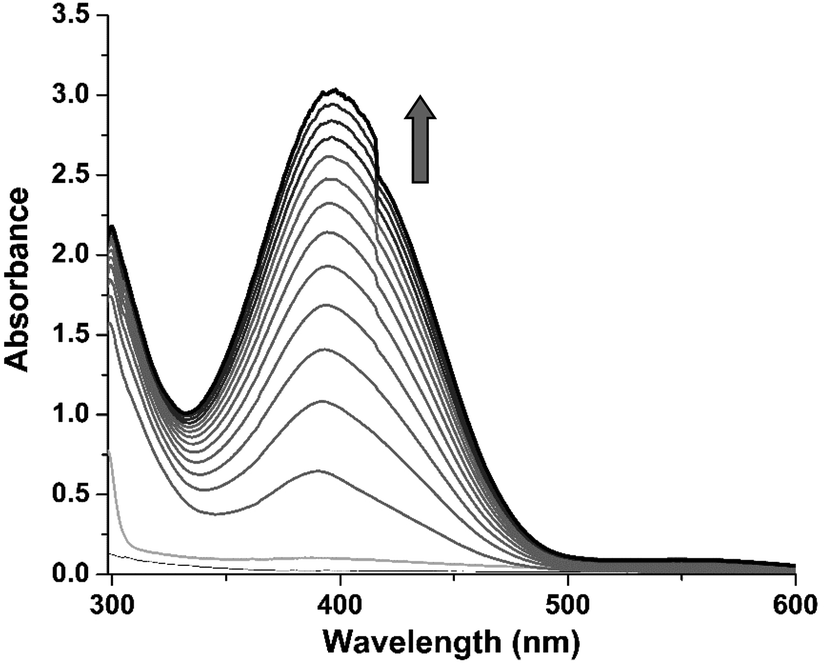
3,5-Di-tert-butyl catechol (3,5-DTBC) is widely used as a model substrate for catecholase activity experiment due to its low redox potential. The catalytic oxidation of 3,5-DTBC into 3,5-di-tert-butyl quinone (3,5-DTBQ) by complex 1 was followed spectrophotometrically by monitoring the increase in the absorption band at 400 nm characteristic to the formation of product, 3,5-DTBQ. For this purpose, 1 × 10−4 M of complex solution was treated with 100 equivalents of 3,5-DTBC (1 × 10−2 M) in methanol solvent at 25 °C under aerobic condition. The oxidation was monitored for about 2 h during which a new absorption band at 400 nm corresponding to the formation of 3,5-DTBQ was observed (cf. Fig. 3).
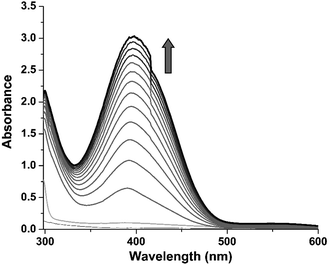 |
| | Fig. 3 The time-dependent UV/vis spectra of the formation of 3,5-DTBQ after addition of 100 equivalents of 3,5-DTBC to a methanolic solution of complex 1 at 25 °C; arrow mark indicates the increase in the characteristic absorbance band (from light gray to dark grey) of 3,5-DTBQ at 400 nm. | |
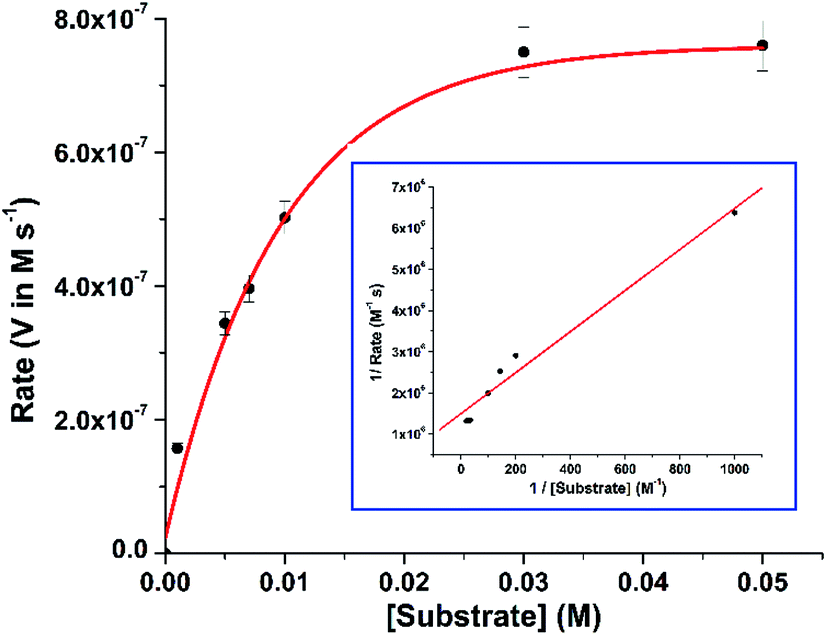
In order to find out the reaction rate between the complex and 3,5-DTBC, a time dependent kinetic experiment was performed using initial rate method. For this purpose, a series of methanolic solution of 3,5-DTBC of varying concentrations [0.1 × 10−2 M to 5 × 10−2 M] were prepared keeping the concentration of complex constant at 1 × 10−4 M. The rate dependence on substrate concentration showed typical hyperbolic curvature (cf. Fig. 4) and the results were analyzed using the Michaelis–Menten equation of enzyme kinetics. Lineweaver–Burk plot (double reciprocal plot) produced reasonable straight-line fitting (R2 value of around 98%) (cf. Fig. 4, inset) and the Michaelis binding constant (KM), the maximum velocity (Vmax) and the turnover number (kcat) were calculated using the equation: 1/V = (KM/Vmax) × 1/[S] + 1/Vmax.
