
New developments in RiPP discovery, enzymology and engineering
Manuel
Montalbán-López†
 a,
Thomas A.
Scott†
b,
Sangeetha
Ramesh†
cd,
Imran R.
Rahman†
e,
Auke J.
van Heel†
f,
Jakob H.
Viel†
f,
Vahe
Bandarian
g,
Elke
Dittmann
h,
Olga
Genilloud
i,
Yuki
Goto
j,
María José
Grande Burgos
k,
Colin
Hill
l,
Seokhee
Kim
m,
Jesko
Koehnke
n,
John A.
Latham
o,
A. James
Link
p,
Beatriz
Martínez
q,
Satish K.
Nair
der,
Yvain
Nicolet
s,
Sylvie
Rebuffat
t,
Hans-Georg
Sahl
u,
Dipti
Sareen
v,
Eric W.
Schmidt
w,
Lutz
Schmitt
x,
Konstantin
Severinov
y,
Roderich D.
Süssmuth
z,
Andrew W.
Truman
aa,
Huan
Wang
ab,
Jing-Ke
Weng
ac,
Gilles P.
van Wezel
ad,
Qi
Zhang
ae,
Jin
Zhong
af,
Jörn
Piel
*b,
Douglas A.
Mitchell
*cdag,
Oscar P.
Kuipers
*f and
Wilfred A.
van der Donk
*deagah
a,
Thomas A.
Scott†
b,
Sangeetha
Ramesh†
cd,
Imran R.
Rahman†
e,
Auke J.
van Heel†
f,
Jakob H.
Viel†
f,
Vahe
Bandarian
g,
Elke
Dittmann
h,
Olga
Genilloud
i,
Yuki
Goto
j,
María José
Grande Burgos
k,
Colin
Hill
l,
Seokhee
Kim
m,
Jesko
Koehnke
n,
John A.
Latham
o,
A. James
Link
p,
Beatriz
Martínez
q,
Satish K.
Nair
der,
Yvain
Nicolet
s,
Sylvie
Rebuffat
t,
Hans-Georg
Sahl
u,
Dipti
Sareen
v,
Eric W.
Schmidt
w,
Lutz
Schmitt
x,
Konstantin
Severinov
y,
Roderich D.
Süssmuth
z,
Andrew W.
Truman
aa,
Huan
Wang
ab,
Jing-Ke
Weng
ac,
Gilles P.
van Wezel
ad,
Qi
Zhang
ae,
Jin
Zhong
af,
Jörn
Piel
*b,
Douglas A.
Mitchell
*cdag,
Oscar P.
Kuipers
*f and
Wilfred A.
van der Donk
*deagah
aDepartment of Microbiology, University of Granada, Spain
bInstitute of Microbiology, ETH Zürich, Zürich, Switzerland. E-mail: jpiel@ethz.ch
cDepartment of Microbiology, University of Illinois at Urbana-Champaign, Urbana, IL, USA. E-mail: douglasm@illinois.edu
dCarl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL, USA. E-mail: vddonk@illinois.edu
eDepartment of Biochemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA
fDept. of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, The Netherlands. E-mail: o.p.kuipers@rug.nl
gDepartment of Chemistry, University of Utah, Salt Lake City, UT, USA
hInstitute of Biochemistry and Biology, University of Potsdam, Germany
iFundación MEDINA, Granada, Spain
jDepartment of Chemistry, Graduate School of Science, The University of Tokyo, Bunkyo, Tokyo, Japan
kDepartment of Microbiology, University of Jaén, Spain
lAPC Microbiome Ireland, School of Microbiology, University College Cork, Cork, Ireland
mDepartment of Chemistry, Seoul National University, South Korea
nSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK
oDepartment of Chemistry and Biochemistry, University of Denver, Denver, CO, USA
pDepartments of Chemical and Biological Engineering, Chemistry, and Molecular Biology, Princeton University, Princeton, NJ, USA
qDept. of Technology and Biotechnology of Dairy Products, Dairy Research Institute-IPLA, Consejo Superior de Investigaciones Científicas-CSIC, Villaviciosa, Spain
rCenter for Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, Urbana, IL, USA
sUniversité Grenoble-Alpes, CEA, CNRS, IBS, Metalloproteins Unit, Grenoble, France
tLaboratory Molecules of Communication and Adaptation of Microorganisms-MCAM (UMR 7245 CNRS-MNHN), National Museum of Natural History, Paris, France
uDepartment of Pharmaceutical Microbiology, University of Bonn, Bonn, Germany
vDepartment of Biochemistry, Panjab University, Chandigarh-160014, India
wDepartment of Medicinal Chemistry, University of Utah, Salt Lake City, UT, USA
xInstitute of Biochemistry, Heinrich Heine University Düsseldorf, Duesseldorf, Germany
yWaksman Institute, Rutgers University, Piscataway NJ and Skolkovo Institute of Science and Technology, Skolkovo, Russia
zDepartment of Chemistry, Technische Universität Berlin, Berlin, Germany
aaDepartment of Molecular Microbiology, John Innes Centre, Norwich, UK
abState Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center of Nanjing University, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China
acWhitehead Institute for Biomedical Research, Department of Biology, Massachusetts Institute of Technology, Cambridge, MA, USA
adInstitute of Biology, Leiden University, Sylviusweg 72, 2333 BE Leiden, The Netherlands
aeFudan University, Shanghai, China
afState Key Laboratory of Microbial Resources, Institute of Microbiology, University of Chinese Academy of Sciences, Beijing, China
agDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL, USA
ahHoward Hughes Medical Institute, University of Illinois at Urbana-Champaign, Urbana, IL, USA
First published on 16th September 2020
Abstract
Covering: up to June 2020
Ribosomally-synthesized and post-translationally modified peptides (RiPPs) are a large group of natural products. A community-driven review in 2013 described the emerging commonalities in the biosynthesis of RiPPs and the opportunities they offered for bioengineering and genome mining. Since then, the field has seen tremendous advances in understanding of the mechanisms by which nature assembles these compounds, in engineering their biosynthetic machinery for a wide range of applications, and in the discovery of entirely new RiPP families using bioinformatic tools developed specifically for this compound class. The First International Conference on RiPPs was held in 2019, and the meeting participants assembled the current review describing new developments since 2013. The review discusses the new classes of RiPPs that have been discovered, the advances in our understanding of the installation of both primary and secondary post-translational modifications, and the mechanisms by which the enzymes recognize the leader peptides in their substrates. In addition, genome mining tools used for RiPP discovery are discussed as well as various strategies for RiPP engineering. An outlook section presents directions for future research.
 Manuel Montalbán-López | The authors of this community review are a multidisciplinary and multinational group of scientists that are all working on the biosynthesis, discovery, and engineering of RiPPs. Oscar Kuipers, Wilfred van der Donk, Jörn Piel, Douglas Mitchell and members of their laboratories were the coordinating authors of the review. Manuel Montalbán-López, Auke J. van Heel, Jakob Viel and Oscar Kuipers (top row, left-to-right) have investigated RiPP biosynthesis in bacteria with a focus on the microbiology and engineering of lanthipeptides. Thomas Scott and Jörn Piel (second row, 1st and 2nd) work on the discovery and biosynthesis of chemically distinct, bioactive RiPPs. Sangeetha Ramesh and Douglas Mitchell (second row, 3rd and 4th) focus on the enzymology and discovery of new RiPP classes and develop new bioinformatic tools. Imran Rahman and Wilfred van der Donk (bottom row, left to right) investigate the enzymology, discovery and engineering of RiPPs. |
1 Introduction
In 2013, a community effort of scientists working on the biosynthesis of ribosomally synthesized and post-translationally modified peptide (RiPPs) resulted in a comprehensive review that described the commonalities of the many natural products generated from ribosomal peptides.1 Since the first recognition of RiPPs as a major group of natural products, many advances have been reported on the understanding of their biosynthesis, engineering of their structures, and discovery of new members. These exciting developments resulted in the First International Conference on RiPPs that was held in April 2019 in Granada, Spain, with Oscar Kuipers as Chair and Manuel Montalbán-López, Jörn Piel, Wilfred van der Donk, and Auke J. van Heel comprising the Organizing Committee. The data presented, in addition to the large volume of new publications, stimulated discussion of a new community effort involving the conference participants. This resulting review outlines the main concepts and overall biosynthetic logic of RiPPs, but will not discuss findings that predate 2013 or RiPP bioactivities for which we refer to previous work.1,2 This review will also not comprehensively list all examples of genome mining for new RiPPs and will only focus on examples of new compound classes and new post-translational modifications.The current work will first discuss new classes of RiPPs that have been discovered (Section 2). In some cases, these were known compound classes that were not previously recognized to be generated from a ribosomally produced precursor peptide; in other cases, these RiPPs consist of new natural product classes that had not been previously reported. After the overview of new RiPPs, we will cover new developments in the understanding of biosynthetic enzymes that introduce the primary, class-defining post-translational modifications (Section 3), followed by a separate section on substrate recognition and leader peptide removal (Sections 4). Section 5 discusses enzymes that install secondary, compound-specific modifications sometimes referred to as tailoring reactions.3
As anticipated, the avalanche of available genomic data has not abated since 2013. As such, tools have been developed to help mine the formidable amount of information for the discovery of new RiPPs. Section 6 of this review discusses both the new bioinformatic programs that have been advanced as well as their use for the discovery of new RiPPs.
Because of their genetically encoded precursor peptides, RiPPs are particularly well-suited for synthetic biology approaches to generate novel structures. The 2013 review forecasted the many intriguing possibilities with respect to bioengineering directions and in the intervening time, many of the tantalizing prospects have been realized and other new ideas have been explored. Section 7 of this review will discuss the advances in synthetic biology from making hybrid molecules to introducing novel functionalities, from engineering high-throughput selection systems to employing new bioactivities, and from using in vitro translation and post-translational modification systems to incorporation of non-canonical amino acids. We trust that this review will help stimulate another period of sustained growth and discovery in the RiPP field.
1.1 Brief overview of RiPP biosynthesis
RiPP precursor peptides are typically composed of multiple segments (Fig. 1A). The core region (or core peptide) is where the post-translational modifications (PTMs) take place. A leader and/or a follower peptide provides binding affinity for many of the PTM enzymes and is removed at some point during the biosynthesis. In addition, the leader peptide (LP) can serve as an allosteric effector to activate the biosynthetic enzymes, the presence of the leader/follower peptide often keeps the maturing peptide inactive, and in some cases, the LP is required to ensure that the PTMs are carried out in the correct order.4,5 For cases where the core peptide undergoes multiple types of PTMs, different parts of the leader/follower peptide can be recognized by different biosynthetic enzymes. The term recognition sequence (RS) has been introduced to identify the specific sequences that are required for a specific enzyme to carry out its chemical transformation on the core peptide. For some RiPPs discussed herein, such as the cyanobactins, dikaritins, orbitides, amatoxins, cyclotides, and lyciumins (see the respective sections), the precursor peptide carries multiple core peptides and multiple recognition sequences, such that one peptide results in more than one RiPP product (Fig. 1B). For most RiPPs, numbering of the precursor peptides uses negative numbers for the LP, counting backwards from the last residue of the LP, whereas positive numbers are used for the amino acid residues in the core peptides (Fig. 1A). In some cases, for instance cyanobactins, alternative numbering better allows recognition of conserved features of the core peptides.6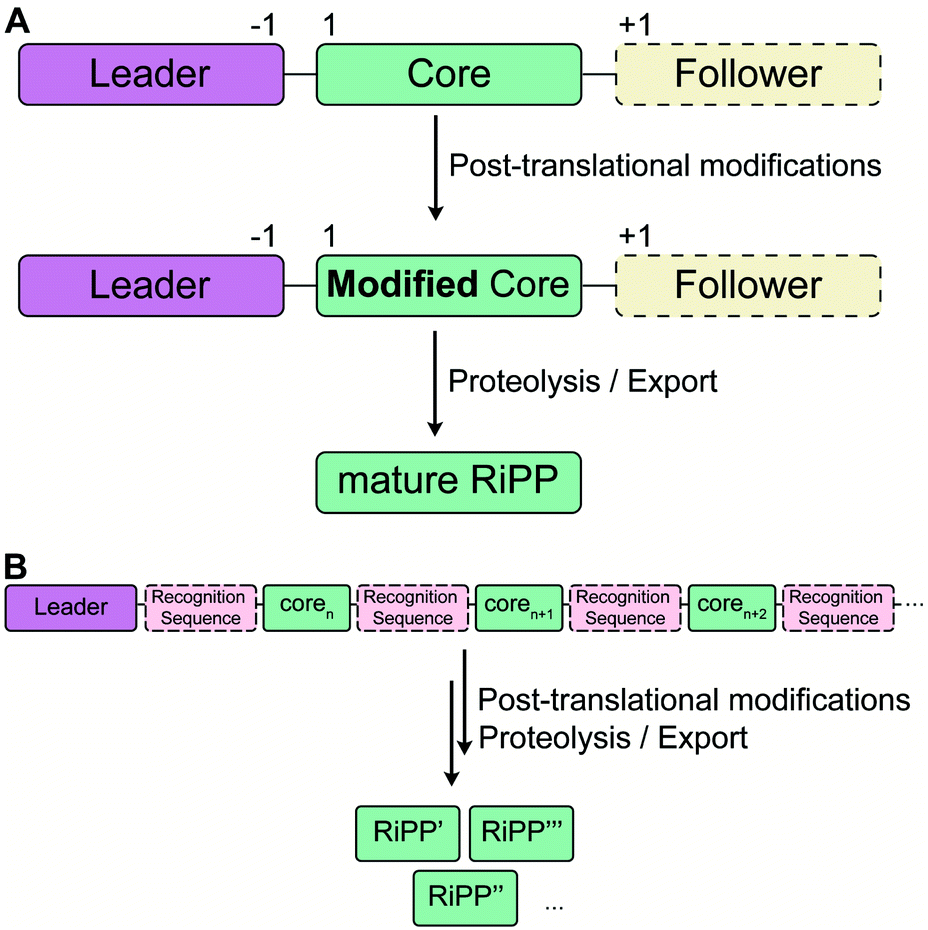 | ||
| Fig. 1 Generic representation of RiPP biosynthesis. (A) Most precursor peptides for RiPPs have two parts, a leader region and a core region. In some cases, a follower peptide is present. The core peptide is post-translationally modified to generate the mature RiPP, which at some stage requires leader/follower peptide removal. (B) For some RiPP classes, the core peptide is flanked on one or both sides by short recognition sequences (RSs) that are important for post-translational modification. These systems often, but not always, have multiple copies of the core peptides, which can be highly diverse in sequence and lead to a group of different RiPP products. | ||
As the number of biosynthetic genes has exploded and many new bioinformatics tools have been developed to identify and annotate them (Section 6), naming of the genes and the corresponding proteins with the 26 letters of the alphabet without creating duplication has become challenging. The American Society for Microbiology uses a specific set of guidelines for gene nomenclature in bacterial genetics.7 Paraphrasing the original definitions, genes are designated by a three-letter, lower-case, italicized locus symbol followed by a single italicized capital letter immediately following the three-letter lower-case locus identification to identify individual associated genes. Unfortunately, our own recommendations in 2013 for RiPP biosynthesis partially ignored this standardization and has led to possible confusion. As the number of genes has rapidly expanded, it has become more and more common to use two (or more) capital letters. For instance (and there are many more examples), for systems covered in this review, RaxST is used in sulfatyrotides (Section 2.11), LanKC is used for class III lanthipeptide dehydratases (Section 3.1.3), BotCD is used for an enzyme in bottromycin biosynthesis (Section 3.7), and AgePT is used for an enzyme in cyanobactin biosynthesis (Section 5.3.7.1). Confusion can arise for instance when a precursor is said to be coexpressed with RaxST and this can be interpreted as co-expression with two proteins (S and T) or co-expression with one protein (ST). This confusion can be avoided in several ways. Some authors have chosen to use numbers instead of letters as the second identifier when running out of letters (e.g. PlpA2, Section 3.14), others have used lower case letters following the first capital letter (e.g. UstYa, Section 3.17), or have used subscript letters (e.g. LanJA; Section 5.1.2). We recommend using such unambiguous nomenclature going forward.
2 New classes of RiPP natural products
With the advent of new sequencing techniques, the number of genome sequences has exploded. Researchers have used this genomic information to connect previously known natural product classes with orphan biosynthetic gene clusters (BGCs) for which the products were not established. In addition, genome mining approaches have led to the discovery of previously unknown RiPP classes by exploring novel BGCs. This section will provide a brief overview of natural products that have been established to be RiPPs since January 2013 in the approximate order of their first recognition as a RiPP. Their biosynthesis, when known, is covered in Section 3. If the biosynthesis of a new RiPP has not yet been studied, its BGC will be briefly discussed in this section. In the structure drawings in this review, the PTMs that are class-defining are highlighted in yellow, whereas secondary (sometimes also referred to as “ancillary” or “tailoring”) modifications are highlighted in cyan, unless specified otherwise in the figure legend.Table 1 lists all currently known RiPP classes with the corresponding class-defining primary PTM or feature denoted. The field has rapidly expanded since the previous comprehensive review (circa January 2013) with 17 new classes and is likely to expand further in the coming years. We note that future classification of all RiPPs into single classes may prove difficult in some cases as RiPPs are discovered that are made by natural hybrid pathways utilizing combinations of biosynthetic enzymes from multiple compound classes.
| Class | Example | Class-defining PTM(s) or feature (enzyme responsible) |
|---|---|---|
| a Abbreviations: AEP, asparaginyl endoprotease; Dha/Dhb, dehydroalanine/dehydrobutyrine; DUF, domain of unknown function; FAS, fatty acid synthase; LP, leader peptide; MT, methyltransferase; PEARL, peptide aminoacyl-tRNA ligase; PKS, polyketide synthase; POP, prolyl oligopeptidase; rSAM, radical SAM. | ||
| Amatoxins/phallotoxins | Phalloidin | N-to-C cyclization, Cys–Trp crosslink |
| Guanidinotides | Pheganomycin | α-Guanidino acid containing peptides (ATP-grasp) |
| Atropitides | Tryptorubin | Aromatic amino acids crosslinked to give a non-canonical atropisomer |
| Autoinducing peptides | AIP-I | Cyclic ester or thioester |
| Bacterial head-to-tail cyclized peptides | Enterocin AS-48 | N-to-C cyclization (DUF95 & ATP-grasp) |
| Borosins | Omphalotin | Amide backbone N-methylation (N-MT), N-to-C cyclization (POP) |
| Bottromycins | Bottromycin A1 | Macrolactamidine (YcaO) |
| Cittilins | Cittilin A | Biaryl and aryl–oxygen–aryl ether crosslinks (P450) |
| ComX | ComX168 | Indole cyclization and prenylation |
| Conopeptides | Conantokin G | Peptides produced by cone snails |
| Crocagins | Crocagin A | Indole-backbone cyclization |
| Cyanobactins | Patellamides | N-terminal proteolysis (PatA protease) |
| Cyclotides | Kalata B1 | N-to-C cyclization, disulfide(s) (AEP) |
| Dikaritins | Ustiloxin | Tyr-Xxx ether crosslink (UstY) |
| Epipeptides | YydF | D -Amino acids (rSAM) |
| Glycocins | Sublancin 168 | S, O-glycosylation of Ser/Cys |
| Graspetides | Microviridin J | Macrolactones/lactams (ATP-grasp) |
| Lanthipeptides | Nisin | (Methyl)lanthionine, labionin |
| Lasso peptides | Microcin J25 | Macrolactam with threaded C-terminal tail (Asn synthetase homolog) |
| Linaridins | Cypemycin | Dhb, no lanthionines |
| Linear azol(in)e-containing peptides (LAPs) | Microcin B17 | Cys, Ser, or Thr derived azol(in)es (YcaO) |
| Lipolanthines | Microvionin | C-terminal labionin/avionin containing peptide and N-terminal FAS/PKS segment |
| Lyciumins | Lyciumin A | Pyroglutamate, Trp–Gly crosslink |
| Streptide | Streptide | Trp–Lys crosslink (rSAM) |
| Methanobactins | Methanobactin | Oxazolones (DUF692) |
| Microcin C | Microcin C | Aminoacyl adenylate or cytidylate with a phosphoramidate linkage (ubiquitin E1 homolog) |
| Mycofactocin | Mycofactocin | Val–Tyr crosslink (rSAM) |
| Orbitides | Cyclolinopeptide A | N-to-C cyclization; no disulfides |
| Pantocins | Pantocin A | Glu–Glu crosslink (PaaA) |
| Pearlins | Thiaglutamate | aa-tRNA derived (PEARL) |
| Proteusins | Polytheonamide | Nitrile hydratase LP |
| Pyrroloquinoline quinones | PQQ | Glu–Tyr crosslink (rSAM) |
| Ranthipeptides | Freyrasin | Sulfur-to-non-Cα thioether crosslink (rSAM) |
| Rotapeptides | TQQ | Oxygen-to-α-carbon crosslink |
| Ryptides | RRR | Arg–Tyr crosslink (rSAM) |
| Sactipeptides | Subtilosin | Sactionine crosslink (rSAM) |
| Spliceotides | PlpA | β-Amino acids (rSAM) |
| Sulfatyrotides | RaxX | Tyrosine sulfation |
| Thioamitides | Thioviridamide | Backbone thioamide (YcaO) |
| Thiopeptides | Thiostrepton | [4 + 2] cycloaddition of two Dha |
| Thyroid hormones | Triiodothyronine | Iodination of Tyr, excised from thyroglobulin |
2.1 Thioamitides
Thioviridamide was first reported in 2006.8 The structure of this apoptosis inducer produced by Streptomyces olivoviridis contains a C-terminal 2-aminovinyl-cysteine (AviCys) structure previously found in lanthipeptides and linaridins, hinting at its RiPP origin. However, the most unique part of the structure of thioviridamide and related compounds like JBIR-140 (Fig. 2)9 are the thioamide bonds in the peptide backbone. In 2013, the BGC of thioviridamide was reported demonstrating that the compound is indeed a RiPP.10 Since then, studies on its biosynthesis (see Section 3.5) have facilitated genome mining approaches that have uncovered other thioamide containing peptides.9,11–14 The term thioamitides is suggested to describe this family of compounds that have thioamides as the class-defining feature.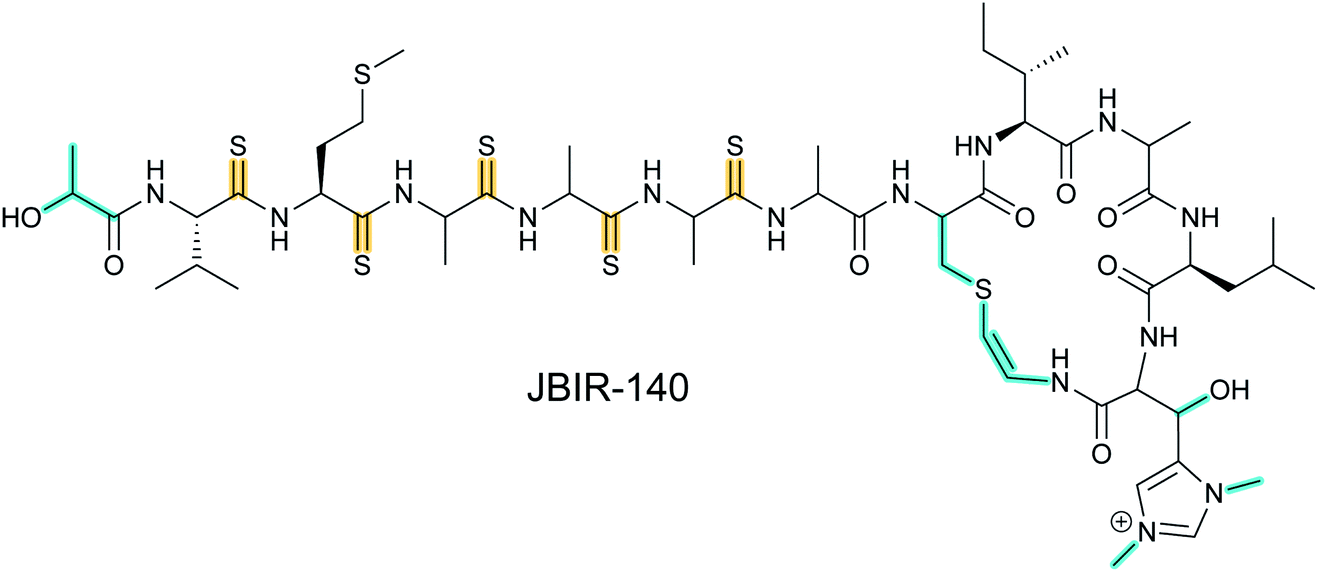 | ||
| Fig. 2 Structure of JBIR-140. Primary PTMs are highlighted in yellow and secondary PTMs are highlighted in cyan. The stereochemistry has been determined for some of the stereocenters.9 | ||
2.2 Dikaritins
Ustiloxins are produced by Ustilaginoidea virens, a pathogenic fungus affecting rice plants. In 2014, ustiloxin B was reported to be a RiPP, the first characterized example in Ascomycetes (Fig. 3A).15 Subsequent studies showed that the phomopsins are also made from a ribosomally synthesized precursor peptide (Fig. 3B),16 and genome mining uncovered new groups of compounds such as asperipin-2a (Fig. 3C).17 The term dikaritins has been suggested to describe the family of fungal compounds that are made by similar biosynthetic pathways involving crosslinks between a phenolic group derived from Tyr and the side chains of various other amino acids (see Section 3.17).16 Like many cyanobactins (Section 3.6), microviridins (Section 3.15), amatoxins (Section 3.18), orbitides, cyclotides (Section 3.19), and other plant-derived RiPPs (Section 2.9), dikaritin precursor peptides contain multiple core peptides that are located in repetitive sequence motifs.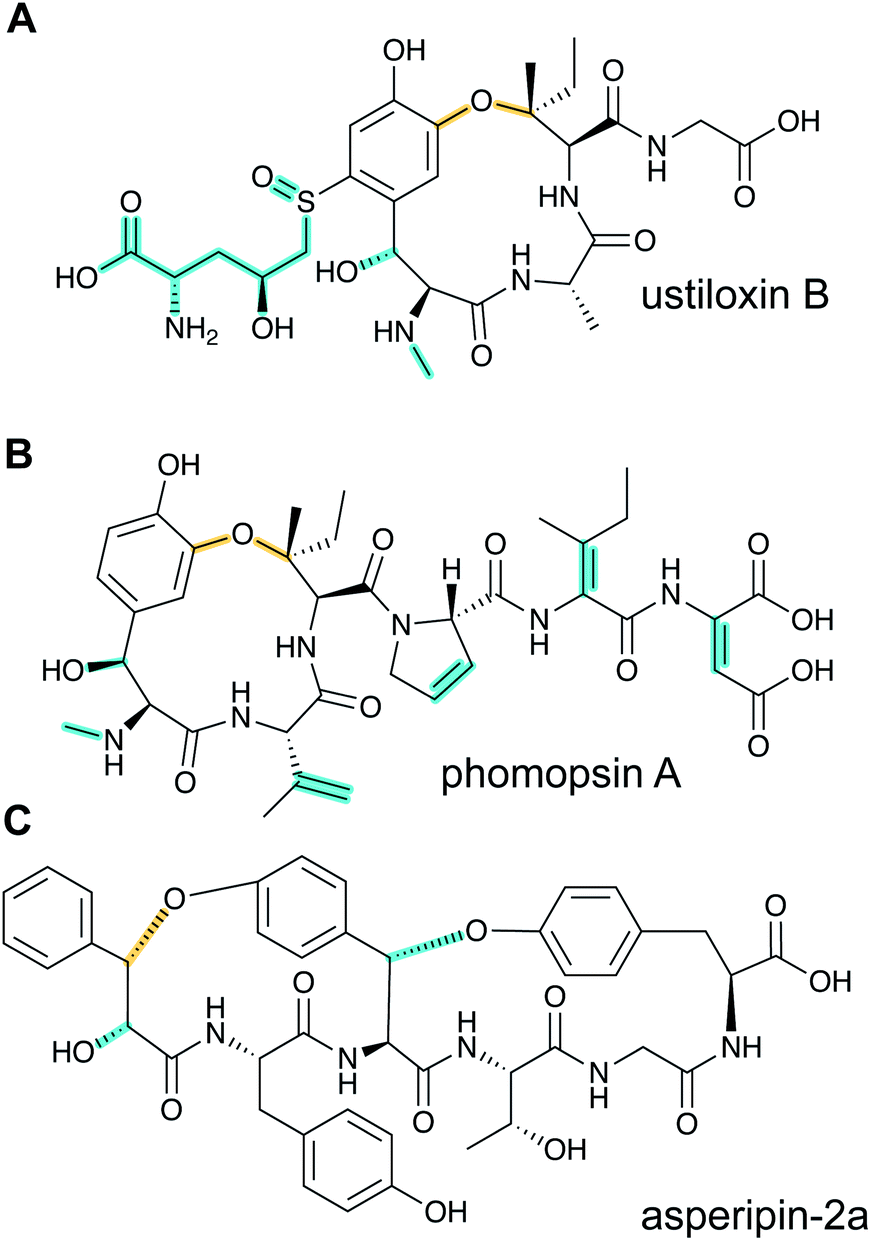 | ||
| Fig. 3 Structures of representative dikaritins. (A) Ustiloxin B. (B) Phomopsin A. (C) Asperipin-2a; the stereochemistry is proposed based on NMR data.18 Primary PTMs are highlighted in yellow and secondary PTMs are highlighted in cyan. | ||
2.3 Guanidinotides
Pheganomycin consists of the non-proteinogenic amino acid (S)-2-(3,5-dihydroxy-4-hydroxymethyl)phenyl-2-guanidinoacetic acid connected to a peptide that contains a tert-Leu (Fig. 4A). The compound is produced by Streptomyces cirratus and has activity against mycobacteria. In 2015, the peptide was reported to be ribosomally synthesized with the non-proteinogenic amino acid appended in a PTM process.19 Two different pheganomycin core peptides are present in the precursor peptide (Fig. 4B) resulting in a group of congeners. We suggest the name guanidinotides to describe any additional compounds that feature α-guanidino acids attached to ribosomal peptides. | ||
| Fig. 4 (A) Structure of pheganomycin and its congeners. Primary PTMs are highlighted in yellow and secondary PTMs are highlighted in cyan. (B) Sequence of the precursor peptide of the pheganomycins. The two core sequences are underlined. | ||
2.4 Mycofactocin
The occurrence of mycofactocin in mycobacteria as a unique redox cofactor with a ribosomal origin was predicted in 2011.20 A core structure termed premycofactocin has been suggested based on biochemical studies of the biosynthetic enzymes (Fig. 5) (Section 3.21),21 and oligoglycosylated derivatives have been detected in cells.22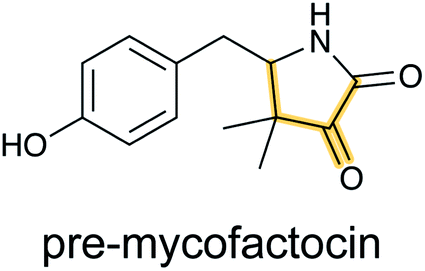 | ||
| Fig. 5 Structure of pre-mycofactocin (see Section 3.21). Further PTMs such as oligoglycosylation of the phenol that are currently not completely elucidated take place to furnish the final structure.22 | ||
2.5 Streptides
Streptoccocci use various signaling molecules for intercellular communication. One such molecule of unknown structure was reported to be cyclic and derived from a gene-encoded peptide.23 In 2015, the structure of the compound was revealed (Fig. 6A), showing that it contained a post-translationally installed Lys–Trp crosslink, and the term streptide was introduced for this group of streptococcal molecules. Gene deletion studies and biochemical experiments confirmed that it was a member of the RiPP class and related molecules have been discovered by genome mining in subsequent studies, some of which have undergone additional bond formation (e.g.Fig. 6B).24 Streptide BGCs are positively regulated by quorum sensing and are widespread in streptococcal genomes.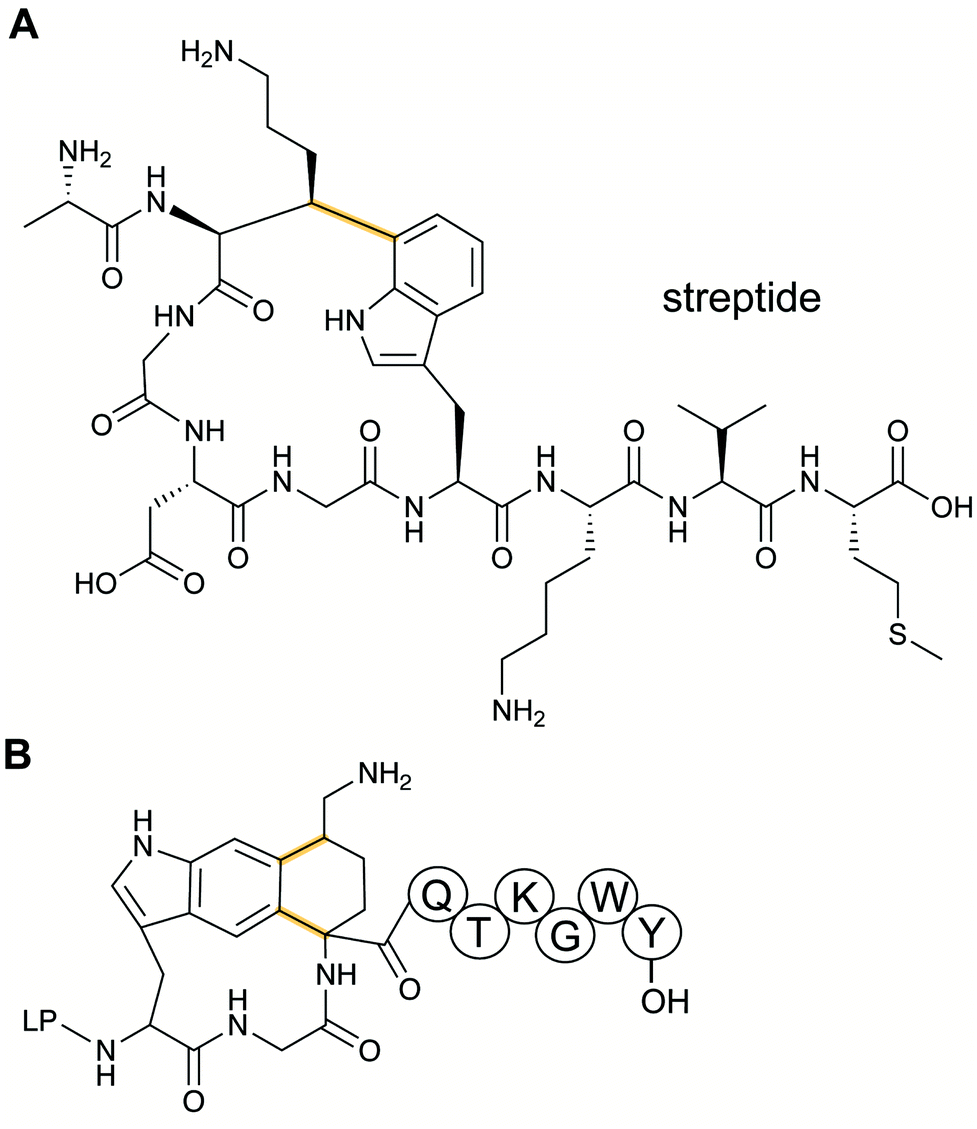 | ||
| Fig. 6 Structures of (A) streptide, and (B) streptide-like RiPP with an additional crosslink; LP = leader peptide. | ||
2.6 Borosins
N-Methylation of backbone amides is a commonly found structural feature in non-ribosomal peptides but it had been absent from RiPPs until the discovery of the omphalotins produced by fungi (Fig. 7).25,26 As described in Section 3.16, the omphalotins are a unique type of RiPP in that the substrate is attached to the enzyme that introduces the N-methylations. The term borosin is used for the family of fungal products that are made by this unusual biosynthetic strategy.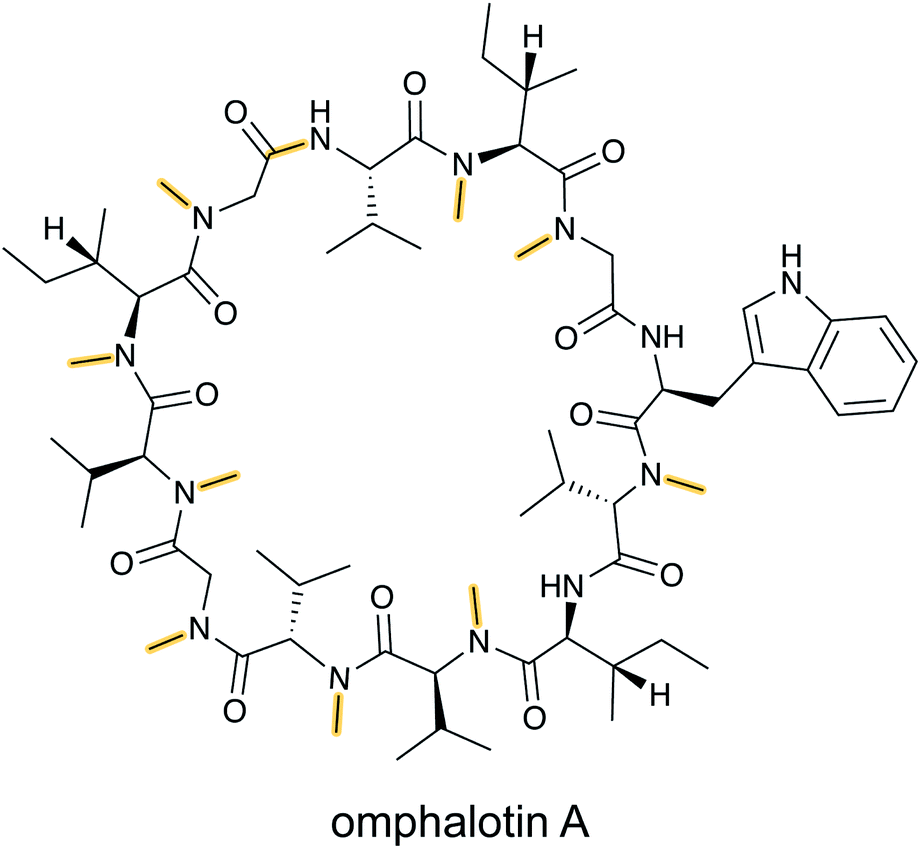 | ||
| Fig. 7 Structure of omphalotin A illustrating the class-defining N-methylations and head-to-tail cyclization in yellow. | ||
2.7 Crocagins
Metabolome mining of the myxobacterium Chondromyces crocatus Cm c5 resulted in the discovery of the crocagins, which are amongst the smallest RiPPs as they are formed from three amino acids of a precursor peptide (Fig. 8).27 The BGC was identified by genome sequencing and targeted gene inactivation in the producer strain. Crocagin A was shown to bind to the carbon storage regulator protein CsrA and as such inhibits CsrA binding to its target RNA.27 | ||
| Fig. 8 (A) Structure of crocagin A. (B) BGC for crocagin A. (C) Sequence of the precursor peptide with the core peptide in red. In the absence of other characterized crocagins, all PTMs that form the heterocyclic central structure are currently class-defining and are shown in yellow. The secondary modification (i.e. N-terminal methylation) is shown in cyan. | ||
2.8 Epipeptides