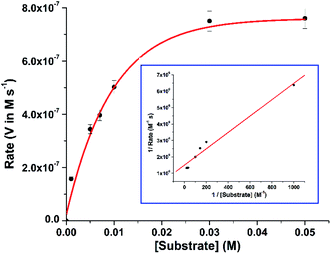 |
| | Fig. 4 Rate vs. substrate concentration plot for complex 1. (Inset) The Lineweaver–Burk plot. | |
The kinetic parameters obtained for 1 were compared to those of some previously reported mono- and bis(μ-phenoxo)-bridged dicopper(II) complexes and listed in Table 2. It is evident that complex 1 exhibited superior catecholase activity over related dicopper(II) complexes46–50 of reduced Schiff base ligands derived from N-(2-hydroxybenzyl)-amino acids (cf. Table 2, entry 1–8). This might be attributed to the structural change in the coordination environment of 1 where each Cu(II) ion is hexacoordinated (against the pentacoordination environment around Cu(II) ion in the latter complexes) and the additional coordination provided by pyridyl group (having σ-donor and π-acceptor properties) has positive influence on the reactivity. Comparing with the reactivities of mono(μ-phenoxo)-bridged dinuclear copper(II) complexes26,27,30 (cf. Table 2, entry 9–14), it is to be noted that in complex 1, double bridging phenoxyl groups may influence the catecholase reactivity in two different ways: (I) they can sterically interact with the incoming catechol substrate leading to decrease in reactivity; (II) they can facilitate the reactivity by participating in the proton-transfer process and stabilizing the CuIICuII ↔ CuICuI catalytic cycle. In order to unravel the role of bridging phenoxyl groups of complex 1 in catechol oxidation, theoretical calculations have been performed and discussed in the following section.
Table 2 Kinetic parameters of bis(μ-phenoxo)-bridged dicoppelr(II) complexesa
| Entry |
Complex |
Vmax (in M s−1) |
KM (in M) |
kcat (in h−1) |
| H2Sab4 = N-(2-hydroxybenzyl)-4-aminobutyric acid; H2Sbal = N-(2-hydroxybenzyl)-β-alanine; H2 Sae = N-(2-hydroxybenzyl)-aminoethanesulfonic acid; H2Sam = N-(2-hydroxybenzyl)-aminomethanesulfonic acid; H2MeSch12 = 2-[(2-hydroxy-5-methylbenzyl)amino]cyclohexane-1-carboxylic acid; H2MeSch11 = 1-[(2-hydroxy-5-methylbenzyl)amino]cyclohexane-1-carboxylic acid; H2MeScp11 = 1-[(2-hydroxy-5-methylbenzyl)amino]cyclopentane-1-carboxylic acid; HDEMP = 2-[[2-(diethylamino)ethylamino]methyl]phenol; L22 = 2,6-bis(((E)-2-(piperazin-1-yl)ethylimino)methyl)-4-methylphenol; L23 = 2,6-bis(((E)-2-(pyrrolidin-1-yl)ethylimino)methyl)-4-methylphenol; L21 = 2,6-bis((E)-2-(3-morpholinopropylimino)methyl)-4-methylphenol; L1-OH = 2,6-[N-methyl-N-(2-pyridylethyl)aminomethyl]phenol. Ref. 50. Ref. 47. Ref. 26. Ref. 27. Ref. 30. |
| 1 |
[Cu2(papy)2(CH3OH)2] (1) |
2.0 × 10−4 |
2.97 × 10−4 |
7200 |
| 2 |
[Cu2(Sab4)2(H2O)2]·0.5H2Ob |
1.93 × 10−4 |
8.7 × 10−3 |
3800 |
| 3 |
[Cu2(Sbal)2(H2O)2]b |
6.5 × 10−6 |
10.4 × 10−3 |
1287 |
| 4 |
[Cu2(Sae)2]·2H2Ob |
1.63 × 10−4 |
4.3 × 10−3 |
4612 |
| 5 |
[Cu2(Sam)2(H2O)2]·H2Ob |
0.41 × 10−4 |
5.4 × 10−3 |
1140 |
| 6 |
[Cu2(MeSch12)2(H2O)2]c |
1.05 × 10−4 |
4.3 × 10−3 |
3120 |
| 7 |
[Cu2(MeSch11)2(H2O)2]c |
0.36 × 10−4 |
14.6 × 10−3 |
1080 |
| 8 |
[Cu2(MeScp11)2(H2O)2]c |
0.34 × 10−4 |
15.4 × 10−3 |
1003 |
| 9 |
[Cu2(DEMP)2(ClO4)2]d |
2.6 × 10−6 |
3.3 × 10−3 |
93.6 |
| 10 |
[Cu2(DEMP)2(OH)](ClO4)d |
3.89 × 10−6 |
4.6 × 10−3 |
233.4 |
| 11 |
[Cu2(H2L22) (OH) (H2O)-(NO3)](NO3)3·2H2Oe |
9.0 × 10−4 |
2.3 × 10−3 |
32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
| 12 |
[Cu2(L23) (OH) (H2O)2](NO3)2e |
4.0 × 10−4 |
7.6 × 10−3 |
14400 |
| 13 |
[Cu2(L21) (N3)3]e |
8.0 × 10−4 |
7.0 × 10−4 |
28800 |
| 14 |
[Cu2(L1-O) (μ-OH)][ClO4]2f |
0.76 × 10−4 |
11.17 × 10−3 |
5470 |
2.6 DFT study and mechanism of catechol oxidation
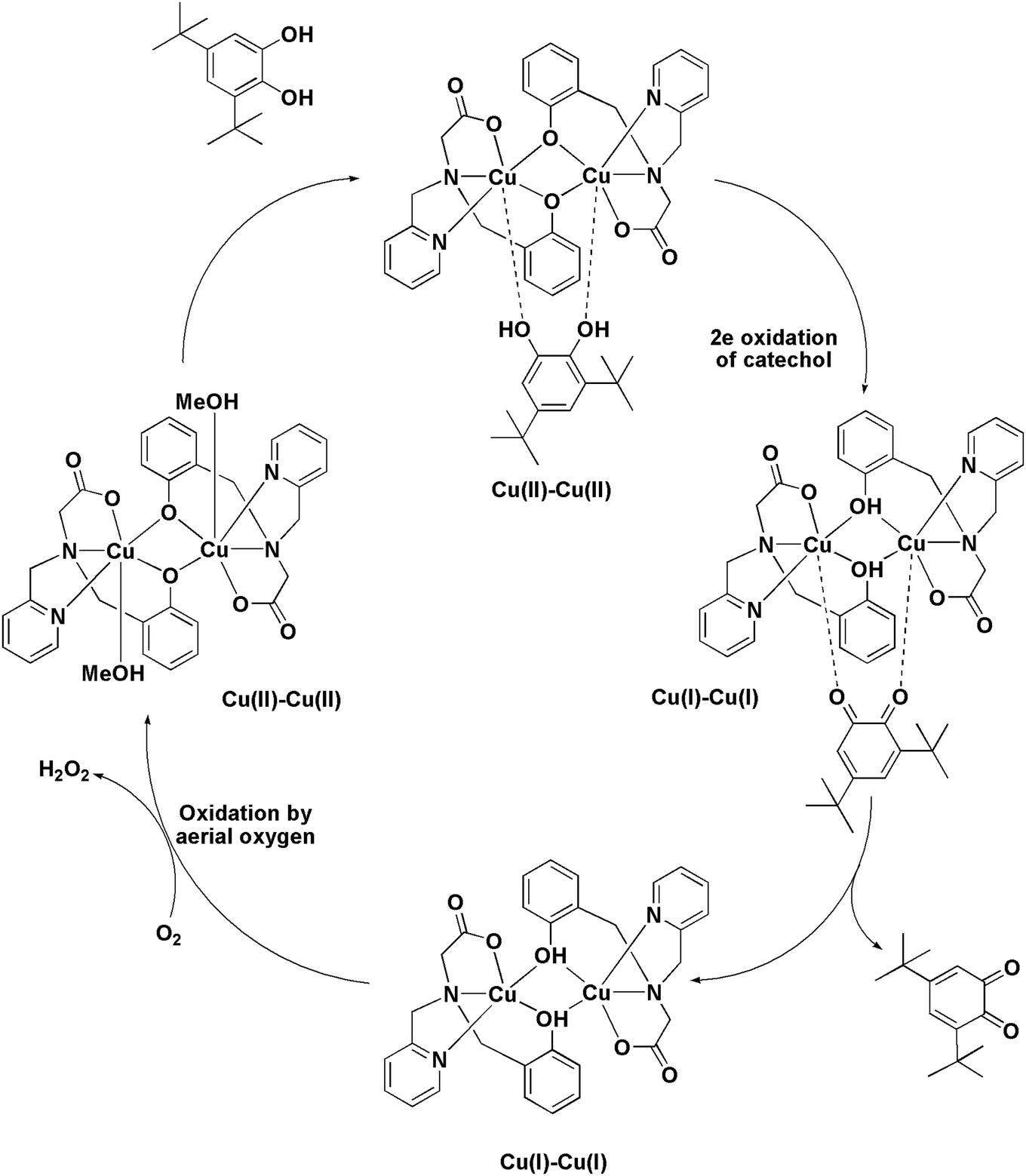
The ESI-MS analysis of 1 in CH3CN has shown that the complex retained its dinuclear entity in the solution. Therefore, based on the experimental results we have proposed a catalytic cycle (shown in Scheme 3) involving dicopper(II) centers. The catalytic cycle consists of three main steps: (I) the catechol substrate displaces the solvent molecules (CH3OH) and binds to Cu(II) centers in a bridging mode to yield [Cu2(papy)2(catecholate)] complex. This is followed by (II) intramolecular electron transfer to two Cu(II) ions leading to two-electron oxidation of catechol to form ortho-quinone coordinated to the two Cu(I) ions. Finally, (III) ortho-quinone is released and dicopper(I) species is oxidized by atmospheric oxygen to regenerate dicopper(II) species and the catalytic cycle continues. Formation of H2O2 has been detected by means of the molybdate-accelerated I3− assay (cf. Fig. S5, ESI†).