Several new mechanisms of introducing D-amino acids into ribosomally synthesized peptides have been discovered in the past six years (see Sections 3.12 and 3.13). The most general method is epimerization by radical S-adenosyl methionine (SAM) epimerases as they could in principle epimerize the α-center of the 19 chiral proteinogenic amino acids. Such radical SAM epimerases were discovered in the biosynthesis of proteusins,28 where epimerization is part of a much larger set of PTMs (see Section 3.12). More recently, peptides that appear to only undergo epimerization, with no other PTMs, have been described that also require radical SAM proteins, but from a different subfamily.29 These peptides were named epipeptides (e.g.Fig. 9).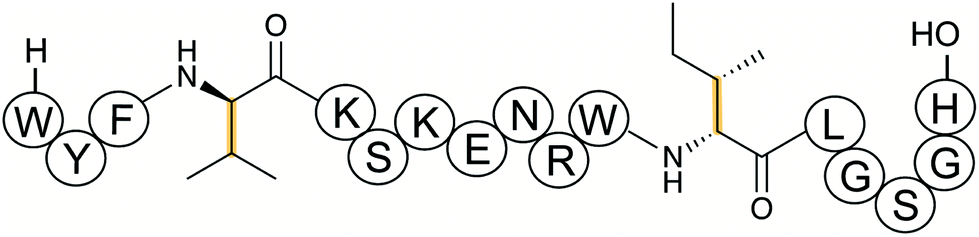 | ||
| Fig. 9 Structure of an epimerized peptide from Bacillus subtilis. Yellow indicates the class-defining, epimerization. Note on the peptide representation in this review: a free N-terminus is depicted as H- and a free carboxyl terminus as –OH. | ||
2.9 Lyciumins
Lyciumins are protease-inhibiting cyclic peptides originally isolated from the Chinese medicinal plant Lycium barbarum (goji berry) (Fig. 10A).30 Current members are characterized by crosslinks between the indole nitrogen of Trp to the α-carbon of a Gly within the peptide while also carrying a pyroglutamate residue at the N-terminus. Lyciumin precursor genes encode both a BURP domain (BURP is an acronym for the four initially identified examples of the domain: BNM2, USP, RD22, and PG1β)31 and repetitive lyciumin core peptide motifs (Fig. 10B).32 Heterologous expression of the lyciumin precursor gene alone is sufficient to produce the derived lyciumins in tobacco.32 The attachment of the BURP domain to the precursor peptide suggests that, like the borosins (Section 2.6), one or more of the PTMs might be catalyzed by a protein connected to the substrate. Like some other RiPPs (cyanobactins, graspetides, cyclotides, dikaritins, orbitides, and pheganomycin),1 the precursor peptides of lyciumins contain multiple core peptides (Fig. 10B). Genome mining suggests that lyciumin chemotypes are widespread in flowering plants and will likely have considerable structural diversity.32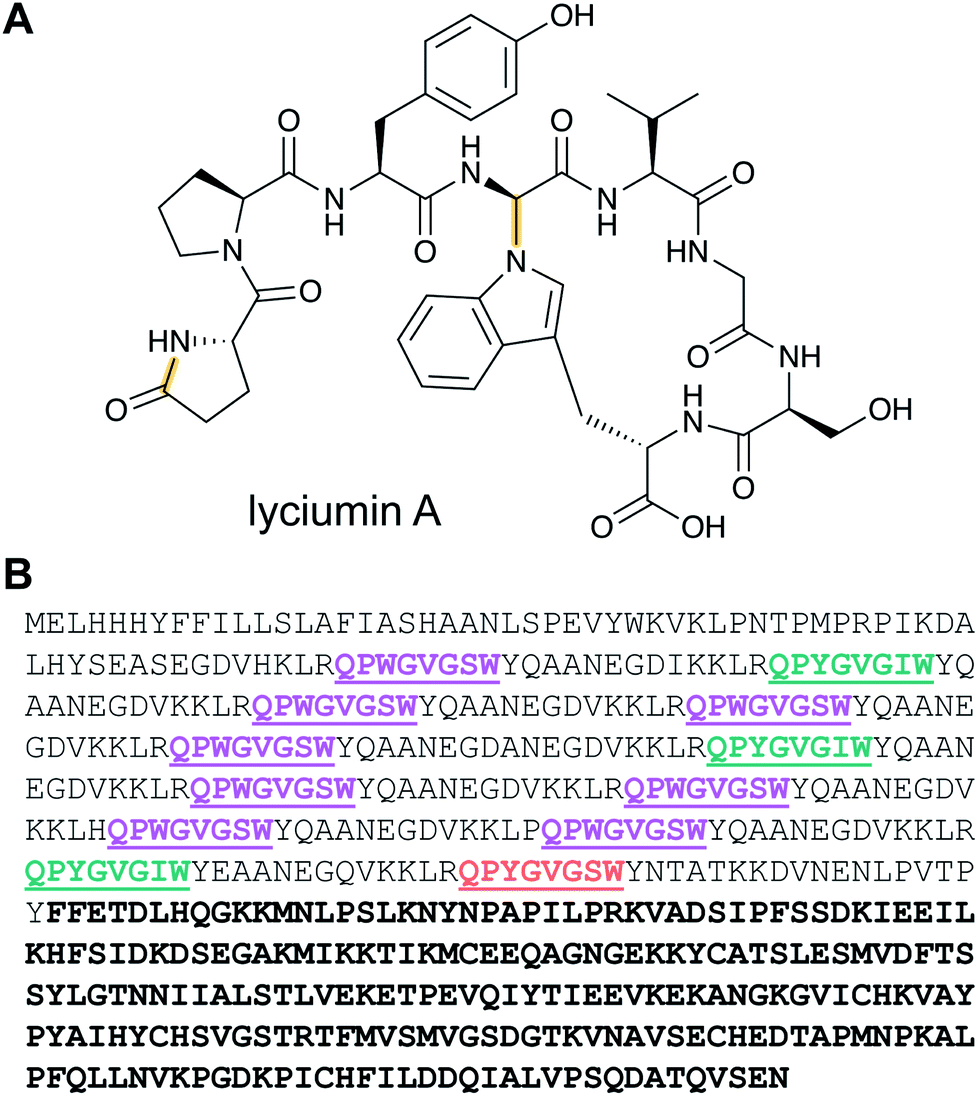 | ||
| Fig. 10 (A) Structure of lyciumin A. The class-defining modifications are in yellow. (B) Precursor peptide with core peptides underlined and colored according to unique core peptide sequences. The C-terminal bold sequence indicates the BURP domain. | ||
2.10 Lipolanthines
Lanthipeptide biosynthetic genes had been previously observed near polyketide synthase genes,33 but whether the encoded proteins indeed cooperate to make a hybrid natural product was unclear until the structure of microvionin was reported34 (Fig. 11). This molecule constitutes the first example of a lipopeptide where the peptide part is ribosomally synthesized. The compound was isolated from a culture of Microbacterium arborescens in a bioactivity-guided screen for antimicrobial natural products. In addition to the unusual lipidation, microvionin contains a decarboxylated analog of labionin termed avionin (Fig. 11). The relationship between avionin and labionin is analogous to that between lanthionine and the AviCys motif found in lanthipeptides and other RiPPs (e.g.Fig. 2). Genome mining studies demonstrated that BGCs for lipidated class III lanthipeptides are widespread and the name lipolanthines was introduced for this distinct group of RiPPs.34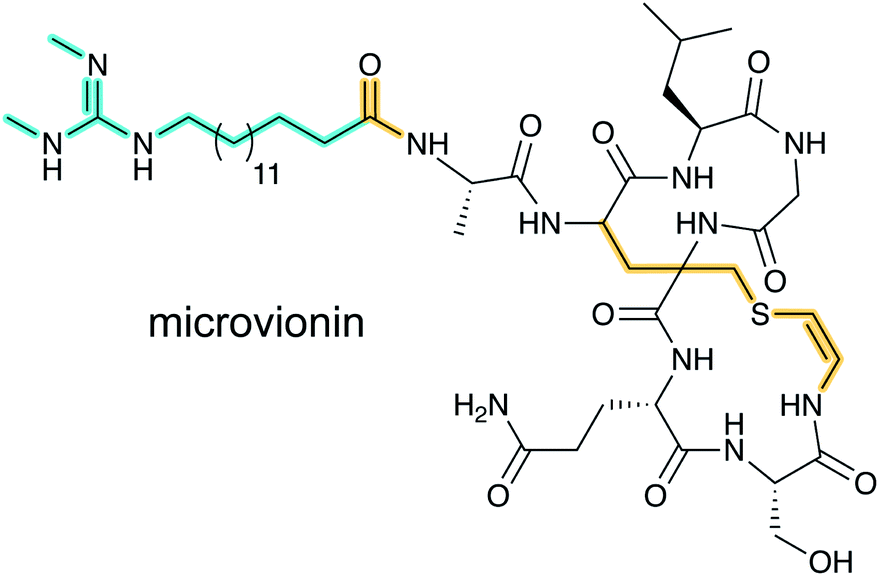 | ||
| Fig. 11 Structure of microvionin. The avionin (decarboxylated labionin) is highlighted on the right, and together with N-terminal lipidation forms the class-defining PTM for lipolanthines; the lipid structure (cyan) can vary and is not class-defining. | ||
2.11 RaxX and PSY1 sulfatyrotides
The recognition of bacterial biomolecules by plant cell surface receptor kinases commonly mediates resistance against pathogens. In rice, one of these kinases is XA21 that helps detect the Gram-negative pathogen Xanthomonas oryzae pv. oryzae (Xoo). The Xanthomonas-derived activator of XA21 is a peptide termed RaxX (Fig. 12A).35 Xoo strains evading XA21-mediated immunity carry a RaxX mutation that replaces a Tyr moiety, suggesting this residue is important for recognition of wild-type RaxX. Notably, this Tyr is the site for a post-translational modification resulting in side-chain O-sulfation, carried out by the sulfotransferase RaxST encoded in the rax locus (Fig. 12B). While Tyr sulfation is common in eukaryotes, RaxST is the first known prokaryotic sulfotransferase involved in post-translational modification. The RaxX peptide was shown to be produced from a 60-amino acid precursor peptide.35 The precursor has a 38-amino acid LP that ends in a double Gly motif that is often found in RiPPs (see Section 4.4.1). Moreover, the RaxX BGC (Fig. 12B) contains a peptidase-containing ATP-binding cassette transporter (PCAT) that removes the LP (see Section 4.4.1 for discussion of PCATs). RaxX resembles an 18-residue plant peptide PSY1 that is also sulfated on Tyr and serves as a hormone (Fig. 12C). It was therefore suggested that RaxX is a hormone mimic that benefits Xoo by interfering with host cellular processes.36,37 PSY1 is also ribosomally synthesized as a 75-amino acid precursor peptide that is not only sulfated but also glycosylated.38 Thus, both PSY1 and RaxX are RiPPs. Corroborating the virulence factor hypothesis, a synthetic 13-residue sulfated RaxX-derived peptide showed similar effects on root growth in Arabidopsis and rice as PSY1.36 Similar effects were also observed for completely matured RaxX, which was recently identified in Xoo extracts as a 21-residue peptide representing the proteolytically released, sulfated RaxX core peptide.35 These insights on a functionally unusual RiPP might lead to applications in plant protection. In support of such avenues, exogenously applied sulfated RaxX peptide suppresses infection by virulent Xoo strains in rice.39 Since the term sulfatides is already used for glycolipids,40 we suggest the term sulfatyrotides for RiPPs where Tyr sulfation is the primary modification.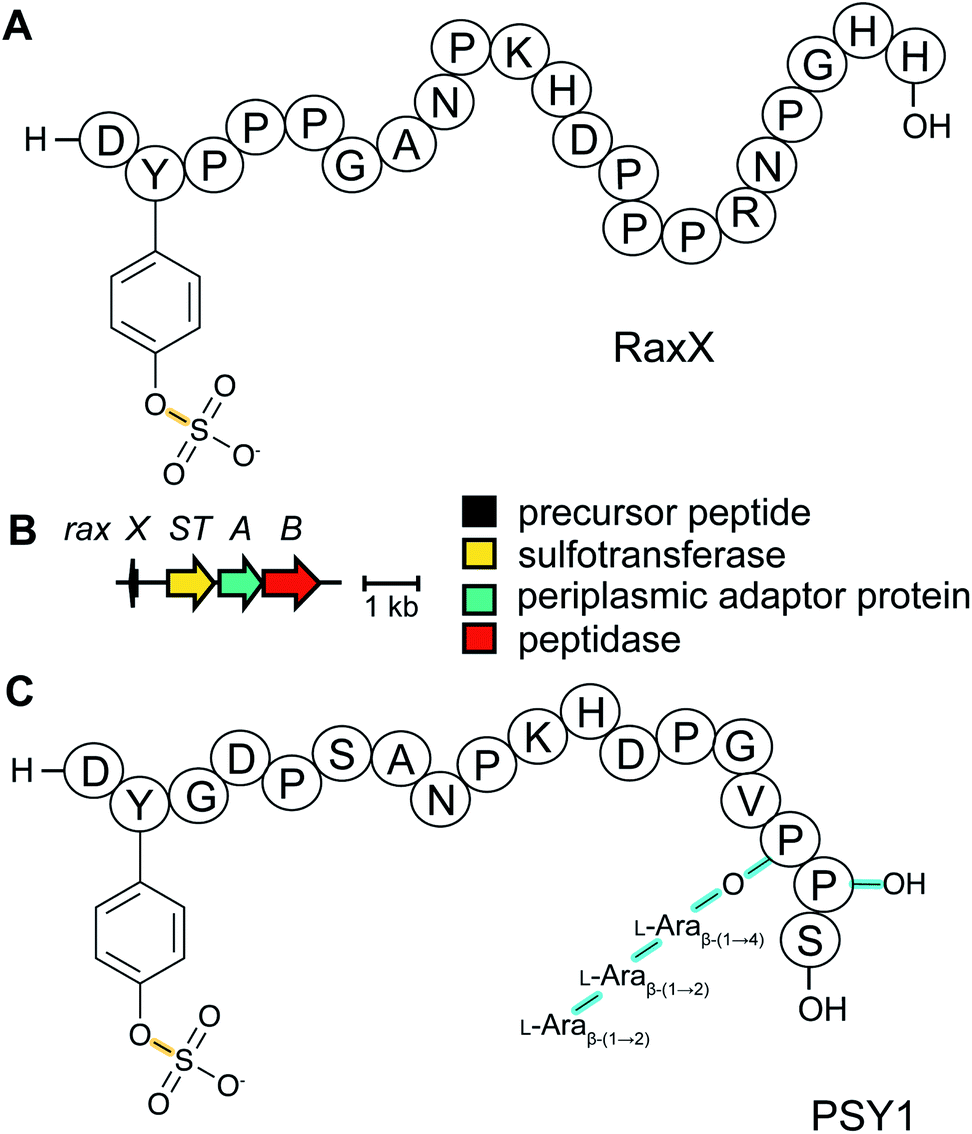 | ||
| Fig. 12 (A) Structure of bacterial RaxX. (B) BGC for RaxX production. (C) Structure of the plant peptide PSY1. The arabinose chain is attached to (4R)-hydroxyPro. A second hydroxylated Pro is indicated as Pro-OH (the C-terminal OH reflects a free carboxylic acid; see Fig. 9). Tyr sulfation (yellow) is the class-defining PTM for sulfatyrotides. | ||
2.12 Spliceotides
All of the compound groups discussed thus far were either known to belong to the RiPP group of natural products prior to investigation of their biosynthesis, or they were discovered by bioassay-guided isolation. In 2018 and 2019, several examples were reported of completely new families of RiPPs that were bioinformatically discovered through profiling of genes encoding enzymes that are divergent from known RiPP BGCs. One such example is the report of a fundamentally new PTM reaction that converts a dipeptide motif into a β-peptide (Fig. 13).41 During this process, part of one amino acid is excised leading to the name spliceotides for the natural products formed by this unusual reaction (Section 3.14).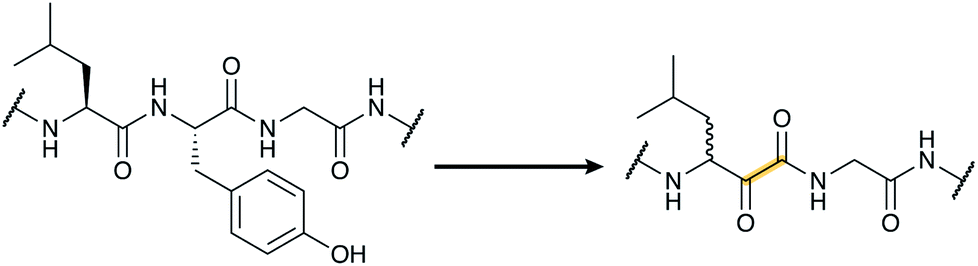 | ||
| Fig. 13 Example of a splicease reaction. The class-defining α-oxoamide backbone bond is shown in yellow. Stereochemistry at the preceding Leu α-carbon has not yet been defined. See Section 3.14 for further details. | ||
2.13 Ranthipeptides
Another example of a RiPP group that was identified bioinformatically before the molecule was characterized is the ranthipeptides (radical non-α-carbon thioether peptides).42,43 Lanthipeptides and sactipeptides are RiPP families with thioether crosslinks (Sections 3.1 and 3.9). For the sactipeptides, the intramolecular linkages are sulfur to α-carbon thioethers (sactionine) and include diverse amino acids. Sactionines are introduced by radical SAM enzymes, whereas for lanthipeptides the thioether linkages are to the β-carbons of dehydrated Ser/Thr residues. A bioinformatic search for homologs of the radical SAM enzymes that introduce the thioether crosslinks in sactipeptides resulted in the discovery of a large family of unique proteins. The substrates encoded next to these radical SAM enzymes were previously referred to as SCIFFs (for![[s with combining low line]](https://www.rsc.org/images/entities/char_0073_0332.gif) ix
ix ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ys
ys ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) n
n ![[f with combining low line]](https://www.rsc.org/images/entities/char_0066_0332.gif) orty-ive residues).42 Characterization of these radical SAM-dependent pathways demonstrated they did not result in sactionines but instead generated crosslinks to the β or γ-carbons of a variety of amino acids.43 Examples of ranthipeptides are freyrasin produced by Paenibacillus polymyxa that contains six thioether links to the β-carbon of Asp residues (S-Cβ) (Section 3.10) and thermocellin that contains a thioether cross-linked to the γ-carbon of Thr (Fig. 14A). A β-thioether linkage was also introduced by a homolog of the radical SAM enzyme (NxxcB) that generates the Lys–Trp crosslink in streptide (Section 2.5). The resulting product features a thioether linkage between a former Cys and Asn (Fig. 14B).44
orty-ive residues).42 Characterization of these radical SAM-dependent pathways demonstrated they did not result in sactionines but instead generated crosslinks to the β or γ-carbons of a variety of amino acids.43 Examples of ranthipeptides are freyrasin produced by Paenibacillus polymyxa that contains six thioether links to the β-carbon of Asp residues (S-Cβ) (Section 3.10) and thermocellin that contains a thioether cross-linked to the γ-carbon of Thr (Fig. 14A). A β-thioether linkage was also introduced by a homolog of the radical SAM enzyme (NxxcB) that generates the Lys–Trp crosslink in streptide (Section 2.5). The resulting product features a thioether linkage between a former Cys and Asn (Fig. 14B).44
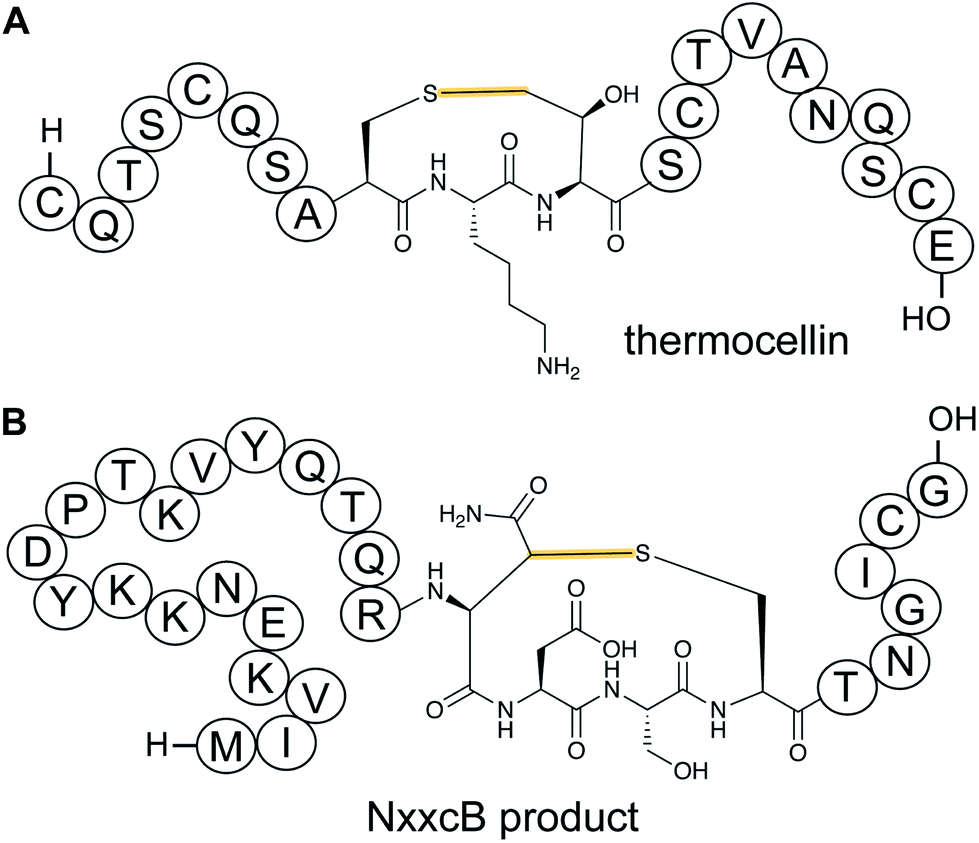 | ||
| Fig. 14 (A) Structure of ranthionine in thermocellin (shown is most likely a biosynthetically immature structure).43 (B) Ranthipeptide featuring a Cys–Asn crosslink.44 H- denotes a free N-terminus and –OH a free carboxylic acid C-terminus. The unifying feature of ranthipeptides is a thioether linkage formed from a Cys thiol to a non-α-carbon of another amino acid (yellow) in a radical-mediated process. | ||
2.14 Ryptides, rotapeptides, and darobactin
Genome mining for additional radical SAM enzymes regulated by quorum sensing in Streptococci resulted in the discovery of enzymes that install different intramolecular linkages than those found in streptide (Section 2.5). Examples are C–C crosslinks between Arg and Tyr in ryptides (Fig. 15A)45 and ether crosslinks between the side chain oxygen of Thr and the α-carbon of Gln in rotapeptides (Fig. 15B).46 Thus, it is clear that crosslink formation by radical SAM enzymes greatly expands the methods available for macrocyclization of RiPPs. Indeed, a recent bioassay-guided discovery effort resulted in the identification of darobactin from the nematode symbiont Photorhabdus khanii HGB1456. Darobactin contains both Lys–Trp and Trp–Trp crosslinks, including an ether linkage between the C7 indole of Trp1 and the β-carbon of Trp3 (Fig. 15C).47 Genome sequencing demonstrated the molecule was a RiPP generated from a 58-amino acid precursor peptide and the BGC contains a gene (darE) encoding a radical SAM enzyme belonging to the SPASM48 subfamily (Fig. 15D). SPASM is an acronym based on the founding members involved in the maturation of subtilosin, pyrroloquinoline quinolone (PQQ), anaerobic sulfatase-maturing enzyme, and mycofactocin. DarE shares only modest sequence similarity to StrB, the enzyme that installs a Trp–Lys C–C crosslink in streptide (Section 3.11).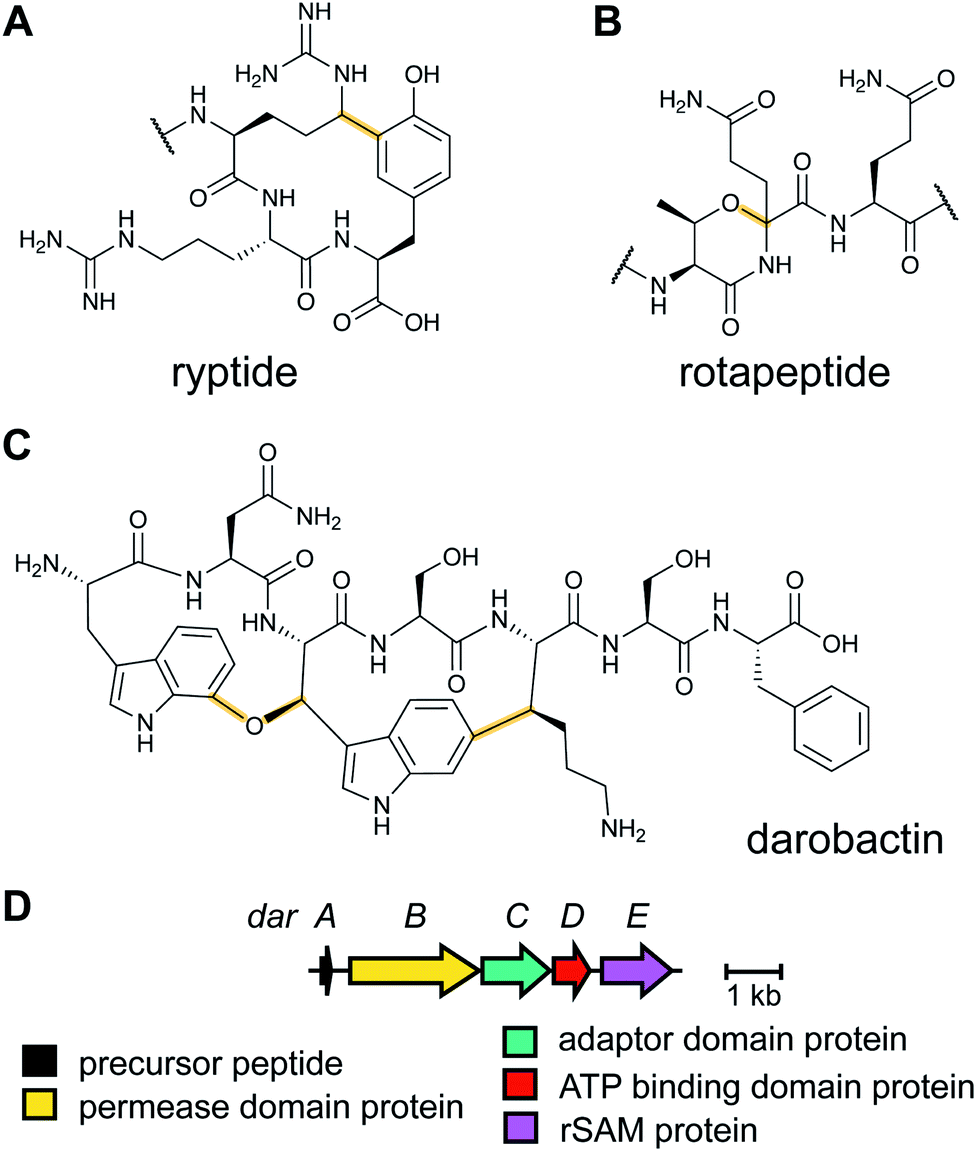 | ||
| Fig. 15 Structures of bacterial peptides that are macrocyclized by radical SAM proteins. (A) Arg–Tyr crosslink in ryptides. (B) Thr–Gln crosslink in rotapeptides. (C) Lys–Trp and Trp–Trp crosslinks in darobactin. (D) Darobactin BGC. | ||
2.15 Pearlins
A final example of a new RiPP family that was initially noted bioinformatically is the pearlins. Class I lanthipeptide dehydratases transfer glutamate from glutamyl-tRNA to the side chain of Ser/Thr in their precursor peptide substrates followed by glutamate elimination (Section 3.1.1).49 Genome mining revealed divergent dehydratase homologs that lacked the glutamate elimination domain. These proteins uniquely add amino acids from amino acyl-tRNA to the C-terminus of a precursor peptide.50 Thus, these enzymes were named peptide aminoacyl-tRNA ligases (PEARLs). The identity of the initially added amino acid can vary. In the currently characterized examples, peptide extension is followed by further post-translational modification of the newly appended amino acid to either generate new structures such as 3-thiaglutamate, or previously known natural products such as the ammosamides (Fig. 16).50,51 To underscore the common biosynthetic logic, the products of these pathways are now termed pearlins. The pearlins do not have a class-defining PTM. Instead they are unified by a common non-ribosomal aminoacyl-tRNA dependent extension of a ribosomal peptide (Section 3.24).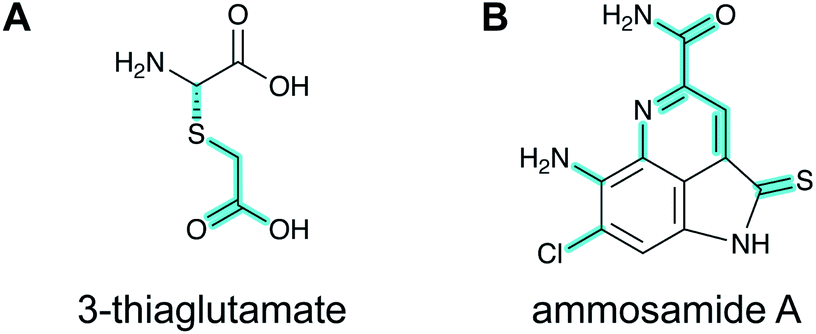 | ||
| Fig. 16 Structures of two pearlins made by post-translational modification of scaffold peptides. The class-defining PTM of pearlins is initial aminoacyl-tRNA-dependent extension of a ribosomal peptide. The appended amino acid is then further modified, and in the last biosynthetic step, the initially generated peptide bond undergoes peptidolysis. Thus, all PTMs in the final structure are secondary modifications (cyan). | ||
2.16 Atropitides
Tryptorubin A (Fig. 17A) was isolated in 2017 from Streptomyces sp. CLI2509, which was in turn isolated from the bracket fungus Hymenochaete rubiginosa.52 The compound contains crosslinks between aromatic side chains resulting in a rigid, polycyclic structure. A recent total synthesis effort showed the compound can exist in two different non-interchangeable forms with identical chemical connectivity, but where the two isomers are not related by the common stereochemical definers: point chirality, E/Z geometry across double bonds, or axial chirality (canonical atropisomerism). The authors suggested the two forms of the molecule be termed non-canonical atropisomers. Importantly, the synthetic efforts demonstrated that only one of these non-canonical atropisomers is produced in nature.53 Although the initial biosynthetic origin was speculated to be a non-ribosomal peptide synthetase (NRPS),52 the more recent study suggests that the compound is a RiPP, and that the crosslinks are likely introduced by oxidative cyclization involving a cytochrome P450 enzyme (Fig. 17B). We suggest the name “atropitides” for any subsequently discovered compounds that are similarly biosynthesized and display non-canonical atropisomerism.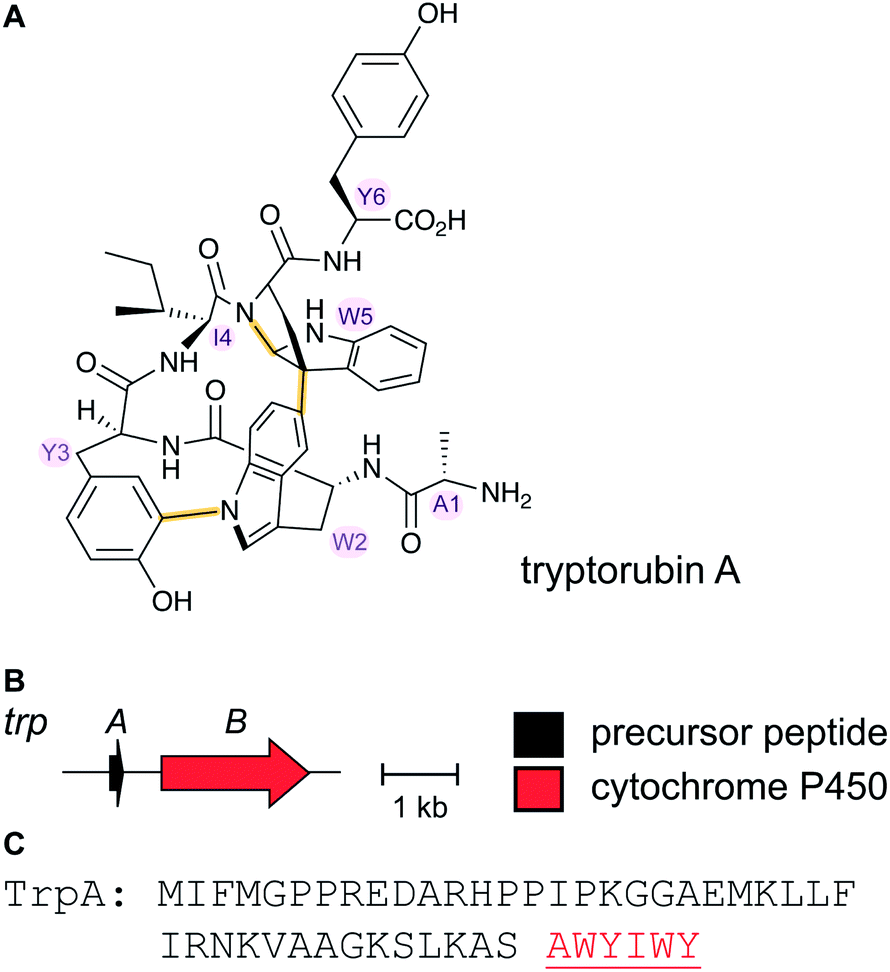 | ||
| Fig. 17 (A) Structure of the natural isomer of tryptorubin A. In this isomer, the Ile4–Trp5 “peptide bridge” lies above the Trp2–Tyr3 crosslink. In the non-natural isomer, this bridge is located below the Trp2–Tyr3 crosslink. For details see ref. 53. Both Trp2–Tyr3 and Tyr3–Trp5 crosslinks are considered class-defining and thus are highlighted in yellow. (B) BGC for tryptorubin A and (C) the sequence of the precursor peptide with the core peptide in red. | ||
2.17 Lanthidins, class V lanthipeptides
The antibiotic cacaoidin (Fig. 18A) is the first reported member of the lanthidins, a new RiPP subfamily with structural elements found in lanthipeptides and linaridins.54 Cacaoidin was isolated from a strain of Streptomyces cacaoi and bears an unprecedented N,N-dimethyl lanthionine (NMe2Lan) that is not found in known lanthipeptides. N-terminal bis-N-methylation has been reported for linaridins (e.g.Fig. 27B) and LAPs (Section 5.3.5), but these RiPP families lack lanthionines. The molecule also combines other unusual structural features, such as O-glycosylation of Tyr with a 6-deoxygulopyranosyl-(rhamnopyranose) disaccharide and several D-amino acids including D-2-aminobutyric acid (Abu) (Fig. 18A). The cacaoidin BGC could not be identified by any prediction software tool and was mapped in the region adjacent to the core peptide structural gene (Fig. 18B). The cluster shows low homology with those of other lanthipeptides or linaridins and suggests an alternative RiPP biosynthetic pathway. Since linaridins are characterized by dehydrobutyrine (Dhb) residues (see Section 3.3), which are not present in cacaoidin, but lanthipeptides are characterized by lanthionine, which is present in cacaoidin, we propose that lanthidins are class V lanthipeptides that are made via a biosynthetically distinct pathway since the BGC does not contain genes for class I–IV lanthionine synthases (Section 3.1).55 Very recent genome mining efforts identified additional class V lanthipeptides, which are referenced here, but not further discussed as they appeared after finalization of this review.56,57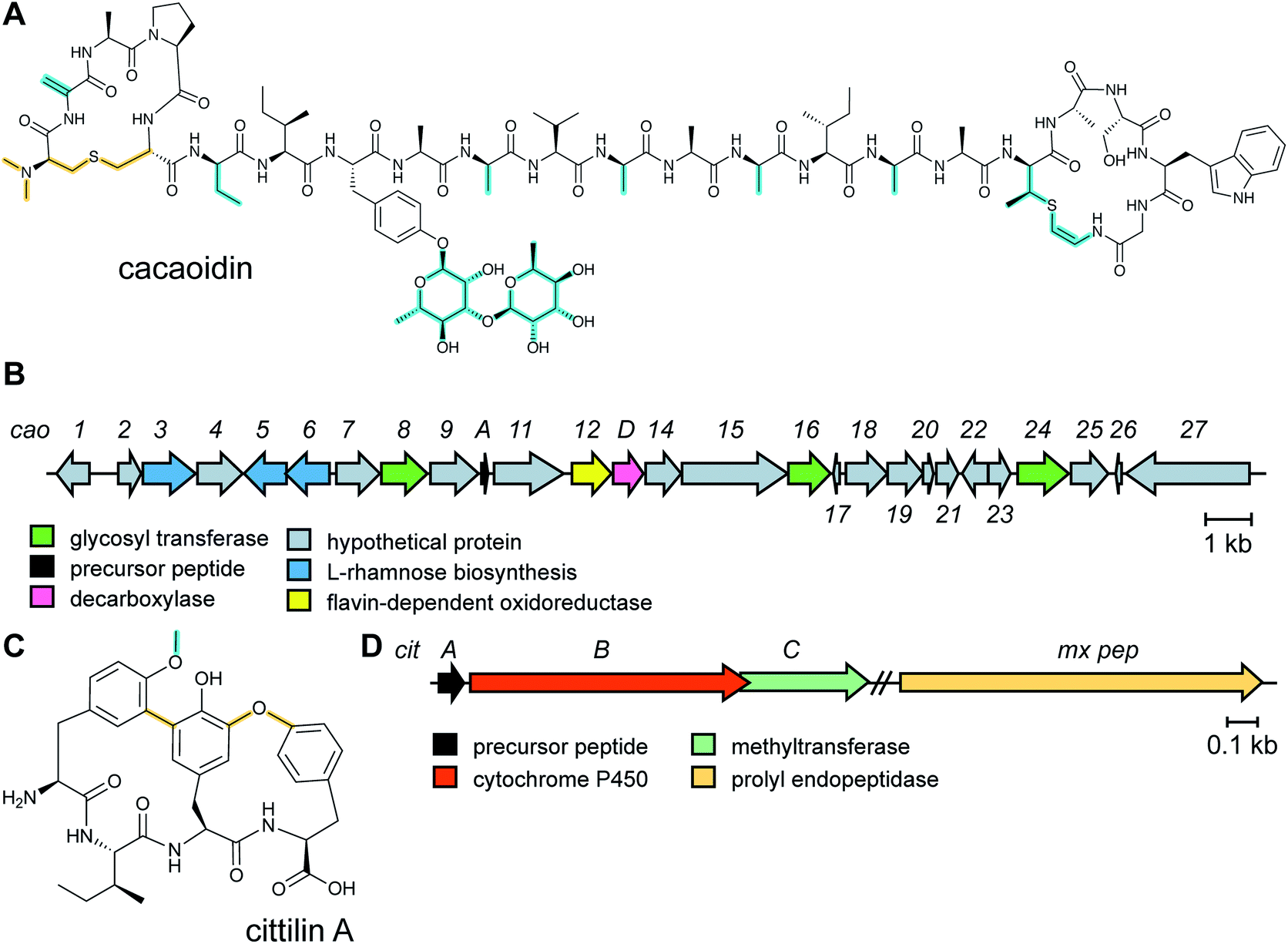 | ||
| Fig. 18 (A) Structure of the class V lanthipeptide cacaoidin. The stereochemistry of the lanthionine is proposed. The class-defining PTMs are yellow. Secondary PTMs are cyan. At present the dimethylated N-terminal lanthionine is present in all known members and is colored yellow. It may be that over time this PTM may turn out not to be class -defining. (B) BGC for cacaoidin biosynthesis. (C) Structure of cittilin A. (D) BGC for cittilin A. | ||
2.18 Cittilins
The cittilins were first reported after submission of this review and will not be discussed in detail. Produced by Myxococcus xanthus, the cittilins are bicyclic tetrapeptides with biaryl and aryl–oxygen–aryl ether crosslinks (Fig. 18C).58 They are made from a 27-amino acid precursor peptide encoded in a BGC that also contains a cytochrome P450 (CitB) and a methyltransferase (CitC). A prolyl endopeptidase that removes the leader peptide is encoded outside of the BGC.3 Advances in the understanding of biosynthetic enzymes that install primary post-translational modifications
As noted in Table 1, RiPP families are typically defined by characteristic PTMs called primary modifications. Secondary, compound-specific modifications are introduced by tailoring processes, which complete the biosynthetic pathway. In the past six years, our understanding of the enzymes that catalyze the primary modifications has been greatly improved for previously known RiPPs. Furthermore, many of the enzymes that produce the newly discovered RiPP families discussed in Section 2 have been investigated and will also be discussed in the following section.3.1 Lanthipeptides: introduction and biosynthesis
Lanthipeptides contain the β-thioether crosslinked bis amino acids lanthionine (Lan) and methyllanthionine (MeLan, Fig. 19A). These structures are formed from initial dehydration of Ser and Thr residues to the corresponding dehydro amino acids dehydroalanine (Dha) and dehydrobutyrine (Dhb), respectively (Fig. 19A). Subsequent conjugate addition of Cys thiols to these dehydroamino acids results in Lan and MeLan. Prior to 2013, the stereochemistry of the Michael-type addition was believed to always result in D-(Me)Lan in which the α-stereocenter of the former Ser/Thr residue had D-stereochemistry, but in the intervening years it has become clear that the stereoselective protonation of the enolate intermediate can take place from either the Re or Si face. Intriguingly, protonation of the enolate is not confined to a single face in a particular peptide, because both DL and LL stereochemistry can result within the same core peptide.59–61 Recent studies have also shown that lanthipeptides are encoded in a much wider range of organisms than previously anticipated including archaea and bacteroidetes.62–64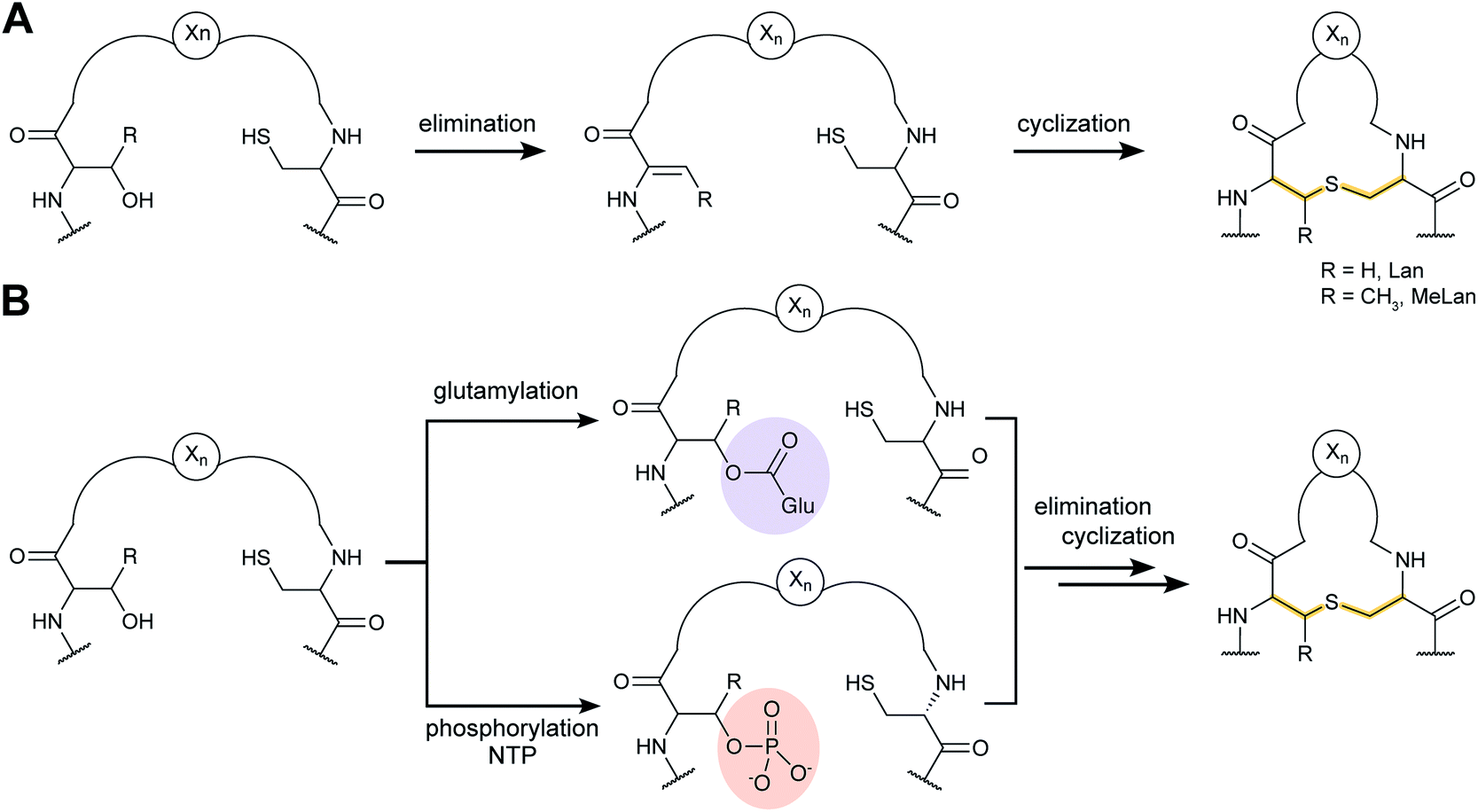 | ||
| Fig. 19 (A) Post-translational modifications resulting in lanthionine and methyllanthionines from Ser/Thr and Cys. (B) Two different mechanisms for dehydration utilizing either Glu-tRNA or NTP for activation of the Ser/Thr side chain hydroxyl groups prior to elimination of Glu/phosphate. | ||
Currently five classes of lanthipeptides have been reported that differ in the biosynthetic enzymes that carry out (Me)Lan formation. New knowledge reported on these enzymes is briefly discussed in the following sections for class I–IV. The recent discovery of the lanthidins (Section 2.17) revealed the latest independent pathway to lanthionines (class V),54,56,57 which at present is not yet well understood.
3.1.1.1 Class-defining enzyme: aminoacyl-tRNA dependent dehydratase. In vitro reconstitution of the biosynthesis of class II–IV lanthipeptides had been reported prior to 2013,1 but reconstitution of class I biosynthesis had proven recalcitrant. The explanation for this difficulty came when it was revealed that dehydration by the LanB dehydratases (InterPro families IPR006827 and IPR023809) during class I lanthipeptide biosynthesis requires glutamyl-tRNA, a co-substrate that was not anticipated (Fig. 19B).49 The discovery of the requirement of glutamyl-tRNA had been foreshadowed by targeted mutagenesis of the NisB enzyme, the LanB involved in nisin biosynthesis, which resulted in glutamylation of Ser and Thr residues in the precursor peptide, NisA, during heterologous expression in Escherichia coli.65 Reconstitution of the dehydration activity in vitro required the macromolecular fraction of E. coli cell extract, and the activity was lost upon treatment with RNAse. Subsequent in vitro biochemical studies confirmed the requirement of glutamyl-tRNA and demonstrated the need for glutamyl-tRNA synthetase and illustrated that NisB does not amino acylate uncharged tRNA. The discovery that the LanB dehydratases use glutamyl-tRNA also opened up new opportunities to study the biosynthesis of other RiPPs that use homologous enzymes as discussed in Section 3.2.
A crystal structure of the dehydratase NisB bound to the LP of its substrate NisA (Protein Databank, PDB: 4WD9) demonstrated the enzyme contained three domains (Fig. 20A).49 One domain (InterPro family IPR006827) catalyzes the transfer of glutamate from the tRNA to the peptide substrate. Then, the glutamylated peptide is translocated to another domain (InterPro family IPR023809) in which the glutamate is eliminated to form Dha/Dhb. A ping-pong type mechanism in which the glutamyl group is first transferred from the glutamyl-tRNA to the enzyme prior to transfer of the glutamyl moiety to the Ser/Thr in NisA was ruled out by mutagenesis. The LP-binding domain in NisB49 (termed the RiPP precursor Recognition Element,66 RRE) will be discussed further in Section 4.1.1. Structure elucidation with a non-reactive analog of glutamylated NisA (PDB: 6M7Y) identified the elimination active site of NisB (Fig. 20B), while the glutamyl transfer active site is inferred based on a structure of a biosynthetic enzyme that carries out Glu-tRNA dependent glutamylation of Ser residues during thiopeptide biosynthesis (see Section 3.2).67 Covalent attachment of the LP to the RRE demonstrated that catalysis can take place with the LP occupying only a single binding site ruling out models in which the LP needs to move during catalysis.68
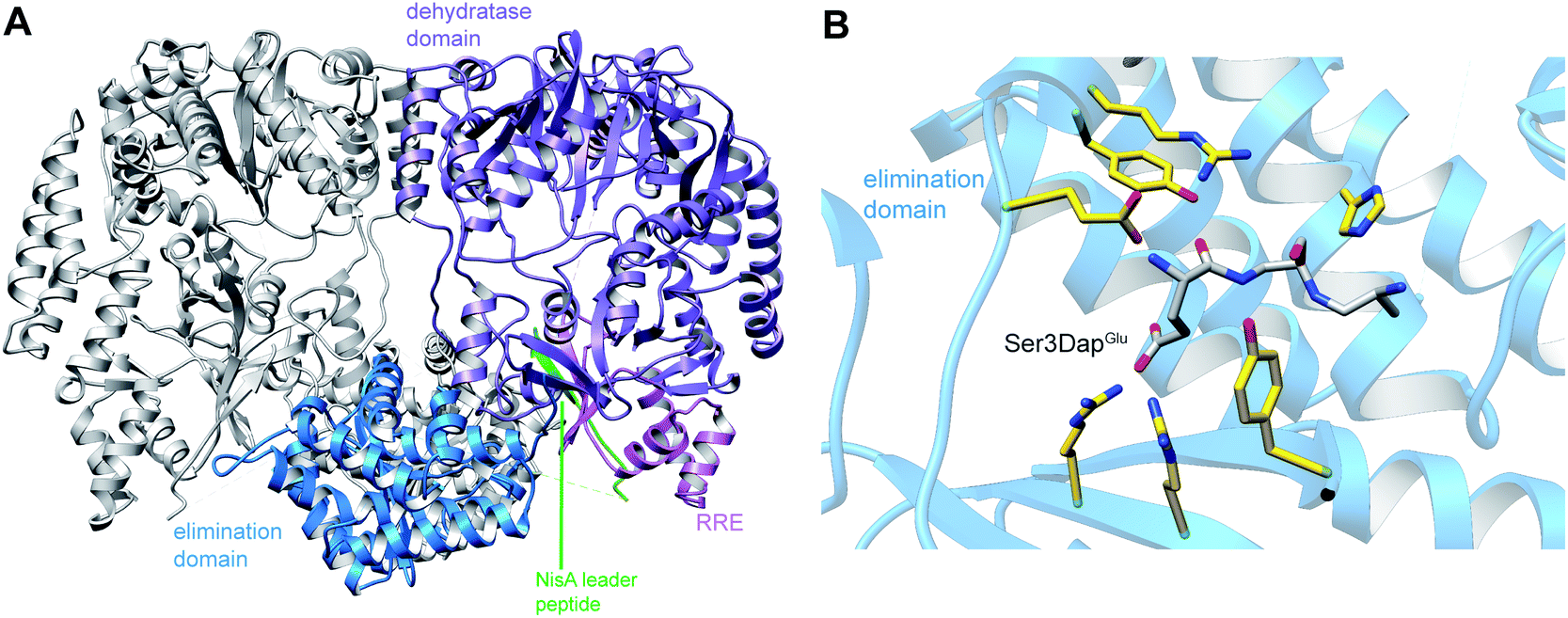 | ||
| Fig. 20 (A) Crystal structure of the NisB homodimer with the NisA LP bound (PDB: 4WD9).49 Only one monomer is colored for clarity. (B) Glu elimination active site with a non-reactive amide-linked Glu bound (PDB: 6M7Y).67 Two Arg residues recognize the Glu side chain of the glutamylated substrate peptide. | ||
Studies with LanB enzymes from different bacterial phyla have shown that the nucleotide sequence in the acceptor stem of the tRNAGlu is critical for the enzyme to accept the cognate glutamyl-tRNA as substrate,69,70 an observation that explains difficulties in heterologous expression of many class I lanthipeptides. Some RiPP BGCs, such as those of the antifungal pinensins63 or the thiopeptides,71 contain split LanB dehydratases (see Section 3.2) in which glutamylation and elimination are carried out by separate polypeptides.
Compared to advances in understanding the mechanism of dehydration and LP recognition, the mechanism of cyclization in class I lanthipeptides by LanC enzymes (InterPro family: IPR033889) remains to be investigated in detail. Cyclization has been shown to be reversible for both class I and class II systems,72 but the mechanism of catalysis is not well understood. Several studies reported prior to 2013 suggested that complex formation involving the dehydratase LanB, the stand-alone cyclase LanC, the LanA substrate and the LanT transporter is important for the overall PTM process.1 Both LanB (Section 4.1.1)49 and LanC73 are believed to engage the same region of the NisA LP. The stoichiometry in these solution complexes has been suggested to involve one substrate per NisB dimer,74 unlike the co-crystal structure of NisB with NisA where the stoichiometry is 1 NisB dimer: 2 NisA peptides. Thus, complex formation appears to result in a rearrangement that is currently not yet understood at the molecular level.
3.1.2.1 Class-defining enzyme: ATP dependent LanM synthetase. Class II lanthipeptides are biosynthesized by a bifunctional lanthipeptide synthetase (LanM, InterPro family: IPR017146). Activation of the Ser/Thr side chains for dehydration in class II lanthipeptides is achieved by phosphorylation followed by phosphate elimination (Fig. 19B). The cyclization step is catalyzed by a domain that is homologous to the LanC cyclase enzymes in class I lanthipeptide biosynthesis. The first X-ray structure of a bifunctional LanM enzyme was reported for CylM,75 the synthetase that makes both the small and large subunits of the virulence factor, enterococcal cytolysin, produced by Enterococcus faecalis.59 Surprisingly, the crystal structure showed that the dehydratase domain is structurally similar to mammalian lipid kinases (Fig. 21A). A distinct domain termed the kinase activation (KA) domain holds the activation loop in an active conformation and provides two residues that previously had been shown to be critical for phosphate elimination (Fig. 21B). The structure also explains why ADP must be present for phosphate elimination as phosphorylation and phosphate elimination occur in the same active site (Fig. 21B). No LP binding site could be identified in the structure, but recent hydrogen–deuterium exchange mass spectrometry data suggest that the LP may bind at a site made up of loops connecting the capping helices (see Section 4.1.3).76,77
 | ||
| Fig. 21 (A) Crystal structure of the class II lanthipeptide synthetase CylM (PDB: 5DZT).75 The dehydration domain architecture is similar to that of eukaryotic lipid kinases. The kinase activation domain (KA) holds the activation loop in a defined conformation. (B) Phosphorylation active site showing typical features of protein kinases as well as the two residues from the KA domain (Thr and Arg) that are involved in β-elimination of the phosphate group to form the dehydro amino acids. | ||
Detailed kinetic studies on the process of multiple dehydrations and cyclizations that turn a linear peptide into a polycyclic product have suggested that for enzymes that make a single natural product, substrate and enzyme have co-evolved to result in a well-defined order of catalytic events. The cyclizations become increasingly facile as the compound nears its final cyclization state.78–80 In contrast, enzymes that have multiple substrates with diverse core peptides are much less efficient in general and the cyclizations become slower as the peptide matures.78 Thus, a model has been proposed in which highly substrate tolerant enzymes with multiple physiological substrates are inherently slower as they have not co-evolved with one substrate to achieve efficient catalysis. In turn, this allows these substrate tolerant enzymes to generate many different products. The manner by which a single cyclization active site can convert highly divergent substrates into defined polycyclic topologies has been hypothesized to be determined by the sequence of the precursor peptides and not the enzyme.78,81,82 This model is also supported by studies that show that for some lanthipeptides the stereochemistry of cyclization is determined by the peptide substrate sequence and not the enzyme.59,60
3.1.3.1 Class-defining enzyme: NTP-dependent trifunctional LanKC synthetase. Class III lanthipeptides are generated by trifunctional enzymes (LanKC) that contain lyase, kinase (InterPro family: IPR000719), and cyclase domains (InterPro family: IPR007822). The lyase and kinase domains have clear sequence similarity with the corresponding domains in class IV lanthipeptide synthetases (Section 3.1.4), but the cyclase domain is unique amongst lanthipeptide cyclization enzymes, as it lacks the zinc binding residues. Another unusual aspect of class III lanthipeptides is that the cyclization domains form lanthionine or labionin (Lab) or both. Labionins are products of tandem conjugate additions (Fig. 22A), and the factors that determine Lan or Lab formation are not yet understood. Detailed investigation of the CurKC enzyme involved in curvopeptin biosynthesis demonstrated a C-to-N terminal directionality of processing.83
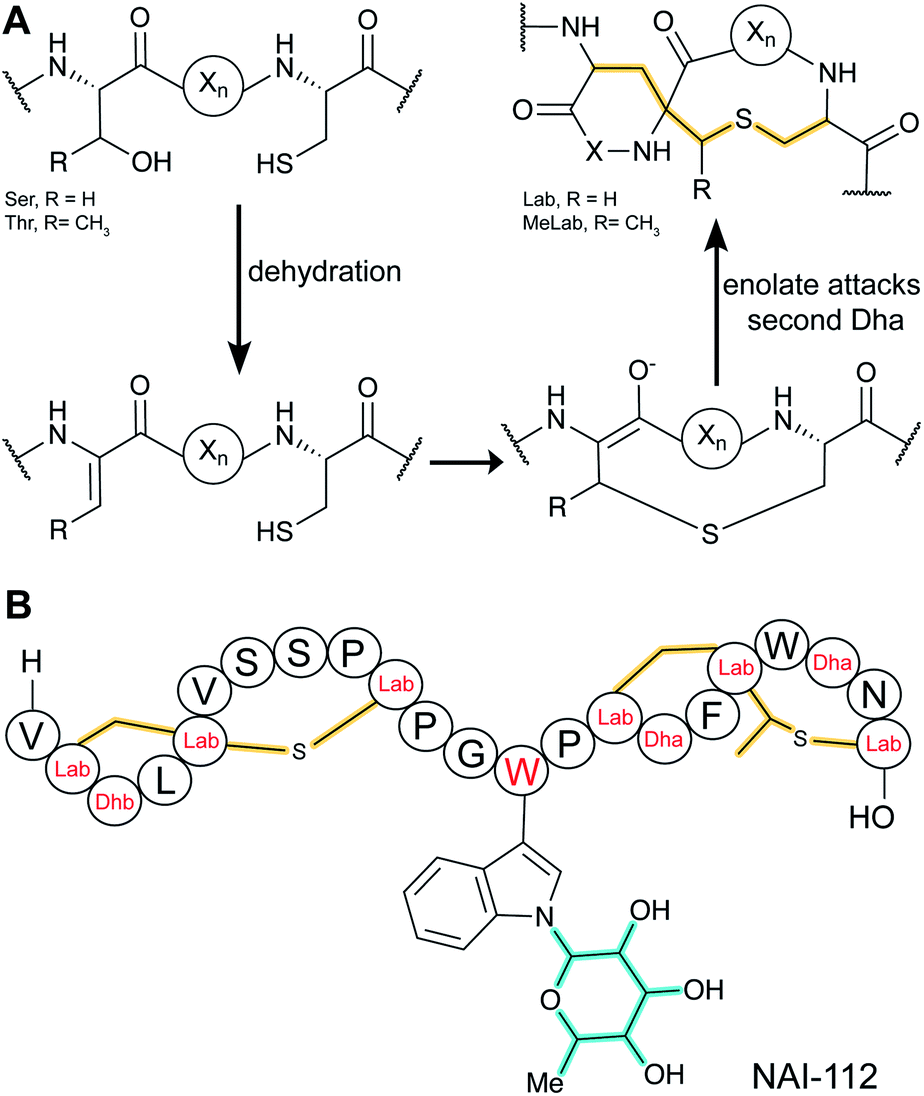 | ||
| Fig. 22 (A) Formation of labionin and methyllabionin crosslinks highlighted in yellow. (B) Structure of class III lanthipeptide NAI-112 featuring a MeLab. Also unique in RiPPs is the N-glycosylation of the Trp indole nitrogen, a secondary modification (cyan). H- denotes a free N-terminus and –OH denotes a free carboxyl C-terminus. | ||
Whereas before 2013 only Lab structures had been reported, in 2014 the first example of a methyllabionin (MeLab) was found in NAI-112, which also is unusual in that it is glycosylated on Trp (Fig. 22B).84 At present the stereochemistry at the additional chiral center of MeLab is unknown. The BGC of NAI-112 was identified and contains a gene for a glycosyltransferase. An interesting variant of class III lanthipeptides was discovered in which the N-terminus of a Lab-containing peptide was adorned with a fatty acid (see Section 3.1.5).34 This class of compounds has been termed lipolanthines.
3.1.4.1 Class-defining enzyme: NTP dependent trifunctional LanL synthetase. Like class III lanthipeptides, class IV lanthipeptides are generated by trifunctional enzymes called LanL that contain lyase, kinase, and cyclase domains (same InterPro families as LanKC: IPR000719 + IPR007822). At present only venezuelin and its congeners have been characterized, although genome sequences and synthetic biology studies suggest that other ring topologies can also be made by LanL-containing gene clusters.62,85 Recent biochemical experiments showed that the LP of the precursor peptide binds to the kinase domain and that the dehydration process for venezuelin-like compounds occurs in an ordered and directional manner, from N-to-C terminus.86,87
3.1.5.1 Class-defining enzymes: combination of PKS/FAS and class III lanthipeptide synthetases. During an antibiotic screening study, microvionin with activity against Staphylococcus aureus was discovered in extracts of a Microbacterium arborescens culture.34 Structure elucidation revealed a peptide with two new features comprising an AviCys-Lab moiety named avionin and an N-terminal N,N′-bismethylated guanidine fatty acid (Fig. 11). The family was termed “lipolanthine” because of its resemblance to non-ribosomal lipopeptides. The BGC pinpointed by searching for the core sequence of microvionin, encodes class III lanthipeptide machinery as well as ten additional genes that were proposed to generate and attach the guanidino lipid moiety (Fig. 23A). These include genes encoding a putative 3-oxoacyl-ACP synthase (MicF) and an acyl carrier protein (MicACP), suggesting a hybrid fatty acid synthase-RiPP pathway. Avionin formation was studied in vitro showing that for sulfur bridge formation to occur, MicD, which catalyzes an oxidative decarboxylation of the C-terminal Cys residue, must be present. Bioinformatic analyses identified in actinomycete genomes five additional BGCs with gene sets related to that of the microvionin cluster. However, these actinomycete BGCs contain a gene encoding a putative type I PKS gene with an NRPS loading module instead of fatty acid synthase genes, suggesting two independent pathways to the guanidinylated moiety or a related structure. This hypothesis was confirmed by isolating the peptide product from Nocardia terpenica, named nocavionin. Thus, the RiPP segment of the lipolanthines are formed by previously reported features of lanthipeptide machinery: C-terminal decarboxylation by a LanD enzyme previously found in class I and II lanthipeptides (InterPro family: IPR003382; Section 5.9), and dehydration and cyclization by a class III lanthipeptide synthetase to arrive at the novel avionin structure (Fig. 23B). A follow-up study revealed 80 additional BGCs that were divided into four subtypes depending on the type of PKS or NRPS and on the presence or absence of a LanD.88 LanKC enzymes encoded in BGCs that also encode a LanD were shown to be dependent on the LanD for labionin synthesis suggesting enzyme complex formation and/or allosteric regulation.88 Biochemical insights into how the linkage of the FAS/PKS/NRPS product to the RiPP segment is formed have not yet been reported.
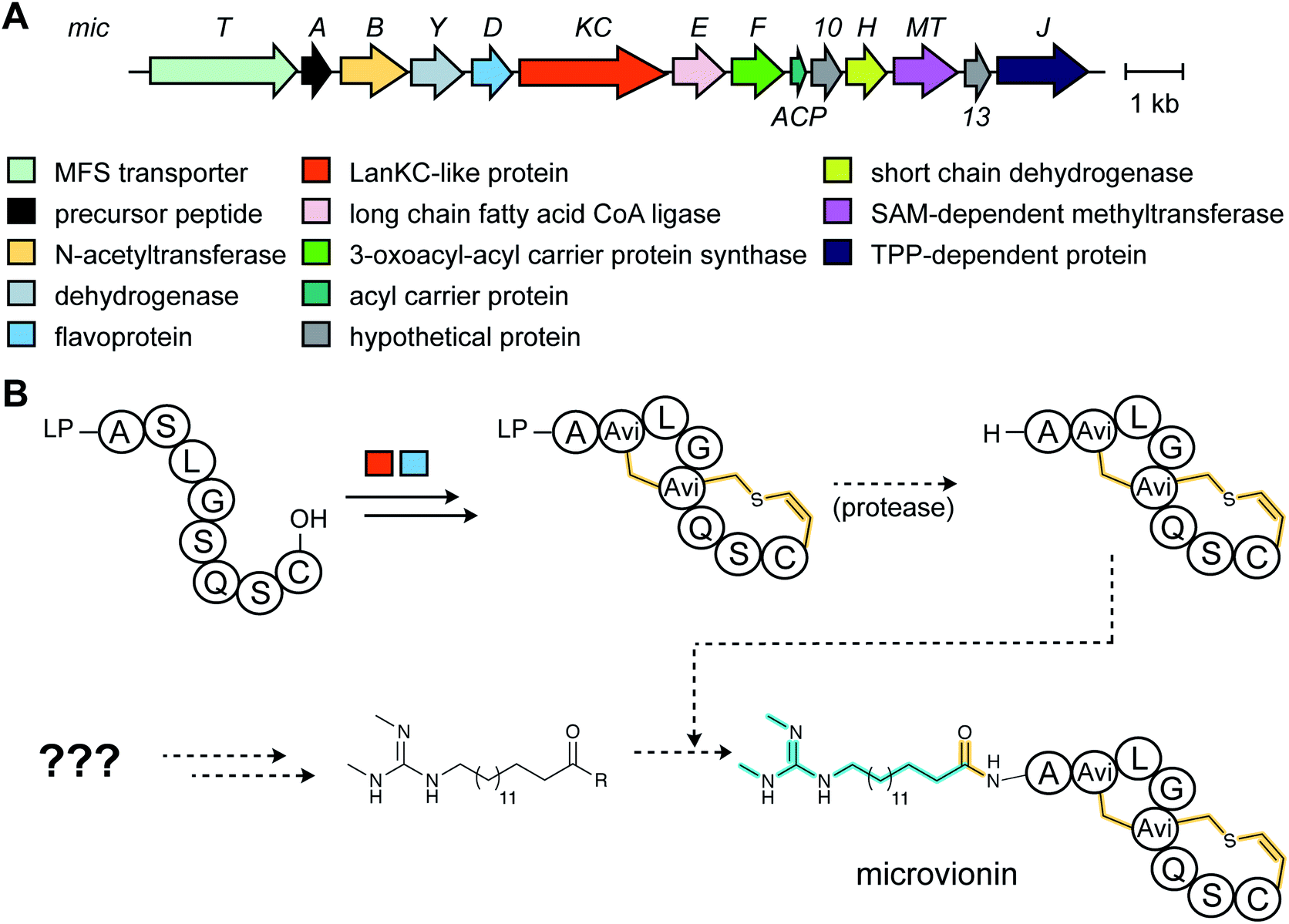 | ||
| Fig. 23 (A) BGC of microvionin. MFS, major facilitator superfamily; TPP, thiamine pyrophosphate. (B) Proposed biosynthetic pathway to microvionin. A ribosomal peptide is post-translationally modified by decarboxylation of the C-terminal Cys and subsequent cyclization (Fig. 22A) to generate avionin. After LP removal, a fatty acid is attached to the liberated N-terminus. Class-defining PTMs are yellow while secondary PTMs that can vary in structure are cyan. | ||
3.2 Thiopeptides: introduction and biosynthetic pathway
Although thiopeptide biosynthesis had been studied at the genetic level,1 in 2013 none of the core biosynthetic steps had been reconstituted in vitro. At a minimum, thiopeptide biosynthesis requires installation of thiazoles, dehydroamino acids, and cycloaddition of two dehydroamino acids. Biochemical studies on the biosynthesis of thiomuracin demonstrated a strict order of events in which the combined action of a cyclodehydratase (TbtG, YcaO InterPro family: IPR003776) and dehydrogenase (TbtE; InterPro family: IPR020051) (see Section 3.4) with a designated LP-binding protein (TbtF) (see Section 4.1.1) results in the introduction of six thiazoles in an ordered, but non-directional process (Fig. 24).71,89 Only after installation of these thiazoles does the dehydratase act on the peptide. For micrococcin P1 biosynthesis, genetic studies suggest that prior to dehydratase activity, one additional PTM has to take place, the oxidative decarboxylation of a C-terminal Thr (Section 5.9).90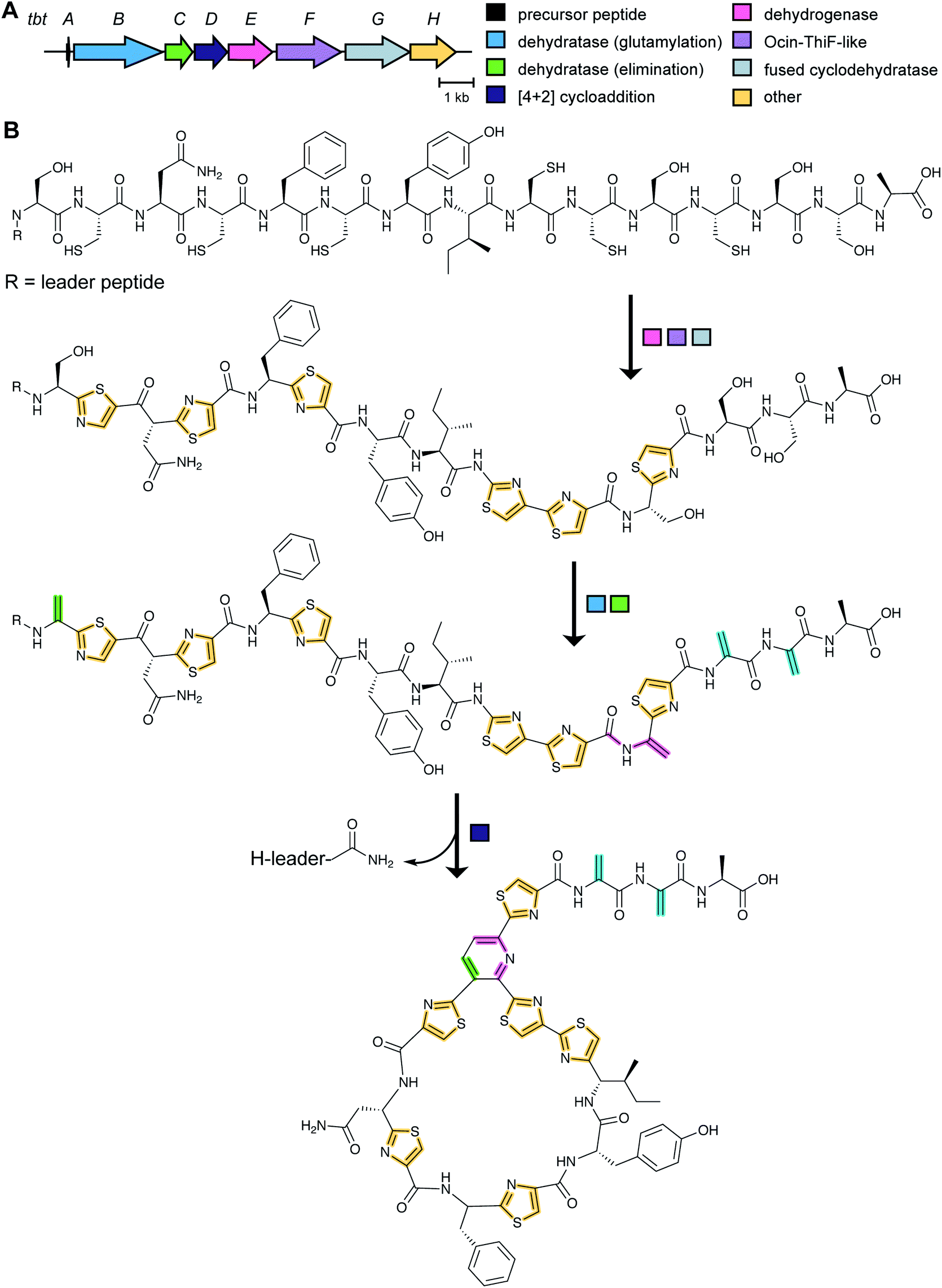 | ||
| Fig. 24 (A) BGC of the thiopeptide thiomuracin. (B) Biosynthetic pathway to the core structure of thiomuracin showing the obligatory order of thiazole formation, dehydration, and cyclization. The LP is eliminated by TbtD as a C-terminal carboxamide. The thiazoles (yellow) and the 6-membered nitrogen-containing heterocycle (green/purple) are the class-defining PTMs for thiopeptides. The pyridine in the final structure is color-coded to illustrate the origin of the atoms that come from two dehydroalanines of the preceding intermediate. | ||
Thiopeptide BGCs were known to contain a split dehydratase reminiscent of LanB lanthipeptide dehydratases. When the structure of the latter was reported (see Section 3.1.1), it became clear that the split genes in thiopeptide BGCs encode separate enzymes for Ser glutamylation and elimination to form Dha. The glutamylation enzyme TbtB from thiomuracin biosynthesis (InterPro family: IPR006827) required the use of tRNAGlu from the producing organism Thermobispora bispora, as Glu-tRNA from E. coli was not accepted.71 TbtC (InterPro family: IPR023809) was then demonstrated to eliminate the glutamate to introduce four Dha residues in an ordered and directional process.89 Mutagenesis studies and co-crystallization with a non-reactive analog of Glu-tRNA identified the glutamyl transfer active site of TbtB (Fig. 25).67 Although TbtB contains a LP-binding motif, catalysis is not LP-dependent. Instead, TbtB specifically binds to the hexathiazole-containing core peptide.71,89 The functional origin for using a split dehydratase rather than the fused version found in lanthipeptide biosynthesis is not presently clear. Split dehydratases are not limited to thiopeptides, as they are also involved in the biosynthesis of the LAP goadsporin91 (Fig. 97) and the pinensin family of class I lanthipeptides.63
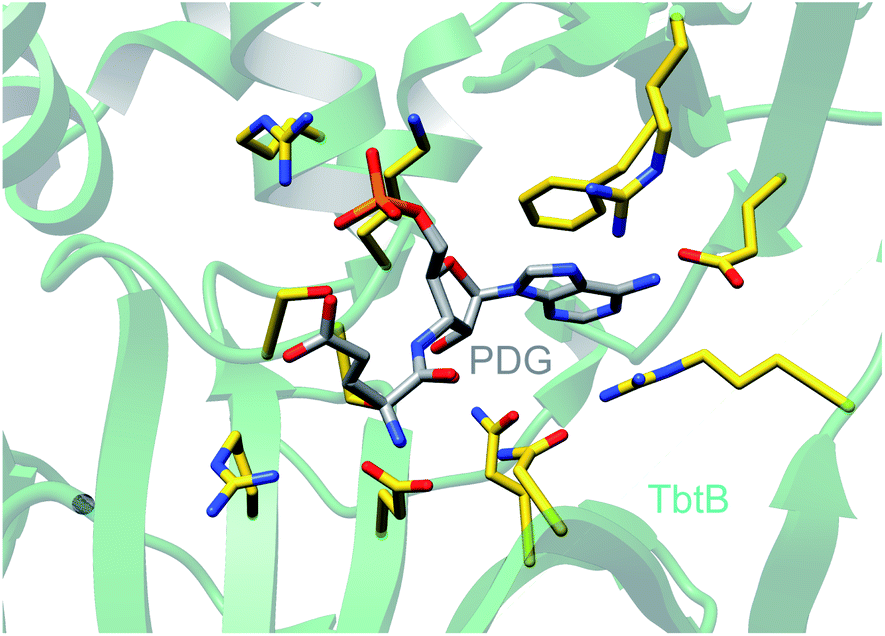 | ||
| Fig. 25 Crystal structure of the glutamylation enzyme TbtB containing 5′-phosphoryl-desmethylglutamycin (PDG), an analog of AMP with an amide-linked glutamate attached to the 3′-position of the ribose (PDB: 6EC8).67 | ||
As noted above, a minimalistic thiopeptide biosynthetic pathway requires an RRE, a cyclodehydratase (YcaO), a dehydrogenase, a dehydratase and a [4 + 2] cycloaddition enzyme. Many thiopeptide BGCs encode multiple copies of one or more of these canonical proteins, sometimes as various fusions of these proteins. Insights into the function of these at first glance redundant proteins was provided for the biosynthesis of sulfomycin produced by Streptomyces viridochromogenes.92 Its biosynthetic gene cluster encodes two YcaO-domain containing proteins SulC and SulD. Heterologous expression in E. coli showed that SulC in complex with the didomain protein SulB (containing RRE and ocin-ThiF domains) converts Cys2 and Thr9 in the precursor peptide to the corresponding thiazoline and methyloxazoline, respectively.92 These azolines are oxidized to the corresponding azoles by a SulEFG complex that has a non-canonical make-up of fusion proteins (containing RRE, flavin-dependent dehydrogenase, and E1-like adenylase, see Section 3.4).92 Subsequently, SulD in close cooperation with SulEFG converts Thr5, Cys7, and Ser12 to the corresponding azoles. The authors show that a complex is formed by SulDEFG,92 something that is likely a staple of many RiPP biosynthetic pathways.93 The study of the sulfomycin biosynthetic enzymes illustrates that many different protein combinations may have evolved in thiopeptide biosynthetic pathways to achieve the same overall transformation as the more simple BGCs.
A crystal structure of PbtD (an ortholog of TbtD involved in GE2270A biosynthesis) with a product analog bound (PDB: 5W99) provides some insight into the overall cyclization process,97 but individual amino acids that catalyze the various steps involved in the [4 + 2] cycloaddition reaction and subsequent water and LP elimination have not yet been identified. The structure of TbtD did show that, although the N-terminal amino acids of the LP are not required for catalysis, they do clearly bind to the LP-binding site (Fig. 26).89 The reaction catalyzed by TbtD is believed to involve first tautomerization of one Dha to the corresponding iminol, followed by the actual [4 + 2]-cycloaddition, elimination of water from the hemiaminal, and finally elimination of the leader peptide to form the pyridine (Fig. 26C). The first support of this order of events was obtained with a synthetic substrate analog in which several thiazoles were substituted by oxazoles resulting in a slow substrate. With this analog, the hemiaminal intermediate appears to build up as indicated by MS data. Acidic hydrolysis of the intermediate allowed spectroscopic characterization of the resulting diketone (Fig. 26C).98 Thiopeptides undergo a plethora of compound-specific secondary PTMs, some of which have been investigated since 2013 and will be discussed in Section 5.
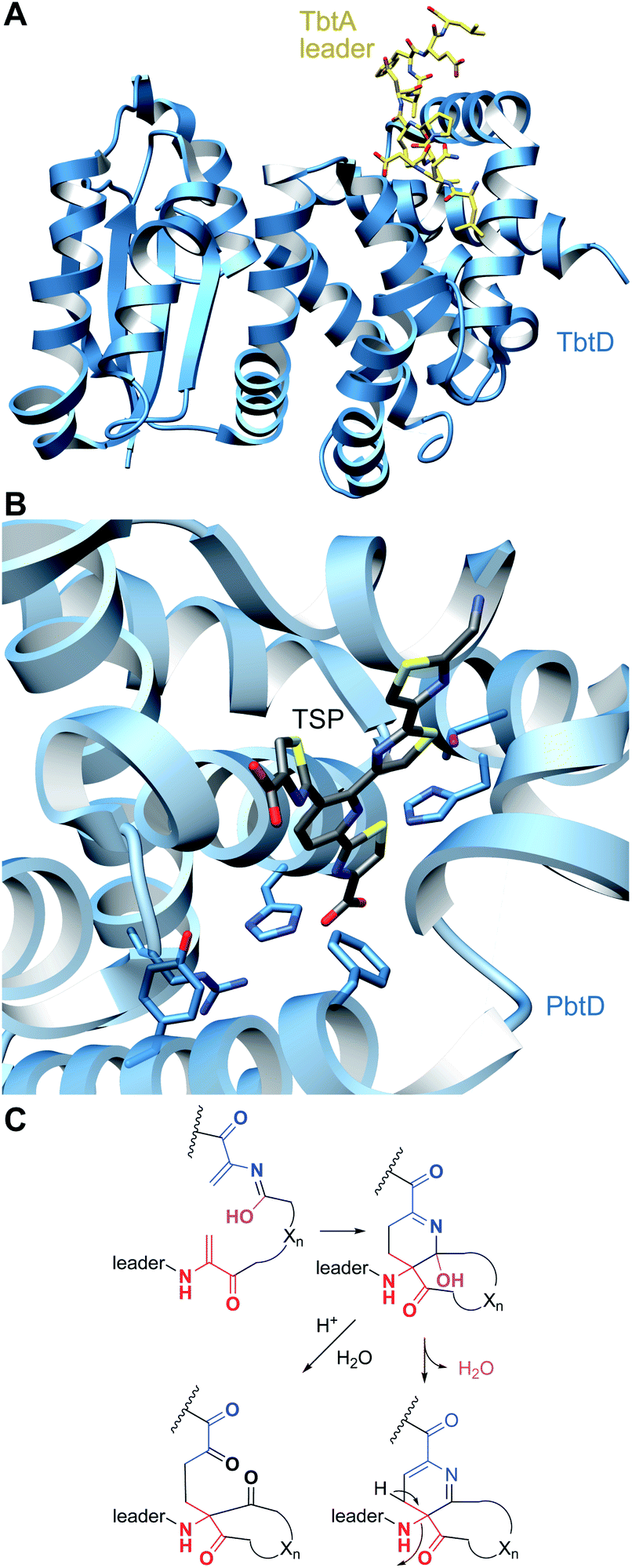 | ||
| Fig. 26 (A) Crystal structure of TbtD, the enzyme that catalyzes the [4 + 2]-cycloaddition in thiomuracin biosynthesis, bound to the LP of its substrate (PDB: 5WA4).97 (B) A tri-substituted pyridine (TSP) product analog bound in the active site of the orthologous enzyme from the GE2270A biosynthetic pathway (PDB: 5W99). (C) Proposed mechanism of pyridine formation by initial [4 + 2]-cycloaddition to generate a hemiaminal intermediate. Subsequent dehydration and LP elimination yield the pyridine product. The hemiaminal has been intercepted with a slow substrate and was hydrolyzed to the diketone.98 | ||
3.3 Linaridin biosynthesis
Linaridins (linear arid peptides) constitute a moderately sized RiPP class characterized by the presence of dehydrobutyrine (Dhb) on linear, ribosomal peptides (Fig. 27).99 Initially classified as lanthipeptides, owing to the presence of Dhb and AviCys moieties, the absence of lanthipeptide-like dehydratases and the distinct route predicted to form AviCys established linaridins as a separate class of RiPPs.100 Thus far, five linaridins, namely cypemycin, grisemycin, legonaridin, mononaridin, and salinipeptin have been characterized, which exhibit features such as Dhb, N-terminal dimethylation (and N-oxidation), AviCys, L-allo-isoleucine, D-amino acids, and dimethylimidazolidin-4-one.100–104 The only PTM common to all characterized linaridins is the dehydration of Thr to Dhb, which represents the class-defining modification for linaridins. Being a relatively underexplored class of RiPPs, the Dhb-installing enzyme(s) remain unknown, although the most likely candidates are homologs of CypH or the related split LegH/LegE protein. Mutagenesis studies performed on the cypemycin precursor CypA in a heterologous host revealed that the putative dehydratase is capable of dehydrating both Ser and Thr, although Thr is processed more efficiently.105 The dehydratase activity was shown to be site-specific, as substituting a highly conserved Ser to Thr (S16T) gave no product. The biosynthetic enzymes tolerate changes made to increase the solubility of cypemycin, revealing their promiscuous nature that could potentially be capitalized upon for bioengineering efforts.105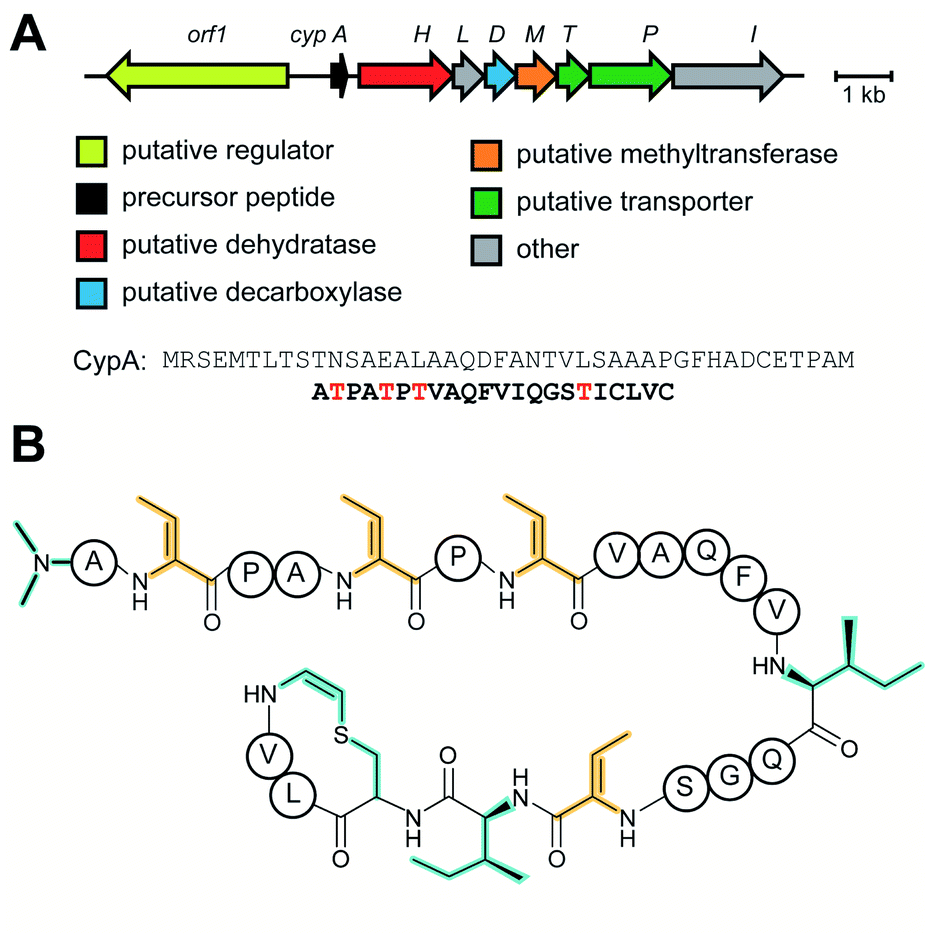 | ||
| Fig. 27 Linaridin BGC and structure. (A) BGC and precursor peptide sequence of the linaridin cypemycin. The core peptide is in bold with sites of dehydration in red. (B) Structure of cypemycin. Unlike other RiPPs for which the C-terminal AviCys ring is generated from Cys and Ser, for cypemycin this structure is formed from two Cys residues by a process that is currently unresolved. Class-defining PTMs are yellow and secondary PTMs are cyan. The stereochemistry at the α-carbon of the AviCys macrocycle has not been definitively established. | ||
3.4 Linear azol(in)e-containing peptides (LAPs): introduction and biosynthesis
![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif) inear
inear ![[a with combining low line]](https://www.rsc.org/images/entities/char_0061_0332.gif) zol(in)e-containing
zol(in)e-containing ![[p with combining low line]](https://www.rsc.org/images/entities/char_0070_0332.gif) eptides (LAPs) comprise a class of RiPP characterized by the presence of thiazol(in)e and/or (methyl)oxazol(in)e heterocycles, resulting from the backbone cyclodehydration of Cys, Ser, and Thr residues (Fig. 28).1 Although azol(in)e heterocycles are frequently observed in other classes of RiPPs (i.e. thiopeptides, Section 3.2; azol(in)e-containing cyanobactins, Section 3.6; and bottromycins, Section 3.7), the presence of other class-defining post-translational modifications set these latter compounds apart from LAPs. LAP biosynthesis begins with the ribosomal synthesis of a precursor peptide, comprised of N-terminal leader and C-terminal core regions.1 Azole installation (Fig. 28C) on the core peptide proceeds in two steps by the action of a trimeric synthetase, as exemplified by studies on the microcin B17 pathway.106 In the first step, a member of the YcaO superfamily (InterPro families IPR003776 and IPR019938) associates with a partner protein belonging to the E1-ubiquitin activating (E1-like) superfamily and installs azolines in an ATP-dependent manner. In roughly half of all known LAP BGCs, the E1-like and YcaO proteins are fused as a single protein, highlighting the importance of their collective effort in azoline installation.107 Select azolines are subsequently oxidized to azoles by a flavin mononucleotide (FMN)-dependent dehydrogenase, when encoded in the BGC. The LP is typically removed by a protease and the mature LAP is exported from the producing cell. Other ancillary modifications such as methylation (Section 5.3.5), acetylation (Section 5.9.1), and dehydration (e.g. goadsporin,91 Section 3.2) may further elaborate the structure of LAPs.108,109 Much of the mechanistic information underlying azoline installation stems from studies on cyclodehydratases involved in the biosynthesis of azol(in)e-containing cyanobactins (as the biosynthesis proceeds via generation of a LAP-like intermediate) and LAPs themselves. While YcaOs involved in RiPP biosynthesis have been reviewed elsewhere in great detail,110 this section will focus on the current state of knowledge about the cyclodehydratases (also called heterocyclases) and dehydrogenases in the LAP and cyanobactin pathways.
eptides (LAPs) comprise a class of RiPP characterized by the presence of thiazol(in)e and/or (methyl)oxazol(in)e heterocycles, resulting from the backbone cyclodehydration of Cys, Ser, and Thr residues (Fig. 28).1 Although azol(in)e heterocycles are frequently observed in other classes of RiPPs (i.e. thiopeptides, Section 3.2; azol(in)e-containing cyanobactins, Section 3.6; and bottromycins, Section 3.7), the presence of other class-defining post-translational modifications set these latter compounds apart from LAPs. LAP biosynthesis begins with the ribosomal synthesis of a precursor peptide, comprised of N-terminal leader and C-terminal core regions.1 Azole installation (Fig. 28C) on the core peptide proceeds in two steps by the action of a trimeric synthetase, as exemplified by studies on the microcin B17 pathway.106 In the first step, a member of the YcaO superfamily (InterPro families IPR003776 and IPR019938) associates with a partner protein belonging to the E1-ubiquitin activating (E1-like) superfamily and installs azolines in an ATP-dependent manner. In roughly half of all known LAP BGCs, the E1-like and YcaO proteins are fused as a single protein, highlighting the importance of their collective effort in azoline installation.107 Select azolines are subsequently oxidized to azoles by a flavin mononucleotide (FMN)-dependent dehydrogenase, when encoded in the BGC. The LP is typically removed by a protease and the mature LAP is exported from the producing cell. Other ancillary modifications such as methylation (Section 5.3.5), acetylation (Section 5.9.1), and dehydration (e.g. goadsporin,91 Section 3.2) may further elaborate the structure of LAPs.108,109 Much of the mechanistic information underlying azoline installation stems from studies on cyclodehydratases involved in the biosynthesis of azol(in)e-containing cyanobactins (as the biosynthesis proceeds via generation of a LAP-like intermediate) and LAPs themselves. While YcaOs involved in RiPP biosynthesis have been reviewed elsewhere in great detail,110 this section will focus on the current state of knowledge about the cyclodehydratases (also called heterocyclases) and dehydrogenases in the LAP and cyanobactin pathways.