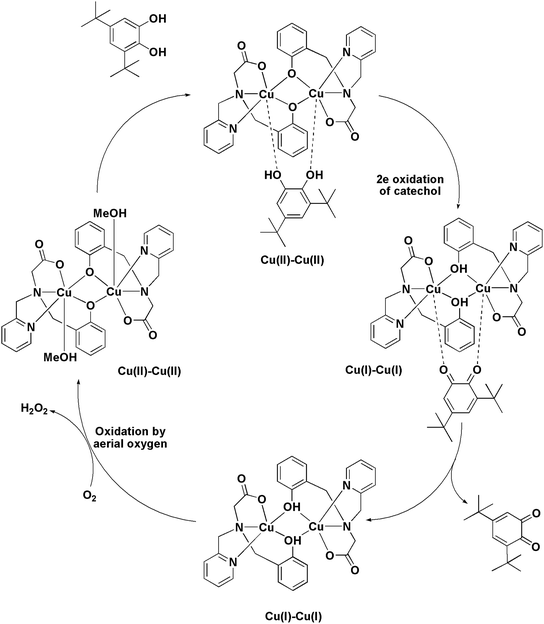 |
| | Scheme 3 The proposed mechanism of catechol oxidation by [Cu2(papy)2(CH3OH)2] (1). | |
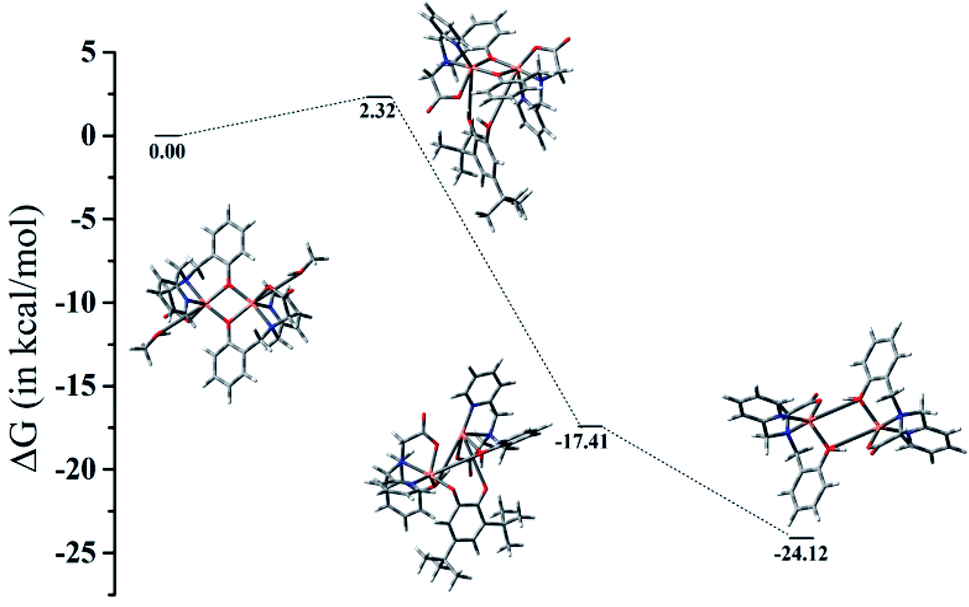
The computed energetic profile consisting of free energies for product formation through the substrate binding and conversion to quinone-like product is shown in Fig. 5. From the free energy profile, we observe that substrate binding to the Cu(II)–Cu(II) catalyst raises the energy by ∼2 kcal mol−1 (energy barrier for the product formation). The oxidation of catechol substrate to quinone lowers the energy by ∼17 kcal mol−1 from the reference point (Cu dimer energy set at 0 kcal mol−1). During this conversion, we observe that the oxidation state change of copper from Cu(II) to Cu(I) is mediated by abstraction of protons from catechol by the bridging phenoxide groups. Following this, the elimination of the product takes place with an energy lowering by ∼7 kcal mol−1 (i.e., lowering by ∼24 kcal mol−1 from the reference point), leading to the formation of a Cu(I)–Cu(I) dimer complex. These findings indicate that a minimum energy pathway for the formation of the quinone-form of the catechol substrate would form via usage of the Cu(II)–Cu(II) dimer catalyst. The proton abstraction by the bridging phenolate groups is preferred over that of the solvent (methanol) as the latter raises the energy by more than 300 kcal mol−1, whereas the former brings down the energy by ∼17 kcal mol−1, leading to a subsequent stable product formation (absolute Gibbs free energies and coordinates of the complexes are given in ESI†).