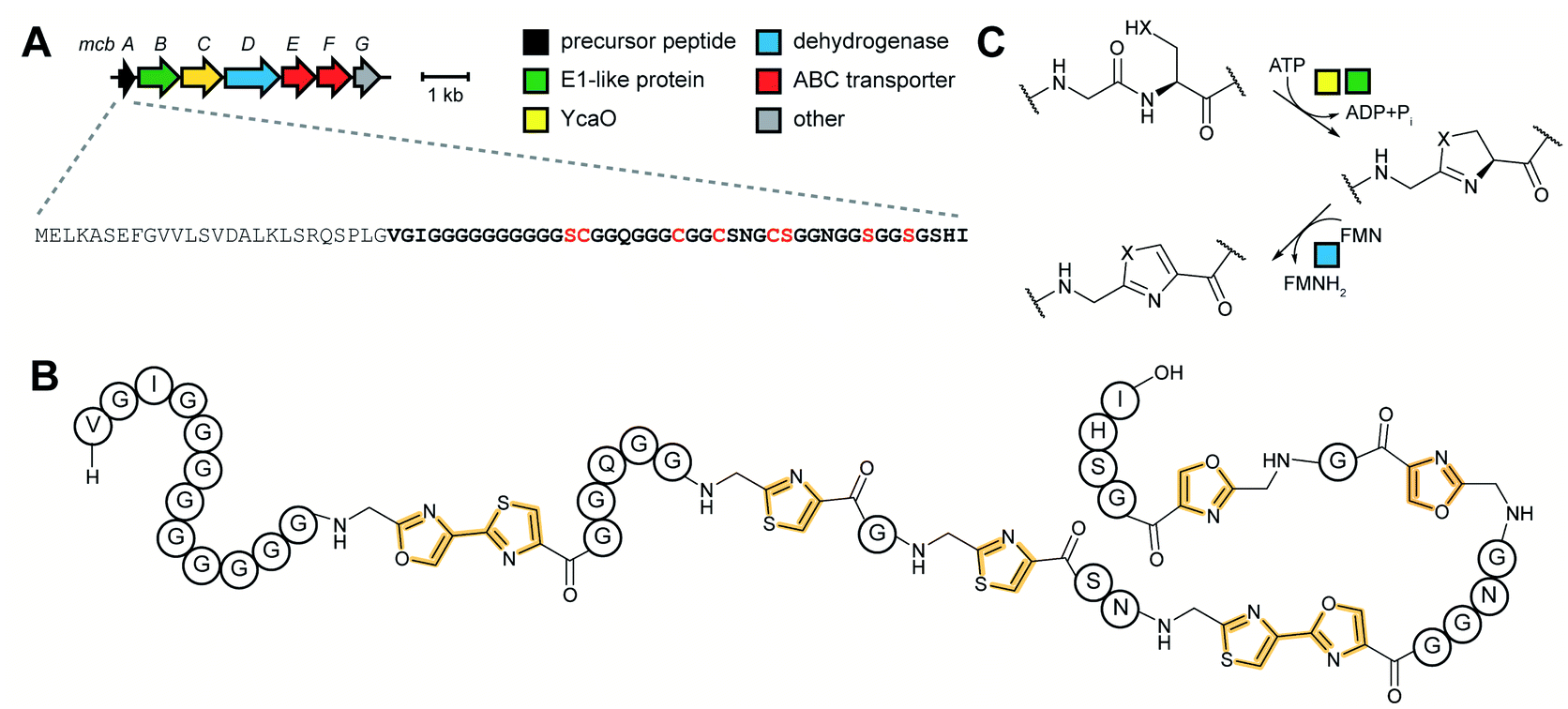 | ||
| Fig. 28 LAP BGC and structure. (A) BGC and precursor peptide sequence of the LAP microcin B17. The core peptide is in bold and residues that are modified are in red. (B) Structure of microcin B17. The class-defining azole heterocycles are yellow. (C) Biosynthetic scheme for azole installation in LAPs. | ||
 | ||
| Fig. 29 Mechanistic model for YcaO-catalyzed cyclodehydration. | ||
In contrast, TruD, a cyclodehydratase from the trunkamide (an azol(in)e-containing cyanobactin; see Section 3.6) pathway, was reported to produce AMP and pyrophosphate (PPi) as byproducts when supplied with PatE2 (a precursor peptide from a related biosynthetic pathway), indicative of an adenylation mechanism.114 The crystal structure of TruD (PDB: 4BS9) (Fig. 30A) gave the first insight into an azoline-forming cyclodehydratase, which was a dimer composed of three domains. Domains 1 and 2 share structural homology with BalhC and MccB, an adenylase involved in microcin C biosynthesis (Section 4.1.3); however, TruD lacks the conserved ATP-binding loops seen in MccB. Domain 3 of TruD, which is homologous to BalhD, constituted a novel fold characterized by a large, negatively charged, shallow cleft. Although not much information regarding substrate recognition and nucleotide binding could be obtained from the TruD crystal structure, biochemical data ruled out a kinase-type mechanism for TruD.114 A nucleotide-bound crystal structure of LynD (PDB: 4V1T) (Fig. 30B), another cyanobactin cyclodehydratase, revealed that the α-phosphate of the enzyme-bound ATP is obstructed from the peptide substrate, making the previously described adenylation mechanism less likely for heterocyclization.115 With biochemical data supporting an adenylation mechanism and crystallographic data disfavoring an adenylation mechanism, the ATP chemistry utilized by cyanobactin cyclodehydratases remained unclear. More recent studies using nonhydrolyzable ATP analogs in the heterocyclization reactions catalyzed by LynD and MicD (a related cyclodehydratase) provided clarity regarding the mechanism.116 Both LynD and MicD were reported to produce heterocycles on the peptide substrate, PatE2K, upon reaction with ATP analogs harboring a nonhydrolyzable α–β phosphate bond (AMP-CPP and AMP-NPP). However, heterocyclization was not observed when the reaction was carried out using ATP analogs with a nonhydrolyzable β–γ phosphate bond (AMP-PCP and AMP-PNP). Moreover, HPLC analysis of the reaction catalyzed by a MicD fusion enzyme (with an N-terminally fused leader peptide) on a leaderless variant of the peptide substrate revealed accumulation of ADP before decomposing to AMP. Together, these data established that cyclodehydration proceeds via a kinase mechanism. Production of AMP and PPi as reaction byproducts has been proposed to occur as a result of a disproportionation or transphosphorylation reaction between Pi and ADP catalyzed by the enzyme.116
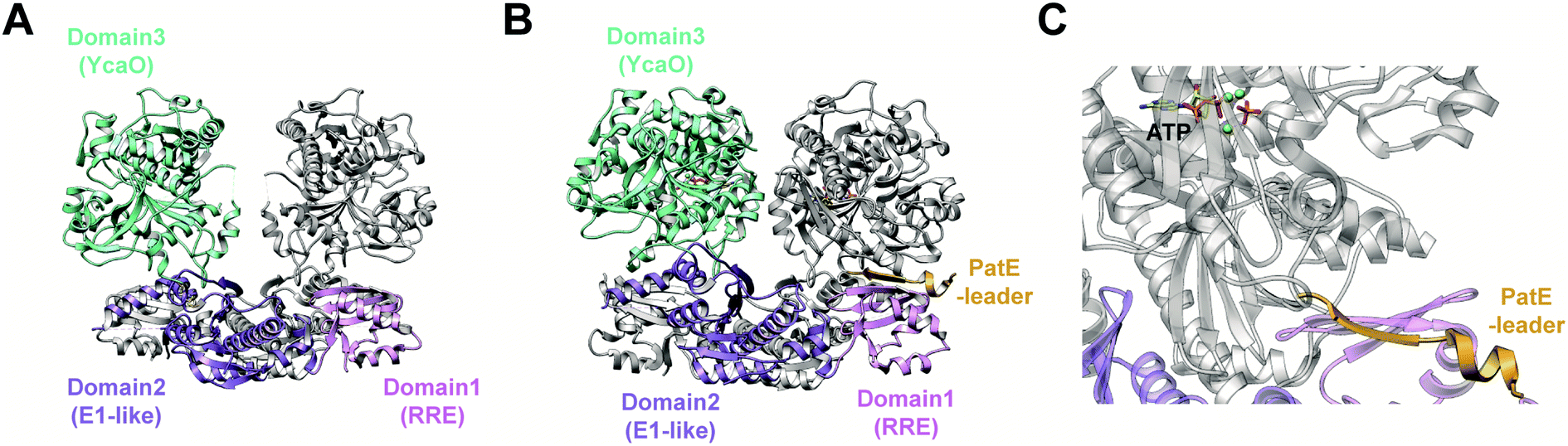 | ||
| Fig. 30 Cyanobactin cyclodehydratases. (A) Structure of the TruD dimer (PDB: 4BS9). (B) Nucleotide and substrate bound structure of the LynD dimer (PDB: 4V1T). In panels (A) and (B), monomer 1 is colored by domain and monomer 2 is gray. (C) Binding of substrate PatE2 and ATP. RRE: RiPP precursor recognition element (Section 4.1.1). | ||
A nucleotide-bound crystal structure was obtained for an orphan YcaO from E. coli (denoted as Ec-YcaO; PDB: 4Q86) (Fig. 31).117 Although not yet firmly associated with a function, information obtained from the crystal structure of Ec-YcaO provided valuable insights into understanding the reactions catalyzed by YcaOs in RiPP biosynthesis. Ec-YcaO comprises a novel ATP-binding fold, which is conserved among all characterized YcaOs to date.114,115,117–119 Ala variants of the ATP-binding residues in BalhD predicted through sequence alignment with Ec-YcaO exhibited an increased KM for ATP and were verified to be vital for BalhD cyclodehydration activity in vitro, suggesting that ATP utilization is a universal feature of YcaOs.117 Fittingly, thioamide-forming YcaOs (Section 3.5) and macrolactamidine-forming YcaOs (Section 3.7), also require ATP to process their cognate substrates.118–121 Structural alignment of cyanobactin cyclodehydratases TruD and PatD (azoline-forming YcaOs) with Methanocaldococcus jannaschii YcaO (Mj-YcaO, thioamide-forming enzyme, PDB: 6PE3) revealed that the C-terminal carboxylate in TruD/PatD (and not Mj-YcaO) extends into the active site, possibly serving as a base that deprotonates the β-nucleophile of Thr/Ser/Cys. The C-terminal carboxylate is presumed to be kept in the extended conformation due to an abundance of Pro residues, often found as a PxPxP motif, which is conserved among ∼90% of the bioinformatically identified azoline-forming YcaOs.119 Indeed, alterations at the C-terminus of BalhD and McbD abolishes cyclodehydration activity.117
 | ||
| Fig. 31 Structure and ATP-binding residues of Ec-YcaO. (A) Structure of nucleotide-bound dimeric Ec-YcaO (PDB: 4Q86).117 Monomer 1 is colored by ATP-binding domain (green) and tetratricopeptide repeat domain (purple) with monomer 2 in gray. (B) ATP-binding residues of Ec-YcaO are shown as gold sticks colored according to heteroatoms. AMP is shown as gray sticks colored according to heteroatoms. | ||
While certain YcaO proteins catalyze cyclodehydration, the E1-like partner protein serves as a potentiator and regulator of YcaO. This enhancement was initially demonstrated using the Balh pathway, where BalhC accelerates the rate of azoline installation by BalhD by nearly 1000-fold.112 Omission of BalhC from the reaction led to a non-stoichiometric consumption of ATP, which otherwise was tightly coupled to azoline installation;112 further, the order of ring formation in the absence of BalhC was dysregulated.113 Fluorescence polarization studies revealed that BalhC engages the LP of BalhA (independently of BalhD), with a KD commensurate with that of the full-length precursor peptide, establishing its role in substrate recognition and presenting the core to BalhD.117 This idea is further corroborated by the fact that the N-terminal winged helix-turn-helix (wHTH) domain of BalhC shares structural homology with similar domains found in TruD,114 LynD,115 McbB,122 and NisB49 (a lanthipeptide dehydratase, Section 3.1.1) known to interact with the leader region of their corresponding precursor peptides. This wHTH motif, now recognized as the RiPP precursor recognition element (RRE), is a common feature employed by more than half of all known prokaryotic RiPP classes to engage their respective LPs (Section 4.1.1).66 In addition to binding the RRE, Phe8 and Leu12 of the substrate McbA were shown to occupy two hydrophobic pockets in McbB1-dom1 and in the McbB1-dom1/McbB2-dom2 interface in the McbBCD synthetase complex (Section 4.1.1),122 rationalizing the importance of an Fxxx(L/I/V) motif in the leader peptide of several LAP precursor peptides previously observed to be critical to bind to synthetases.123 In certain LAP BGCs, such as the heterocycloanthracins (HCA), a divergent member of the E1-like superfamily, termed the F-protein (InterPro family: IPR022291), is responsible for substrate recognition.124In vitro studies on the HCA cluster from Bacillus sp. Al Hakam showed that HcaF (Ocin-ThiF-like domain) is essential for azoline formation catalyzed by HcaD (E1-like protein fused to the YcaO). The N-terminal domain of the fused cyclodehydratase, HcaD, lacks the RRE and indeed does not bind the peptide substrate. Instead, it brings the peptide substrate-bound HcaF and YcaO domain into proximity for catalysis.124
3.5 Thioamitides: introduction and biosynthesis
Thioamitides, a name coined in this review, are RiPPs characterized by thioamide(s) in place of amides in the peptide backbone. The founding member of the family, thioviridamide, was reported as an apoptosis inducer with a multitude of post-translational modifications, including five backbone thioamides, a β-hydroxy-N1,N3-dimethylhistidinium, AviCys, and an N-terminal pyruvyl moiety to which an acetone moiety was added during isolation.11,126,127 Since the discovery of the thioviridamide BGC (Fig. 32A) in 2013,128 several groups have employed genome mining strategies to uncover the diversity of the family, leading to the characterization of several thioviridamide-like compounds that inhibit growth of various cancer cell lines (Fig. 32B).9,11–13 These thioamitides each feature three or four backbone thioamides. A putative function has been assigned for each gene in the thioviridamide BGC; however, only the FMN-dependent cysteine decarboxylase TvaF has been biochemically characterized to date.129 TvaH, a YcaO homolog, has been proposed to install the class-defining thioamide modifications during thioviridamide biosynthesis.128 TvaI, a TfuA-like protein (InterPro family: IPR012924) cooccurs with TvaH in all identified thioviridamide BGCs and has been proposed to support thioamidation, although its precise role remains undefined. The characterization of additional thioviridamide-like compounds has revealed that the number of thioamides currently varies from three to five, and identified further rare post-translational modifications, such as Phe hydroxylation and reduction of the N-terminal pyruvyl moiety in thioalbamide.11 | ||
| Fig. 32 Thioamitide BGC and structure. (A) Two thioamitide BGCs: thioholgamide (tho) and thioviridamide (tva) and the sequence of the ThoA precursor peptide with the residues converted to thioamides in red. (B) Structure of thioholgamides. Class-defining PTMs are shown in yellow and secondary PTMs in cyan. | ||
 | ||
| Fig. 33 Proposed mechanism for thioamidation catalyzed by YcaOs. | ||
Approximately 10% of methanogens lack an identifiable tfuA in their genome. TfuA-independent YcaOs from Methanocaldococcus jannaschii (Mj-YcaO) and Methanopyrus kandleri (Mk-YcaO) efficiently catalyzed thioamide installation on a synthetic 11-mer McrA substrate that contained five residues upstream and downstream of the modified Gly.118 Nucleotide-bound crystal structures were obtained for Mk-YcaO (PDB: 6CI7), which displayed an overall architecture similar to LynD (azoline-forming YcaO; PDB: 4V1T) and Ec-YcaO (orphan YcaO; PDB: 4Q86) (Section 3.4). While the nucleotide-binding residues are largely conserved across these proteins, the active site region of Mk-YcaO is different from the azoline-forming YcaOs, hinting towards a divergent substrate-binding site or reorganization upon substrate binding.118 More recently, Mj-YcaO was co-crystallized with a synthetic 11-mer peptide substrate derived from the MCR α-subunit (Mj-McrA).119Mj-YcaO bound to Mj-McrA (PDB: 6PEU) (Fig. 34) represents the first reported structure of any YcaO protein bound to its peptide substrate and permitted definitive identification of residues involved in peptide binding. This study also revealed that when presented with an 11-mer peptide variant containing a Cys residue near the site of modification, Mj-YcaO was capable of forming a thiazoline.119 Indeed, many of the active site residues identified in Mj-YcaO are highly conserved among divergent YcaO proteins, including members catalyzing azoline formation (Section 3.4) and macrolactamidine formation (Section 3.7), in addition to thioamide formation.114,115,117–121Mj-YcaO, however, lacks a basic residue for initial Cys deprotonation and cyclodehydration activity was observed only when a surrogate base was supplied in the assay. This cyclodehydration activity further supports the idea that all YcaO proteins modify their substrates via a shared mechanism and that it is the identity of the nucleophile attacking the amide carbonyl carbon that determines the chemical outcome. For thioamidating YcaOs, the nucleophile derives from a yet unidentified source of sulfide, which attacks the target amide bond to yield a tetrahedral oxyanion intermediate (Fig. 33). Following attack of ATP and release of ADP, the resulting phosphorylated thiolate intermediate collapses to release the thioamidated product and phosphate as a byproduct. The role of TfuA-like proteins in thioamidation is presently unclear and possible roles include activation of YcaO, regulation of ATP usage by YcaO, and/or assisting in the delivery of sulfide equivalents.131
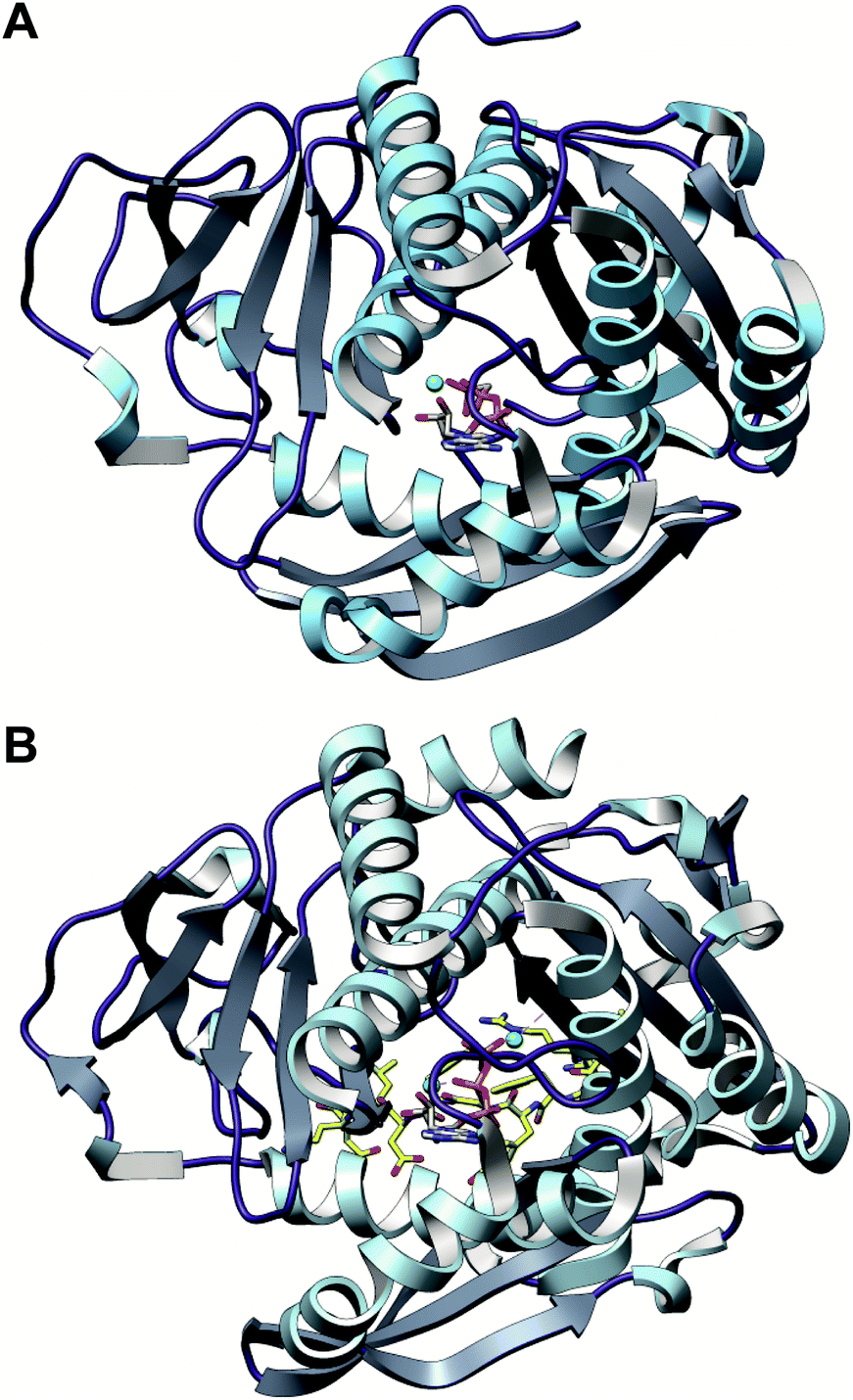 | ||
| Fig. 34 Structures of thioamide-installing YcaOs. (A) Nucleotide-bound structure of Mk-YcaO (PDB: 6CI7). (B) Nucleotide- and peptide-bound structure of Mj-YcaO (PDB: 6PEU). Structures in panels (A) and (B) are colored by secondary structures. Nucleotide in panel (A) and (B), and peptide substrate in panel (B) are shown as gray and gold sticks, respectively, and colored according to heteroatom. | ||
Evidence for the involvement of the YcaO/TfuA pair in RiPP thioamidation stems from a recent study on thiopeptides (Section 3.2), another RiPP class that is known to feature thioamides; however, thioamidation of thiopeptides is a rare, secondary modification. During a recent bioinformatic expansion of the thiopeptide family, a novel thiopeptide, saalfelduracin, featuring a single thioamide was isolated and characterized.132 Similar moieties have also been reported in Sch 18640 and thiopeptin; however, their biosynthetic origin was unknown. Genome sequencing of the native producers of these thiopeptides allowed identification of their putative BGCs, all of which encode YcaO–TfuA protein pairs, akin to the saalfelduracin and thioviridamide BGCs. Chromosomal insertion of ycaO and tfuA from the Sch 18640 BGC into a thiostrepton producer (lacks thioamides), resulted in the detection of a metabolite identical to Sch 18640. Thioamidation was dependent on the presence of TfuA and YcaO.132 Recently, a new RiPP-mining tool, RIPPER (Section 6.3) was employed to locate thioamidated RiPPs and revealed that YcaO–TfuA gene pairs are widespread among many uncharacterized BGCs in Actinobacteria.14 Heterologous expression of a member of one such BGC enabled isolation and initial characterization of the thiovarsolins, presumed shunt metabolites from an enigmatic RiPP family that contain a single thioamide moiety installed in a (A/G)PR tripeptide that is repeated five times in the VarA precursor peptide from Streptomyces varsoviensis. Isolation of thioamidated GPR and APR peptides indicates that thioamidation occurs multiple times on a single precursor peptide. Deletion of the YcaO and TfuA encoding genes from the BGC abolished production of the thioamidated products, further corroborating their role in thioamide installation. Co-expression of the precursor peptide, YcaO and TfuA genes in Streptomyces coelicolor was sufficient to observe thioamidated peptides.14
3.6 Cyanobactins: introduction and biosynthesis
Cyanobactins (Fig. 35) are diverse RiPP natural products produced by symbiotic and free-living cyanobacteria.1 Although they were described earlier as N-to-C macrocyclized peptides, examples of linear cyanobactins have surfaced,133 expanding the structural diversity of this family. Cyanobactin biosynthesis (Fig. 35) begins with the ribosomal generation of the precursor peptide, called the E peptide. In contrast to precursor peptides from most other RiPP classes that feature a single core peptide, cyanobactin precursor peptides are organized into multiple cassettes. Each cassette bears a core peptide that forms the mature cyanobactin flanked by highly conserved N-terminal and C-terminal recognition sequences (RSII and RSIII, respectively) that guide the biosynthetic enzymes. The core sequences in the precursor peptide are hypervariable and give rise to chemically diverse products. The order of post-translational tailoring has been established for the patellamide (Fig. 35) and trunkamide pathways.134 When present, a YcaO orthologous to the LAP cyclodehydratase recognizes RSI (present only in cyanobactin BGCs harboring YcaO) in the precursor peptide and converts Cys/Ser/Thr residues to azoline heterocycles (see Section 3.4). The protease A then recognizes and cleaves C-terminal to RSII in the precursor peptide, liberating the LP. The protease/macrocyclase G identifies RSIII, cleaving N-terminal to this sequence, and in some examples, catalyzing N-to-C cyclization of the core peptide to form the mature cyanobactin. Other PTMs, such as oxidation of azolines, N and O-methylation, and prenylation, have been observed in cyanobactins (Sections 5.8 and 5.9).135 PatA homologs are the only biosynthetic enzymes common to all predicted cyanobactin BGCs, as exemplified by genome mining studies.133 This renders PatA and its orthologs as the class-defining enzyme for cyanobactins.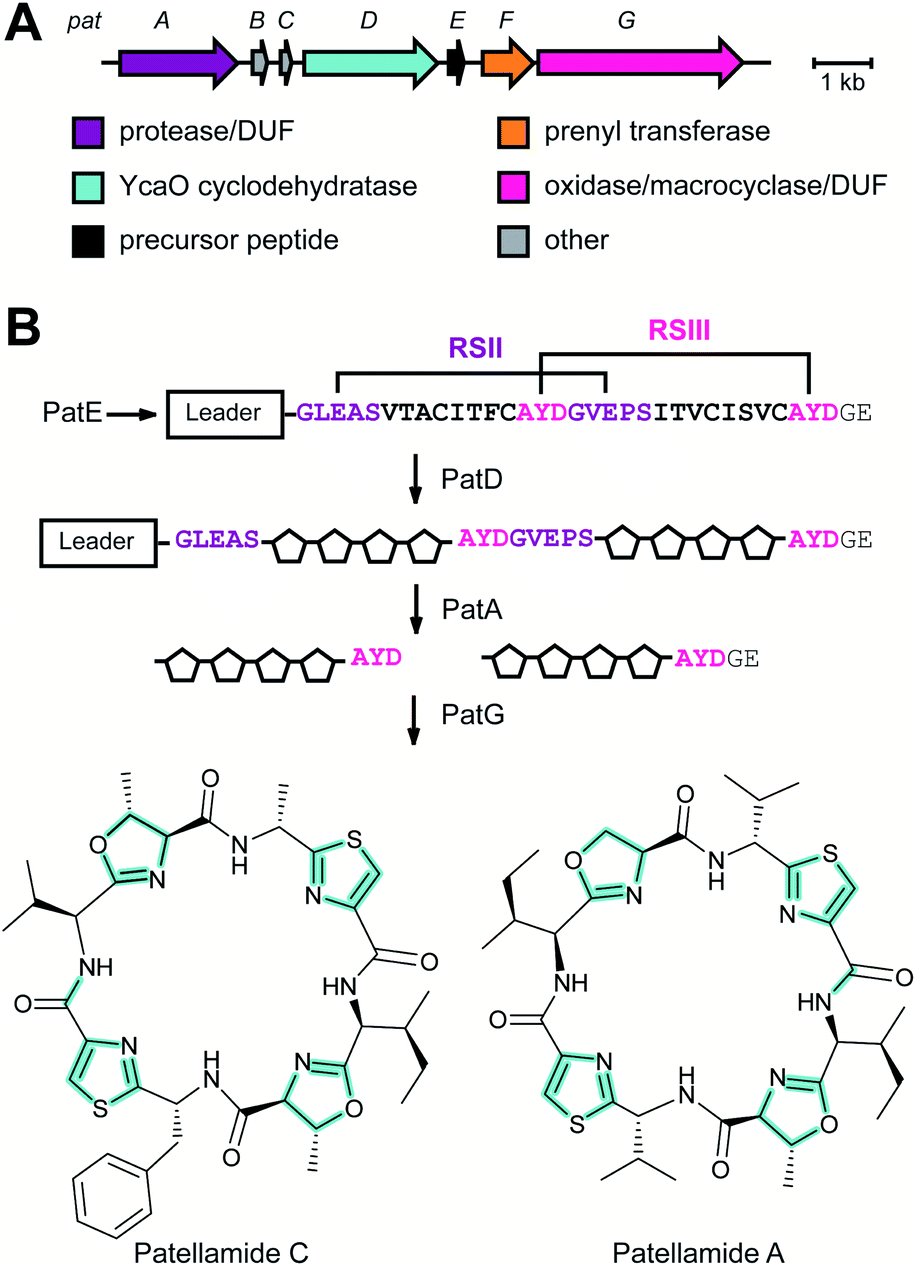 | ||
| Fig. 35 Cyanobactin biosynthesis. (A) BGC of the cyanobactin patellamide. (B) Patellamide biosynthesis beginning with the ribosomal synthesis of the PatE precursor peptide and culminating in the synthesis of two cyanobactins, patellamide A and C. PatD catalyzes cyclodehydration of Cys, Ser, and Thr residues, then PatA cleaves C-terminal to RSII, and PatG cleaves N-terminal to RSIII and cyclizes the peptide. Because PatA is the class-defining enzyme, azol(in)e formation and head-to-tail cyclization are secondary PTMs (cyan). | ||
 | ||
| Fig. 36 Structure of cyanobactin proteases. (A) PatA (PDB: 4H6V) protease domain. (B) PagA (PDB: 4H6W) protease domain. Figures in panel (A) and (B) are colored by secondary structures. Both PatAPro and PagAPro exhibit a subtilisin-like protease fold. (C and D) Catalytic triad of PatAPro and PagAPro, respectively. | ||
3.7 Bottromycins: introduction and biosynthesis
Bottromycins are peptide antibiotics recognized for their potent growth inhibitory activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant enterococci (VRE).1 They are relatively short peptides (8-mers) and feature an array of modifications including the class-defining macrolactamidine, a thiazole, Asp esterification, epimerization, decarboxylation, and β-methylation of Phe, Pro, and two of the three Val residues (Fig. 37). Their ribosomal origin and BGCs were identified by four groups in 2012 and are well conserved among several Streptomyces spp.138–141 While biosynthetic studies have been conducted on several orthologous BGCs, this section will use the Streptomyces bottropensis gene nomenclature shown in Fig. 37A for simplicity. The bottromycin precursor peptides stand unique among other RiPP classes in that they exhibit an inverted RiPP paradigm, where the core peptide precedes a C-terminal follower peptide. Bottromycin BGCs encode two YcaO proteins, three radical SAM methyltransferases, an O-methyltransferase, an α/β hydrolase, an amidohydrolase, a cytochrome P450 enzyme, a leucyl-aminopeptidase, a transporter, and a transcriptional regulator. Roles for each of these proteins have been proposed.138–141 Gene deletion studies conducted on various bottromycin-producing strains assigned the role of Asp methylesterification to the O-methyltransferase and β-methylation of Phe, Val, and Pro to the three class B radical SAM methyltransferases found in the BGC.138,141 Biochemical studies showed that the leucyl-aminopeptidase BmbK removes the N-terminal Met142,143 and confirmed that BmbB catalyzes O-methylation of Asp.143 An untargeted metabolomics study carried out on mutants of the BGC in Streptomyces scabies proposed an order for the post-translational modifications in bottromycin biosynthesis as detailed in Fig. 37. This study implicated the YcaO proteins, BmbD and BmbE, in thiazoline and macrolactamidine installation, respectively, which was subsequently confirmed via detailed biochemical studies.143 The α/β hydrolase BmbG was shown to be responsible for the epimerization of the Asp residue (Section 5.1.3).144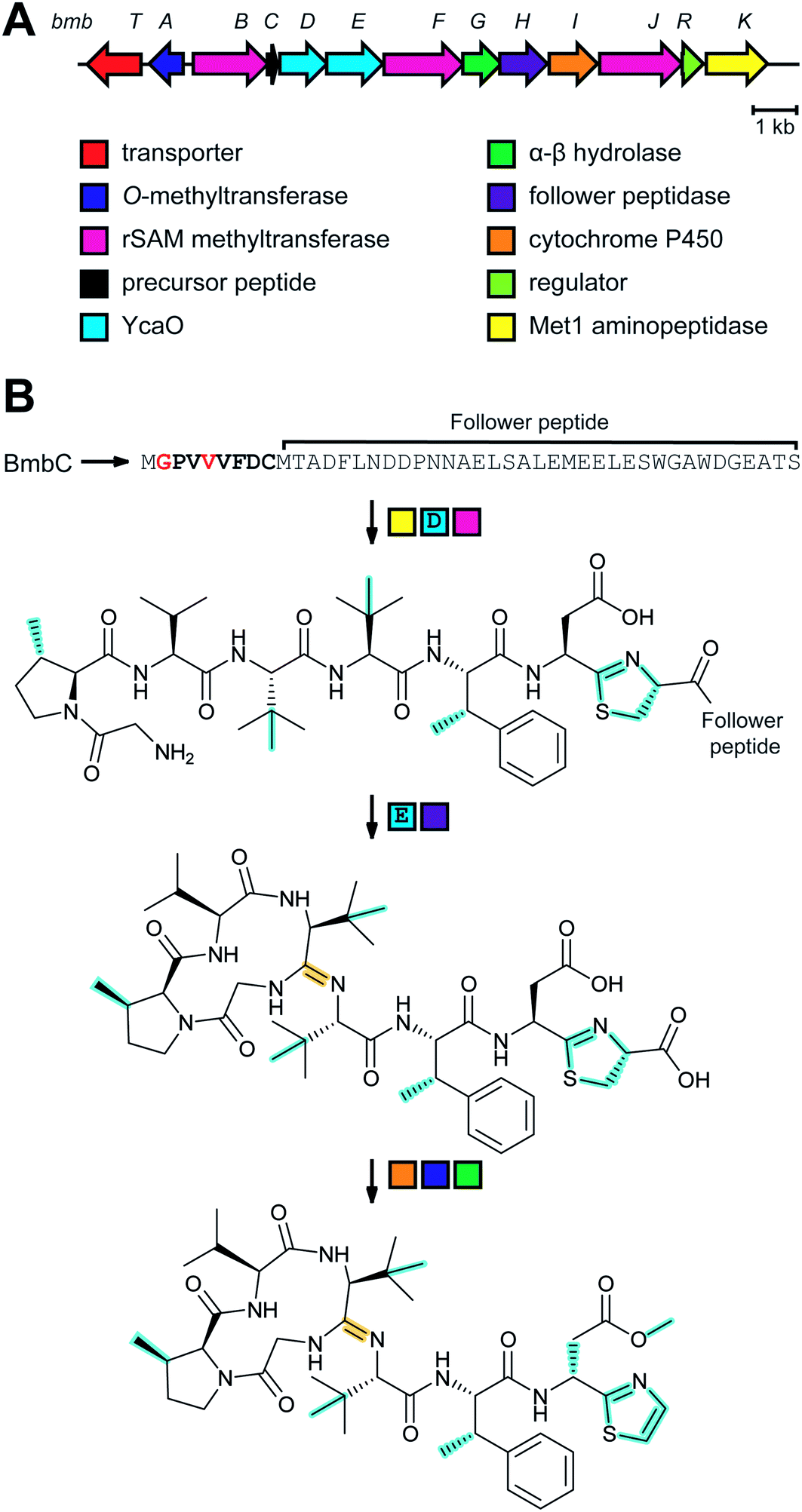 | ||
| Fig. 37 Bottromycin biosynthesis. (A) Bottromycin BGC in Streptomyces bottropensis. (B) Proposed order of steps for bottromycin biosynthesis.143,144 In the first three steps, in no particular order, the N-terminal Met is removed by BmbK, thiazolines are installed by BmbD, and Val, Pro and Phe are C-methylated by BmbBF. Then the class-defining macrolactamidine is formed by BmbE, the follower peptide is removed by BmbH, and the Asp residue is epimerized by BmbG. In the final two steps, BmbI is thought to oxidize the thiazoline to a thiazole by oxidative decarboxylation and the Asp is methylated by BmbA. | ||
Macrolactamidine-forming YcaOs are the first YcaOs reported to form larger ring systems and use nitrogen nucleophiles.120 The BmbE ortholog-catalyzed reaction was optimal at a pH of 9.5, where most of the N-terminus is deprotonated and available as a nucleophile for macrolactamidination, while also retaining enzymatic stability.120 Prolonged reaction times led to opening of the macrolactamidine, which was dependent on the presence of the macrolactamidine-forming YcaO and ATP. This reversibility was explained by the finding that the in vivo reaction requires the presence of an additional protein, i.e. BmbG (alternative name BotAH).143 Indeed, in vitro studies later showed that the BmbG ortholog acts as a gatekeeper during bottromycin biosynthesis by removing the follower peptide C-terminal to the thiazoline, essentially preventing reopening of the macrolactamidine.145 Recently, the YcaO protein KlpD was shown to install a six-membered amidine ring, three thiazolines, and one oxazoline during the maturation of a RiPP named klebsazolicin.146 Therefore, KlpD is a rare example of a YcaO known to accept oxygen, sulfur, and nitrogen nucleophiles.
As macrolactamidine-forming YcaOs are not known to harbor RREs, studies then focused on understanding how these enzymes identify their substrates. Systematic C-terminal trimming of precursor peptides was performed, given that bottromycins use a follower peptide as opposed to a LP. These variants were assessed for stability in the presence of the YcaO by employing thermal shift assays and revealed that precursor peptide residues 30–39 are the most important for the peptide-protein interaction.120 This conclusion is also supported by studies on an orthologous pathway, which identified residues 1–34 as the minimal substrate required for biosynthesis.121 Macrolactamidine-forming YcaOs are generally tolerant to changes in the core region, in vitro. However, they strictly install the macrolactamidine on the fourth core residue as evidenced by the inability to alter ring size.121 Macrolactamidine-forming YcaOs harbor the conserved ATP-binding motifs observed among other YcaOs that are essential for activity as evidenced by the lack of processing by Ala variants.110,117,120,121 In contrast to the azoline-forming, E1-dependent YcaOs involved in LAP and cyanobactin biosynthesis (Sections 3.4 and 3.6, respectively), macrolactamidine-forming YcaOs do not contain a Pro-rich C-terminus. The C-terminus is relatively basic and moderate truncations of the enzyme are tolerated as opposed to the azoline-forming YcaOs.119,121 A bioinformatic survey of YcaO proteins found in NCBI predicted that macrolactamidine-forming stand-alone YcaOs are more prevalent than previously appreciated.121
3.8 Lasso peptides: introduction and biosynthesis
Lasso peptides are RiPPs characterized by a unique [1]rotaxane conformation resembling that of a threaded lariat (Fig. 38)1 with more than 70 family members isolated thus far. The lasso fold consists of a macrolactam bond between the N-terminus of the peptide and the side chain carboxylate of Asp/Glu at positions 7–9 of the core peptide.1,147 The remaining C-terminal portion of the peptide is threaded through the macrolactam and is often held in position by bulky amino acid residues (termed steric lock or plug residues), by disulfide linkages, or sometimes by both. This conformation endows many lasso peptides with remarkable stability against proteases and heat,1 although a few temperature-sensitive lasso peptides have been reported.148–150 In addition to their stability, lasso peptides span a wide-range of potentially useful bioactivities including antimicrobial, anticancer, and antiviral activities.151–153 Like other RiPPs, lasso peptide biosynthesis begins with the ribosomal synthesis of the precursor peptide (A) composed of leader and core regions. Lasso peptides are formed from a two-step sequence catalyzed by two enzymes, a cysteine protease homologous to transglutaminase (B protein), and a macrolactam synthetase (lasso cyclase) homologous to asparagine synthetase (C protein) that likely form a complex.1 In approximately one-third of bioinformatically predicted lasso peptide BGCs,154 the B protein is fused to the C-terminus of the RRE domain.155 Although ATP has been reported to be required for processing by fused B proteins,156 proteolysis by split B proteins does not require ATP.157,158 The RRE domain-containing protein is referred to as the E or B1 protein, while the peptidase is termed the B or B2 protein. Either way, the RRE is responsible for binding the leader region of the precursor peptide66,157,159–161 while the peptidase performs LP cleavage.157 The lasso cyclase then installs the macrolactam linkage when the C-terminal tail is threaded in an ATP-dependent manner.156,157 Two studies independently reported in vitro reconstitution of the lasso peptide encoded in the Thermobifida fusca genome, termed fusilassin or fuscanodin.150,158 In addition to the class-defining macrolactam, ancillary modifications such as phosphorylation (Section 5.6), acetylation (Section 5.9.1), methylation (Section 5.3.7), deimination (Section 5.5), hydroxylation (Section 5.7.4.1), and epimerization (Section 5.1.3) have been observed on lasso peptides.152,154,162–164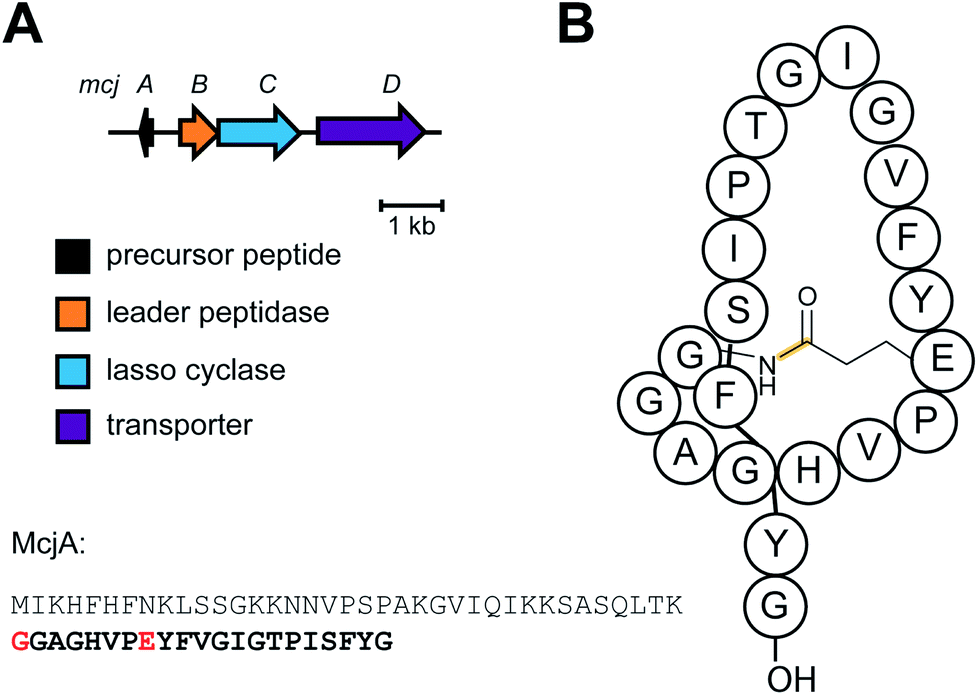 | ||
| Fig. 38 Lasso peptide BGC and structure. (A) BGC of the lasso peptide microcin J25 (MccJ25). The MccJ25 precursor peptide sequence is shown. The core peptide is shown in bold and residues forming the macrolactam linkage are shown in red. (B) Cartoon representation of MccJ25; OH reflects a free carboxyl C-terminus. The class-defining macrolactam linkage is highlighted in yellow. | ||
Until genome mining was used to prioritize discovery efforts, all lasso peptides isolated possessed either Gly (Class II) or Cys (Class I) at position 1.1 Recent discoveries have uncovered lasso peptides harboring Ala, Ser, Leu, Asp, Tyr, and Trp at position 1.154,158,168–171 An extensive bioinformatic investigation of the lasso peptide biosynthetic space facilitated by the genome mining tool RODEO (Section 6.2), identified ∼3000 lasso peptides in prokaryotes, as of August 2018.154,158 The precursor peptides predicted in this study feature all canonical amino acids at position 1 except Pro. A majority of the bioinformatically predicted lasso peptides feature Gly at position 1, in line with its prevalence among known lasso peptides. Akin to many other RiPP classes, the RRE domain governs interactions with the precursor peptide during lasso peptide biosynthesis (Section 4.1.1).
3.9 Sactipeptides: introduction and biosynthesis
Sactipeptides (Fig. 39) are RiPPs typified by sulfur-to-α-carbon thioether (i.e. sactionine) linkages between a donor Cys and an acceptor residue. Thus far, six sactipeptides, namely subtilosin A, thurincin H, thuricin CD (two peptide products), sporulation killing factor (SKF), and huazacin have been characterized from various Bacillus species, most of which exhibit narrow-spectrum growth inhibitory activity towards other Firmicutes.1,43 After ribosomal synthesis of the sactipeptide precursor, sactionine linkage(s) are installed by a radical S-adenosylmethionine (rSAM) enzyme (InterPro family: IPR007197). The initial demonstration of this activity was with AlbA, which acts on the subtilosin precursor, SboA, in a LP-dependent manner.172 The subtilosin BGC also encodes proteases and transporters, which are predicted to convert the thioether(s)-bearing precursor peptide into mature subtilosin.172 The rSAM “sactisynthases” that install the class-defining thioether bridges belong to a large enzyme superfamily known to catalyze a multitude of difficult reactions in both primary and secondary metabolism.173–175 Despite the wide range of reactions catalyzed, rSAM enzymes are unified by a common mechanism. rSAM enzymes house a [4Fe–4S] cluster in their active site where three of the four Fe atoms are coordinated by three Cys in a conserved CxxxCxxC (where x is any amino acid) motif.173 The fourth “unique” Fe is ligated by the α-carboxylate and α-amine moieties of SAM. Under reducing conditions, the [4Fe–4S]+ cluster transfers an electron to reductively cleave SAM to Met and a 5′-deoxyadenosine radical (5′-dA˙). This 5′-dA˙ then abstracts an unactivated hydrogen atom from the substrate which transforms to the product or initiates a reaction cascade depending on the type of rSAM enzyme.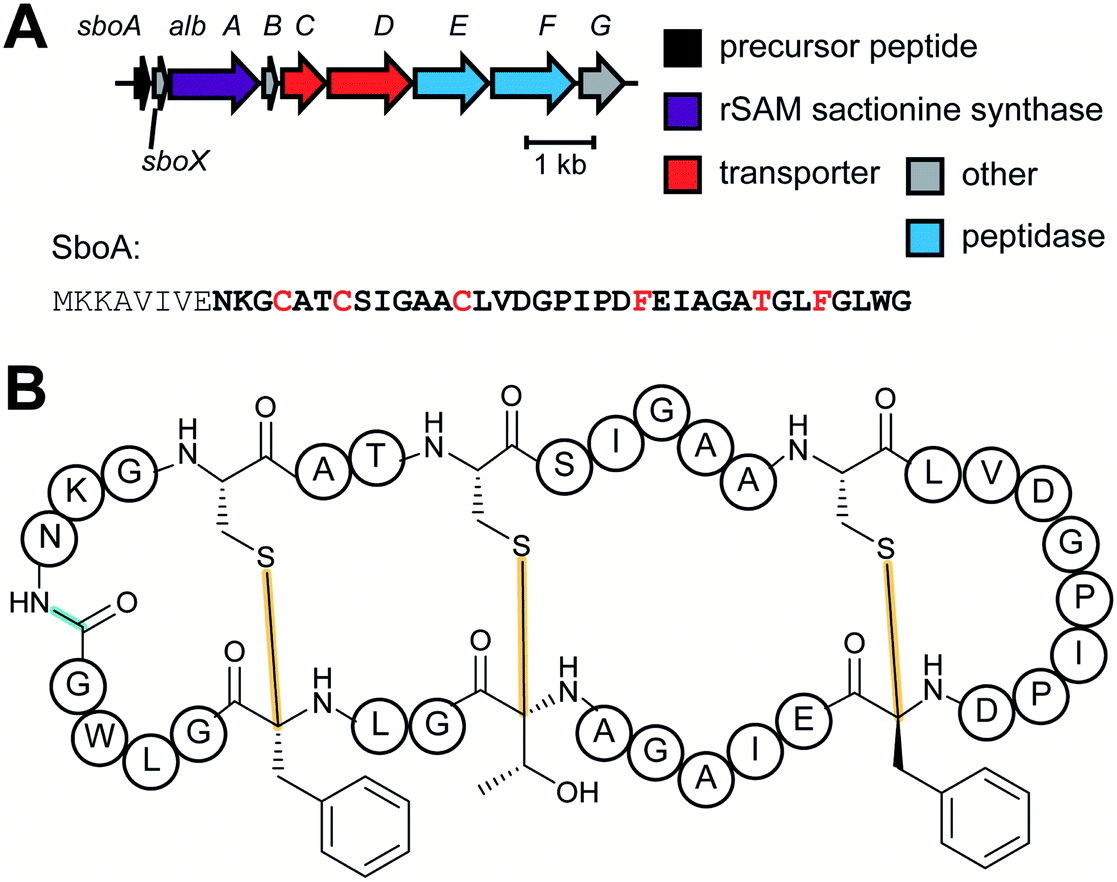 | ||
| Fig. 39 Sactipeptide BGC and structure. (A) Subtilosin A BGC and precursor peptide sequence. The core peptide and the residues involved in the class-defining sactionine modification are in bold and red, respectively. (B) Structure of subtilosin A. Sactionine linkages are in yellow. The secondary macrolactam modification is highlighted in cyan. | ||
AlbA and SkfB have been reported to harbor two [4Fe–4S] clusters (including the canonical rSAM iron-sulfur cluster), as evidenced by UV-vis and electron paramagnetic resonance (EPR) spectroscopy;172,176 however, a subsequent study showed a total of three [4Fe–4S] clusters in AlbA.179 The canonical [4Fe–4S] cluster involved in the reductive cleavage of SAM was corroborated by a lack of SAM cleavage activity by AlbA and SkfB variants lacking the Cys in the conserved CxxxCxxC motif. Changes made to Cys residues predicted (based on sequence alignments) to coordinate the auxiliary [4Fe–4S] cluster(s), prevented thioether formation in both the subtilosin and SKF pathways, despite retaining SAM cleavage activity.172,176 These AlbA variants when complexed with the substrate SboA also failed to display the characteristic UV-vis absorption shift observed with wild-type AlbA and SboA, or a leaderless SboA variant, which the authors attribute to a direct interaction between the AISC and the core peptide of SboA.172 Based on this, a radical mechanism for thioether installation by rSAM enzymes was proposed (Fig. 40). According to this mechanism, the 5′-dA˙ formed by the reductive cleavage of SAM abstracts a hydrogen atom from the α-carbon of the acceptor amino acid in the precursor peptide, thus generating a carbon-centered radical. This radical then forms the thioether bond with the thiyl radical generated by the transfer of an electron from the donor Cys to the AISC for which one of the sulfur ligands derives from the donor Cys. The now reduced AISC either transfers the electron to the oxidized primary [4Fe–4S] cluster or via an external agent to regenerate the enzyme for another round of catalysis.172 A polar mechanism has also been put forth (Fig. 40), where the carbon-centered radical intermediate undergoes oxidation to form an N-acyliminium intermediate, while the electron is captured by the auxiliary [4Fe–4S] cluster. This N-acyliminium intermediate can then be attacked by the thiolate of the donor Cys to form the thioether.179 Evidence for H-atom abstraction was provided by two independent studies on the subtilosin and SKF pathways. In one study, the authors used an SboA variant that contained residues 27–35 linked to residues 1–6 by an amide bond with the acceptor Phe deuterated uniformly. When this substrate was treated with AlbA, deuterium was incorporated into 5′-dA suggesting that the H-atom abstracted by 5′-dA˙ was from the acceptor Phe.179 However, this observation did not prove that the abstracted H-atom was exclusively from the α-carbon of the acceptor. The first definitive evidence for this mechanism leveraged the substrate promiscuous nature of SkfB.180 These researchers showed that upon reaction with SkfB, deuterium was transferred from the substrate SkfA into 5′-dA only when the acceptor Ala was labeled at both the α and β positions and not when Ala was labeled at the β position alone. Later studies used a radical clock (cyclopropylglycine) inserted in place of the acceptor Met in SkfA to show quenching of the generated substrate radical via ring opening in the presence of SkfB. A catalytically inactive variant of SkfB that lacks the twitch domain still catalyzed ring opening in the substrate analog, suggesting that the AISC was not essential for the initial abstraction of the hydrogen atom from SkfA.181 This finding, along with studies using AlbA variants that lack the AISC, points towards a role in thioether installation, but the precise role played by the auxiliary clusters remains unclear.172,176,179,181
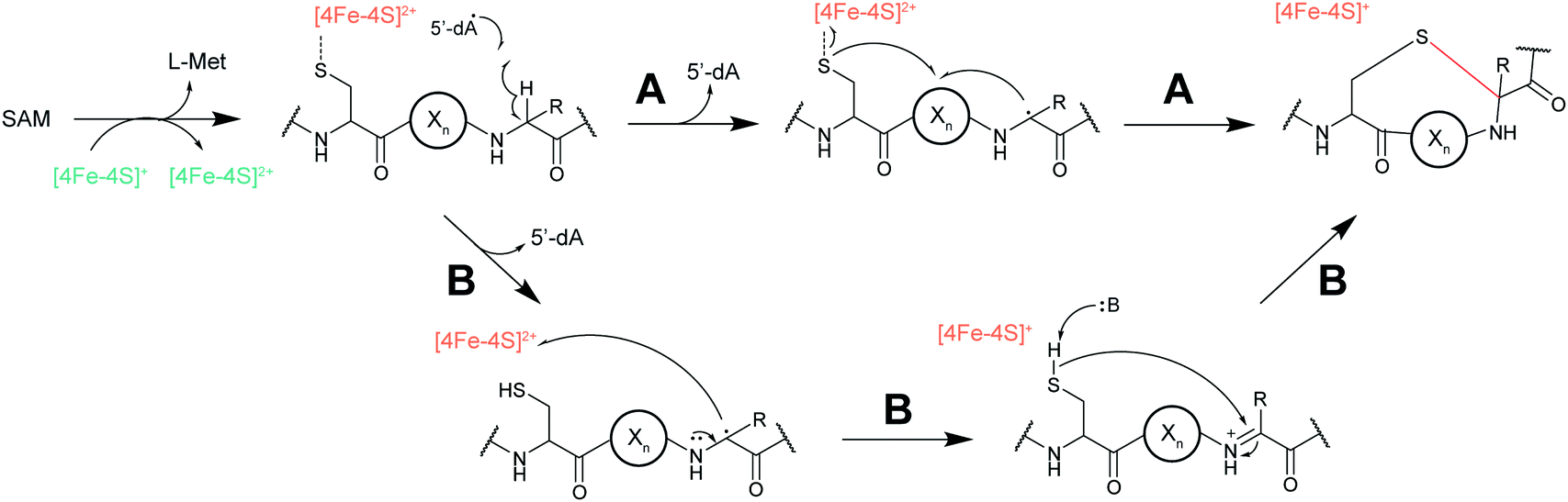 | ||
| Fig. 40 Proposed mechanistic schemes for rSAM mediated sactionine installation in sactipeptides. (A) Radical mechanism. (B) Polar mechanism. Reduction of the rSAM cluster and oxidation of the AISC returns the enzyme to a catalytically active state. The first Fe–S cluster (teal) resides in the SAM-binding domain and is used for the reductive cleavage of SAM and generation of the 5′-dA radical. The second Fe–S cluster (orange) resides in the auxiliary domain and is believed to coordinate the peptide substrate through Cys ligation. | ||
The only sactionine-forming rSAM enzyme that has been structurally characterized thus far is SkfB. SAM-bound SkfB (PDB: 6EFN) displays an overall modular architecture composed of a core rSAM domain flanked by an N-terminal RRE (Section 4.1.1) and the C-terminal AISC (twitch) domains (Fig. 41).178 The rSAM domain is folded into a partial (β/α)6 triose phosphate isomerase (TIM) barrel, characteristic of rSAM enzymes where the β-sheets line the inside of the barrel and α-helices flank the exterior. The SAM-binding [4Fe–4S] cluster resides in the loop region following the β1 strand in the core domain. The structure also revealed a combination of side chain and backbone interactions involved in orienting SAM in a catalytically active position.178 Similar to that observed in other RiPP biosynthetic enzymes, the peptide-binding RRE domain is formed by a three-stranded antiparallel β-sheet followed by three helices, and is fused to the rSAM domain.66,178 Although substrate-bound crystal structures of SkfB are not available, the sactisynthase involved in thurincin H biosynthesis lacking the N-terminal 89 residues (comprising the RRE) is incapable of forming sactionines despite exhibiting SAM cleavage activity.182 The C-terminal AISC (twitch) domain of SkfB, formed by a three-stranded antiparallel β-sheet followed by two α-helices, coordinates a single [2Fe–2S] cluster, which was further confirmed by Mössbauer spectroscopy.178 This finding was in contrast to an earlier study that suggested the AISC of SkfB was a [4Fe–4S] cluster.176 An overlay of the conserved Cys within the SkfB twitch domain with that of BtrN, another rSAM-enzyme that uses an auxiliary [4Fe–4S] cluster, revealed that SkfB is capable of accommodating an [4Fe–4S] cluster.178 Analysis of enzymatic activity in the context of Fe–S cluster content is expected to elucidate their role in catalysis.
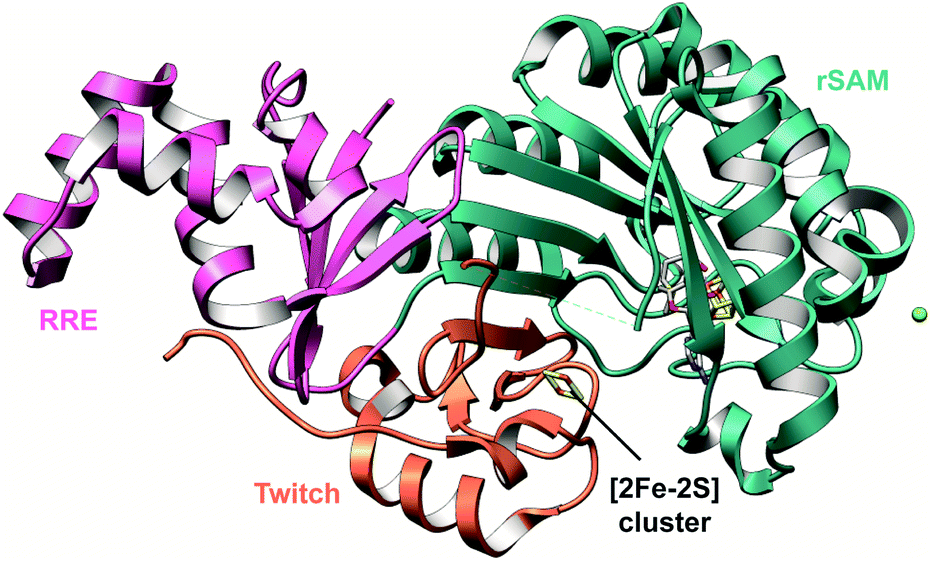
3.10 Ranthipeptides: introduction and biosynthesis
A bioinformatic investigation of the rSAM superfamily identified a group of Cys-rich peptides, primarily from the Clostridium genus, called SCIFFs (ix ys n orty-ive residues).42 These peptides were encoded next to a gene encoding a rSAM enzyme and thus were predicted to be substrates for post-translational modification. Although no mature SCIFF-derived natural product has ever been isolated, purified rSAM enzymes from predicted SCIFF-containing BGCs, CteB and Tte1186, installed a single thioether linkage between a donor Cys and an acceptor Thr on cognate precursor peptides, CteA and Tte1186a, in vitro.183,184 A more recent bioinformatic analysis conducted on rSAM maturases predicted to install thioether linkages, including sactisynthases and SCIFF maturases, revealed that CteB (and Tte1186) is more closely related to QhpD (Interpro family: IPR023886) than to any sactisynthases.43 QhpD is involved in the maturation of quinohemoprotein amine dehydrogenase (QHNDH), an enzyme that catalyzes the oxidative deamination of aliphatic and aromatic amines.185 QhpD installs two β-thioethers and one γ-thioether linkage between Cys and Asp/Glu residues in the small subunit of QHNDH, QhpC (Interpro family: IPR015084).185 In contrast to sactionines, which readily fragment under standard collision-induced dissociation (CID) conditions, no inter-ring fragmentation by CID was reported for the SCIFF-derived products,183,184 which is reminiscent of lanthipeptide-like behavior. Quantum mechanical calculations performed on model, isomeric peptides that were linked by either a sactionine or a β-thioether revealed that the β-thioether linked isomer was more stable, providing a possible explanation for the differential behavior of SCIFF-derived products vs. sactipeptides under CID conditions.43 Together, these data suggested that the thioethers formed on CteA and Tte1186a were not α-linked. Indeed, in vivo studies using a CteA variant labeled with deuterium at the α and β positions of Thr showed a mass loss of 2 Da upon co-expression with CteB, demonstrating that the modification yielded a γ-thioether.43 Thus, thermocellin (modified CteA),43,183 NxxC peptide (featuring one β-thioether between Cys and Asn),44 and freyrasin (featuring 6 β-thioethers between 6 Cys and 6 Asp residues)43 now form a new class of RiPPs termed ranthipeptides (
![[n with combining low line]](https://www.rsc.org/images/entities/char_006e_0332.gif) on-α
on-α  peptides) (Fig. 14 and 42). Ranthipeptides are distinguished from other thioether-containing RiPPs, such as lanthipeptides that display lanthionines (β-thioethers) installed by Michael-like nucleophilic addition (Section 3.1), and sactipeptides that include sactionines installed by rSAM enzymes (Section 3.9).43,44
peptides) (Fig. 14 and 42). Ranthipeptides are distinguished from other thioether-containing RiPPs, such as lanthipeptides that display lanthionines (β-thioethers) installed by Michael-like nucleophilic addition (Section 3.1), and sactipeptides that include sactionines installed by rSAM enzymes (Section 3.9).43,44
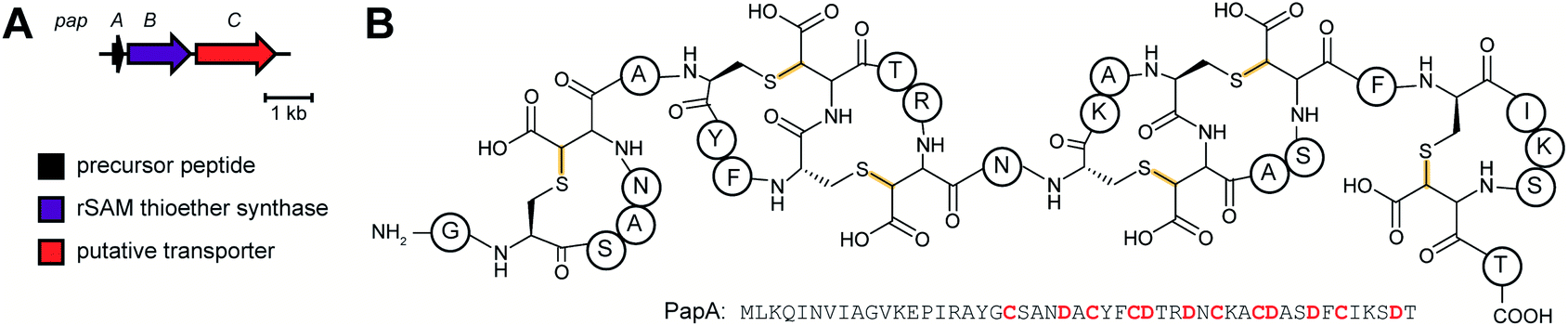 | ||
| Fig. 42 Ranthipeptide BGC and structure. (A) BGC of the ranthipeptide freyrasin. (B) Structure and precursor peptide sequence of freyrasin. The class-defining β-thioether linkages in the structure are yellow and the post-translationally modified Cys/Asp residues in the primary sequence are shown in red. | ||
CteB, originally proposed to be a sactisynthase, is the first ranthipeptide maturase and thioether-forming rSAM enzyme to be structurally characterized. The crystal structure of CteB (PDB: 5WGG) was obtained with bound SAM and a truncated version of the substrate CteA comprising residues 1–21 (CteA1–9 and 21 were resolved in the structure). This structure revealed three distinct domains consisting of the canonical rSAM domain flanked by an N-terminal RRE and a C-terminal AISC (SPASM) domain (Fig. 43).183 The rSAM domain displays the classic (α/β)6-TIM barrel fold and features the common [4Fe–4S] and SAM-binding motifs. The RRE domain interacts with CteA in a manner similar to that observed in other substrate bound crystal structures of RiPP modifying enzymes (Section 4.1.1).49,66,115,183 Fittingly, a PapB variant lacking the RRE domain was incapable of installing thioethers on the precursor peptide PapA when heterologously expressed in E. coli.186 The AISC domain of CteB houses two auxiliary [4Fe–4S] clusters. One of the clusters (auxiliary cluster II) is coordinated by four Cys in the SPASM domain, whereas auxiliary cluster I features an open coordination site which is bound to Cys21 of CteA.183 The presence of an open coordination site on auxiliary cluster I and its proximity to CteA suggests that the auxiliary cluster may be involved in the activation of the donor Cys thiol. With a computational model of CteB bound to full-length CteA, the authors showed that the donor Cys ligated to the [Fe–S] cluster and the acceptor Thr are indeed in sufficient proximity for thioether formation.183
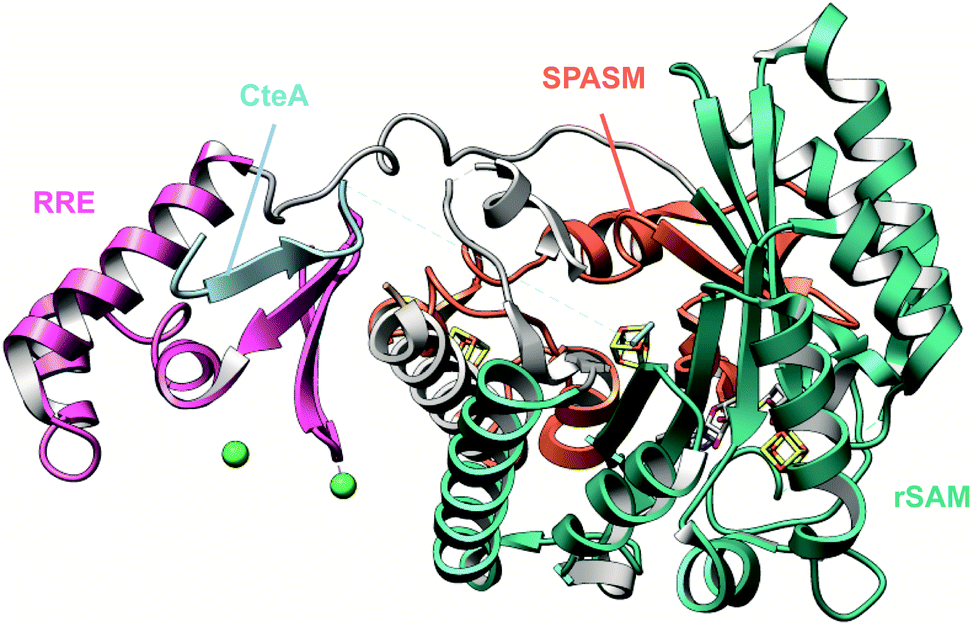 | ||
| Fig. 43 Structure of the ranthipeptide maturase CteB.187 CteB (PDB: 5WGG) exhibits a tripartite structure composed of rSAM, RRE and SPASM domains. The structure is colored according to domains. The [Fe–S] clusters and Ca2+ ions are shown as sticks and spheres, respectively. | ||
Substrate scope has been studied for two ranthipeptide maturases, NxxcB and PapB.44,186 Since ranthipeptide maturases are believed to use a homolytic mechanism for non-α thioether installation, the acceptor residues could theoretically be any amino acid with a side chain. Indeed, replacement of Asn20 involved in β-thioether formation in NxxC peptide with Gln, Asp, Glu, and Ala resulted in thioether bearing peptides with varying yields, indicating the ability of these enzymes to accept non-natural acceptor residues.44 However, PapB failed to install a β-thioether crosslink when the acceptor Asp was changed to Ala or Asn in a heterologous host.186 PapB installed a γ-thioether crosslink when an acceptor Asp was changed to Glu, similar to QhpD which generates both β- and γ-thioethers in QhpC.185,186 This observation also suggests that the carboxylate moiety in the acceptor residue may play a role in substrate recognition. PapB requires three intervening residues between the acceptor and donor residues for efficient thioether installation, but does not exhibit a preference for the identity of these residues. PapB also does not exhibit interdependency on ring-installation order.186
3.11 Streptide and rotapeptides: introduction and biosynthesis
Streptides are macrocyclic RiPPs characterized by C–C crosslinks between Lys and Trp residues, produced mainly by Streptococcal species (Section 2.5).188 They were initially identified as cyclic peptides in Streptococcus thermophilus LMD 9 culture supernatants at high cell densities in response to a novel quorum-sensing system involving the Rgg transcriptional regulator upon binding of a pheromone, SHP (hort ![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif) ydrophobic eptide).189,190 The precise macrocyclic linkage found in streptide, a covalent bond between Cβ of Lys and the aromatic C7 of Trp on a 9-mer peptide (Fig. 44), was elucidated in 2015 using a combination of high-resolution MS/MS, and multi-dimensional NMR analyses.188 A recent total chemical synthesis of streptide confirmed an R stereochemical configuration for the C3 position of Lys2.191 A locally encoded rSAM enzyme, StrB, was shown to be essential for streptide maturation with subsequent in vitro studies showing that C–C formation activity of StrB was LP-dependent. The modified product is proposed to be processed by StrC, possibly in coordination with other proteases, to produce the mature streptide product.188
ydrophobic eptide).189,190 The precise macrocyclic linkage found in streptide, a covalent bond between Cβ of Lys and the aromatic C7 of Trp on a 9-mer peptide (Fig. 44), was elucidated in 2015 using a combination of high-resolution MS/MS, and multi-dimensional NMR analyses.188 A recent total chemical synthesis of streptide confirmed an R stereochemical configuration for the C3 position of Lys2.191 A locally encoded rSAM enzyme, StrB, was shown to be essential for streptide maturation with subsequent in vitro studies showing that C–C formation activity of StrB was LP-dependent. The modified product is proposed to be processed by StrC, possibly in coordination with other proteases, to produce the mature streptide product.188
 | ||
| Fig. 44 Streptide BGC and structure. (A) BGC and precursor peptide sequence for streptide. The core peptide is shown in bold, and Lys and Trp residues crosslinked in streptide are shown in red. (B) Structure of streptide. The class-defining C–C crosslink is shown in yellow. | ||
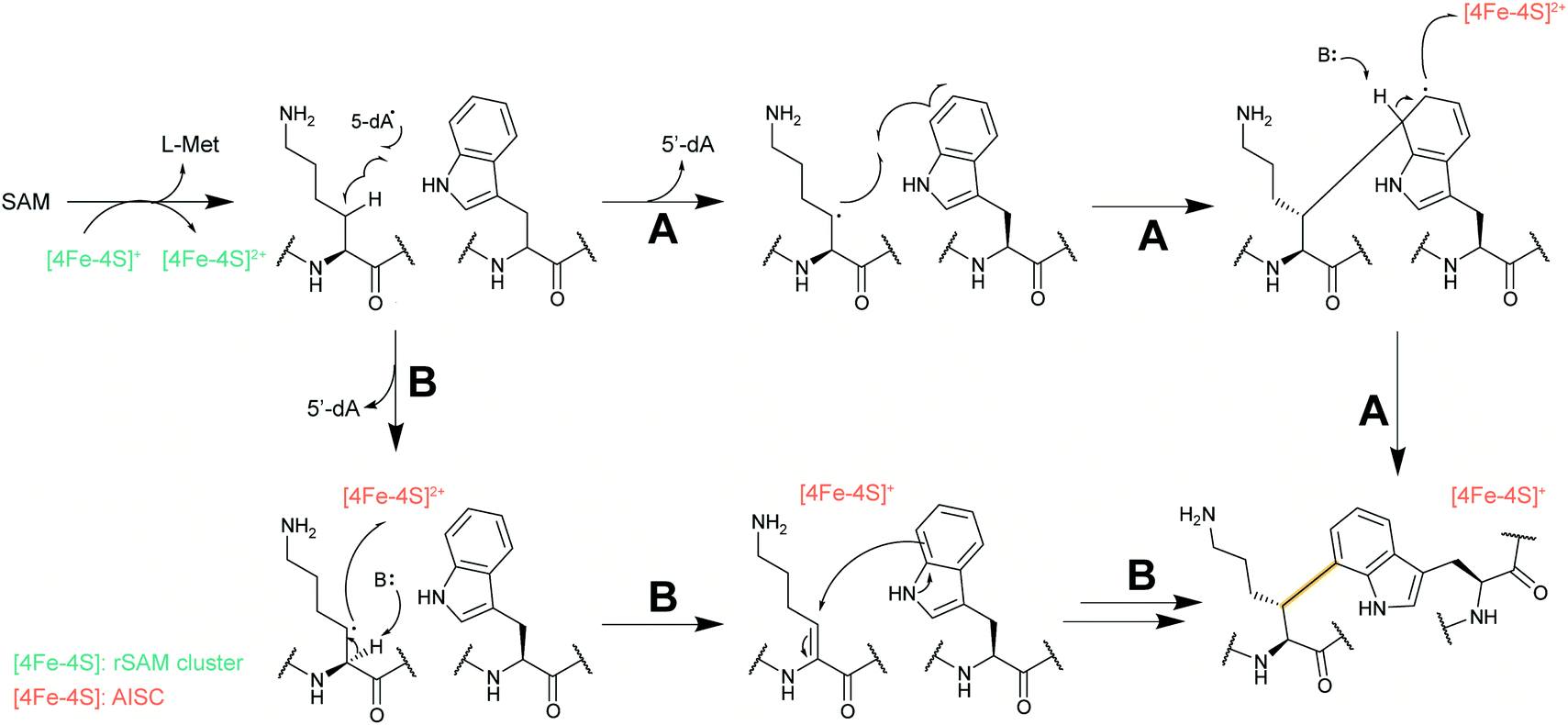 | ||
| Fig. 45 Proposed mechanistic scheme for rSAM-mediated C–C crosslinking in streptides. (A) Homolytic mechanism. (B) Heterolytic mechanism. In both mechanisms, transfer of an electron from the AISC to the SAM-binding cluster, directly or via an external agent, returns the enzyme to a catalytically competent state. | ||
Several crystal structures of SuiB were reported in 2017, which enhanced the understanding of streptide biosynthesis.193 SuiB, like CteB and SkfB (Sections 3.9 and 3.10), consists of an RRE-rSAM-SPASM tridomain architecture (Fig. 46). The N-terminal RRE domain adopts the wHTH-motif observed among other RiPP modifying enzymes to engender binding to their respective precursor peptides (Section 4.1.1).66,193 Unexpectedly, the SuiA-bound structure of SuiB revealed that the leader region of SuiA binds primarily through the bridging region that houses the first auxiliary [4Fe–4S] cluster rather than the RRE domain. These interactions are mediated by hydrogen bonding between the well-conserved LESS motif in the leader region of SuiA and the bridging region, which are expected to orient the core peptide in the active site for catalysis (Section 4.1.1, Fig. 70).193 Although it is possible that the RRE domains in SuiB could be vestigial, large scale conformational changes were observed in the RRE domain upon SuiA binding, which support interactions undetected in the crystal structure. The snapshot of the enzyme captured in the crystal structure plausibly represents a state where the RRE has released the precursor peptide upon reaching a certain stage of the catalytic cycle.193
 | ||
| Fig. 46 Structure of the streptide maturase SuiB bound to SuiA (PDB: 5V1T).193 SuiB exhibits a triple domain architecture akin to other RiPP modifying rSAM enzymes. The structure is colored according to domain and the [Fe–S] clusters are shown as sticks. | ||
Studies performed in the Sui and Aga pathways have revealed the promiscuous nature of the rSAM enzymes involved in streptide biosynthesis.192 SuiB and AgaB accept both SuiA and AgaA as substrates, suggesting that small C-terminal extensions and deletions are tolerated. Changes made to the Lys2 and Trp6 residues involved in the crosslinking are not generally tolerated, however a substrate containing the Trp analog W6Bzt (3-benzothienyl-Ala) reacted with a ∼4-fold reduction in rate. Changes made to the Asp in the conserved streptide motif “KGDGW” were also tolerated, with a 3-fold rate reduction. These studies revealed the tolerant nature of streptide synthases to accept natural and non-natural amino acids, which could be explored to generate unnatural streptide analogs.192 Although the Cβ of Lys is modified in streptide, removal of the ε-amino group of Lys is not tolerated, as evidenced by lack of modification on a norleucine variant. As a result, two roles were proposed for Lys2 in streptide biosynthesis. (1) Lys2 provides hydrogen bonding or ionic contacts to orient the substrate at the active site and, (2) Lys2 serves as a Brønsted base in the proposed catalytic mechanism.192 These hypotheses were tested in a subsequent study using AgaA Lys2 analogs that varied either in the side chain length or the pKa.194 If the ε-amino group of Lys was important for binding, then changes to side chain length, while retaining the amine, would diminish turnover. Alternatively, if the Lys2 served as a Brønsted base, then replacement of the Lys ε-amino group with –OH and –SH, that vastly increase the pKa yet retain H-bonding interactions, would negatively affect enzymatic activity. The enzymatic turnover was observed to be more dependent on the side chain length rather than the pKa of the side chain, supporting an anchoring role. The increased turnover observed in the NH2-group bearing analogs compared to the thiol- and hydroxy-bearing analogs suggests that the interaction may occur through salt-bridges.194 The crystal structure of SuiB into which crosslinked SuiA was modeled revealed a potential base (Glu319) within 5 Å of SuiA-Trp(6) that could be involved in the deprotonation step of the proposed mechanism.193 The E319A variant reduced kcat by ∼8-fold.194 However, the role played by Glu319, whether a catalytic base or an aid in the relay of protons from the active site, remains to be elucidated.
A bioinformatic survey conducted in streptococcal genomes using rSAM enzymes that occur in the context of a Rgg/Shp quorum sensing system revealed several groups of short peptides each with conserved motifs.24In vitro treatment of a synthesized precursor peptide from one such group, termed the WGK cluster, with the cognate rSAM enzyme from Streptococcus ferus, produced a novel streptide featuring a tetrahydro[5,6]benzindole moiety generated through the installation of two C–C crosslinks between Lys and Trp residues (Fig. 14).24 The mechanism employed by the corresponding rSAM enzyme, WgkB, to install two C–C linkages, is incompletely understood.
3.12 Proteusins and aeronamides: introduction and biosynthesis
Proteusins are a family of mostly genome-predicted195 bacterial peptides. Proteusin BGCs are noteworthy for their highly variable gene sets encoding maturation enzymes.Until recently, the only known proteusins were the polytheonamides from the uncultivated sponge symbiont ‘Entotheonella factor’. Polytheonamides are remarkably complex cytotoxic peptides carrying numerous modifications including D-amino acids, C-methylations, and N-methylations (Fig. 47) that induce the formation of a membrane-inserting β-helix pore. By heterologous expression, functions of all polytheonamide biosynthetic enzymes were characterized.28,196 PoyD represents a new family of radical SAM epimerases (see Section 5.1) and introduces 18 D-amino acid residues through irreversible L-to-D epimerization. Polytheonamide displays an alternating D/L pattern that is important for β-helix formation. The methyltransferase PoyE was shown to N-methylate eight Asn side chains.197 The timing of this modification was studied at different intervals of coexpression in E. coli, revealing a complex pattern of largely C-to-N-terminal processing.28N-Methylation occurs on epimerized Asn residues, but in E. coli, PoyE was also able to recognize L-Asn and Gln units on non-natural peptide variants. For natural polytheonamides, N-methylated residues are arranged in a linear fashion on the β-helix and are connected by amide hydrogen bond bridges that stabilize the secondary structure.198–200 Based on this pattern, extended artificial polytheonamide-based peptide variants were created with an inserted, Asn-containing repeat, which were N-methylated up to ten times.28 Coexpression studies in E. coli assigned the putative Fe(II)-2-ketoglutarate-dependent enzyme PoyI as responsible for hydroxylation of Val and N-Me-Asn side chains (four in total), but it was also able to recognize His in core peptide variants.28
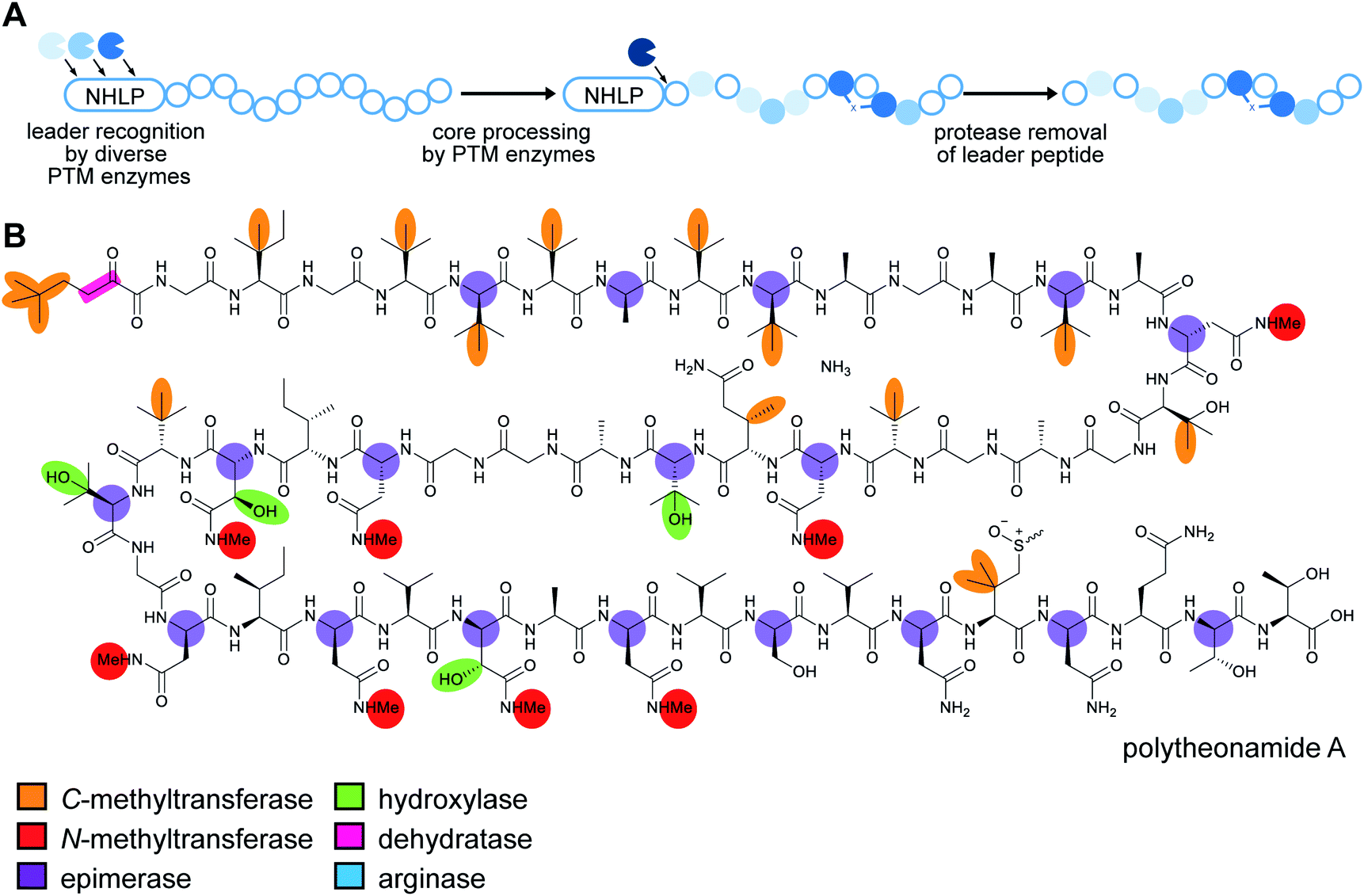 | ||
| Fig. 47 Proteusin biosynthetic logic and structure of polytheonamide A. (A) Overall biosynthetic pathway. (B) Structure of polytheonamide A. Since proteusins do not have class-defining PTMs (the NHLP is the class-defining feature, Table 1), a color-coded key identifies the diverse modifications that occur during the maturation process. NHLP, nitrile hydratase-like LP. | ||
Seventeen additional modifications in polytheonamides are C-methylations that occur on unactivated sp3 carbons of diverse amino acid residues within the PoyA core peptide. Likely candidates for catalyzing these methylations were PoyB and PoyC,28 which are both putative class B rSAM methyltransferases (MTs) that are characterized by the presence of an additional N-terminal cobalamin (Cbl)-binding domain.174,201 Indeed, confirmation of their role in PoyA polymethylation in heterologous expression experiments could only be achieved by switching from E. coli to a rhizobial host capable of producing Cbl (Section 5.3.2).28
Molecular dynamics calculations were performed on polytheonamide variants suggesting that the extended hydrogen bond clamp formed by precisely aligned N-methylated Asn are of major importance for the stability of the β-helix.200 Since six amino acid residues correspond to one helix turn, an NX5N repeat in the core sequence is necessary to align multiple Asn residues on the polytheonamide nanotube. This insight was used in a genome mining strategy to identify further candidates for polytheonamide-like BGCs in sequenced genomes.202 Searching for BGCs containing a gene combination for an epimerase, an N-methyltransferase, and a precursor with NX5N core repeats identified orphan BGCs in diverse sources. These comprise another sponge metagenome (a gamma-proteobacterium),203 a marine single-cell genome (Verrucomicrobia), a deep-rock metagenome (alpha-proteobacteria), and the culturable beta-proteobacterium Microvirgula aerodenitrificans from a wastewater purification community.202 For the latter organism, trials to identify a matching compound in cultures failed even after studies on a constructed reporter strain revealed conditions under which the biosynthetic genes are expressed. Products were ultimately accessed by introducing a His6-tagged precursor variant into the producer, isolating the uncleaved modified precursor, and releasing the modified core peptide in vitro with the purified peptidase of the pathway. This strategy provided the new polytheonamide congener aeronamide A (Fig. 48A), a potent cytotoxin carrying one dehydration, 21 D-residues, five N-methylated Asn residues, and seven C-methylations, with only traces of side products. Core peptide switching experiments confirmed the remarkable efficiency of M. aerodenitrificans in generating hypermodified peptides, highlighting its potential as a synthetic biology chassis.202 A core peptide from the deep-rock biome was converted to “polygeonamides” carrying up to 23 D-amino acids (the highest number observed to date) and multiple further modifications.
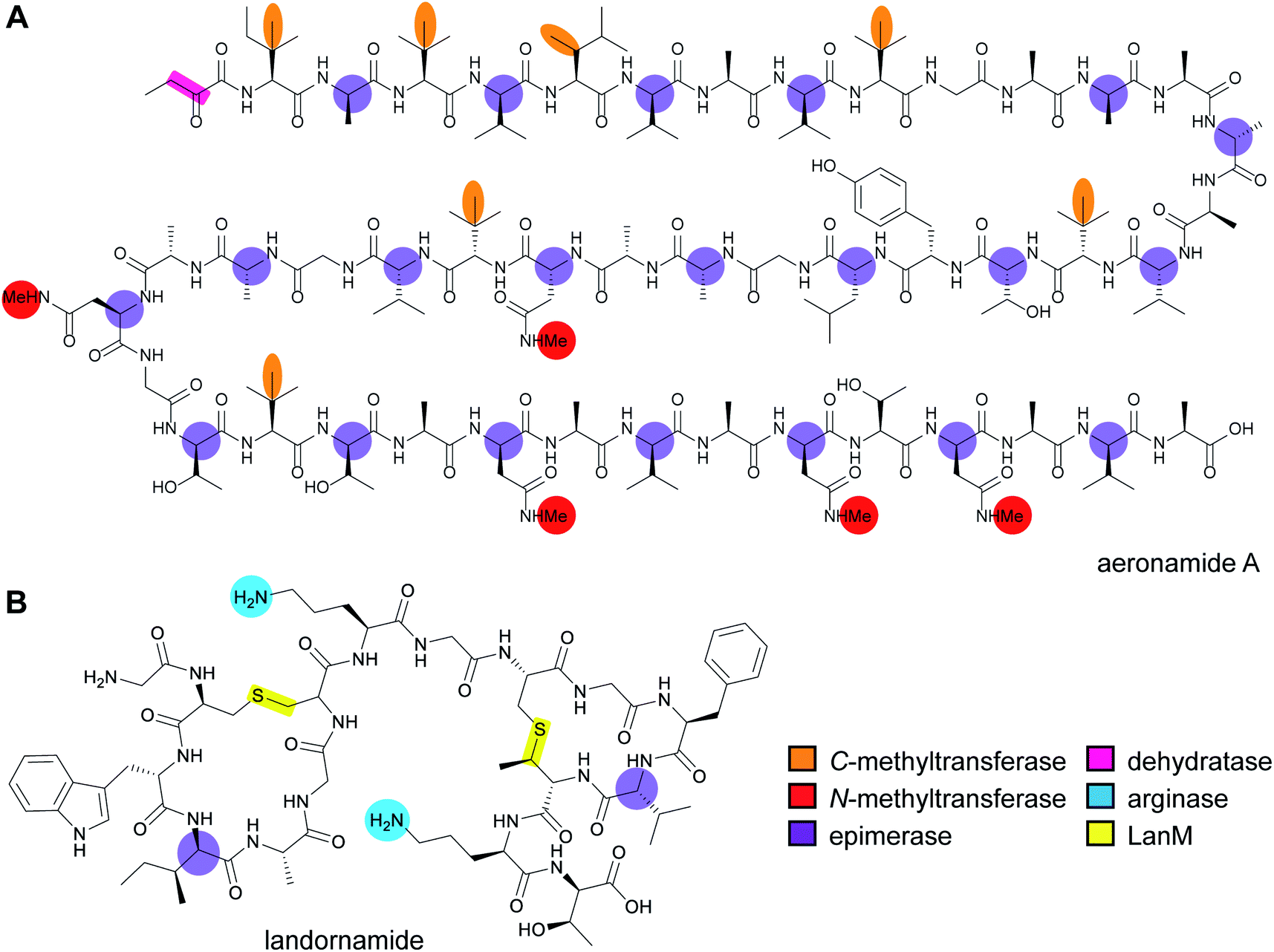 | ||
| Fig. 48 Structures of (A) aeronamide A and (B) landornamide. Since proteusins do not have class-defining PTMs (Table 1), the color coding is based on the enzymes that install each modification. | ||
Polytheonamide-type BGCs represent only a small fraction of the architecturally diverse proteusin gene clusters detected in bacterial genomes.195,204 Cyanobacteria are particularly enriched in proteusin BGCs, suggesting major potential for the discovery of structurally unusual bioactive natural products. One example is landornamide (Fig. 48B; see Section 5.4).
3.13 Epipeptides: introduction and biosynthesis
Epipeptides were discovered based on work showing that a peptide assigned to the yyd RiPP BGC of Bacillus subtilis induces a two-component system that regulates stress response.205 The yyd cluster (Fig. 49A) encodes, in addition to the precursor YydA, a single predicted maturase, YydG, with homology to rSAM enzymes of a previously unassigned family, but the nature of the modification remained unknown in the initial study. This function was recently identified in vitro using purified enzyme and a truncated version of the precursor peptide YydA.29 A mixture of isobaric peptides was obtained that carried deuterium at an Ile and/or a Val site when conducting the assay in a D2O-based medium. Further labeling experiments located the site of initial hydrogen abstraction as the α-carbon, and the nature of the conversion was identified as L-to-D-epimerization using synthetic peptide standards. The study also showed that replacement of a Cys residue in YydG by Ala resulted in an altered product profile with peptides containing ruptured backbone bonds at either side of the α-carbon. These and other data suggested the Cys as the source of the backside hydrogen introduced during epimerization of the substrate (Fig. 49B).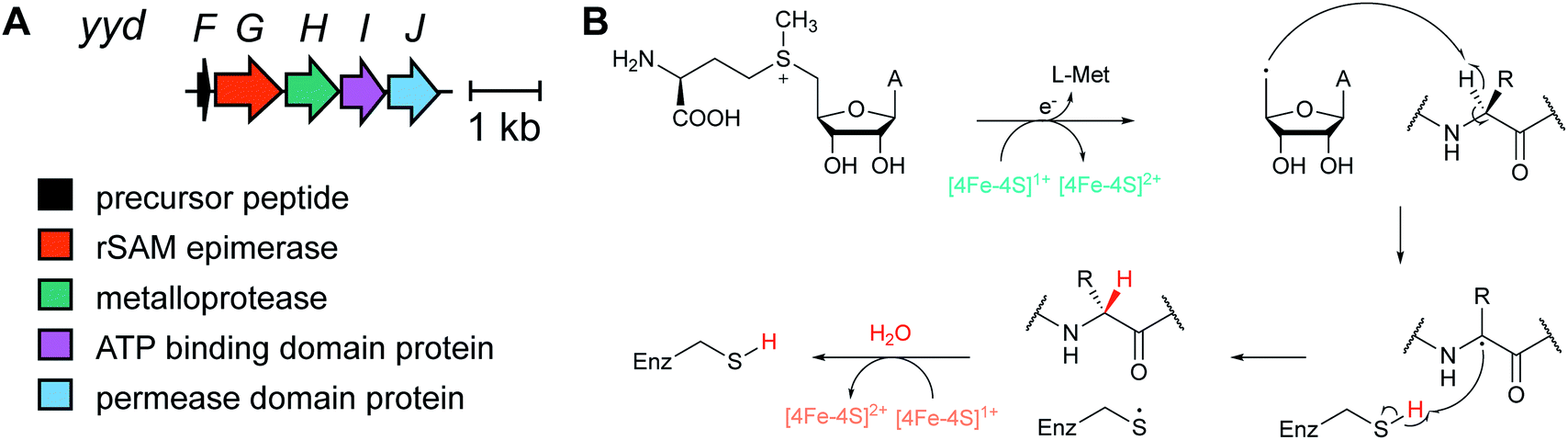 | ||
| Fig. 49 (A) BGC of yyd. (B) Proposed mechanism of YydG epimerization. YydG catalyzes substrate Cα hydrogen atom abstraction, resulting in the loss of amino acid residue stereochemistry. An active site Cys residue then donates a hydrogen atom to the resulting carbon-centered radical intermediate, resulting in an epimerized substrate. The AISC is believed to quench the resulting thiyl enzyme radical to regenerate the Cys H-atom donor residue. PoyD-type enzymes (Section 5.1.1) are thought to use a similar overall mechanism. | ||
3.14 Spliceotide biosynthesis and class-defining enzyme: radical SAM splicease
PlpX-type spliceases were discovered in genome studies aimed at characterizing the function of orphan rSAM genes present in several bacterial RiPP BGCs.41 The plp cluster (Fig. 50A) produces an as-yet unknown natural product and is located in the genome of the cyanobacterium Pleurocapsa. It encodes, among other proteins, one proteusin-like (NHLP) and two Nif11-type precursor peptides (see Section 4.4.1), a PoyD-like epimerase, and the protein pair PlpXY, of which PlpY is a small accessory protein with distant similarity to RREs (Section 4.1.1) that is required for PlpX-mediated maturation. Coexpression of plpXY with either of the two Nif11 precursor genes (plpA2 and plpA3; Fig. 50B) revealed an unusual modification resulting in an α-keto-β-amino residue (from hereon referred to as ketoamide) moiety, i.e., featuring a backbone that is extended by one carbon unit. Comparison of the peptide with the precursor gene sequence suggested that loss rather than addition of core peptide atoms resulted in this difficult-to-rationalize modification. The biosynthetic origin was established by incorporation studies using isotopically labeled precursors. These experiments revealed PlpA3 underwent an unprecedented peptide backbone splicing reaction, in which the rSAM enzyme removes all atoms of a C-terminal Tyr unit except the amide carbonyl (Fig. 50C). Further experiments suggested considerable biotechnological potential of this transformation. It was shown that at least two ketoamide moieties can be introduced into the same core peptide, that codon exchange provided access to a range of β-ketoamides (Gly, Ala, Val, Leu, Ser, Pro, Gln), and that the full-length core peptide was not needed for modification (in the case of PlpA3, truncation to 11 residues was possible).41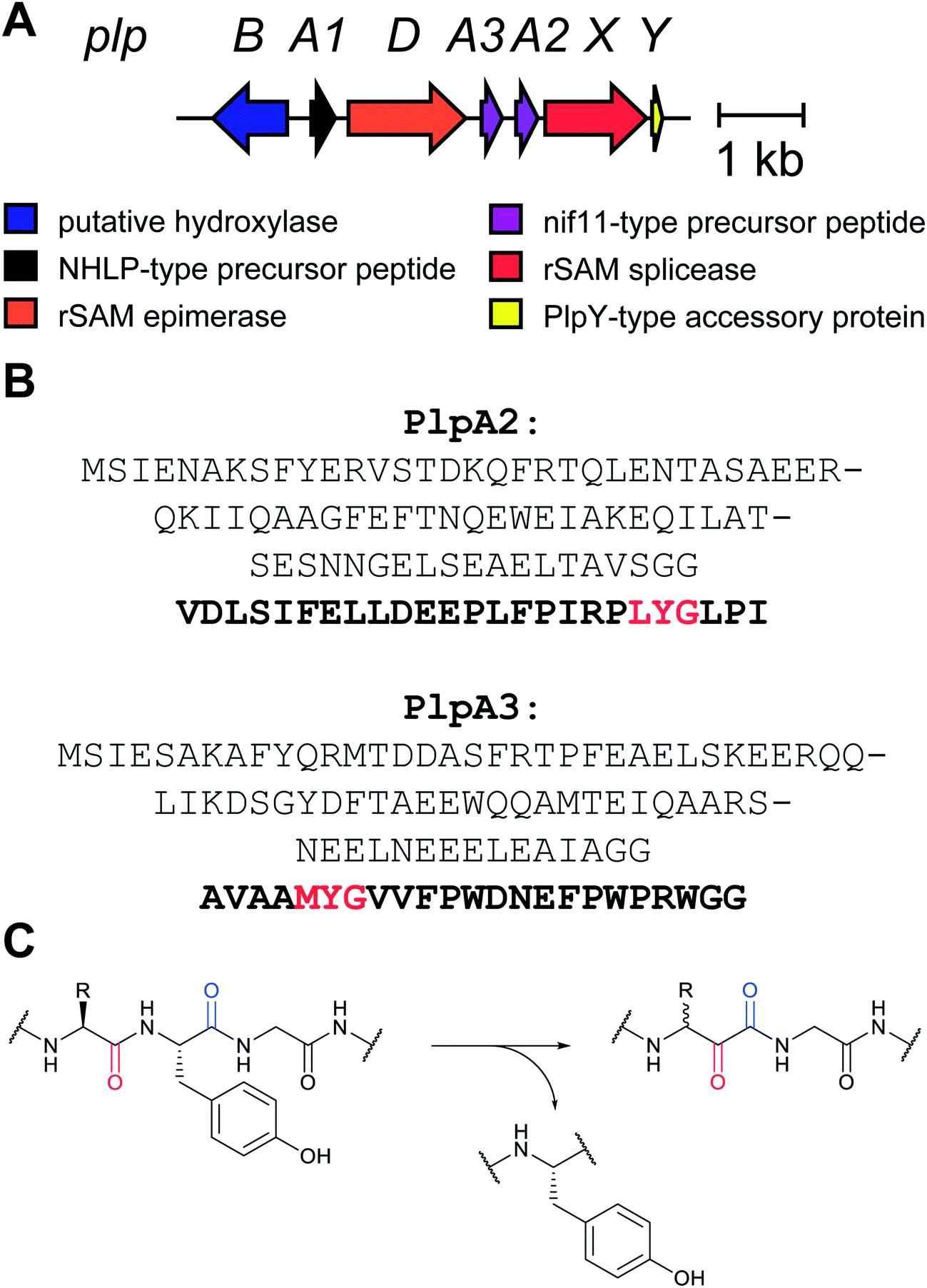 | ||
| Fig. 50 (A) The plp BGC. (B) Precursor peptides with the core peptide in bold and the spliced sequence in red. (C) Splicease-mediated tyramine excision and α-keto-β-amino acid generation. One tyramine equivalent is excised from the peptide backbone, which is subsequently reconnected to introduce a ketoamide moiety and extend the backbone by one carbon unit. The exact structure of the excised product has not yet been identified. | ||
3.15 Graspetides including microviridins: introduction and biosynthesis
Microviridins (Fig. 51) are compounds characterized by a cage-like structure, resulting from two lactone linkages between the side chain carboxyl group of Asp/Glu and the hydroxyl group of Ser/Thr, and a lactam linkage between the δ-carboxyl group of Glu and the ε-amino group of Lys. Microviridins inhibit serine proteases such as trypsin, chymotrypsin, subtilase, elastase, and the 20S proteasome, and have been recognized for their potential use in the treatment of pulmonary emphysema and some cancers.1 Their biosynthesis was elucidated using in vitro and in vivo studies by two independent groups.206,207 The lactones and lactams are sequentially installed on the precursor peptide by two dedicated ATP-grasp ligases, MvdD/MdnC and MvdC/MdnB (InterPro families: IPR026446 and IPR026439), that are homologous to the RimK ATP-grasp ligases involved in the maturation of the ribosomal protein S6.206,207 The cyclized precursor is then processed by a yet-unidentified leader peptidase. The liberated cyclized core peptide is typically acetylated at the N-terminus by a locally encoded N-acetyltransferase (Section 5.9.1) to yield the mature microviridin.206,207 Aside from canonical microviridins, the structurally related peptide, marinostatin, consisting of two macrolactones in a microviridin-like core motif TxRxPSDxDE has also been identified in Alteromonas sp. B-10–31.1 | ||
| Fig. 51 Graspetide BGC and structure. (A) BGC and precursor peptide sequence of the graspetide microviridin J. The N-acetyl transferase is located elsewhere in the genome of the producer. The core peptide is in bold. The residues involved in lactone and lactam modifications are in purple and pink, respectively. (B) Structure of microviridin J with the class-defining PTMs in yellow and secondary PTMs in cyan. | ||
Recently, the non-microviridin ATP-grasp-modified RiPPs plesiocin and thuringinin were produced via heterologous expression.208,209 Plesiocin possesses four hairpin-like bicyclic repeats formed by two intramolecular ester linkages on a conserved TTxxxxEE (x is any amino acid) motif.208 Thuringinin features three hairpin-like bicyclic motifs introduced by two ester linkages on a conserved TxxTxxxExxDxD core motif.209 While plesiocin is a serine protease inhibitor akin to the microviridins, the bioactivity of thuringinin remains unknown.1,208,209 Recent genome mining studies have revealed the existence of a plethora of topologically diverse RiPPs modified by ATP-grasp ligases and therefore have been described as omega-ester containing peptides (OEPs).209–211 While the term OEP is suitable to describe microviridins, plesiocins, and thuringinins, all of which feature an ATP-grasp ligase mediated macrolactone linkage, it may not be appropriate for molecules lacking the macrolactone but exhibiting the macrolactam linkage (also installed by ATP-grasp ligases). A more appropriate name for this RiPP class would be “graspetides”, which is reflective of the enzyme that installs the class-defining macrolactam and/or macrolactone modifications. Therefore, we reclassify microviridins, plesiocins, and thuringinins as specific members of the newly formed graspetide family and recommend usage of graspetide to address ribosomal peptides that feature macrolactones and/or macrolactams installed by ATP-grasp ligases.
 | ||
| Fig. 52 Structure of MdnA bound MdnC dimer (PDB: 5IG9).212 Monomer 1 is colored by domain and monomer 2 is shown in gray. | ||
Studies on MvdC and MvdD (MdnB and MdnC orthologs) have revealed that both enzymes permit limited variation in the core peptide.213 One pot reactions using constitutively activated MvdD and MvdC generated microviridin libraries targeted at serine proteases (Section 7.2).215 Recent bioinformatic analysis of 174 BGCs distributed across Proteobacteria, Cyanobacteria, and Bacteroidetes revealed a far greater diversity in graspetide precursors than previously appreciated, thus holding promise for even more substrate tolerant pathways.216 A distributive ATP-grasp ligase from Anabaena, AMdnC, for instance, processed three cores on the cognate precursor peptide, AMdnA, in vitro.217 This enzyme also produced a doubly macrolactonized peptide as the only product upon reaction with the non-cognate precursor peptide, MdnA. Furthermore, AMdnC also processed engineered variants of AMdnA containing up to four core peptides and installed up to eight macrolactones, highlighting the promiscuous nature of AMdnC and its potential use for the generation of graspetide analogs. The fourth “quasi” core peptide, obtained from the region between the first and second core peptide of AMdnA, was not always processed by AMdnC, and its role during the maturation process is unclear.217AMdnC follows an overall non-strict N-to-C directionality while possessing a strict order within each core peptide, installing the larger lactone first followed by the smaller lactone.217 The cluster also harbors AMdnB, a homolog of MdnB, and whether or not it processes AMdnA and lactonized AMdnA, is yet to be seen. It remains undetermined if the mature natural product is an exceptionally large graspetide with multiple core-sequence repeats or if the final compound is cleaved into separate, smaller microviridin-type products.
The multi-core architecture of plesiocin has been leveraged to impart dual functionality to the mature product by generating a variant that inhibits both trypsin and chymotrypsin.210 Leu3 in the TTLAIGEE core motif was identified to be the major determinant governing specificity to chymotrypsin which was then replaced with an Arg in the fourth core peptide to imbue anti-trypsin activity. TgnB, the thuringinin maturase, installs macrolactones in a distributive manner on TgnA.209,217 However, unlike MdnC and AMdnC, TgnB installs the smaller inner lactone first followed by the outer lactone.207,209,217 TgnB also exhibits selectivity for the acidic residues involved in crosslinking. The inner lactone requires a Glu while the outer lactone requires an Asp, which are also conserved among all predicted thuringinin precursor peptides.209
3.16 Borosin biosynthesis and class-defining enzyme: amide N-methyltransferase
The borosin RiPP family is a group of cyclic peptides distinguished by the presence of α-N-methyl groups, which are introduced by SAM-dependent N-methyltransferases (N-MTs).25,26,218 These borosin N-MTs are fused as N-terminal domains to their respective substrate core peptides (Fig. 53A) and catalyze the iterative, autocatalytic N-methylation of peptide residues, representing the defining enzymatic feature of the borosin family. Additional amino acid residues are encoded in a follower region, located C-terminally of the core peptide (Fig. 53A) that is proposed to perform a similar role as LP sequences in directing recognition between PTM enzymes and their substrates.26 It has been suggested that the covalent attachment of the substrate to the enzyme, also seen for the lyciumins (Section 2.9), may increase the local concentration of the very hydrophobic (and thus insoluble) core peptide.219 | ||
| Fig. 53 (A) Sequence of the N-MT for the biosynthesis of omphalotin A illustrating the enzymatic domain (green), LP (blue), core peptide (orange), and follower peptide (grey). (B) Crystal structure of OphA-DeltaC6 (PDB: 5N0Q)220 with the clasp domain of monomer A in light blue, the remainder of monomer A in dark blue, and monomer B in green. | ||
The best-studied member of the borosins is omphalotin A (Fig. 7), which is produced by the basidiomycete fungus Omphalotus olearius and is a potent nematicide (LD50 of 2 μg mL−1) that is active against the plant pathogen Meloidogyne incognita.221,222 Omphalotin A is derived from OphMA, which was discovered by two groups concurrently by mining the O. olearius genome for a gene candidate encoding the omphalotin peptide scaffold.25,26 Electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) analysis of full-length OphMA expressed in E. coli resulted in the detection of a range of multiply methylated species, with up to 11 methylations reported. Such mass shifts were not detected in a variant product lacking the C-terminal omphalotin-encoding region. Further tandem MS and electron-transfer dissociation (ETD) fragmentation analyses unambiguously localized nine methylation sites to nitrogens in omphalotin peptide bonds, with two-additional methylations located C-terminally to the core peptide.25 Observed methylations extend from the N- to the C-terminus suggesting specific directionality for the MT activity. Similar experiments using a fungal heterologous expression host also detected an 11-fold methylated product, and co-expression with the associated protease, OphP, generated products with up to nine methylations, consistent with the published omphalotin A structure.26 Based on homology with other homodimeric SAM-dependent MTs, it was proposed that residues Tyr98 and Ser129 might be located within the OphMA SAM-binding pocket. Consistent with this hypothesis, the substitution of either residue with Ala abolished OphMA methylation activity. OphMA was shown to likely associate as a homodimer, raising the question of whether monomer automethylation is intra- or intermolecular. To distinguish between the two possibilities, inactive OphMA variants Y98A and S129A were transiently co-expressed with a catalytically active enzyme carrying a core peptide variant.25 The mutants were shown to harbor up to ten α-N-methylations, supporting an intermolecular autocatalytic activity for OphMA.