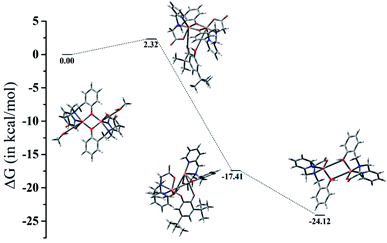 |
| | Fig. 5 Gibbs free energy profile showing minimum energy pathway of product formation. | |
3 Conclusion
In summary a new bis(μ-phenoxo)-bridged dicopper(II) complex, 1 of the tripodal ligand, H2papy has been synthesized and characterized. X-ray structure analysis of 1 has revealed that the Cu⋯Cu separation is 3.208 Å, lying in the desired range of 2.9–3.2 Å required for catechol coordination. The Cu–O–Cu bridging angle is found to be relatively large (96.46(4)°). The large out-of-plane shift of the phenyl rings in 1 is associated with the hexacoordination environment around each Cu(II) center. Magnetic susceptibility measurement has shown that two Cu(II) centers are weakly ferromagnetically coupled via the bis(μ-phenoxo)-bridging unit and may be correlated to the large out-of-plane shift and the anti conformation of the phenyl rings. The catechol oxidation activity is investigated using this complex and it has been observed that the catalytic efficiency of 1 is superior compared to the related bis(μ-phenoxo)-bridged dicopper(II) complexes. The DFT study has revealed that bridging phenoxo groups facilitate the two-electron oxidation of catechol by accepting protons from incoming catechol and stabilizing the Cu(II)–Cu(II) ↔ Cu(I)–Cu(I) catalytic cycle in the due course. It is to be mentioned that the exogenous pyridyl donors may also promote the catechol oxidation by 1 as the related bis(μ-phenoxo)-bridged dicopper(II) complexes of reduced Schiff base ligand, N-(2-hydroxybenzyl)-amino acid (without any pyridyl side arm) are found to be less efficient compared to 1.
4 Experimental sections
4.1 Materials
Cu(OAc)2·2H2O and 3,5-DTBC were purchased from Sigma Aldrich and used as received. CH3OH was purchased from Merck and used as received. Tetrabutylammonium percholorate was obtained from Sigma Aldrich. H2papy was prepared following a literature method.52
4.2 Physical measurements
1H NMR spectra were recorded on a Brucker Advance DPX 500 MHz spectrometer, and SiMe4 was taken as internal standard. IR spectra were obtained using a Perkin-Elmer 783 spectrophotometer. A Perkin-Elmer 240C elemental analyzer was used to collect micro analytical data (C, H and N). ESI-MS spectra were recorded on a micro mass Q-TOF mass spectrometer (serial no – YA263). UV/vis spectroscopy and kinetic measurement were performed using a Shimadzu 2450 spectrophotometer. Cyclic voltammetry was carried out in a 0.1(M) Bu4NClO4 solution using a three electrode configuration (platinum working electrode, auxillary electrode and Ag/AgCl reference electrode and methanol as a solvent) and using CH instruments 700E model. E1/2 for the ferrocinium–ferrocene couple under the experimental condition was 0.665 V.
4.3 Synthesis of [Cu2(papy)2(CH3OH)2] (1)
To a methanolic solution (5 mL) of H2papy (80 mg, 0.2941 mmol), a methanolic solution (15 mL) of Cu(OAc)2·2H2O (58.82 mg, 0.2941 mmol) was added dropwise and the mixture was stirred for about 2 h at room temperature. The resulting deep green solution was filtered and kept for slow evaporation. After few days deep green crystals of 1 were collected. Yield: 80 mg (88% per Cu-atom). ESI-MS: m/z 682.9042 [M]+. Anal. calcd (%): C 52.52, H 4.96, N 7.66. Found (%): C 52.11, H 5.08, N 7.38. UV/vis (CH3OH) λ (nm) (ε in M−1 cm−1): 402 (4850), 667 (2740).