In order to determine sequence requirements for methylation, OphMA variants were also examined in vivo. Removal of the C-terminal follower sequence still resulted in methylation, albeit incomplete, and introduction of a TEV protease cleavage site upstream of the core peptide sequence was also tolerated.25 The activity of the catalytic domain of an OphA homolog from Dendrothele bispora, dbOphMA, was also unaffected by the absence of the follower peptide sequence, for which it had no measurable affinity in fluorescence polarization experiments.223 The dbOphMA N-MT domain was shown not to modify free substrate peptide variants, suggesting N-methylation likely occurs because of the covalent linkage of the catalytic domain to its cognate substrate and homodimer formation. It is postulated that the follower peptide sequence functions instead as a recognition element for the subsequent action of the cluster-encoded peptidase that liberates the mature borosin product. The substitution of various dbOphMA core peptide residues with Pro led to reduced or abolished methylation activity, suggesting OphMA homologs struggle to accept residues that are conformationally challenging to bind. However, replacing the OphMA and dbOphMA core peptides with sequences resembling N-methylated peptides of non-ribosomal origin (e.g. cyclosporine A) did result in productive methyl transfer, albeit incompletely with respect to their natural product equivalents and with shifts in the native pattern of methylation.25,223 This observed lack of substrate tolerance is thought to be due to steric hindrance at the nitrogen atom caused by the presence of non-native amino acids larger than Val/Thr in the core sequences tested. Together these results, and a subsequent study on core peptide variants that led to mixtures of products,219 suggest limited substrate flexibility concerning both the core and flanking sequences among OphA homologs.
Recent crystallographic studies of truncated OphMA and dbOphMA variants and active site mutants have revealed that OphMA forms a complex catenane structure in which homodimers intertwine to methylate each other's C-termini (Fig. 53B).220,223 The N-MT domain is separated from the core sequence in each cis monomer by a ‘clasp’ domain that wraps around the adjacent subunit's N-MT domain to position the core peptide into the other subunit's active site. Mutation of residues identified as being responsible for substrate peptide binding significantly reduced or abolished methylation activity, confirming their catalytic significance.220 Combined with quantum-mechanical calculations and in vitro experiments, the current working mechanistic proposal for OphMA activity begins with deprotonation of the amide bond by a water molecule or enzyme base (Fig. 54). A tyrosine residue is suitably positioned for this function.220,223 The deprotonated nitrogen atom of the resulting imidate intermediate is then well-positioned for SN2 attack on the SAM methyl group. S-adenosyl homocysteine (SAH) is produced as a byproduct of this reaction and is thought to then be displaced by another molecule of SAM to enable a subsequent amide methylation.220,223 The side chains of the core peptide do not form specific interactions with the enzyme, possibly providing another explanation why the substrate is covalently attached to the protein.220
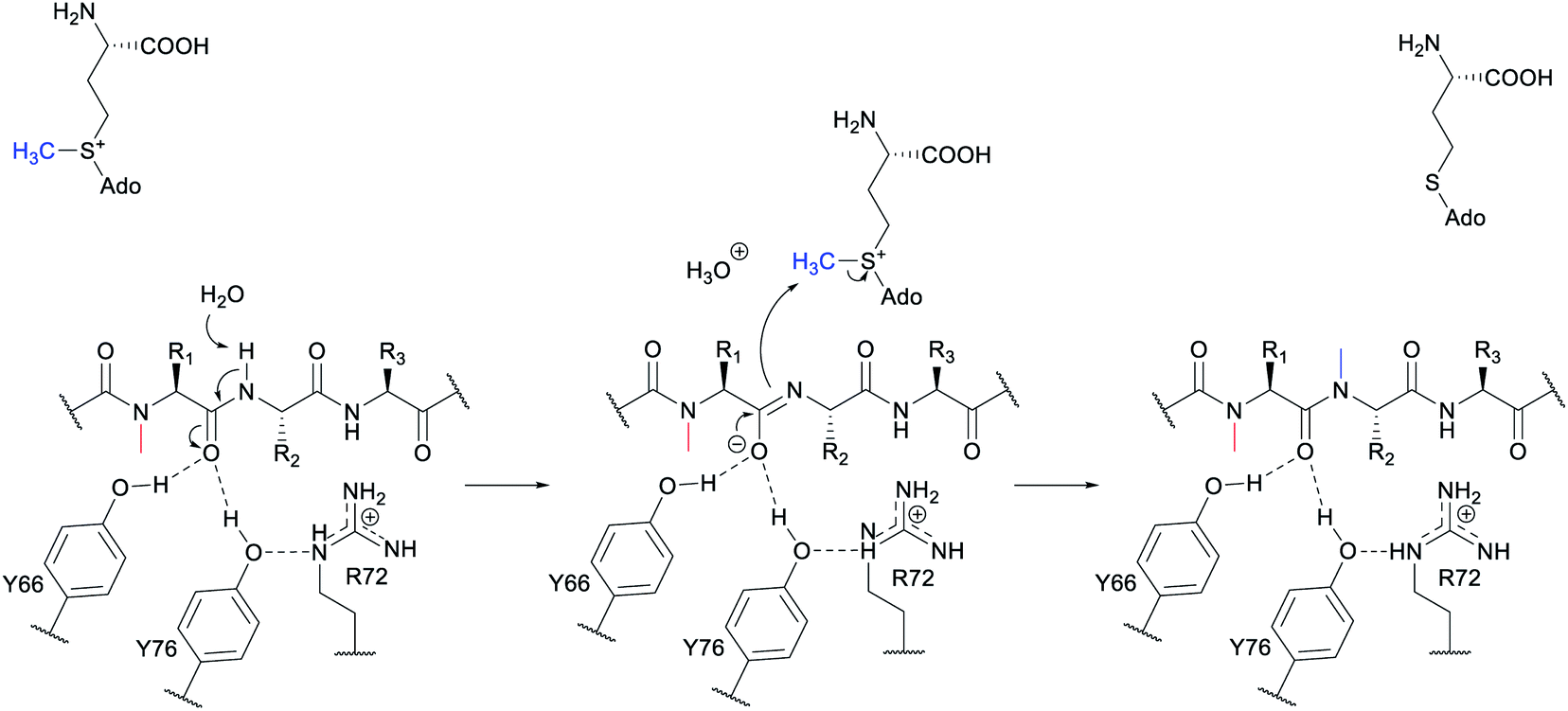 | ||
| Fig. 54 Proposed mechanism of OphMA methylation. A substrate amide proton is removed by a water molecule acting as a base. The resulting imidate is stabilized by H-bonding to Tyr66 and Tyr76.220 Arg72 is hypothesized to stabilize the tyrosinate that results from proton removal, suggesting the existence of an imidic acid intermediate. Subsequent SN2 attack on the SAM methyl group by the imidate completes the transformation. | ||
3.17 Dikaritins: introduction and biosynthesis
Prior to 2014, fungal RiPPs were restricted to the amatoxins produced by Basidiomycetes (Section 3.18). The discovery that ustiloxins are RiPPs provided the first example in Ascomycetes.224 The ustiloxin B biosynthetic gene cluster was identified based on the concurrent expression of contiguous genes in the genome of Aspergillus flavus, which had not been known to produce ustiloxins. Analysis of the BGC demonstrated a precursor peptide UstA with 16 repeats of a core peptide that corresponded to the sequence of ustiloxin B (Fig. 55A and B).15 These core peptides are flanked by repeats that are likely recognition sequences, and UstA also contains a signal peptide that likely sends it to the endoplasmic reticulum and the Golgi apparatus, after which the peptide is thought to be processed by the endoproteinase Kex2 at the C-terminal side of a Lys–Arg sequence (Fig. 55B).225 In addition to the precursor peptide, the BGC contains 14 genes (Fig. 55A). Gene disruption experiments supported the association of many of these genes with ustiloxin production, with the two proteins that have no characterized homologs, UstYa and UstYb (Protein family: PF11807; DUF3328; InterPro family: IPR021765), being required for production.17,226 Sequence alignment of more than 200 UstY homologs that were all found in aspergilli suggested N-terminal transmembrane helices.17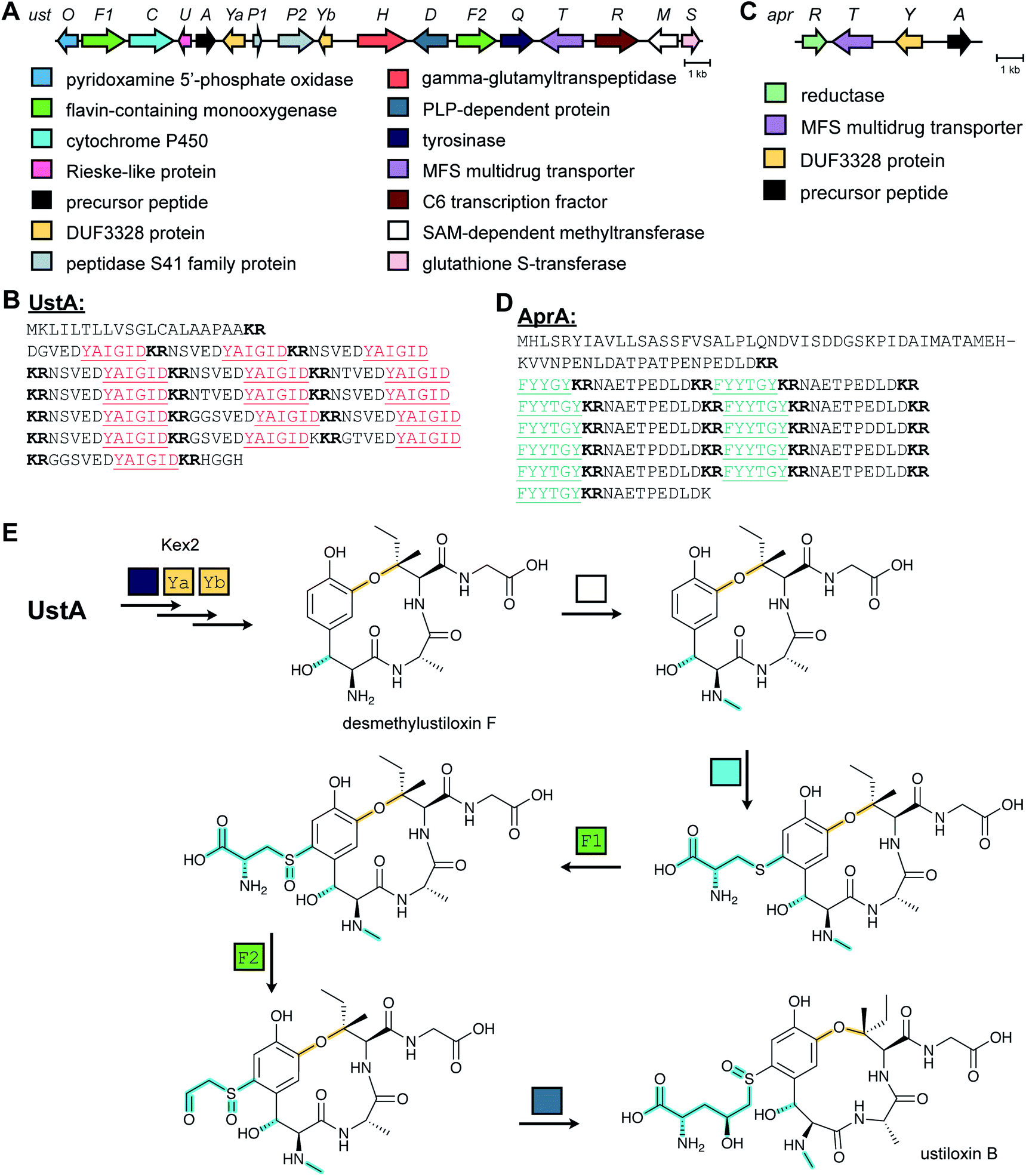 | ||
| Fig. 55 (A) BGC for ustiloxin biosynthesis. (B) Sequence of the ustiloxin precursor peptide showing the LP (top row), the repeats of the core peptide (red) and recognition sequences (black), and the predicted Kex2 protease processing site at the C-terminus of each core sequence (bold). (C) BGC for asperipin 2a. (D) Sequence of the asperipin 2a precursor peptide showing the LP (top 2 rows), the repeats of the core peptide (teal) and recognition sequences (black), and the predicted Kex2 protease processing site at both the N- and C-terminus of each core sequence (bold). (E) Proposed biosynthetic pathway to ustiloxin B. The class-defining PTM is yellow and secondary PTMs are cyan. | ||
Using an algorithm developed by the authors to detect highly repeated peptide sequences, other examples of genes encoding a repeated core peptide with an N-terminal signal peptide were detected in Aspergillus and Fusarium as well as the rice pathogen Ustilaginoidea virens, the producer of ustiloxins A and B.227 Homologs of all genes except those encoding the peptidases UstP and UstH were observed in the BGC for the latter. The UstA precursor peptide in U. virens contains eight core peptide repeats of which five have the sequence YVIG and three have the sequence YAIG, consistent with observed production of both ustiloxins A and B.227
The bioinformatic analysis of fungal genomes also resulted in the discovery of new or proposed compounds.17,228 Precursor peptides containing a signal peptide and repeated core peptides were observed frequently in the neighborhood of homologs of the UstYa and UstYb proteins and as many as 96 ustiloxin precursor peptide/UstY pairs were identified.17 At least 38 different types of repeat sequences were reported that may produce a wide variety of post-translationally modified products. One example from A. flavus was further characterized and the BGC consisting of just four genes (Fig. 55C) was shown to produce the new RiPP asperipin-2a, which contains a paracyclophane structure (Fig. 3).17,18 Interestingly, the core peptides of asperipin-2a are flanked on both sides by Lys–Arg motifs that are believed to be processing sites for the Kex2 protease (Fig. 55D).
 | ||
| Fig. 56 (A–C) Plant-derived compounds with structural homology to asperipin 2a. (D) BGC for phomopsins. (E) Sequence of the phomopsin precursor peptide showing the LP (top 2 rows), the repeats of two different core peptides (green) and recognition sequences (black), and the predicted Kex2 protease processing site at the C-terminus of each core sequence (bold “KR”). | ||
Other fungi produce several other groups of RiPPs that are structurally related to the ustiloxins such as the epichloëcyclins and the phomopsins. Epichloë endophytes of grasses produce cyclic peptides in planta from a ribosomal precursor GigA that, depending on the Epichloë species, has 1–8 repeats of a core peptide flanked by a Lys–Arg sequence.231 At present, the structure of the epichloëcyclin products is not known but MS analysis indicates they are cyclic and contain dimethylated Lys. The cyclization was suggested to occur between a Tyr and an N-terminal Ile. The biosynthetic genes remain unreported. Phomopsins are a group of hexapeptide mycotoxins produced by the pathogenic Ascomycete Phomopsis leptostromiformis and are generated from a BGC that resembles that of the ustiloxins (Fig. 56D). They contain a similar macrocycle as the ustiloxins formed by crosslinking of Tyr and Ile residues. In addition, the phomopsins contain several dehydro amino acids (Fig. 3). Phomopsins derive from a precursor peptide that contains multiple core repeats flanked by a Lys–Arg motif that is converted to the final product(s) (Fig. 56E).16 The precursor peptide PhomA from P. leptostromiformis contains five copies of a sequence that are converted to phomopsin A and three additional copies of a sequence that produce phomopsin P (Fig. 56E). The BGC also contains a tyrosinase (PhomQ), two S41 family proteases (PhomP1,P2), a SAM-dependent methyltransferase (PhomM), a zinc finger type regulator (PhomR), five homologs of the abovementioned UstY protein (PhomYa-e), and several proteins of unknown function (Fig. 56D). At present, the activities of the PhomY proteins are not known but they are likely involved in generation of the four dehydro amino acids and the formation of the macrocycle. The methyltransferase activity was investigated in vitro (see Section 5.3.5).16 The class-defining features of the dikaritins are the ether crosslinks introduced by the UstY homologs.
3.18 Amatoxin biosynthesis and class-defining enzyme
Amatoxins such as α-amanitin are N-to-C cyclized peptides produced by mushrooms (Fig. 57). They also contain Cys–Trp crosslinks (tryptathionine) and various hydroxylations. Amatoxins are produced as a linear precursor peptide that contains a leader peptide, core peptide and C-terminal recognition sequence.1 Several recent studies have provided new insights into the biosynthesis of the amatoxins (Fig. 57). Given that the core peptides for α-amanitin are flanked by Pro residues, the involvement of a prolyl oligopeptidase (POP; InterPro family: IPR001375, IPR002470) had been previously proposed in amatoxin biosynthesis, but cyclization had not been experimentally verified. In vitro activity of a POP from Galerina marginata showed the enzyme catalyzes two reactions during the biosynthesis of α-amanitin.232 The enzyme was expressed in Saccharomyces cerevisiae, and the purified protein first hydrolyzed the 35-amino acid precursor peptide C-terminal to an internal Pro to release a 25-amino acid C-terminal peptide (Fig. 57). Subsequently, the enzyme performed a transpeptidation at a second Pro to generate a cyclic octamer. The two processes were not processive in vitro as the product of the first step was released. Furthermore, synthetic peptides corresponding to the 25-mer and analogs were cyclized. These experiments identified a minimum sequence required for proteolysis at the first Pro, and for cyclization upon proteolysis at the second Pro. Kinetic experiments showed that the enzyme does not have much preference of the full length peptide over the 25-mer. In a follow-up paper, the utility of GmPOPB was demonstrated by testing 127 novel peptide substrates, 100 of which were cyclized (see Section 7.4.1).233 The authors also demonstrated that the activity is specific for the POP investigated (termed POPB) as a second POP encoded in the genome (POPA) did not catalyze cyclization. POPA enzymes are present in all species of Amanita and Galerina whereas POPB was only associated with amatoxin producing species. | ||
| Fig. 57 POPB-catalyzed cyclization of the core peptide during the biosynthesis of α-amanitin. The class-defining PTMs are highlighted in yellow with secondary PTMs highlighted in cyan. | ||
A detailed kinetic analysis of GmPOPB showed that product release is ordered with the cyclic peptide departing before the follower peptide.234 The dissociation of the latter is slow, partially limiting the overall turnover rate. A subsequent crystallographic study provided insights into the various steps and explained why the product of the first step needs to be released prior to rebinding in a different conformation that enables cyclization.235 These observations led to the design of optimized simpler substrates that may be beneficial for biotechnological applications. At present, the mechanisms of tryptathionine formation and side chain hydroxylation of Trp, Pro and Ile have not yet been elucidated. The combination of POP and the unidentified tryptathionine synthase is the class-defining feature of amatoxins.
3.19 Biosynthesis of N-to-C cyclized peptides including cyclotides and orbitides
Head-to-tail cyclized RiPPs include large peptides236,237 and also many cyanobactins from cyanobacteria (Section 3.6), the fungal amatoxins (Section 3.18), and the plant-derived cyclotides and orbitides.1 In recent years, several of the enzymes that catalyze the cyclization step have been experimentally characterized. The Pro oligopeptidase involved in amatoxin cyclization was discussed in Section 3.18, and structures of the cyanobactin macrocylase PatG (Section 3.6) were solved in 2012.238 This section will discuss the newly discovered or newly characterized members of the plant asparaginyl endoprotease family (AEP, C13 protease family; InterPro family: IPR001096) including butelase 1 (ref. 239) and OaAEP1b,240 a cyanobactin macrocyclase from Oscillatoria sp. PCC 6506 (OscGmac),241 an AEP from Canavalia ensiformis (jack bean, CeAEP1) that cyclizes sunflower trypsin inhibitor (SFTI),242 and the orbitide cyclase PCY1.243OaAEP was isolated from the cyclotide-producing plant Oldenlandia affinis and was recombinantly expressed in E. coli, providing the first example of an active cyclization AEP generated by heterologous expression.240 It was demonstrated to cyclize a diverse array of peptides including native kalata substrate precursors, but unlike POPB (Section 3.18) the enzyme did not catalyze the N-terminal cleavage step required for cyclization to take place, suggesting another protease may conduct this step. Use of two enzymes for removing the leader peptide and cyclization seems more common as it is also observed for the patellamides (PatA and PatG, Section 3.6) and orbitides (see below). The catalytic parameters of OaAEP are lower compared to those of butelase 1. Recently a group of more efficient enzymes that demonstrate cyclization activity at both acidic and neutral pH, termed canonical AEP ligases (to distinguish them from AEPs that have pH dependent ligation or hydrolysis activities), was reported.247
PatG (also called PatGmac) is a subtilisin-type serine protease involved in macrocyclization of the cyanobactin patellamide (Fig. 35). Genome mining studies identified a putative cyanobactin gene cluster in the genome of Oscillatoria sp. PCC 6506. Its macrocyclase OscGmac was expressed in E. coli and purified.241 The enzyme showed higher substrate tolerance than PatGmac and was used to cyclize peptides of 6–30 amino acids, including D-amino acids.
Cyclization of the orbitide1 segetalin A is catalyzed by a member of the S9A family of serine proteases, PCY1, isolated from the plant Saponaria vaccaria.243 This family includes the prolyl oligopeptidases (see Section 3.18) involved in head-to-tail cyclization of amatoxins, but PCY1 does not cleave adjacent to Pro residues. As noted above for OaAEP1b, PCY1 also requires an additional protease (OLP1) to remove an N-terminal peptide before it can cyclize a peptide with a free amino terminus (Fig. 58).243 Mutagenesis experiments demonstrated that substrate recognition is governed by an 11–16-residue follower peptide that is removed in the cyclization step.243 The crystal structure of PCY1 (PDB: 5UW3) showed that the enzyme utilizes the cleaved C-terminal follower peptide of the substrate to attain a conformation that promotes macrocyclization.248 This finding explained why the enzyme produces nine different orbitides of different sequence and ring size from substrates that have highly conserved follower sequences with especially high conservation in the C-terminal six residues that were visualized in the structure. Unlike the mostly hydrophobic interactions used for LP recognition in the biosynthesis of class I lanthipeptides, cyanobactins, lasso peptides and PQQ (see Section 4.1.1), the follower peptide/recognition sequence binds to PCY1 via well-defined hydrogen bonds that result in closure of the active site with a large internal cavity that promotes macrocyclization and excludes water to prevent competing hydrolysis.248
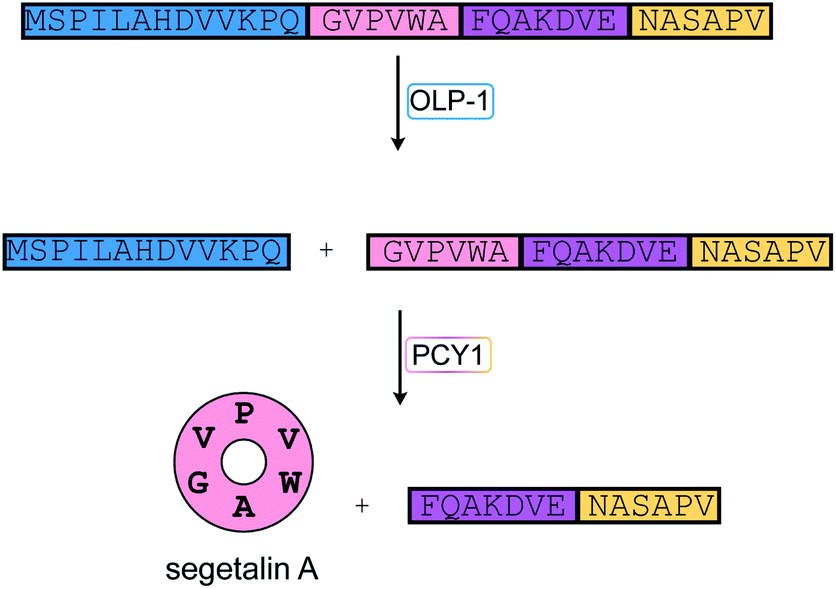 | ||
| Fig. 58 PCY1-catalyzed cyclization of the core peptide during the biosynthesis of the orbitide segetalin A. The endopeptidase OLP1 first removes the LP (blue). The highly conserved 6-amino acid terminal peptide (yellow) makes key interactions with PCY1 in the second proteolytic step to promote intramolecular trapping of the initial acyl-enzyme intermediate by the N-terminal amine. | ||
One of the advantages of PCY1, despite slow turnover rates (∼0.12 s−1 to 1 h−1),243,249 is the high substrate tolerance and the ability to produce small macrocycles (5–9 residues). A structure of an enzyme variant in which the active site Ser had been replaced by Ala showed electron density for part of the core peptide of pre-segetalin B1 in addition to the follower peptide. Additional binding analysis suggested that a shorter C-terminal tail might still result in sufficient affinity for catalysis. Therefore, a minimal substrate follower sequence of just three residues was designed and demonstrated to support macrocyclization.249
In contrast to the advances in the understanding of the cyclization processes of the amatoxins, cyclotides, orbitides, and cyanobactins, this step is still poorly understood for the circular bacteriocins.236,237 Recent genetic studies have investigated the importance of the leader peptide sequence and the potential involvement of specific genes,250,251 but no definitive information regarding the molecular details have emerged. These studies do suggest that leader peptide removal and cyclization are two separate processes.
3.20 Biosynthesis of pheganomycin/guanidinotides
Four enzymes including a type III PKS are known to catalyze the formation of 2-(3,5-dihydroxyphenyl)-2-oxoacetic acid during the biosynthesis of vancomycin.252 Reasoning that this compound is only a few putative steps away from the N-terminal structure of pheganomycin (Fig. 4A), a search for orthologs of these enzymes resulted in the identification of the pheganomycin BGC encoding the PGM9–11 proteins as well as an additional 18 genes nearby (Fig. 59A).19 One of these genes encodes the novel peptide ligase, PGM1, that was shown to catalyze amide bond formation between (S)-2-(3,5-dihydroxy-4-methoxyphenyl)-2-guanidinoacetic acid and a peptide derived from the precursor peptide encoded by pgm2 (Fig. 59B). PGM1 is an ATP-grasp enzyme (defined in Section 3.15) that connects a ribosomally synthesized peptide with the α-guanidino acid. The enzyme was demonstrated to be tolerant to changes in both the amino acid and the peptide allowing preparation of pheganomycin analogs. This substrate tolerance was analyzed by site-directed mutagenesis and structure determination of the enzyme (PDB: 3WVQ and 3WVR).19 PGM4, PGM7, PGM8 and PGM12 were hypothesized to convert 2-(3,5-dihydroxyphenyl)-2-oxoacetic acid to the N-terminal guanidino acid, whereas PGM3, a rSAM methyltransferase, was proposed to C-methylate Val to tert-Leu (see Section 5.3 for similar transformations). Finally, PGM14 may be involved in excision of the core peptide from the precursor PGM2. Thus, pheganomycin was shown to be the first example of a ribosomally synthesized peptide that is subsequently linked to a non-proteinogenic amino acid formed in part by a type III PKS. This discovery was later followed by the reports of other hybrid combinations of RiPP and non-RiPP pathways (e.g. Sections 3.1.5 and 3.24).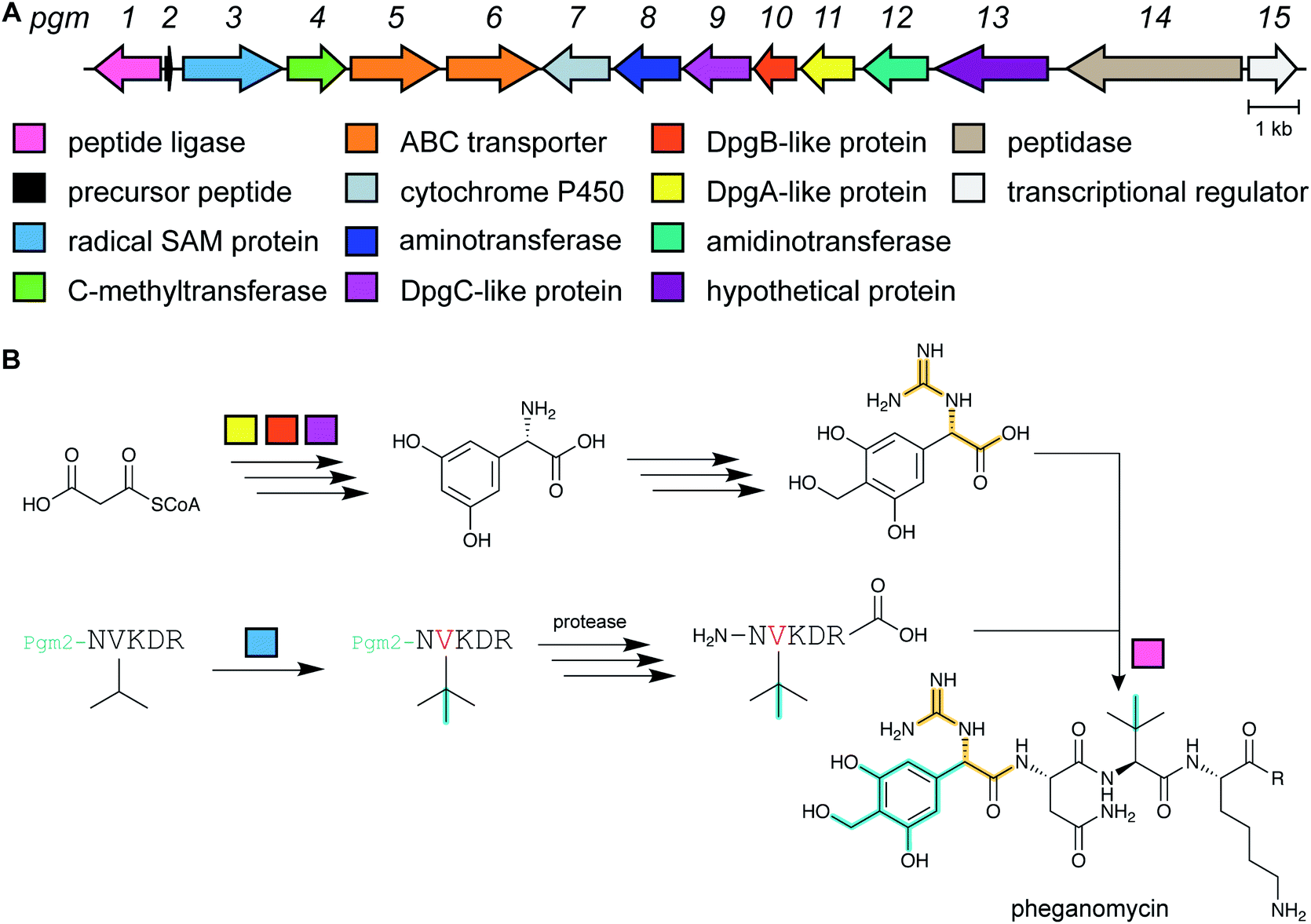 | ||
| Fig. 59 (A) BGC for pheganomycin. (B) Convergent biosynthetic pathway to pheganomycin in which the ATP-grasp ligase PGM1 connects an α-guanidino acid to a ribosomally synthesized peptide that likely is C-methylated in a prior step by a rSAM methyltransferase (PGM3). Class-defining structural features are in yellow; secondary features that may vary are in cyan. | ||
3.21 Biosynthesis of pyrroloquinoline quinone (PQQ) and mycofactocin
In recent years, much progress has been made in the understanding of the biosynthesis of the redox cofactor PQQ. Gene disruption studies demonstrated that five genes are required for PQQ production (Fig. 60A) with two other genes often but not always present in the operon.253 In the first step, Glu and Tyr residues in the precursor peptide PqqA (Fig. 60B) are crosslinked by the rSAM protein PqqE in a reaction that requires the small protein PqqD (RRE domain, see Section 4.1.1) to bind the LP of PqqA.254,255 A two-component heterodimeric protease PqqFG from Methylobacterium extorquens AM1, a protein that is not always encoded in the PQQ BGC, was able to hydrolyze linear PqqA, preferentially hydrolyzing the peptide both N- and C-terminal to Ser residues.256 Previously, a PqqF homolog from Serratia sp. FS14 had been crystallized demonstrating an HxxEH Zn-binding motif.257 After PqqFG catalysis, trimming from the N- and C-termini of the initial products will be required by one or more additional unknown proteases to generate the crosslinked diamino acid that is the postulated substrate of PqqB (Fig. 60C). This protein has sequence homology with β-lactamases but was shown to functionally behave as an iron-dependent hydroxylase that oxidizes the phenol in a series of analogs of the crosslinked structure to a hydroxy orthoquinone (Fig. 60C).258,259 Subsequently, a cyclization step that is postulated to be spontaneous258 provides the last intermediate that is oxidized by PqqC in an eight electron oxidation process that does not require any cofactors.260 Thus, in recent years the enzymatic activities of PQQ biosynthesis have been elucidated, either with authentic substrates or substrate analogs.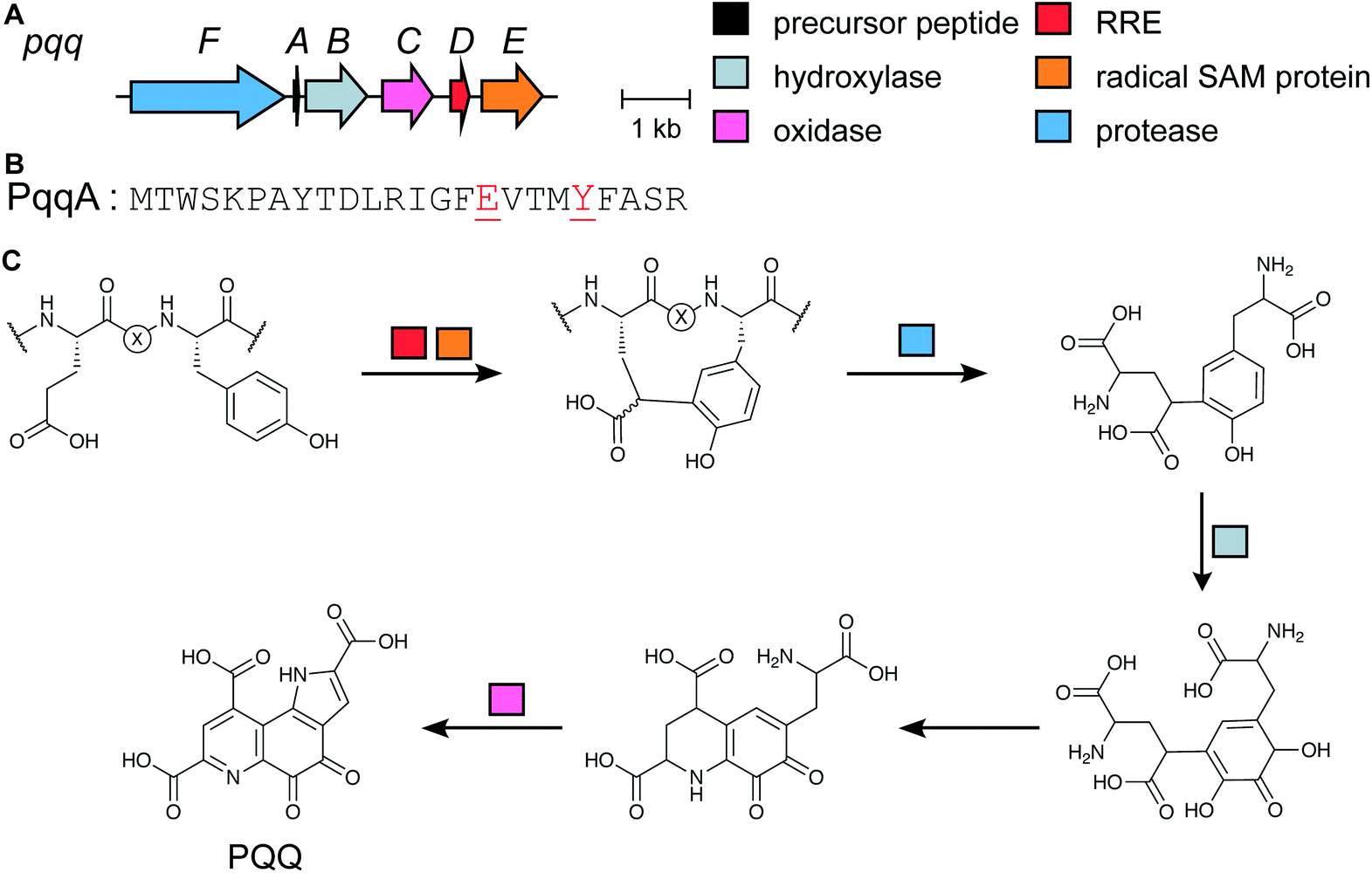 | ||
| Fig. 60 (A) BGC for PQQ in Pseudomonas sp. CMR12a. (B) Precursor peptide sequence with the amino acids that are modified to PQQ colored red. (C) Biosynthetic pathway and structure of PQQ. | ||
Mycofactocin is another RiPP that has been suggested to be a redox cofactor. Its existence was first hypothesized based on a bioinformatic study that showed the co-occurrence of six genes in many bacteria, most notably Mycobacterium sp.20 Furthermore, these genes were only present when members of three families of dehydrogenase genes were also in the genome, leading to the suggestion that the mycofactocin BGC might encode for a redox cofactor that is used by these dehydrogenases (e.g. MtfG, Fig. 61A).20,261 However, the structure of the final product has not yet been determined. Several studies have provided insights into the structure of the final product by investigating the functions of the six biosynthetic genes (mftA-F) (Fig. 61A). The rSAM protein MftC catalyzes the oxidative decarboxylation of the C-terminal Tyr of the precursor peptide MftA (Fig. 61B) in a reaction that also requires MftB for LP binding (RRE, Section 4.1.1).262,263 MftC then catalyzes a second reaction in which the penultimate Val is cyclized onto the initially formed tyramine (Fig. 61C).264 MftE was recently shown to hydrolytically remove the LP generating 3-amino-5-[(p-hydroxyphenyl)-methyl]-4,4-dimethyl-2-pyrrolidinone (AHDP).265 The amino group of AHDP is then oxidized by the FMN-dependent enzyme MftD, a member of the aldolase-TIM barrel fold family, to give pre-mycofactocin (Fig. 61C).21 The molecule was able to oxidize NADH bound to carveol dehydrogenase from M. smegmatis, a known mycofactocin-dependent enzyme, and was competent to support turnover of carveol to carvone. Indeed, its redox properties are not unlike other known peptide-derived redox cofactors,21 and several studies have supported a redox cofactor role for mycofactocin.266–268 At present it is not known how the reduced pre-mycofactocin might be reoxidized for another turnover, but recent metabolomics studies detected both oxidized and reduced glycosylated mycofactocin derivatives in Mycolicibacterium smegmatis.266 Mycofactocin represents another class of RiPP initially discovered bioinformatically (other examples are in Sections 2.12–2.15, 3.14, and 3.24).
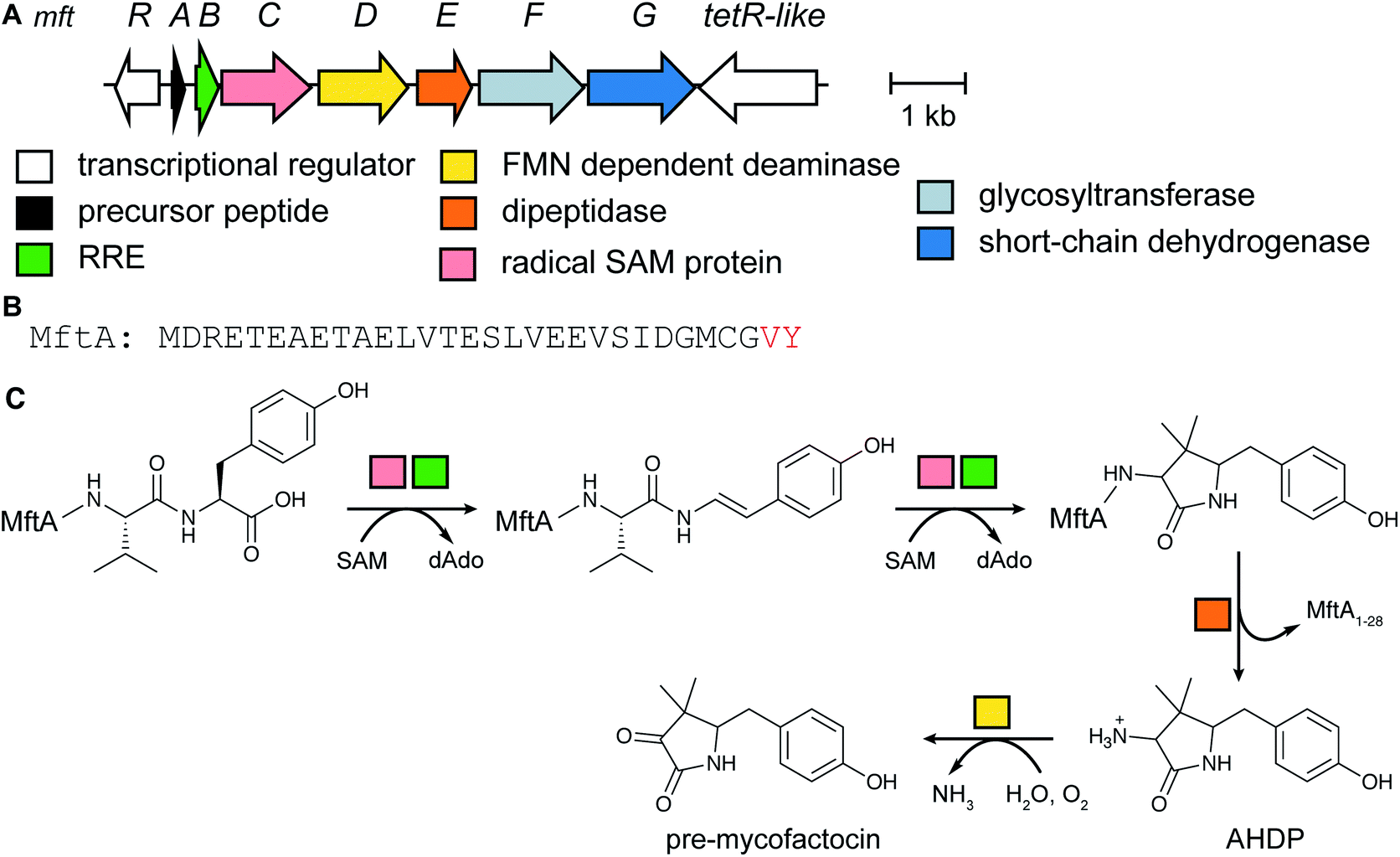 | ||
| Fig. 61 (A) BGC for mycofactocin biosynthesis. (B) Precursor peptide with the amino acids that are turned into mycofactocin highlighted in red. (C) Current understanding of the mycofactocin biosynthetic pathway. The membrane-associated glycosyl transferase MftF may glycosylate the phenol group of pre-mycofactocin.22 | ||
3.22 Biosynthesis of pantocin A
Pantocin A is a small tripeptide inhibitor of L-histidinol phosphate aminotransferase that originates from a Glu–Glu–Asp motif at the center of a 30 amino acid precursor peptide, PaaP. Its characterization was one of the earliest examples of using RiPP production in E. coli to obtain sufficient material for structure elucidation.269 The peptide bears a characteristic bicyclic ring system (Fig. 62).269,270 In addition to a gene involved in host self-immunity (paaC), the paa BGC contains only two biosynthetic genes, paaA which encodes an ATP-dependent enzyme of the ThiF/E1-superfamily, and paaB, encoding an α-ketoglutarate- and iron-dependent oxidase. In vitro reconstitution experiments using PaaA and synthetically prepared PaaP substrate revealed that PaaA catalyzes two dehydrations, a decarboxylation, and is responsible for generating the bicyclic ring system (Fig. 62). The PaaA-catalyzed transformation has been proposed to occur via one of two routes.271 One route begins with condensation of the peptide backbone amide with the activated side chain of Glu16, followed by a Claisen-like condensation with an enolate on the side chain of Glu17. Alternatively, the Claisen-like condensation step between the side chains of the adjacent Glu residues could precede condensation with the backbone amide. Both routes would yield the same bicyclic scaffold. The resulting intermediate iminium/enamine tautomers undergo an oxidative decarboxylation catalyzed by PaaA and/or PaaB, prior to cleavage of the mature product from the peptide backbone.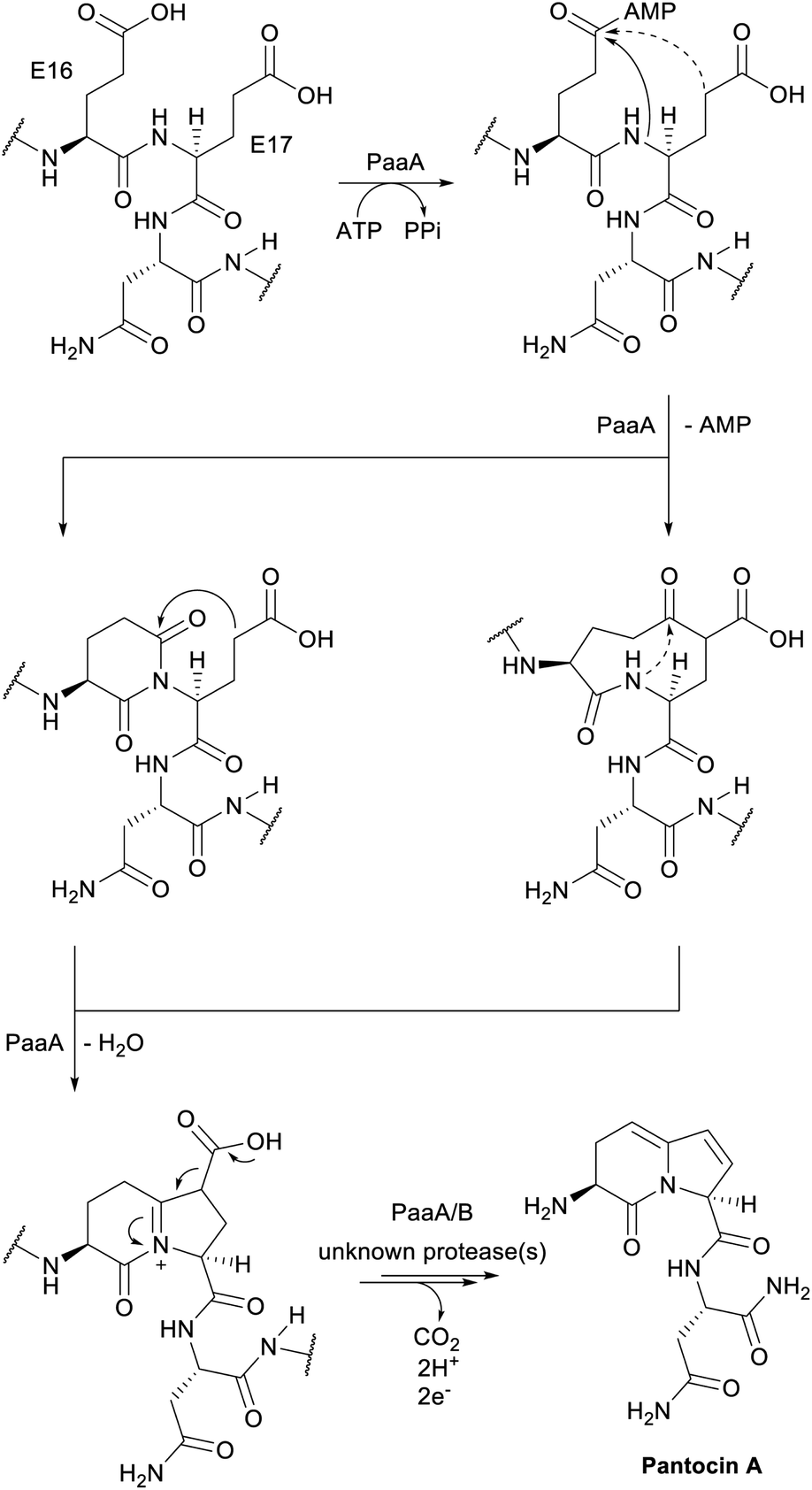 | ||
| Fig. 62 Two possible routes to pantocin A depending on whether condensation of the peptide backbone amide with Glu16 by PaaA preceeds (solid arrows) or succeeds (dashed arrows) condensation between the side chains of Glu16 and Glu17 remains to be elucidated. Both routes yield the same bicyclic core, which is subject to subsequent oxidative decarboxylation. | ||
To probe the functional significance of the N-terminal leader and C-terminal follower sequences for PaaA activity, assays were performed using synthetic truncated PaaP variants.271 While the N-terminal sequence was shown to be essential for PaaA catalysis, removal of the follower still allows the first condensation reaction to occur (by either route described previously). Structural studies have also been conducted for PaaA (PDB: 5FF5) and revealed the presence of an RRE domain (Section 4.1.1) that interacts with PaaP and is believed to direct the core peptide into the enzyme active site. More recently, mRNA display was employed to gain further insights into enzyme-substrate interactions, revealing broad enzyme promiscuity for PaaA outside of its core and predicted binding epitope.272 This work highlights the potential of mRNA display-based techniques for studying RREs, in addition to creating new possibilities for introducing the indolizidinone moiety into synthetic peptide libraries. Critically, the technique also provided insights into the timing of modification, indicating that Glu16, rather than Glu17, is the preferred initial substrate residue. We note that the term pantocin is given to natural products of diverse structures produced by Pantoea agglomerans, some of which may not be RiPPs.270
3.23 Methanobactin biosynthesis
Methanobactins form a family of RiPPs that play a key role in copper homeostasis among methanotrophic bacteria. Common to the structures of all methanobactins are a pair of bidentate ligands, each comprising an oxazolone heterocycle adjacent to an enethiol (or thioamide tautomer), which mediate Cu(I) chelation (Fig. 63A). Although the BGCs encoding these Cu(I)-binding siderophores have been known for many years and are widespread among bacteria,273 the enzymes responsible for installing these characteristic heterocycle-thioamides were only recently characterized from Methylosinus trichosporium OB3b.274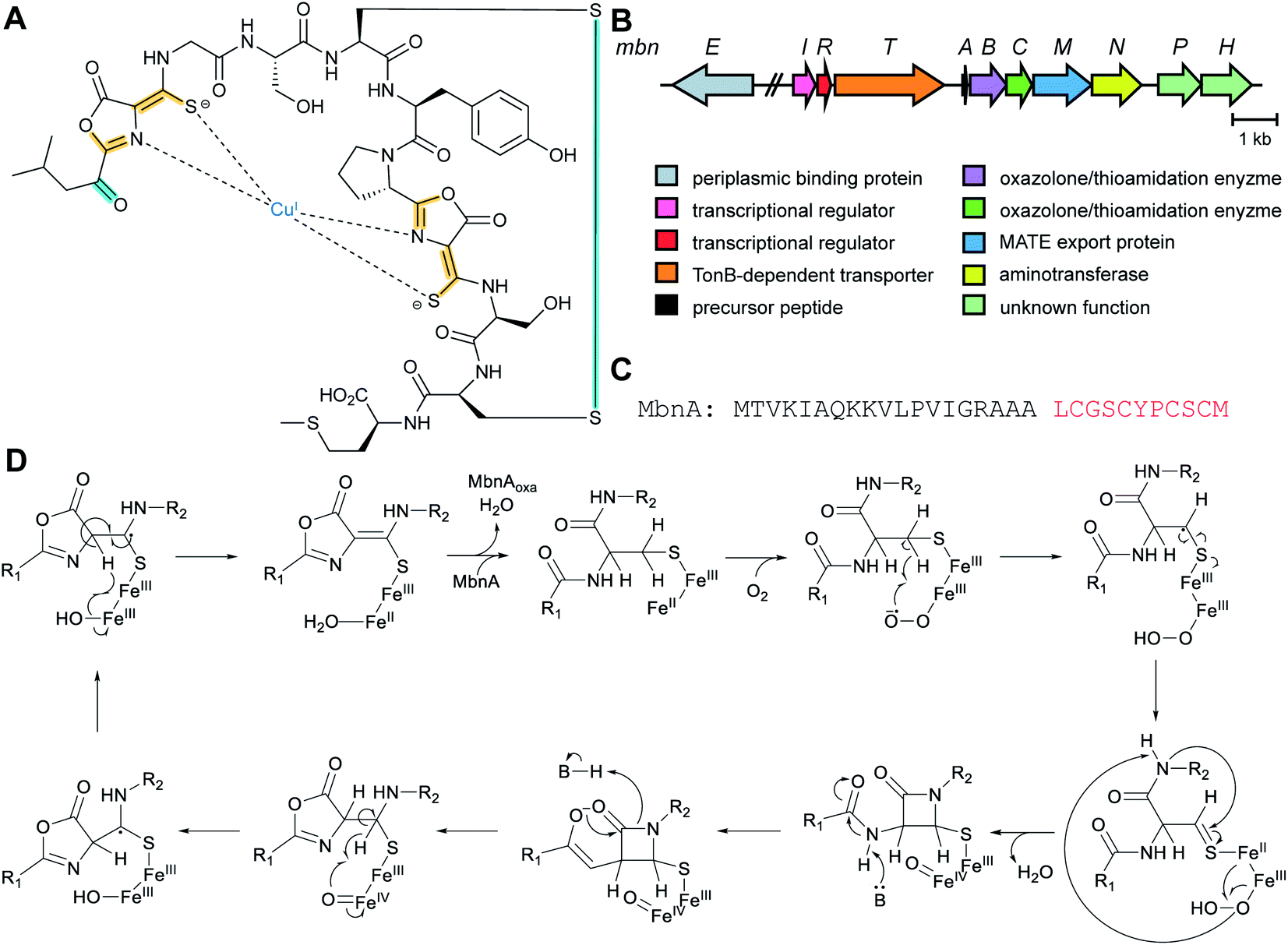 | ||
| Fig. 63 (A) Structure of methanobactin bound to copper(I). (B) BGC of methanobactin from Methylosinus trichosporium OB3b. (C) Sequence of the MbnA precursor peptide. (D) Hypothetical mechanism for MbnBC-catalyzed generation of oxazolone/thioamide moieties on MbnA.274 Class-defining PTMs are in yellow, secondary PTMs in cyan. | ||
It was postulated that the installation of heterocycle-thioamide moieties would require a four-electron oxidation, suggesting the involvement of a redox cofactor. MbnB, a previously uncharacterized protein belonging to DUF692 encoded within methanobactin BGCs, was identified as a potential candidate for this transformation based on low sequence similarity to members of a TIM barrel family that include representatives bearing divalent metal cofactors.275 Heterologous co-expression experiments revealed that MbnB forms a heterotrimeric complex with MbnC, a protein of unknown function, and the precursor peptide MbnA, suggesting a role of MbnBC in biosynthesis. Spectroscopic studies further revealed the presence of a multinuclear Fe (di/tri) cofactor within MbnB,274 consistent with the as-yet unpublished crystal structure from a homolog from Haemophilus somnus 129Pt (PDB: 3BWW) that possesses a diiron cluster for which no activity has yet been reported.
In vitro reconstitution experiments demonstrated that, in the presence of O2, MbnBC reacts with MbnA to produce a product with a mass loss of 4 Da that absorbs strongly at 335 nm. This activity was shown to be dependent on the presence of the predicted multinuclear Fe cofactor, and substitution of the proposed iron ligands abolished MbnB activity. Subsequent MS/MS analysis allowed the mass loss to be localized to the N-terminal Cys21, which is the position of the oxazolone closest to the N-terminus of the final product. Consistent with this result, a C21S substitution in the substrate eradicated peptide modification. MbnBC does not contain any discernable RRE. Utilization of a truncated MbnA variant lacking the leader sequence led to diminished activity, suggesting an alternate mechanism of interaction with the LP. Taken together, these results suggest that the MbnBC homodimer is an oxidase that activates O2 for the cleavage of the three aliphatic C–H bonds on both Cα and Cβ of Cys21 of MbnA, triggering an oxazolone-thiamide-forming four-electron oxidation (Fig. 63B). MbnA processing could be initiated by hydrogen atom abstraction from the Cys21 Cβ atom by a superoxo Fe(III) intermediate.274 The formation of an oxazolone–thioamide moiety via such a metalloenzyme-mediated radical mechanism is unprecedented and presents a novel way of carrying out this chemistry. The mechanism by which MbnBC proceeds to the second residue (Cys27) for modification remains unclear. Another member of the DUF692 family is involved in an equally remarkable transformation involving Cys during pearlin biosynthesis (Section 3.24).
3.24 Pearlin biosynthesis
During the maturation of lanthipeptides and thiopeptides, LanB enzymes catalyze the glutamylation of Ser and Thr residues via a glutamyl-tRNA-dependent mechanism (Sections 3.1.1 and 3.2). Glutamate is subsequently eliminated to generate dehydroamino acid residues (Fig. 19B) that can be further modified.49,71 As part of a bioinformatics survey of LanB-like enzymes, a number of homologs were identified that do not contain a canonical elimination domain, thus originally called small LanBs. One example, TglB, was identified in Pseudomonas syringae pv. maculicola ES4326 that is encoded near a gene encoding a 50-mer peptide (TglA; Fig. 64A).50 Co-expression of these two genes in E. coli resulted in the detection of a new TglA-related mass consistent with condensation with a Cys residue, rather than Glu. Furthermore, MS/MS analysis localized the Cys adduct to the C-terminal Ala of TglA, instead of a canonical ester linkage to Ser as anticipated from lanthipeptide and thiopeptide biosynthesis (Sections 3.1.1 and 3.2). Subsequent in vitro reconstitution experiments using Cys, tRNACys, Cys tRNA synthetase and ATP confirmed that TglB catalyzes the transfer of a Cys to the C-terminus of TglA in a tRNA- and ATP-dependent manner (Fig. 64B). This finding lead to the renaming of small LanBs to minoacyl-t
minoacyl-t![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) NA
NA ![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif) igases (PEARLs). When the same experiment was repeated in a buffer containing H218O, the resulting product contained only a single 18O, and the addition of hydroxylamine to the reaction mixture permitted the trapping of activated TglA as the corresponding hydroxamate. Taken together, these observations indicated a mechanism for TglB in which the TglA C-terminal carboxylate is first activated by phosphorylation, with subsequent amide bond formation to the amino group of Cys-tRNACys (Fig. 64B). tRNACys is subsequently hydrolyzed to generate TglA–Cys. TglB contains a LP binding domain (RRE, Section 4.1.1) and requires only the last 12 amino acids of TglA.276
igases (PEARLs). When the same experiment was repeated in a buffer containing H218O, the resulting product contained only a single 18O, and the addition of hydroxylamine to the reaction mixture permitted the trapping of activated TglA as the corresponding hydroxamate. Taken together, these observations indicated a mechanism for TglB in which the TglA C-terminal carboxylate is first activated by phosphorylation, with subsequent amide bond formation to the amino group of Cys-tRNACys (Fig. 64B). tRNACys is subsequently hydrolyzed to generate TglA–Cys. TglB contains a LP binding domain (RRE, Section 4.1.1) and requires only the last 12 amino acids of TglA.276
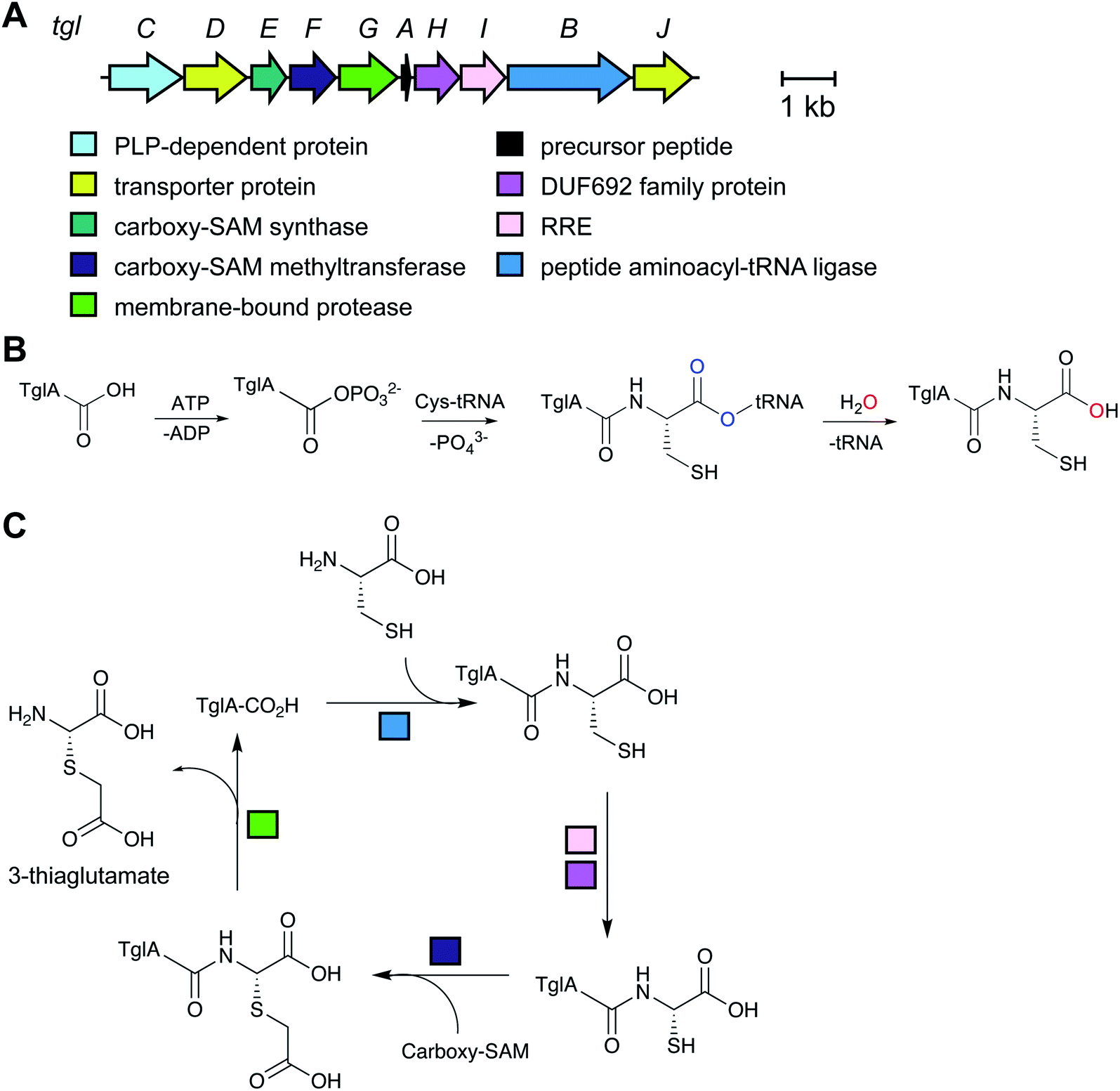 | ||
| Fig. 64 (A) BGC for 3-thiaglutamate. (B) Proposed mechanism of Cys addition to TglA by the PEARL TglB. Oxygen labeling experiments established that the ester bond to the tRNA is hydrolyzed during the reaction. (C) Additional PTMs that lead to 3-thiaglutamate and regenerate TglA. | ||
Further in vivo and in vitro experimentation with other proteins encoded in the tgl BGC revealed that TglH, a DUF692-containing homolog of MbnBC (Section 3.23), and TglI (RRE; Section 4.1.1) catalyze a four-electron oxidative rearrangement that results in excision of the Cβ atom of the appended Cys residue (Fig. 64C).50 TglF, a carboxy-SAM-dependent methyltransferase, subsequently catalyzes thiol carboxymethylation, prior to TglG-mediated proteolytic cleavage to release thiaglutamate. It is not clear whether this unstable compound is an intermediate or the final product of the pathway. Although TglA effectively remains unmodified following these transformations and the pearlins are therefore technically non-ribosomal in origin, a ribosomally synthesized peptide serves as a scaffold for their extension assembly using RRE domains present in TglB, TglI, and TglG,50 and represents a new biosynthetic paradigm.
3.25 Glycocin biosynthesis
Glycocins are glycosylated peptides produced by various Firmicutes, exemplified by glycocin F from Lactobacillus plantarum and sublancin from Bacillus subtilis 168.279 Sugar moieties are attached to Cys, Ser, and/or Thr generating S- or O-glycosides. In some cases, such as thurandacin and glycocin F, a single enzyme glycosylates both a Cys and a Ser.279,280The NMR structure was solved for sublancin 168 revealing a compact structure consisting of two antiparallel α-helices connected by cystine bridges and hydrophic interactions,281 providing a rationale for its proteolytic stability. The structure resembles that of the previously reported glycocin F.279 Successful functional transfer of the sublancin biosynthetic gene cluster into E. coli permitted the rapid generation of sublancin core variants to explore structure–activity relationships.282 These studies identified two residues in helix B as well as the S-glycosyl unit, as important for antibiotic activity. A similar conclusion was drawn regarding the importance of S-glycosylation in glycocin F using synthetic variants.283,284 In addition, variants were tested, in which various different sugar units had been attached by exploiting the substrate tolerance of the S-glycosyl transferases SunS and EntS encoded in the sublancin and enterocin 96 BGCs, respectively.285,286 This exchange of glycosyl moieties exerted unexpectedly little influence on the bioactivity. Further experiments supported earlier evidence that sublancin might target the glucose uptake system.
Utilizing E. coli expression strategies, the function of orphan glycocin BGCs was investigated, motivated by the relatively small number (six) of previously known glycocins. Among 50 potential glycocin BGCs, four from various organisms including a thermophilic Geobacillus sp., an alkaliphilic Bacillus sp., and a Listeria monocytogenes strain yielded new peptides carrying glucose moieties including one, listeriocytocin, containing a diglucosylated Ser.287 Two (mesophilic) Bacillus gene-derived products exhibited good activity against Bacillus cereus. In another study, the new glycocin pallidocin was generated in E. coli by expressing an orphan BGC from the thermophile Aeribacillus pallidus.288 Pallidocin, which is identical to the aforementioned glycocin from Geobacilus sp., features a glucosylated Cys and a predicted dihelical structure and is highly active against related thermophilic Firmicutes and some Bacillus species. Furthermore, co-expression in E. coli of the pallidocin S-glycosyltransferase and export protein with His6-tagged leaderless core peptides that were identified in two other Bacillus genomes, provided two additional examples of antibacterial glycocins.288
4 Engagement and removal of leader peptides and recognition sequences
The modularity of RiPP biosynthesis and the associated plasticity and evolvability of their structures (see Section 7) is accomplished in part through the bipartite nature of the precursor peptide. Typically, RiPP precursor peptides consist of two regions, an N-terminal LP, which serves as the recognition site for some of the biosynthetic enzymes, and a C-terminal core peptide, where the chemical modifications take place.1 In the case of bottromycins and borosins, a peptide region providing substrate recognition is attached to the C-terminus of the core peptide and is thus termed a follower peptide (Fig. 1). Some RiPP pathways, such as those of pantocin and N-to-C cyclized peptides, have leader and follower peptides. The physical separation of substrate recognition from where the modifications are installed results in wide substrate tolerance for many RiPP biosynthetic enzymes. Since 2013, much new information has been obtained regarding the mechanisms of LP recognition, which will be discussed in this section.As noted in Section 1.1, the PTMs in some RiPPs require additional recognition sequences (RSs) beyond the LP that are often found flanking a core peptide. New information regarding the interaction of these often highly conserved RSs with their biosynthetic enzymes has been reported and will also be briefly discussed in this section.
4.1 Leader peptide recognition
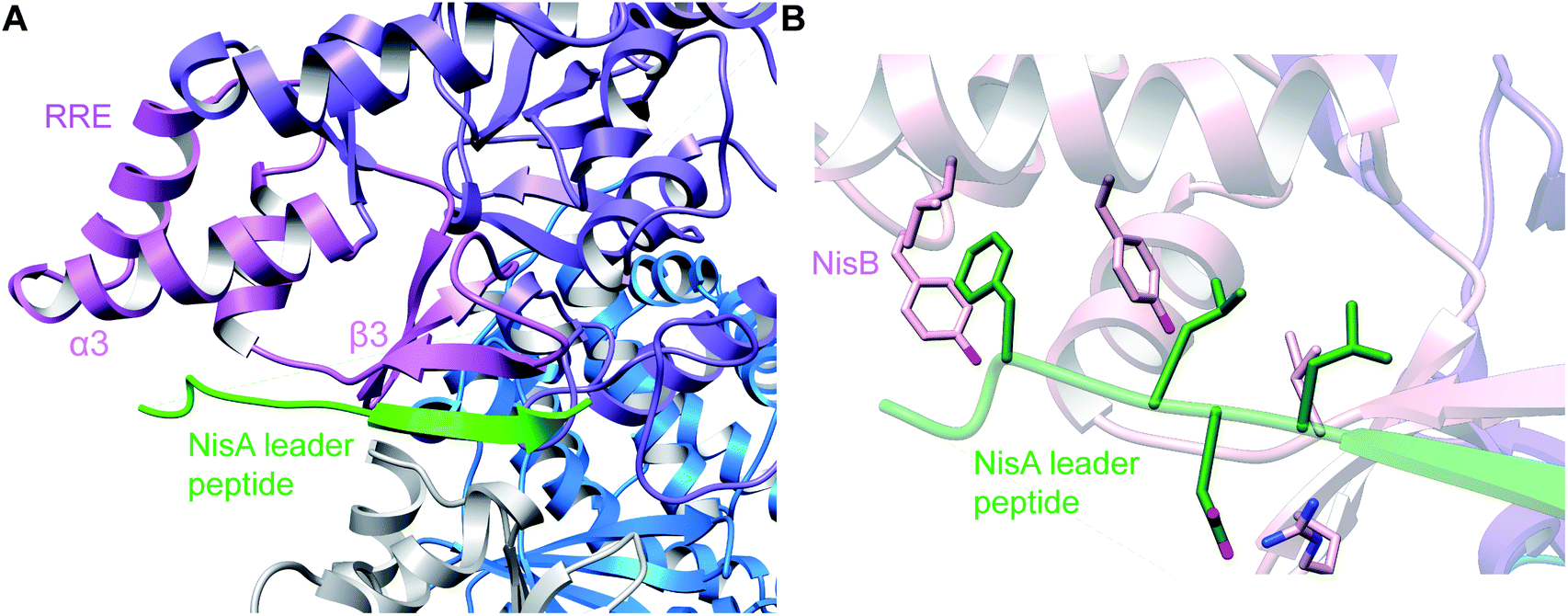 | ||
| Fig. 65 (A) Co-crystal structure of NisB with the dehydrated NisA peptide; only the LP is resolved (PDB: 4WD9).49 The LP binds in antiparallel fashion to β3 of the RRE with further interactions to α3. (B) Interactions between a highly conserved motif on the LP of NisA (the FNLD motif) with pockets and amino acids of the RRE of NisB. The Phe and Leu of the motif insert into hydrophobic pockets and the Asp makes an interaction with an Arg on NisB. An adjacent Leu located C-terminal to the motif provides an additional hydrophobic contact. | ||
Shortly after the publication of the NisA:NisB structure, the complex of LynD, the cyclodehydratase involved in the biosynthesis of the cyanobactin aestuaramide (see Section 3.6), with a minimal substrate PatE′ was reported.115 Residues −18 to −4 of the LP of PatE′ were observed in the structure, where residues Gln(−18) to Ser(−15) adopted a coiled conformation, Ser(−15) to Glu(−11) were involved in a helical turn, Ser(−9) to Ala(−6) formed a β-strand, and Leu(−5) to Gly(−4) adopted a coiled conformation (Fig. 66). The β-strand of the LP of PatE′ again formed an antiparallel interaction to β3 of the binding domain on LynD, and Leu(−13) and Leu(−10) inserted into hydrophobic pockets of α3. In addition, Glu(−11) and Glu(−8) of the LP made salt bridges and hydrogen bonds with LynD (Fig. 66).
 | ||
| Fig. 66 Co-crystal structure of the LP of PatE′ (red) bound to the RRE domain of LynD (PDB: 4V1T).115 The LP binds analogously to how NisA engages with NisB (Fig. 65). | ||
Using a combination of crystallographic data from these co-crystal structures and bioinformatic approaches, it was demonstrated that biosynthetic pathways from many different RiPP classes, including cyanobactins, LAPs, class I lanthipeptides, thiopeptides, microcins, lasso peptides, sactipeptides, PQQ/mycofactocin, proteusins, and bottromycins use the same architectural domain to recognize the LP.66 This RiPP precursor recognition element or RRE, consists of three α-helices (α1–3) and three β-sheets (β1–3) in a wHTH motif. Some RiPP biosynthetic pathways do not have the RRE domain embedded in a biosynthetic enzyme as discussed thus far, but instead encode a discrete RRE. For RiPP biosynthetic enzymes, this motif was first structuraly characterized in PqqD, a discrete RRE-containing protein involved in PQQ biosynthesis (Section 3.21) (Fig. 67). PqqD was later shown to be responsible for binding PqqA, the PQQ precursor peptide,255 and recently an NMR structure of the complex has been determined showing a similar interaction with the LP of PqqA.290 The mode of LP recognition by the RRE has now been elucidated for multiple RiPP biosynthetic enzymes. For instance, biochemical analysis of BalhC and CurC involved in the biosynthesis of LAPs also suggested a central role of the α3/β3 cleft of the RRE,66 and similar findings were reported for the discrete F proteins HcaF and TbtF involved in LAP and thiopeptide biosynthesis, respectively.89,124 Although in most cases, similar binding poses are observed as those described for NisB, LynD, and PqqD, sometimes alternative binding conformations have been reported (see Section 4.1.2).
 | ||
| Fig. 67 Crystal structure of PqqD (PDB: 3G2B).291 The β3 strand and α3 helix that were shown by NMR290 to directly engage the PqqA LP are indicated. | ||
In the case of lasso peptides (Section 3.8), LP binding to an RRE-containing protein was first demonstrated using fluorescence polarization binding assays in the streptomonomicin pathway, where the LP of StmA (precursor peptide) bound to StmE (RRE).66 This study also identified a subset of residues in StmE that mediate the LP interaction. Following this, the RRE from the lariatin biosynthetic pathway (LarB1) was also shown through bilayer interferometry assays to interact with the LP of LarA.159 Photocrosslinking experiments suggested that the RRE may also bind the core region of LarA. A structural model of LarA-RRE binding generated using distance constraints obtained from the crosslinking experiments showed that the peptide bond between the leader and core peptides is solvent-exposed and possibly provides access to the leader peptidase.159 Studies on the paeninodin biosynthetic pathway revealed that in addition to recognizing the PadeA LP, the RRE delivers PadeA to the leader peptidase.157 This conclusion was reached by the lack of cleavage activity on PadeA in the absence of the RRE or in the presence of RRE variants that bind poorly to PadeA. Hydrogen–deuterium exchange MS performed on LP-bound RRE showed decreased hydrogen–deuterium exchange at the α-helices and β3 strand, suggestive of an interaction with the LP. The β1 and β2 strands showed increased exchange consistent with flexibility/exposure after binding. These data, along with mutational studies, support a model for substrate recognition in line with that observed for other RREs, where the LP lies along the α3 helix and interacts with β3 to extend into a four-stranded antiparallel β-sheet.66,157
The recent crystal structures of discrete RRE proteins TfuB1 (also referred to as FusE) and close orthologs (PDB: 6JX3, 5V1V, and 5V1U) bound to cognate LPs support this model.160,161 LP binding was shown to be primarily mediated by hydrophobic interactions with the RRE (Fig. 68). In addition, these structures revealed that Tyr(−17), Pro(−14), and Leu(−12) in a YxxPxL motif, conserved among precursors that harbor a discrete RRE154,159 and shown to be important for substrate recognition by the RRE,158,292,293 fit into hydrophobic pockets formed between the β3 strand and the α-helices.160,161 The inability of RRE variants altered at residues that form these hydrophobic clefts to bind LPs further corroborate these findings.161 Furthermore, insertion of a YxxPxLxxxG motif into the non-cognate precursor peptide to the lasso peptide microcin J25, McjA, turned it into a competent substrate for TbiB2 while the wild-type precursor was not processed. The structure of the FusA LP bound to FusE revealed a hydrophobic patch formed by the LP and the RRE on the β-sheet and is predicted to provide an interaction surface for either the core peptide or leader peptidase for further processing.160
 | ||
| Fig. 68 Co-crystal structures of (A) FusE (TfuB1) and (B) TbiB RRE domains and the LPs of their substrates (blue; PDB: 6JX3 and 5V1V).161 The overall binding mechanism again involves interactions with the β3 strand and α3 helix of the RRE. | ||
McbB involved in the biosynthesis of the LAP microcin B17 (Fig. 28, Section 3.4) also engages its precursor peptide McbA using an RRE. The structure of the McbBCD complex bound to a McbA truncant revealed an unexpected stoichiometry and arrangement, where McbB, McbC, and McbD are present in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio, and the McbB monomers are arranged in a head-to-tail conformation.294 The asymmetric arrangement of McbB monomers allows for different functions of each subunit. The RRE of one of the McbB monomers interacts with the McbA LP by forming a four-stranded antiparallel β-sheet near the α3/β3 cleft similar to the interactions described thus far in this section (Fig. 69). Phe(−19) and Leu(−15) of the LP insert into a hydrophobic cavity in the cleft. Additional interactions were observed between Asp(−12) and Lys(−9) of McbA that interact with the second monomer of McbB by salt bridges.
:1:1 ratio, and the McbB monomers are arranged in a head-to-tail conformation.294 The asymmetric arrangement of McbB monomers allows for different functions of each subunit. The RRE of one of the McbB monomers interacts with the McbA LP by forming a four-stranded antiparallel β-sheet near the α3/β3 cleft similar to the interactions described thus far in this section (Fig. 69). Phe(−19) and Leu(−15) of the LP insert into a hydrophobic cavity in the cleft. Additional interactions were observed between Asp(−12) and Lys(−9) of McbA that interact with the second monomer of McbB by salt bridges.
 | ||
| Fig. 69 Crystal structure of the McbAB2CD complex (PDB: 6GRG).122 (A) Biosynthetic complex containing McbA (cyan), McbB (orange and purple), McbC (green), and McbD (blue) (B) LP (cyan) binding to the RRE of one of the monomers of McbB (purple). | ||
Other RRE domain containing proteins that have been shown to interact with the LPs of their substrates include sactisynthases (Section 3.9),182,183 and RRE domains are also used for recognition of NHLPs. These long LPs (∼90–100 residues) with partial homology to nitrile hydratases are used for the biosynthesis of a variety of RiPPs.195,197 The epimerase OspD involved in proteusin biosynthesis (see Section 5.1.1) contains a C-terminal RRE and mutational analysis of the substrate OspA implicated a stretch of amino acids near the N-terminus that are critical for catalysis.295 This stretch is homologous to other RiPP precursor peptides including class I lanthipeptides, LAPs, and class II lanthipeptides even though the enzymes invoved in the biosynthesis of the latter do not contain RRE domains (see Section 4.1.3).
In the case of MdnC, an ATP-grasp enzyme that installs the first macrocycle in microviridin J (Section 3.15), the LP is bound by a central domain in the protein.212 LP recognition is mainly accomplished through electrostatic interactions involving a conserved Arg in a PFFARFL motif on the MdnA LP (Fig. 71).
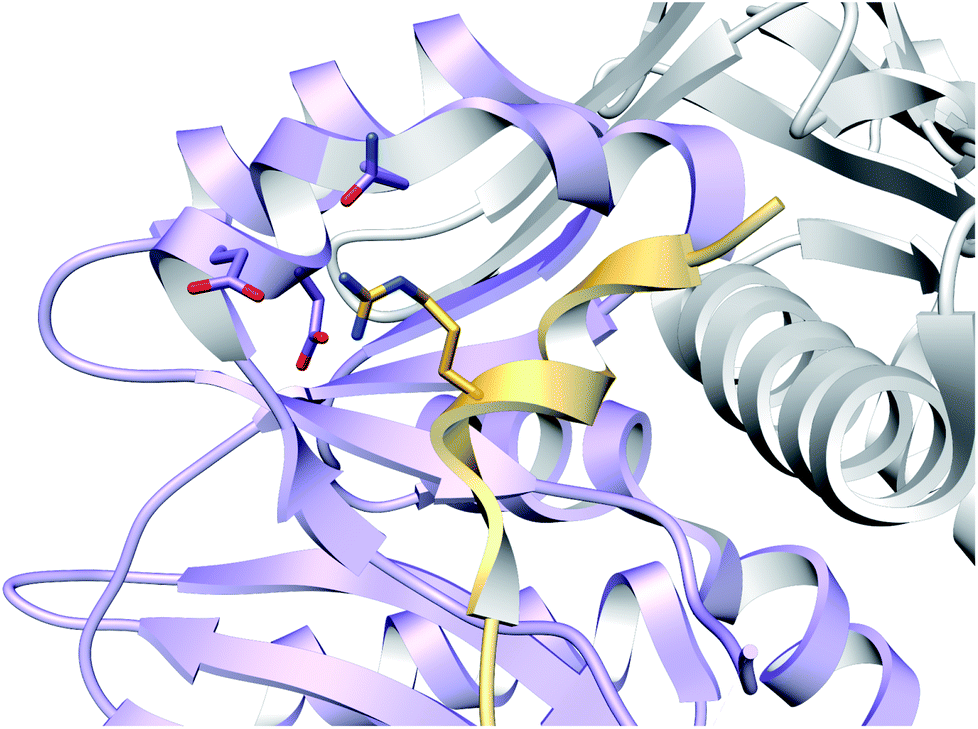 | ||
| Fig. 71 Co-crystal structure of MdnC (purple) and the LP of MdnA (gold) showing substrate recognition via an α-helix in the LP (PDB: 5IG9).212 | ||
In class II lanthipeptide synthetases (LanMs; Section 3.1.2), hydrogen–deuterium exchange mass spectrometry, photocrosslinking studies, and fluorescence polarization measurements identified unstructured loops on the capping domain of the haloduracin β synthetase, HalM2, that may either directly bind to the HalA2 LP, or at least is necessary for HalM2 to enter a conformation to allow for precursor peptide binding (Fig. 72).76,77 For the trifunctional class IV lanthipeptide synthetases (LanLs; Section 3.1.4) composed of lyase, kinase, and cyclase domains, the kinase domain on SgbL is responsible for LP binding based on the available data. Binding and activity studies suggest that a predicted α-helical stretch on the N-terminal region of the substrate SgbA is important for SgbL recognition.86,87 Similarly, NMR data, bioinformatics, and computational predictions suggest that an amphipathic α-helix formed by a conserved region of the leader peptide that positions branched hydrophobic amino acids on one face is also critical for recognition by class III lanthionine/labionin synthetases.88 Structural information is not yet available for LanL enzymes or the related class III LanKC lanthipeptide synthetases (Section 3.1.3).
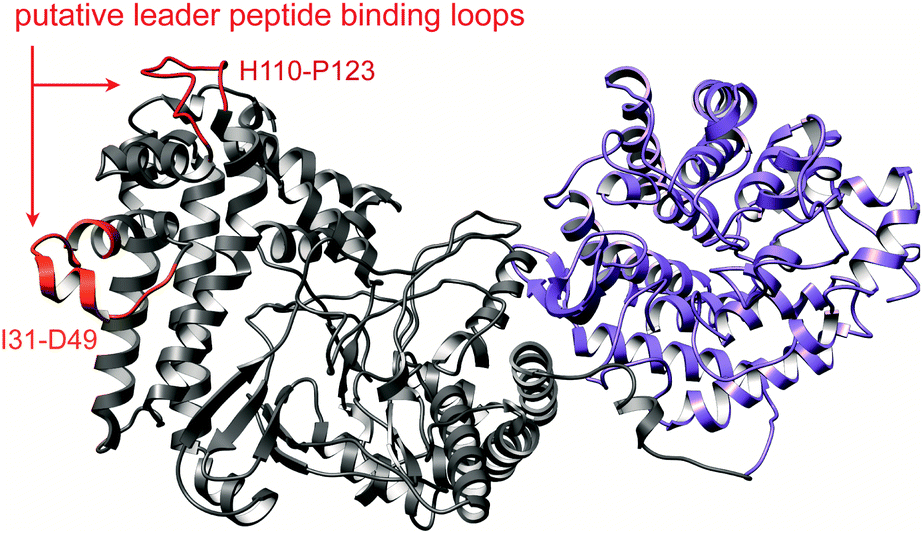 | ||
| Fig. 72 Biophysical and biochemical experiments suggest that the loops connecting the capping helices of the dehydratase domain (grey) of class II lanthipeptide synthetases (LanM proteins) mediate LP binding.76,77 The cyclase domain is shown in purple. | ||
Microcin C (McC, Fig. 73A) is an unusual RiPP because it lacks a LP on its precursor peptide. Therefore, biosynthetic enzymes must use an alternative method for substrate recognition. The N-formylated precursor peptide was shown to be a better substrate than the desformyl analog for MccB, the adenylyltransferase responsible for forming the phosphoramidate linkage in McC.297 Analysis of MccB in complex with N-formyl MccA depicted the N-formyl moiety in a pocket formed by two adjacent monomers of an MccB homodimer (Fig. 73B; PDB: 6OM4) explaining the importance of N-formylation.298
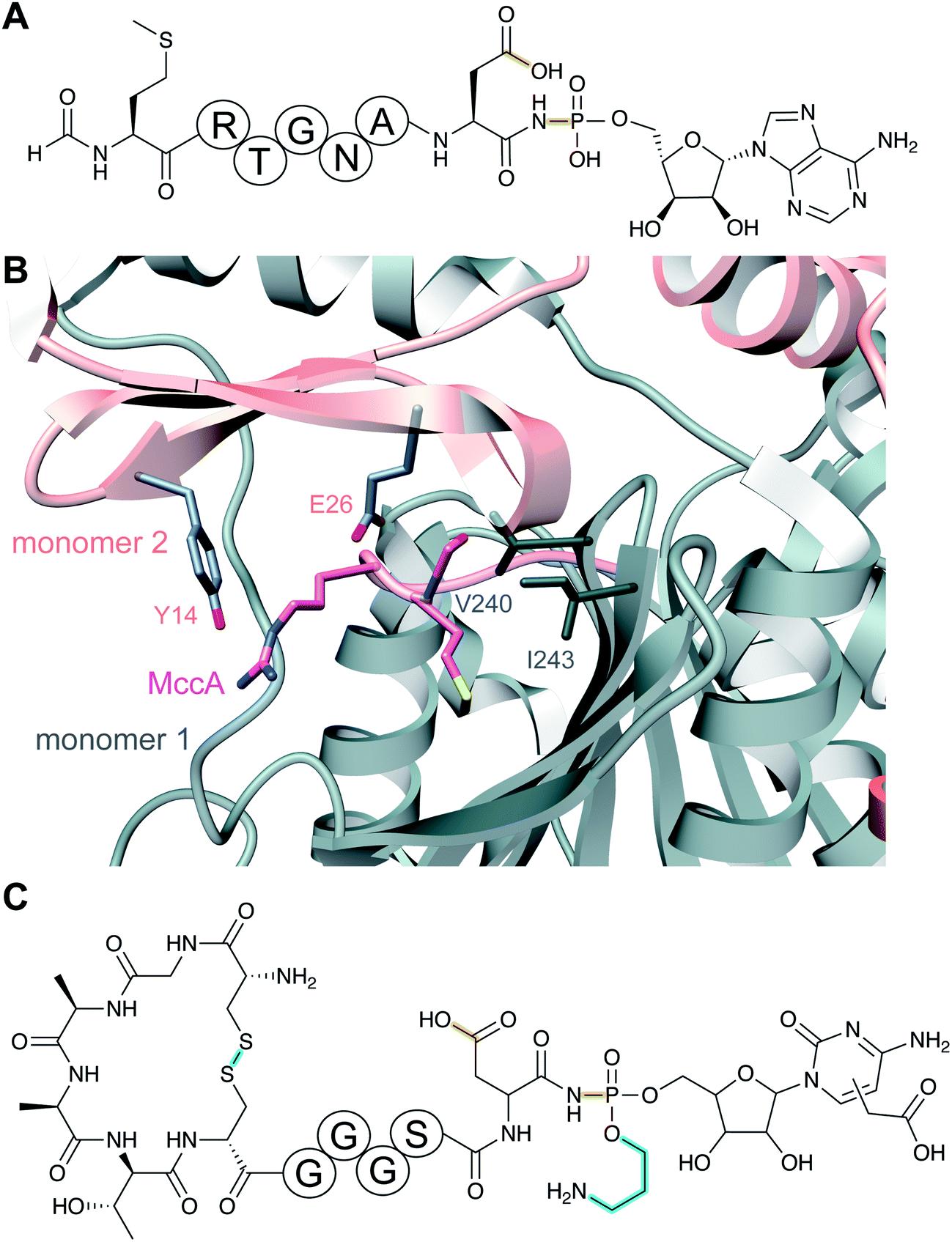 | ||
| Fig. 73 (A) Structure of microcin C. Class-defining PTMs are in yellow, secondary PTMs are in cyan. (B) Substrate recognition by MccB (PDB: 6OM4).298 The two monomers are shown in gray and salmon and the substrate MccA in magenta. The N-formylated N-terminal Met1 and Arg2 are shown in stick format. (C) Structure of a microcin C-like compound from Y. pseudotuberculosis IP32953. | ||
Whereas the heptapeptide microcin C from E. coli is biosynthesized from a precursor lacking a LP, related compounds are made from longer precursor peptides with an N-terminal extension that is released proteolytically. For instance, an 11-amino acid antimicrobial peptide that is modified with a C-terminal carboxymethylated cytosine that is also O-alkylated on the phosphoramidate linkage (Fig. 73C) is generated post-translationally from a 42-amino acid precursor peptide in Yersinia pseudotuberculosis IP32953.299 Removal of the N-terminal peptide is performed by a TldDE-type protease that is also involved in the maturation of microcin B17 (see Section 4.4.3). At present it is not clear whether the N-terminal peptide is a LP in the sense that it is recognized by the MccB-like PTM enzymes, or whether it functions mostly to keep the peptide-nucleotide antibiotic inactive inside the cell. MccB and its orthologs contain an RRE and thus recognition of the N-terminal peptide is likely.
4.2 Molecular interactions of recognition sequences
Compared to the advances in LP engagement by RiPP biosynthetic enzymes, the molecular details of interactions with RSs that flank core peptides (Fig. 1B) is not as well understood. Such RSs are present in the precursor peptides of a variety of RiPPs in bacteria (e.g. cyanobactins, graspetides, pheganomycin), fungi (e.g. amatoxins and dikaritins), and plants (e.g. orbitides, cyclotides, and lyciumins). The best understood RiPP enzymes in this respect are those that catalyze head-to-tail cyclizations in cyanobactin,300–302 amatoxin, and orbitide biosynthesis (Sections 3.6, 3.18, and 3.19, respectively). Recent crystal structures have been solved for the amatoxin cyclase POPB235 and the orbitide cyclase PCY1.248,249 For the latter, well-defined interactions were observed between the enzyme and the highly conserved C-terminal six amino acids (Fig. 74) that keep the follower peptide in the protein and prevent hydrolysis (by water) or another peptide entering the active site (for ligation), thus promoting intramolecular trapping of the acyl-enzyme intermediate to afford cyclized products. The N-to-C cyclization substrates for POPB and PCY1 contain single core peptides and the C-terminal extension are termed follower peptides, but these likely serve an analogous function to RSs in other N-to-C cyclization systems such as cyanobactins that have multiple core peptides (Section 3.6). Although the molecular details of RS engagement for cyanobactin biosynthesis are not well understood on a molecular level, predictive rules that are governed by RSs have been successfully used for engineering purposes (see Section 7).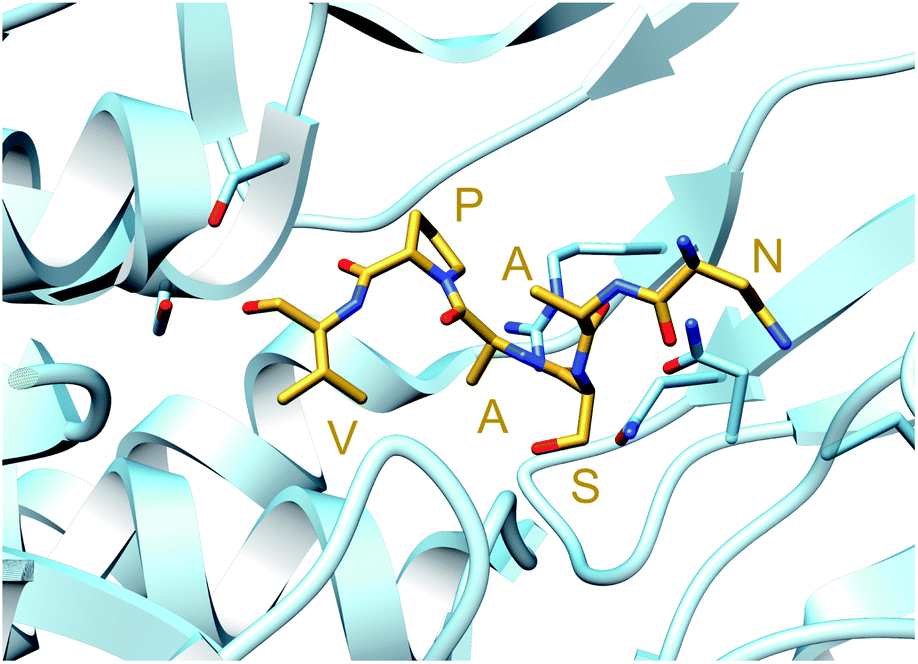
4.3 Summary and outlook on substrate recognition
With the first co-crystal structures of biosynthetic enzymes and their substrates, important new insights have been obtained regarding LP recognition, and bioinformatic approaches have been able to extend these findings to much larger numbers of proteins. Aspects of substrate recognition that are still mostly unresolved include core peptide recognition as only a few structures are available in which electron density of all or part of the core peptide is visible [e.g. the lanthipeptide dehydratase NisB (Fig. 20),67 CteB in ranthipeptide biosynthesis,183 the microcin B17 synthetase (Fig. 69B),122 and the RiPP-related MCR-modifying YcaO that installs thioglycine303]. Furthermore, for biosynthetic pathways in which multiple enzymes act consecutively on a precursor peptide, it is not clear whether the RRE in one protein holds the substrate for other proteins to catalyze their reactions or whether RRE independent substrate recognition motifs are involved. In some cases, it is clear that a LP is not required for catalysis, especially for secondary PTMs. In contrast, in other examples, substrate recognition is mediated by PTMs that are introduced in the core peptide in earlier biosynthetic steps as exemplified by the strictly linear and ordered biosynthetic steps in thiopeptide biosynthesis (Section 3.2).4.4 Leader peptide removal
Removal of the LP is a critical step in RiPP biosynthesis and is often coupled to export of the mature compound. The LP cleaving enzyme varies between different RiPP classes, but common strategies have been recognized as discussed below.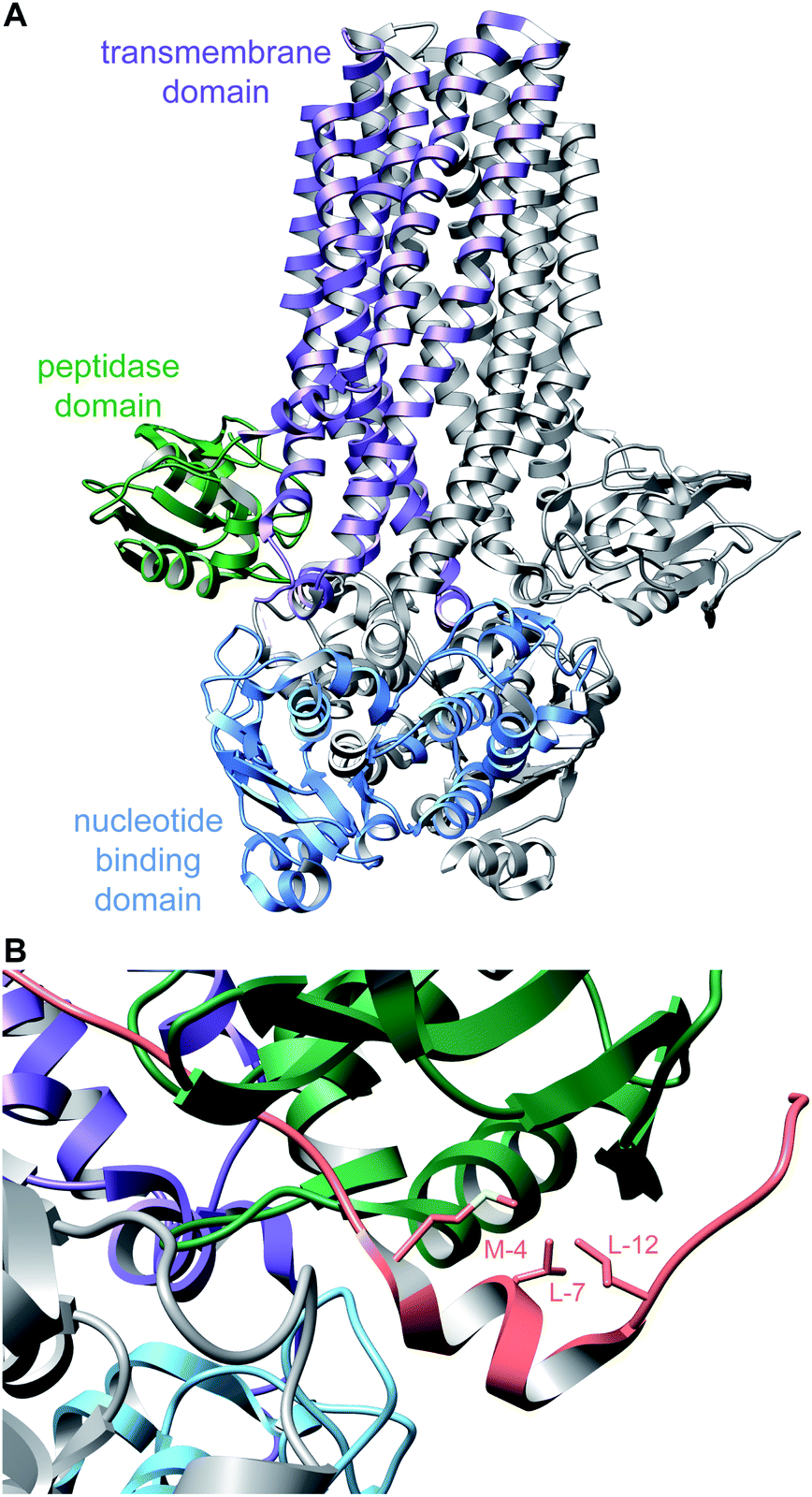 | ||
| Fig. 75 (A) Crystal structure of a PCAT (PDB: 4RY2).305 (B) Cryo-electron microscopy structure of the PCAT in panel (A) with the substrate bound, showing the α-helical fold of the LP segment recognized by the peptidase domain (PDB: 6V9Z).306 | ||
Investigation of the substrate specificity and recognition mechanism of the N-terminal peptidase domain of LahT (LahT150), the PCAT associated with a currently uncharacterized RiPP (Section 5.3.7.1),307 demonstrated tolerance to many substrates.308 The key residues that are important for LP binding are Val(−4), Leu(−7), and Leu(−12), which are conserved in many LPs ending in a double Gly motif. A co-crystal structure using an aldehyde-containing peptide analog showed that these key residues are located on a short α-helix of the LP and insert into a hydrophobic grove in LahT150 (Fig. 76).308 A similar substrate recognition mechanism was more recently shown for a full length PCAT (Fig. 75B). C39-family cysteine proteases are not always encoded as N-terminal domains of transporters and sometimes are found as stand-alone proteins such as PoyH involved in polytheonamide biosynthesis (Section 3.12). Like LahT150, this protease was shown to have broad substrate tolerance and was able to remove LPs from non-cognate RiPP classes.309
 | ||
| Fig. 76 Crystal structure of the protease domain of the PCAT LahT (teal) covalently linked via a Cys in its active site to a peptide aldehyde corresponding to the C-terminal sequence of the ProcA2.8 LP (gold) (PDB: 6MPZ).308 The double Gly residues (positions −1 and −2) are indicated as are two key Leu residues (positions −7 and −12) on an α-helix that insert into hydrophobic pockets to mediate substrate engagement. | ||
Similarly, bifunctional AMS proteins are found in all four classes of lanthipeptides,62 but in some instances, the transport and proteolysis activities are encoded in two different polypeptides. To distinguish the two types of transporters (with and without a protease domain), LanT is used for ABC transporters that lack the protease domain and LanTp was introduced for transporters that contain the N-terminal C39 protease domain.310
In addition to LP removal in bacterial peptides, subtilisin-like serine proteases are necessary for the production of the post-translationally modified (hydroxyPro) plant hormone IDA involved in the abscission process.318 In fungi, the Kex2 Ser proteases (also members of the S8 family) are believed to be involved in processing of the dikaritins (Section 3.17) but the timing is not known. Adding additional complexity, the Kex2 RSs are located at either the N-terminus or C-terminus or both termini of the core sequence, thus the exact nature of the proteolytic processes remains poorly understood.