4.4 X-ray crystallography
The crystal of 1 was immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 123 K. The X-ray diffraction data were collected on a Bruker Kappa Apex II Duo diffractometers using graphite-monochromated Mo Kα radiation (λ = 0.710 73 Å). The measurements were made in the θ range 2.945 to 26.999°. The Denzo Scalepack program packages were used for cell refinements and data reductions. The structure was solved by SUPERFLIP methods,68 using SHELXL - 2013 programs69 with the CrystaMaker70 graphical user interface. Structural refinements were carried out by full-matrix least-square methods on F2 (Table 3).
Table 3 Crystal data and structure refinement for 1
| Empirical formula |
C32H36Cu2N4O8 |
| Formula weight |
731.73 |
| Temperature |
123(2) K |
| Wavelength |
0.71073 Å |
| Crystal system |
Triclinic |
| Space group |
P |
| Unit cell dimensions |
a = 8.5547(3) Å |
α = 113.7330(10)° |
| b = 9.8407(4) Å |
β = 99.453(2)° |
| c = 10.2731(4) Å |
γ = 92.307(2)° |
| Volume |
775.51(5) Å3 |
| Z |
1 |
| Density (calculated) |
1.567 mg m−3 |
| Absorption coefficient |
1.430 mm−1 |
| F(000) |
378 |
| Crystal size |
0.537 × 0.339 × 0.335 mm3 |
| Theta range for data collection |
2.945 to 26.999°. |
| Index ranges |
−10 ≤ h ≤ 10, −12 ≤ k ≤ 12, −12 ≤ l ≤ 13 |
| Reflections collected |
10178 |
| Independent reflections |
3323 [R(int) = 0.0241] |
| Completeness to theta = 25.242° |
98.1% |
| Absorption correction |
Semi-empirical from equivalents |
| Max. and min. transmission |
0.7458 and 0.6281 |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3323/0/210 |
| Goodness-of-fit on F2 |
1.045 |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0251, wR2 = 0.0655 |
| R indices (all data) |
R1 = 0.0259, wR2 = 0.0661 |
| Extinction coefficient |
n/a |
| Largest diff. peak and hole |
0.695 and −0.418 e.Å−3 |
4.5 Kinetic measurement
The increase in the concentration of 3,5-DTBQ has been monitored at 25 °C by UV/vis spectroscopy at 400 nm in quartz cuvettes using a Shimadzu 2450 spectrophotometer. For time dependent kinetic experiment, 2 mL of the substrate solution of different concentrations was placed inside a 1 cm quartz cell and then 60 μL of 5 × 10−3 M standard solution of Cu(II) complex in methanol was added and the increase in absorbance at 400 nm was continuously monitored over time at 25 °C.
4.6 Procedure for detection of H2O2 during the catalytic cycle
To detect the liberation of hydrogen peroxide during the catalytic cycle the reaction mixtures are prepared same as in kinetic experiment. Then after few hours of reaction equal volume of water was added and the mixture was extracted with DCM. The pH of aqueous layer was fixed at 2 with H2SO4. After that 1 mL of 10% KI solution and few drops of 3% ammonium molybdate solution were added to the aqueous layer. In presence of hydrogen peroxide the reaction: H2O2 + 2I− + 2H+ → 2H2O + I2 occurs, and with an excess of iodide ions, the triiodide ion is formed according to the reaction: I2(aq) + I− = I3−. With the addition of an ammonium molybdate solution the reaction is restored instantaneously. Then the formation of I3− can be monitored spectrophotometrically due to the development of the characteristic I3− band (λmax = 353 nm).
4.7 Computational details
In this work, density functional theory (DFT) approach was used to compute the energetics of the involved molecular species to identify the minimum energy pathway. Becke's three parameter hybrid functional71 in combination with the Lee–Yang–Parr correlation function72 was utilized to find the relevant optimized ground state structures. Harmonic frequencies were obtained so as to confirm the true minimum structure (established by the absence of imaginary frequencies). Two different basis sets were used – LANL2DZ73 basis set was used for the transition metal (Cu) whereas 6-31G(d) basis set was used for all the non-metal atoms (C, N, O and H). All calculations were carried out in the gas phase using Gaussian16 software.74
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. M. and U. M. thank the Science and Engineering Research Board (SERB), DST, India for funding via project no. YSS/2015/000304 (to M. M.) and EEQ/2018/000964 (to U. M.). D. M. thanks SERB, DST for a PhD fellowship. A. A. S. thanks Russian Ministry of Science and Education for support (via project no. AAAA-A19-119071190045-0). The authors sincerely thank Dr Serhiy Demeshko and Prof. Franc Meyer of University of Goettingen for collecting the magnetic data of the complex.
References
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS.
- W. Kaim and J. Rall, Angew. Chem., Int. Ed., 1996, 35, 43–60 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
- W. B. Tolman, Acc. Chem. Res., 1997, 30, 227–237 CrossRef CAS.
- M. Fontecave and J.-L. Pierre, Coord. Chem. Rev., 1998, 170, 125–140 CrossRef CAS.
- P. L. Holland and W. B. Tolman, Coord. Chem. Rev., 1999, 190–192, 855–869 CrossRef CAS.
- W. G. Zumft and P. M. H. Kroneck, Adv. Inorg. Biochem., 1996, 11, 193–221 CAS.
- W. G. Zumft, Microbiol. Mol. Biol. Rev., 1997, 61, 533–616 CAS.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS.
- K. D. Karlin and Z. Tyeklar, Bioinorganic Chemistry of Copper, Chapman & Hill, New York, NY, 1993 Search PubMed.
- C. X. Zhang, H.-C. Liang, K. J. Humphreys and K. D. Karlin, in Advances in Catalytic Activation of Dioxygen by Metal Complexes, ed. E. I. Solomon, Kluwer Academic Publishers, Dordrecht, 2003, pp. 79–121 Search PubMed.
- C. Gardemann, C. Eicken and B. Krebs, Acc. Chem. Res., 2002, 35, 183–191 CrossRef PubMed.
- C. Eicken, B. Krebs and J. C. Sacchettini, Curr. Opin. Struct. Biol., 1999, 9, 677–683 CrossRef CAS PubMed.
- C. Eicken, B. Kreb and J. C. Sacchettini, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef PubMed.
- C. Eicken, F. Zippel, K. Buldt-Karentzopoulos and B. Krebs, FEBS Lett., 1998, 436, 293–299 CrossRef CAS PubMed.
- R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211–241 CrossRef.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793–3804 RSC.
- P. Gentschev, N. Moller and B. Krebs, Inorg. Chim. Acta, 2000, 300, 442–452 CrossRef.
- I. A. Koval, D. Pursche, A. F. Stassen, P. Gamez, B. Krebs and J. Reedijk, Eur. J. Inorg. Chem., 2003, 1669–1674 CrossRef CAS.
- M. Merkel, N. Moller, M. Piacenza, S. Grimme, A. Rompel and B. Krebs, Chem.–Eur. J., 2005, 11, 1201–1209 CrossRef CAS PubMed.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC and references therein.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki and E. Schwingel, Inorg. Chem., 2002, 41, 1788–1794 CrossRef CAS PubMed.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348–350 CrossRef CAS PubMed.
- R. E. H. M. B. Osorio, R. A. Peralta, A. J. Bortoluzzi, V. R. de Almeida, B. Szpoganicz, F. L. Fischer, H. Terenzi, A. S. Mangrich, K. M. Mantovani, D. E. C. Ferreira, W. R. Rocha, W. Haase, Z. Tomkowicz, A. dos Anjos and A. Neves, Inorg. Chem., 2012, 51, 1569–1589 CrossRef CAS PubMed.
- C. Fernandes, A. Neves, A. J. Bortoluzzi, A. S. Mangrich, E. Rentschler, B. Szpoganicz and E. Schwingel, Inorg. Chim. Acta, 2001, 320, 12–21 CrossRef CAS.
- A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111–11012 CrossRef CAS.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083–7093 CrossRef CAS.
- A. Banerjee, R. Singh, E. Colacio and K. K. Rajak, Eur. J. Inorg. Chem., 2009, 277–284 CrossRef CAS.
- S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. J. Riley, J. Biol. Inorg Chem., 2008, 13, 499–510 CrossRef CAS PubMed.
- J. Mukherjee and R. N. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS.
- S. Mandal, J. Mukherjee, F. Lloret and R. Mukherjee, Inorg. Chem., 2012, 51, 13148–13161 CrossRef CAS.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526–3536 CrossRef CAS PubMed.
- C. Belle, C. Beguin, I. Gautier-Luneau, S. Hamman, C. Philouze, J. L. Pierre, F. Thomas, S. Torelli, E. Saint-Aman and M. Bonin, Inorg. Chem., 2002, 41, 479–491 CrossRef CAS PubMed.
- D. Sadhukhan, A. Roy, R. J. Butcher, C. J. Gomez Garcia, B. Dede and S. Mitra, Inorg. Chim. Acta, 2011, 376, 245–254 CrossRef CAS.
- P. Comba, B. Martin, A. Muruganantham and J. Straub, Inorg. Chem., 2012, 51, 9214–9225 CrossRef CAS PubMed.
- M. R. Mendoza-Quijano, G. Ferrer-Sueta, M. Flores-Alamo, N. Aliaga-Alcalde, V. Gomez-Vidales, V. M. Ugalde-Saldivar and L. Gasque, Dalton Trans., 2012, 41, 4985–4997 RSC.
- B. Sreenivasulu, Aust. J. Chem., 2009, 62, 978–979 CrossRef and references therein.
- L. Horner and E. Geyer, Chem. Ber., 1965, 98, 2016–2045 CrossRef CAS.
- S. Harmalker, S. E. Jones and D. T. Sawyer, Inorg. Chem., 1983, 22, 2790–2794 CrossRef CAS.
- J. Rall, M. Wanner, M. Albrecht, F. M. Hornung and W. Kaim, Chem.–Eur. J., 1999, 5, 2802–2809 CrossRef CAS.
- S. Adhikari, A. Banerjee, S. Nandi, M. Fondo, J. Sanmartin-Matalobos and D. Das, RSC Adv., 2015, 5, 10987–10993 RSC.
- I. Szilagyi, L. Horvath, I. Labadi, K. Hernadi, I. Palinko and T. Kiss, Cent. Eur. J. Chem., 2006, 4, 118–134 CAS.
- J. Gao, J. H. Reibenspies and A. E. Martell, Inorg. Chim. Acta, 2003, 346, 67–75 CrossRef CAS.
- J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem.–Eur. J., 2002, 8, 247–258 CrossRef CAS.
- J. Ackermann, S. Buchler and F. Meyer, C. R. Chim., 2007, 10, 421–432 CrossRef CAS.
- R. Ganguly, B. Sreenivasulu and J. J. Vittal, Coord. Chem. Rev., 2008, 252, 1027–1050 CrossRef CAS.
- B. Sreenivasulu, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2006, 2656–2670 CrossRef CAS.
- X. B. Wang, J. Ding and J. J. Vittal, Inorg. Chim. Acta, 2006, 359, 3481–3490 CrossRef CAS.
- B. Sreenivasulu, M. Vetrichelvan, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2005, 4635–4644 CrossRef CAS.
- C. T. Yang, M. Vetrichelvan, X. D. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113–121 RSC.
- D. Mukherjee, R. N. Manna, P. Saha, U. Mandal and M. Mitra, J. Mol. Struct., 2021, 1223, 129247 CrossRef CAS.
- M. Mitra, D. Mukherjee, U. Mandal and A. A. Shteinman, J. Coord. Chem., 2018, 71, 3824–3835 CrossRef CAS.
- R. V. Gorkum, J. Berding, D. M. Tooke, A. L. Spek, J. Reedijk and E. Bouwman, J. Catal., 2007, 252, 110–118 CrossRef.
- M. Mitra, R. Singh, M. Pyrkosz-Bulska, M. Haukka, E. Gumienna-Kontecka and E. Nordlander, Z. Anorg. Allg. Chem., 2013, 639(8–9), 1534–1542 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–251 CrossRef CAS.
- P. Chaudhuri, R. Wagner and T. Weyhermuller, Inorg. Chem., 2007, 46, 5134–5136 CrossRef CAS.
- S. Mukhopadhyay, D. Mandal, P. B. Chatterjee, C. Desplanches, J.-P. Sutter, R. J. Butcher and M. Chaudhury, Inorg. Chem., 2004, 43, 8501–8509 CrossRef CAS PubMed.
- K. D. Karlin, Y. Gultneh, T. Nicholson and J. Zubieta, Inorg. Chem., 1985, 24, 3725–3727 CrossRef CAS.
- O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993 Search PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CAS.
- D. Venegas-Yazigi, D. Aravena, E. Spodine, E. Ruiz and S. Alvarez, Coord. Chem. Rev., 2010, 254, 2086–2095 CrossRef CAS.
- T. Kruse, T. Weyhermuller and K. Weighardt, Inorg. Chim. Acta, 2002, 331, 81–89 CrossRef CAS.
- E. Ruiz, P. Alemany, S. Alvarez and J. Cano, J. Am. Chem. Soc., 1997, 119, 1297–1303 CrossRef CAS.
- E. Ruiz, P. Alemany, S. Alvarez and J. Cano, Inorg. Chem., 1997, 36, 3683–3688 CrossRef CAS.
- R. A. Peralta, A. Neves, A. J. Bortoluzzi, A. dos Anjos, F. R. Xavier, B. Szpoganicz, H. Terenzi, M. C. B. de Oliveira, E. E. Castellano, G. R. Friedermann, A. S. Mangrich and M. A. Novak, J. Inorg. Biochem., 2006, 100, 992–1004 CrossRef CAS PubMed.
- R. E. H. M. B. Osorio, R. A. Peralta, A. J. Bortoluzzi, V. R. de Almeida, B. Szpoganicz, F. L. Fischer, H. Terenzi, A. S. Mangrich, K. M. Mantovani, D. E. C. Ferreira, W. R. Rocha, W. Hasse, Z. Tomkowicz, A. de Anjos and A. Neves, Inorg. Chem., 2012, 51, 1569–1589 CrossRef CAS PubMed.
- W. Mazurek, A. M. Bond, K. S. Murray, M. J. O'connor and A. G. Wedd, Inorg. Chem., 1985, 24, 2484–2490 CrossRef CAS.
- L. Platinus and G. Chapius, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- D. C. Palmer, CrystalMaker. CrystalMaker Software Ltd, Begbroke, Oxfordshire, England, 2014 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. Jeffrey Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS, 1H-NMR, FTIR and UV/vis spectra of complex 1, cyclic voltammogram of 1, rates of catecholase activity for 1, DFT calculations are available. CCDC 2068506. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02787e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Ujjwal Mandal*a and
Mainak Mitra
b,
Ujjwal Mandal*a and
Mainak Mitra