For the thiopeptide thiostrepton, the LP removal enzyme was shown to be TsrB (TsrI in an alternative nomenclature).323 TsrB is a member of the epoxide hydrolases that belong to the α/β-hydrolase superfamily. The enzyme functions as a leader peptidase but also catalyzes the nucleophilic attack of the newly formed N-terminus of the core peptide onto an epoxide to form a second macrocyclic ring (see Section 5.7.2).
5 Secondary PTMs: commonalities and differences in accessing molecular diversity
More than ten years ago, it was suggested that RiPPs might rival non-ribosomal peptides in terms of structural diversity.324 Indeed, the past seven years have seen a tremendous expansion in the types of PTMs that are found in RiPPs. In addition to the primary PTMs covered in Section 3 that define each RiPP class, many compounds undergo additional, compound-specific secondary PTMs (sometimes called tailoring reactions analogous to tailoring processes that take place after scaffold formation in other classes of natural products). These tailoring reactions often bestow on the corresponding RiPP higher affinity and selectivity for the target by additional molecular interactions.3 In addition, they can increase environmental and/or metabolic stability, especially by modification of the N- and/or C-termini. In some cases, tailoring reactions can generate a group of congeners from an initially generated molecular scaffold. Such tailoring processes are often slower than the scaffold-forming reactions and a model has been suggested that such systems sacrifice catalytic efficiency for the ability to generate molecular diversity.325 This diversity generating strategy in RiPP biosynthesis complements the alternative approach of hypervariability of core peptide sequences.304,326–328 Both types of pathways result in high molecular diversity at the cost of catalytic efficiency.78,325 This section discusses new tailoring reactions reported since 2013, the molecular mechanisms by which they take place, and highlights differences and commonalities in the various strategies used towards natural diversification of RiPPs. Therefore, the enzymatic processes are organized here by the type of chemical transformation.5.1 Epimerization
Since epimerization is an isobaric modification, deuteration-based methods were developed to allow MS-based characterization. An initial method used a transaminase-deficient E. coli mutant to introduce deuterium labeled amino acids, which undergo exchange of deuterium with hydrogen at epimerized residues during catalysis. Deuterium loss was then localized by tandem MS.196 An improved method, termed orthogonal D2O-based induction system, does not require a transaminase mutant. It involves consecutive expression of the precursor and epimerase genes with a D2O-based medium introduced between the induction of each component.329 With a rapid analytical assay available, the substrate requirements for epimerization were studied more broadly in leader and core peptide isotope exchange studies. The four tested epimerases OspD, PlpD, AvpD, and PoyD exhibited considerable substrate promiscuity and largely introduced related D-amino acid patterns into proteusin core peptides with the same sequence.196,329 Thus, the cyanobacterial enzyme OspD that naturally installs two D-residues in its substrate, generated an alternating polytheonamide-type pattern on the PoyA core peptide. Conversely, patterns were highly distinct for different core peptides epimerized with the same maturase, collectively suggesting that the core sequence largely dictates the product pattern.329 Despite epimerase homologs exhibiting similar regioselectivities for a particular core, major differences existed regarding the order of epimerization. For example, PoyD epimerizes the core peptide of PoyA in a largely C-to-N-directional fashion, while PlpD converted an N-terminal region of PoyA first.329 Correlations between the core sequence and the epimerization pattern, such as position, residue type, or motif, remain obscure.
The activity of proteusin epimerases was also reconstituted in vitro, permitting further mechanistic studies. For PoyD and OspD, a range of simplified precursor variants of the precursor peptide were converted to products, including leaderless versions.330,331 OspD accepted a cyclic version of its core and linear peptides down to five amino acids in length with products carrying the same D-amino acid.331 In these assays, the same directionality was observed as previously reported for in vivo conversions.329 The results suggest significant potential for synthetic applications. Additional insights into the processivity of PoyD were gained by using partially epimerized peptides as test substrates.330 Marked differences regarding reaction rates as well as product patterns were observed for different isomers, showing that initial epimerization events guide downstream reaction paths in vitro. Furthermore, D2O-based labeling experiments and quantification of 5′-deoxyadenosine released by the rSAM enzyme showed that one equivalent of SAM was consumed per epimerized site.330,331 For PoyD, Cys372 was identified as a candidate for donation of the backside hydrogen during epimerization, similar to the findings with YydG (see Fig. 49, Section 3.13).330
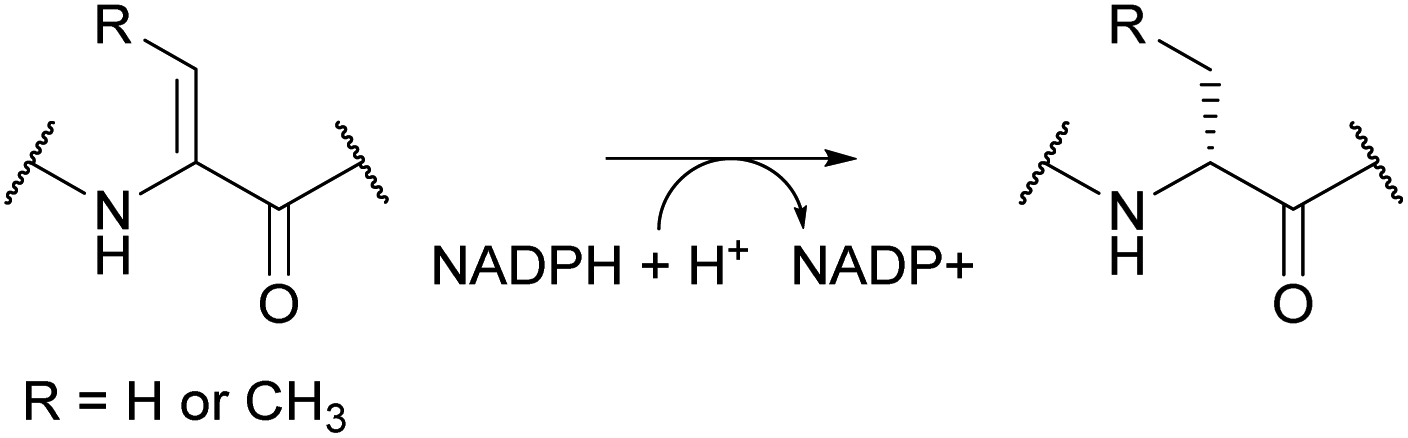 | ||
| Fig. 78 Transformations catalyzed by dehydroamino acid reductases to yield a D-amino acid. | ||
The first in vitro characterization of a LanJ enzyme was conducted on NpnJA, which converts Dha residues to D-Ala in putative peptide substrates (NpnA1–3) of an orphan BGC in Nostoc punctiforme.333 These substrates were predicted to yield linear peptide products as they do not contain Cys and because a lanthipeptide LanM homolog (NpnM) encoded in the BGC is missing several key residues necessary for cyclization activity (Section 3.1.2). Reaction of NpnJA with a range of NpnA3 variants dehydrated by NpnM revealed that NpnJA is selective for the reduction of Dha residues but is capable of introducing D-Ala residues at non-native positions in a LP-independent manner.333 The presence of an N-terminal flanking Asp residue impeded epimerization, consistent with a similar deactivating effect of charged flanking residues reported for LtnJ.332 However, NpnJA tolerated a large number of other flanking residues, demonstrating flexibility beyond the positional specificity of modified residues.333 Successful in vitro reconstitution has also been achieved for the LanJB homolog BsjJB that installs multiple D-Ala residues and a single D-aminobutyrate residue during the maturation of the two-component class II lanthipeptide bicereucin from B. cereus.334
A D-Asp residue is present at position 7 in bottromycin (Fig. 37). Deletion of a P450 gene in the bottromycin BGC yielded an intermediate that was proposed to feature a C-terminal carboxylated thiazoline and an adjacent epimerized Asp.143 Deuterium labelling experiments confirmed that this Asp could undergo non-enzymatic epimerization, which was hypothesized to occur due to the adjacent thiazoline, which lowers the pKa of the aspartate α-proton. Subsequent epimerization was proposed to be prevented by formation of the aromatic thiazole by the P450. However, more recently BotH (equivalent to BmbG), an enzyme in the α/β hydrolase family that lacks the usual catalytic residues, was shown to carry out the epimerization reaction.144 The enzyme acts prior to oxidative decarboxylation that converts the thiazoline at position 8 to a thiazole and also prior to methylation of the side chain of Asp7. Several crystal structures of the enzyme were reported resulting in a proposed mechanism in which the side chain of Asp7 may act as the base that deprotonates at its own Cα.
Epimerized amino acids are also found in conopeptides and peptides from amphibians,340 but the enzymes involved in their formation are currently not known. Similarly, the details of the origin of the D-amino acids in cacaoidin (Fig. 18A) is currently not known. Although its BGC contains a LanJB homolog that may reduce dehydro amino acids, at present the machinery that introduces those dehydro amino acids is not clear.54
5.2 NosL rearrangement during nosiheptide biosynthesis
The thiopeptide nosiheptide bears a distinctive secondary heterocycle that contains an indolic acid moiety derived from L-Trp (Fig. 79A). Mutational analyses demonstrated that two members of the rSAM superfamily, encoded within the BGC, are critical for installing this structural feature. NosL catalyzes a complex rearrangement of L-Trp to 3-methyl-2-indolic acid (MIA), and NosN, which is described in Section 5.3.6, is responsible for methylation of MIA and subsequent macrocyclization to close the secondary heterocycle of nosiheptide. Prior to NosN activity, NosIJK function to transfer MIA to a Cys residue on a linear pentathiazolyl nosiheptide intermediate.341,342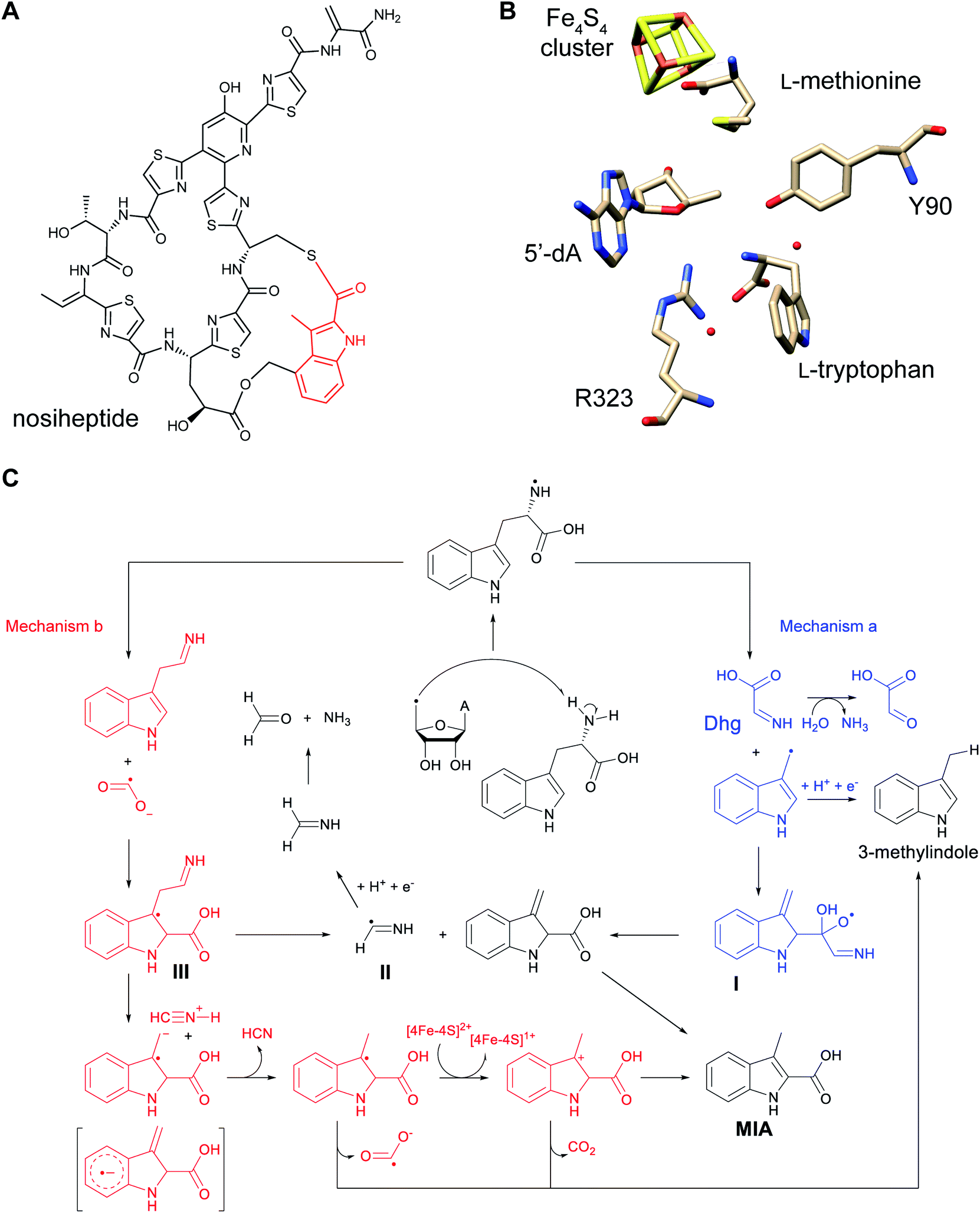 | ||
| Fig. 79 (A) Structure of the thiopeptide nosiheptide. The 3-methyl-2-indolic acid (MIA) fragment is depicted in red. (B) Active site of NosL in complex with its substrate L-Trp. (C) Summary of two mechanistic proposals for the NosL-catalyzed rearrangement. In both mechanisms, 5′-dA˙ abstracts a hydrogen atom from the amino group of Trp. Then, two different β-scission reactions cleave either the Cα–Cβ bond to form dehydroglycine (Dhg) and an indole radical (mechanism a) or the Cα–C1 bond (mechanism b) to generate formyl radical. Subsequent steps to explain the observation of 3-methylindole, Dhg, and formaldehyde in addition to the MIA product are shown. | ||
NosL belongs to the rSAM aromatic amino acid lyase family, which also includes CofH (F420 cofactor biosynthesis), HydG (hydrogenase cluster maturation), and ThiH (thiamine biosynthesis). The latter three enzymes process L-Tyr to produce p-cresol and dehydroglycine (Dhg). NosL has a homolog in the nocathiacin biosynthetic pathway, a thiopeptide that features a similar indolic acid moiety.343 Genetic complementation in addition to in vitro reconstitution experiments established that NosL is responsible for converting L-Trp to MIA.344 In recent years, NosL has been extensively studied, revealing that the reaction involves a remarkable intramolecular migration of the L-Trp carboxylate to the C2 position of the indole ring, the removal of the Cα atom and the amino group, and leaving the former Cβ as a methyl group.344–353
The mechanism by which the enzyme achieves this challenging reaction has been extensively refined as work on NosL has progressed. At first, the prevalence of tryptophanyl and tyrosyl radicals in biology led to the proposal that following canonical homolytic cleavage of SAM, the resulting 5′-dA˙ abstracts a hydrogen atom from the indole nitrogen of L-Trp, rather than from a carbon center.344 However, the determination of a crystal structure of the NosL-L-Trp complex contradicted this proposed H-atom abstraction site and rather suggested that NosL, as well as the tyrosine-lyases HydG and ThiH, would abstract a H-atom from the amino-nitrogen (Fig. 79B).345 This conclusiuon was further supported by studies using various L-Trp and L-Tyr analogs.346,349,354 By analogy with the tyrosine-lyases and based on the early detection of methylindole (MI), in addition to traces of both formaldehyde and glyoxylate as possible shunt products, a mechanistic hypothesis was proposed (Fig. 79C; mechanism a). Like HydG,355 the resulting amino-centered radical was proposed to undergo Cα–Cβ bond scission to yield a methylene radical and Dhg. These two intermediates subsequently recombine to generate a ketyl radical I that undergoes a second β-scission to form 3-methyleneindoline-2-carboxylic acid, which isomerizes to MIA (Fig. 79C). The radical coproduct II may abstract a hydrogen atom from a suitable donor to produce an imine intermediate that is hydrolyzed to yield formaldehyde and ammonia. This mechanism was supported by a number of labelling experiments that allowed tracking of the key atoms in the product. Furthermore, the proposed shunt product 3-methylindole could be accessed by trapping of the methylene radical whereas glyoxylate would be formed as a hydrolysis product of Dhg. However, the subsequent trapping and characterization of the 3-(2-iminoethyl)indoline-2-carboxylic acid radical III as an intermediate of the reaction challenged this initial hypothesis and rather suggested a direct carboxyl radical migration from Cα to the C2 atom of the indole ring, (Fig. 79C, mechanism b).352
Recent biochemical experiments using additional L-Trp analogs further support the involvement of a formyl radical rather than a methylene radical intermediate and additionally demonstrate that cyanide is a coproduct of the NosL-catalyzed reaction, prompting a revision of the proposed mechanism.353 Thus, the initial amino-centered radical undergoes a different β-scission to yield 2-indol-3-yl-ethan-1-imine and a transient carboxyl radical (Fig. 79C; mechanism b). This radical species then adds to the C2′ of the indole to form a 3-(2-iminoethyl)indoline-2-carboxylic acid radical intermediate III, which was experimentally detected.352 Recent structural characterization of NosL in complex with substrate or intermediate analogues, combined with theoretical calculations suggest the carboxyl migration is a two-step β-scission/recombination process.356 The cyanide elimination step is likely heterolytic although the details are still unclear.353,356 The resulting 3-methylindoline-2-carboxylic acid radical intermediate (or its aryl radical anion equivalent in brackets; Fig. 79C) transfers an electron to the NosL [4Fe–4S] cluster to produce a 3-methylindoline-2-carboxylic acid cation. Subsequent loss of either CO2 or a proton yields the experimentally observed products 3-methylindole and MIA, respectively (Fig. 79C). The NosL active site arrangement is such that it is able to exquisitely control the production, orientation and recombination of the carboxyl radical intermediate.356 The reason why in vitro NosL can undergo significant 3-methylindole production rather than MIA is unclear and likely depends on the nature of the reducing agent used to trigger the radical-based reaction. Furthermore, this new mechanism highlights the difference between NosL and tyrosine-lyases, in spite of sharing high sequence and structure similarities. However, the discovery that cyanide is a NosL coproduct unveils an unexpected link to the [FeFe]-hydrogenase maturase HydG, a member of the tyrosine-lyase family.353
5.3 Methylation reactions
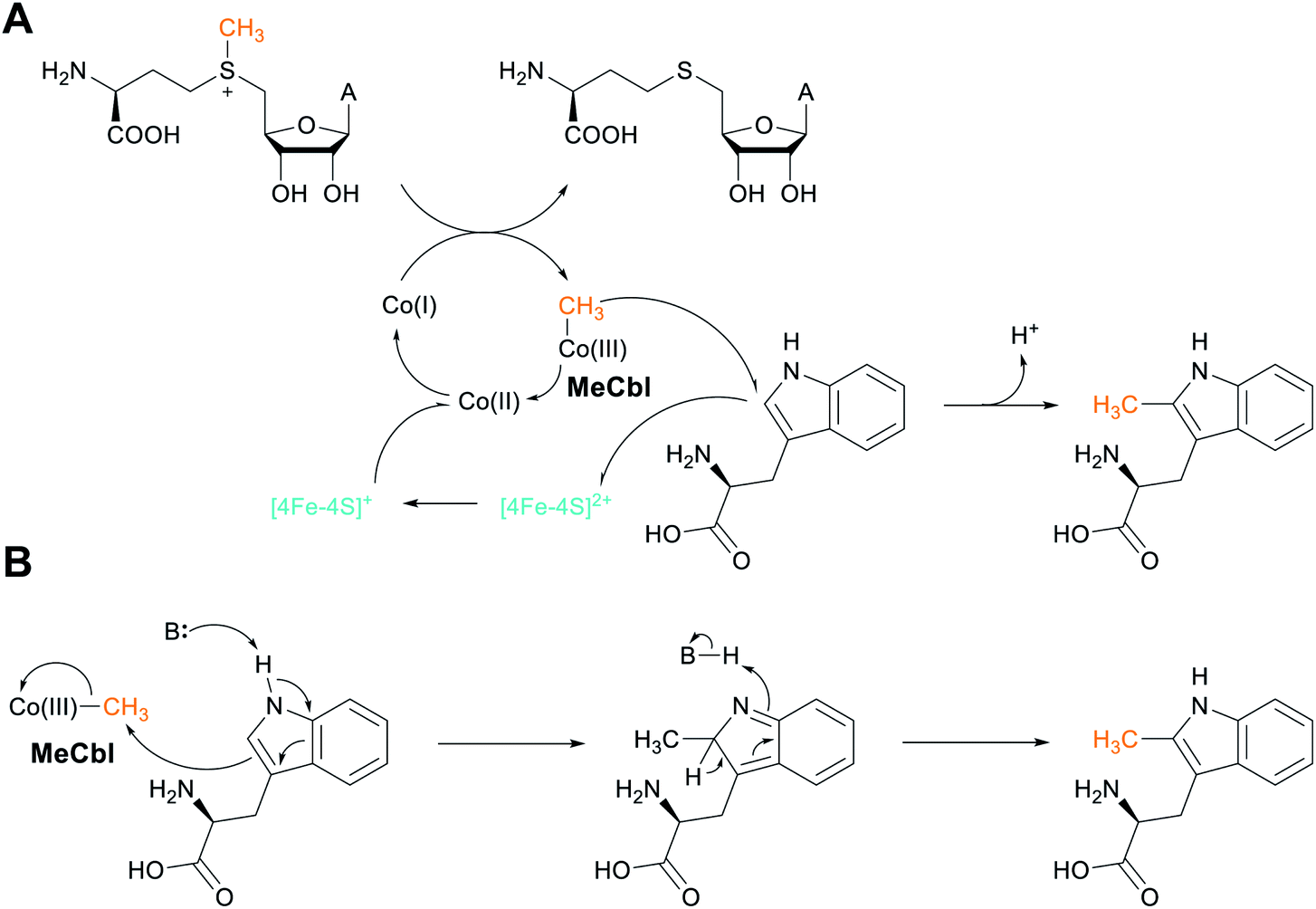 | ||
| Fig. 80 (A) Radical-based mechanism, and (B) heterolytic mechanistic proposal for TsrM-catalyzed C-methylation of L-Trp during the biosynthesis of thiostrepton. | ||
More recent biophysical examination of TsrM has revealed further unusual features of this atypical rSAM enzyme. Analysis of TsrM by Mössbauer and hyperfine sublevel correlation (HYSCORE) spectroscopies confirmed the presence of the [4Fe–4S] cluster, but revealed that neither SAM, nor L-Trp directly coordinate to the cluster.361 Previous work had shown methylcobalamin (MeCbl) to be the physiological cofactor of the enzyme,360 but further spectroscopic analyses have subsequently detected a cob(II)alamin intermediate that is likely present in an uncharacteristic five-coordinate conformation.361 In this instance, the dimethylbenzimidazole ligand to cobalt in cobalamin is replaced by a non-nitrogenous ligand, likely a water molecule.361 These observations prompted an initial proposal for a mechanism in which the MeCbl Co–C bond undergoes homolytic cleavage to generate a methyl radical that subsequently attacks the sp2 C2 of Trp, resulting in a Trp-based radical (Fig. 80A). Loss of an electron to the [4Fe–4S] cluster, followed by deprotonation, would result in 2-MeTrp. Finally, the [4Fe–4S] cluster could then reduce the cob(II)alamin intermediate to cob(I)alamin, a supernucleophilic species that might regenerate MeCbl by attack on the SAM methyl group, and would account for the accumulation of SAH.360 Such a double-displacement mechanism is consistent with the net retention of stereochemistry observed during prior in vivo labeling experiments.362
While this hypothesis has features that are in line with current knowledge of Met synthase and corrinoid/iron-sulfur protein function,360,361 recent work utilizing Trp analogs has indicated a non-radical-based mechanism for TsrM (Fig. 80B).363 This study discovered that the unmodified N1-amine group is crucial for turnover and that analogs substituted with a sulfur or oxygen at this position competitively inhibit methyl transfer. Furthermore, no formation of cob(II)alamin, nor any substrate radicals, was detected during reactions with L-Trp or its analogs, which would argue against homolytic Co–C bond cleavage. Consequently, an alternate mechanism based on SN2 chemistry was proposed in which the C2 of Trp is activated by N1 deprotonation, which induces the π electrons to attack the MeCbl methyl group, resulting in the production of cob(I)alamin and a methylated Trp intermediate. Subsequent C2 deprotonation and N1 reprotonation would generate the final product.363 The question remains as to why a rSAM enzyme has evolved to catalyze a reaction that does not seem to require the reductive cleavage of SAM, but from a chemical reactivity viewpoint, heterolytic chemistry to methylate a nucleophilic position in an indole appears more logical than radical chemistry.
The activity of PoyB and PoyC is determined by features within the PoyA core peptide, with PoyC acting on residues 1–21 and PoyB on residues 23–49. PoyC was demonstrated to also catalyze up to three of the previously proposed four C-methylations of an N-terminal Thr residue, which is dehydrated by the class II lanthipeptide dehydratase PoyF. Together, these modifications are necessary for establishing the polytheonamide N-terminal tert-butyl group (Fig. 47B) that enhances membrane insertion and bioactivity of these pore-forming cytotoxins.28
In vitro experiments using a synthetic truncated PoyA-1–15 peptide substrate variant have provided further insights into PoyC catalysis, resulting in the formation of a singly-methylated product at L-Val14, with SAH and 5′-dA detected as byproducts.364 Conversion of this substrate residue to the D-Val configuration leads to unmethylated product, indicating that methylation precedes epimerization at this position during polytheonamide maturation. Substitution of the Cys residues present within a non-canonical CX7CX2C [4Fe–4S]-binding motif to Ala resulted in a PoyC variant incapable of substrate methylation, and SAH and 5′-dA byproducts were no longer detected. The AX7AX2A variant also lost the ability to load MeCbl, suggesting a critical role for the [4Fe–4S] cluster in Cbl methylation. Consistent with this observation, when the reaction was performed using CD3-SAM, MeCbl was converted to CD3-Cbl, indicating that PoyC-bound MeCbl is recycled from SAM during catalysis.364 A mechanism has been proposed to explain the observations (Fig. 81).
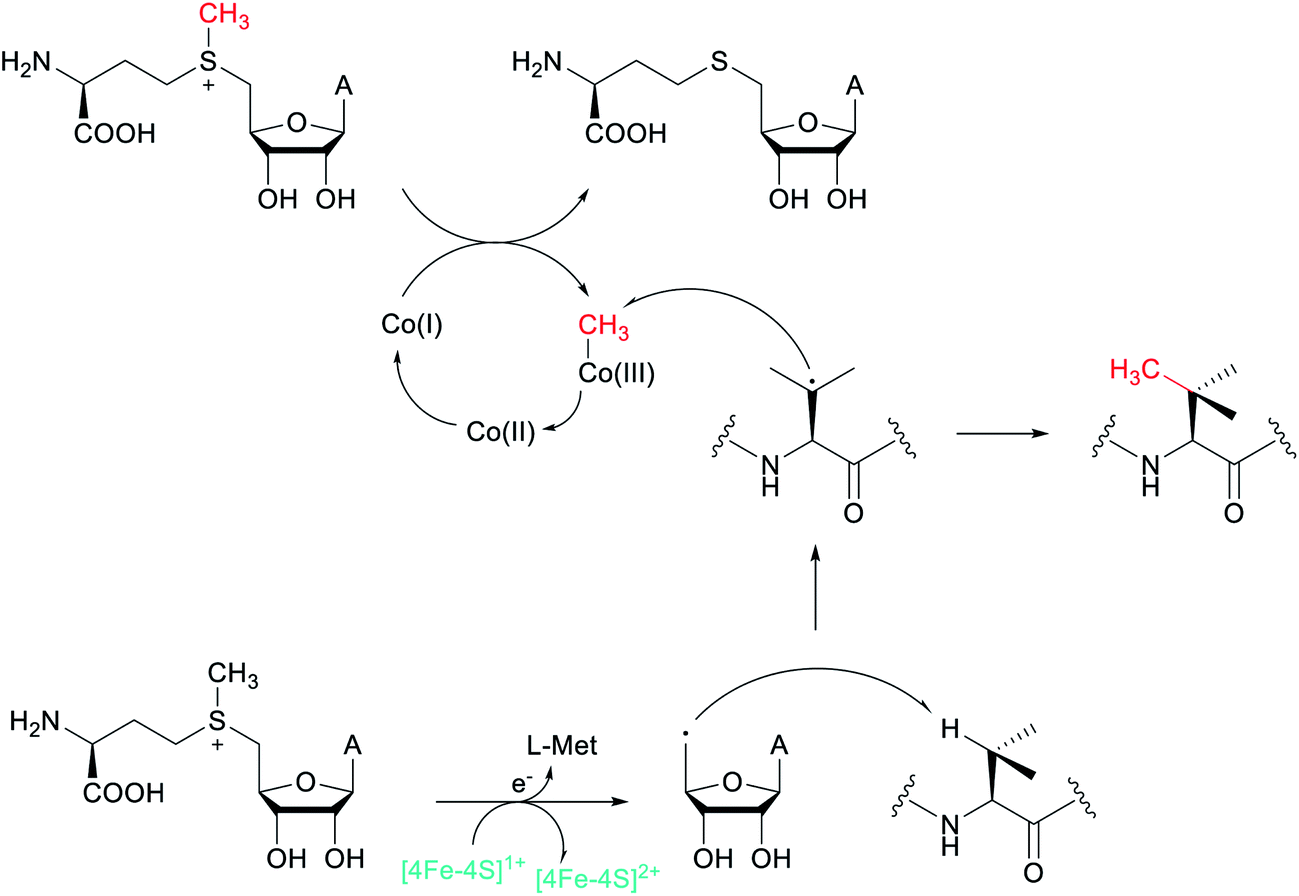 | ||
| Fig. 81 Proposed mechanism of cobalamin-dependent C-methylation catalyzed by PoyB and PoyC illustrated for Val methylation. Reductive cleavage of SAM leads to 5′-dA˙ that abstracts a hydrogen atom from Val in the substrate peptide. The resulting substrate radical then attacks the methyl group of MeCbl forming cob(II)alamin, which is reduced and methylated by SAM to regenerate MeCbl. | ||
Similar class B rSAM MTs are also involved in the bottromycin biosynthetic pathway (Section 3.7), where mutagenesis experiments have confirmed their involvement in Cβ methylations on Pro2, Val4/5, and Phe6.138,141,365 However, to date in vitro characterization studies have not yet been reported for these enzymes.
To obtain further insights into the TbtI reaction, five Cys residues conserved among TbtI orthologs were replaced with Ala.368 Only Ala variants of the three Cys residues that supply ligands for [4Fe–4S] cluster coordination resulted in inactive enzyme, whereas substitution of the remaining Cys residues had no adverse effects on TbtI activity. The observation that no additional Cys were required for activity precluded a class A-type rSAM MT reaction in which the SAM methyl group is first transferred to a protein Cys residue.174 Experiments in which the enzyme was treated with 14CH3-SAM provided further support against the possibility that the methyl group might be transferred to a different residue within the protein.
Further in vitro experimentation detected the canonical rSAM reaction products, namely 5′-dA and SAH, at near-stoichiometric quantities to the methylated product, and these were formed at similar rates.368 When TbtI was supplied with CD3-SAM, ∼90% of the 5′-dA produced contained a single deuterium, suggesting that the 5′-dA˙ produced by the homolytic cleavage of SAM may abstract a hydrogen from a second molecule of SAM that serves as the methyl donor. Indeed, in a subsequent set of experiments utilizing CD3-SAM, an ion was detected by MS that was consistent with the transfer of a CD2H group to Thz4 in the TbtA hexazole peptide. Since the enzyme does not transfer a methyl group, the term methylase may be more appropriate than methyl transferase.
Based on the combined data, a mechanism was proposed (Fig. 82) that begins in a canonical fashion with the [4Fe–4S]-mediated reductive cleavage of SAM1 to generate 5′-dA˙ and Met. 5′-dA˙ then abstracts a hydrogen atom from the methyl group of SAM2 to generate a methylene radical intermediate. Such a SAM-derived radical is unprecedented in rSAM enzymology. This SAM-derived radical is proposed to add to the electrophilic Thz4 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond to generate a resonance-stabilized substrate radical intermediate (X, Fig. 82), triggering subsequent deprotonation from the β-position by an active site base and the elimination of SAH. Glu105 has been suggested as a candidate active-site residue for deprotonation, because it is conserved in HemN (Glu145), and when substituted with Ala, resulted in a TbtI variant with severely reduced activity. Deprotonation would form a second resonance-stabilized radical intermediate (Y) with partial restoration of thiazole aromaticity. Reduction of Y, a possible additional function of the [4Fe–4S] cluster, would lead to formation of the resonance-stabilized anion Z (Fig. 82, mechanism a). Protonation of Z by the conjugate acid of an active site base, possibly Glu105, would complete the transformation.
C bond to generate a resonance-stabilized substrate radical intermediate (X, Fig. 82), triggering subsequent deprotonation from the β-position by an active site base and the elimination of SAH. Glu105 has been suggested as a candidate active-site residue for deprotonation, because it is conserved in HemN (Glu145), and when substituted with Ala, resulted in a TbtI variant with severely reduced activity. Deprotonation would form a second resonance-stabilized radical intermediate (Y) with partial restoration of thiazole aromaticity. Reduction of Y, a possible additional function of the [4Fe–4S] cluster, would lead to formation of the resonance-stabilized anion Z (Fig. 82, mechanism a). Protonation of Z by the conjugate acid of an active site base, possibly Glu105, would complete the transformation.
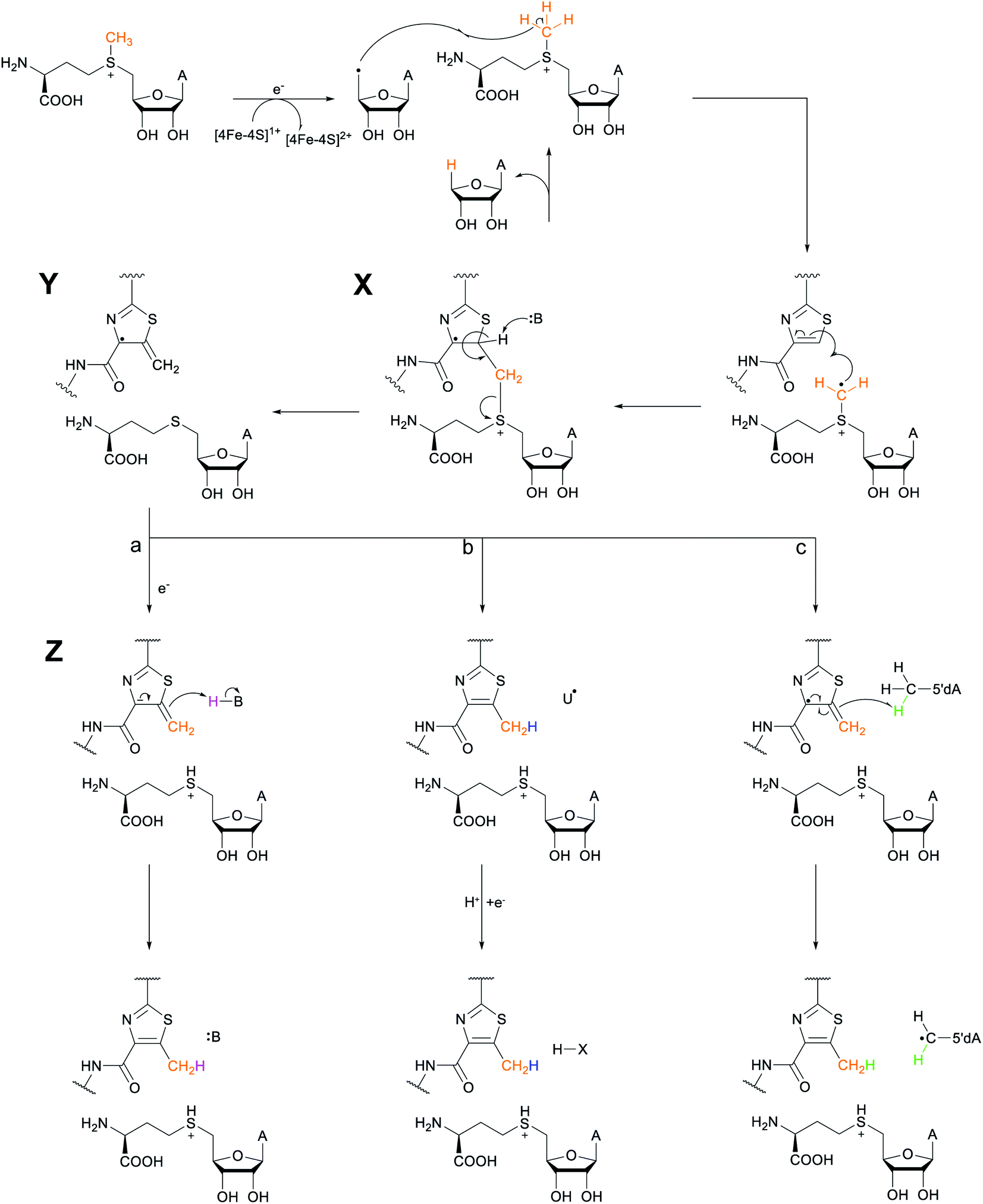 | ||
| Fig. 82 Proposed mechanisms for C-methylation of thiazole by the rSAM protein TbtI. In all three mechanisms, 5′-dA˙ generated from one SAM molecule abstracts a hydrogen atom from the methyl group of another SAM molecule, and the resulting methylene radical adds to the electrophilic thiazole to form radical X. Upon elimination of SAH, three different mechanisms may convert the intermediate radical Y to the methylated thiazole product. In mechanism b, U is an unidentified redox active amino acid of TbtI. | ||
A plausible variation on the mechanism involves the abstraction of a hydrogen atom from a redox active amino acid (H-U) in the TbtI active site (mechanism b). The resulting amino acid would be subsequently reduced in preparation for another turnover. Alternatively, the allylic radical Y could abstract a hydrogen atom from the methyl group of 5′-dA (mechanism c), but in vitro experiments using SAM deuterated at the 5′ position and methyl group effectively ruled out this possibility. Although additional biochemical and structural studies are required to further elucidate the TbtI mechanism, the data so far represent a fundamentally different use of SAM for this class of enzymes, reflecting their exceptional catalytic diversity.
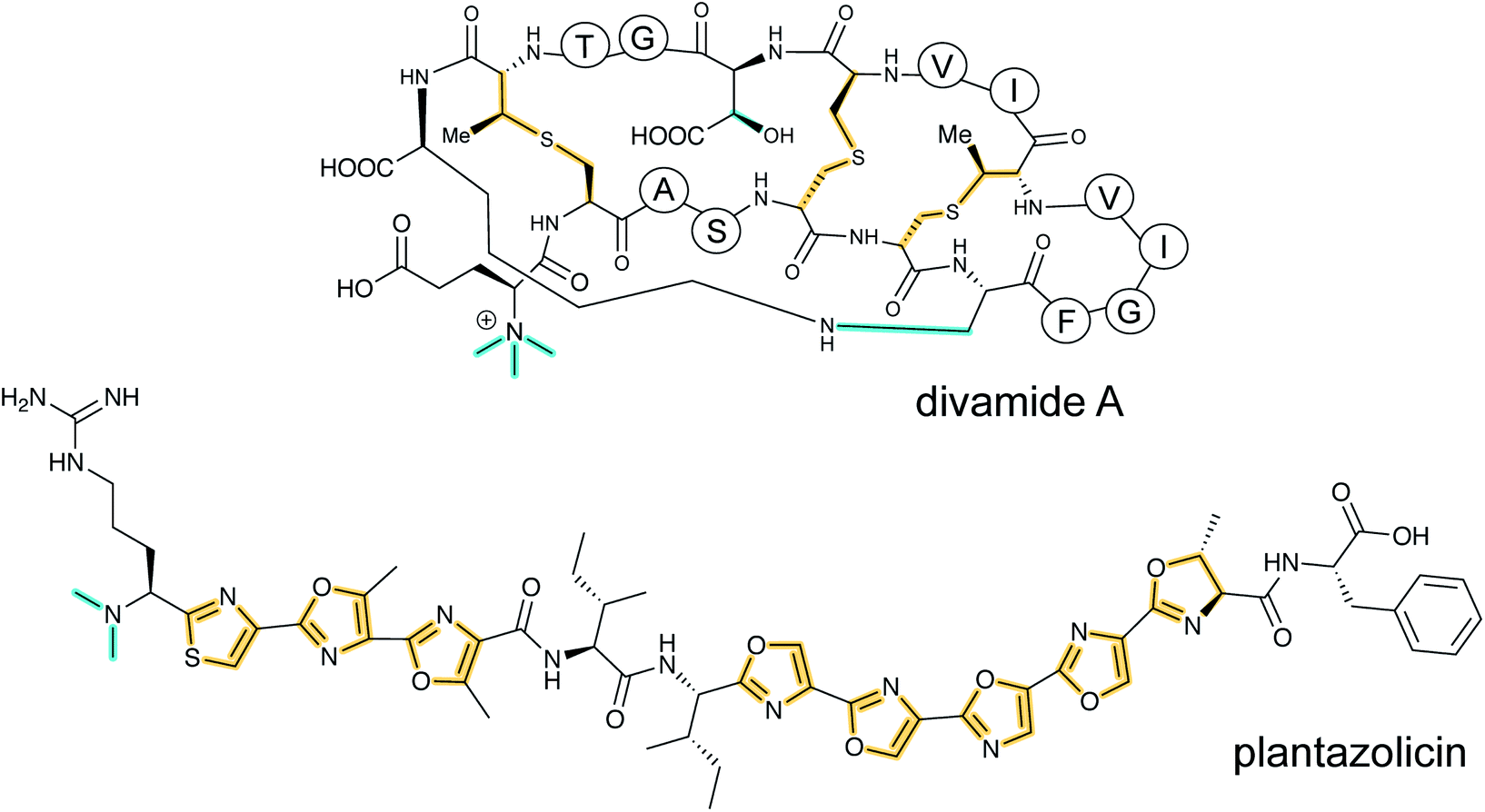 | ||
| Fig. 83 Structures of the N-methylated RiPPs divamide A and plantazolicin. Divamides are cinnamycin/duramycin-like class II lanthipeptides and plantazolicin is a LAP. For the cypemycin structure, see Fig. 27. Class-defining PTMs are yellow while secondary PTMs are cyan. | ||
The methyltransferase for the LAP plantazolicin (Section 3.4), termed BamL or PznL, as well as the ortholog BpumL from a related BGC encoded by Bacillus pumilus, were reconstituted in vitro. For a range of offered peptides with N-terminal Arg, but lacking heterocyclized moieties, both enzymes were highly selective and methylated only azole-bearing desmethyl-plantazolicin.377 In contrast, a range of synthetic heterocyclized plantazolicin analogs with truncated C-termini were all accepted, including a minimalistic Arg–Cys-based dipeptide with one thiazole moiety.378,379 The crystal structures of BamL and BpumL, which also included versions with bound truncated substrates (PDB: 4KVZ, 5DLY, 5DM0, 4KWC, and 5DM1), provided a rationale for this selectivity.377 They revealed a plantazolicin-binding cleft with a hydrophobic tunnel harboring the azoles units. Constrictions in the narrow cleft provide an architecture that only allows access of heterocyclized substrates with planar azole moieties. The active-site pocket is sufficiently long to accommodate about the first five residues of the substrate. In agreement, KM values showed a tendency to increase with decreasing peptide length.
Unlike these LAP biosynthetic enzymes, a study on the pathway of the linaridin cypemycin380 (Section 3.3) showed that its methyltransferase CypM converted structurally diverse peptides that include open-chain as well as ring-containing molecules, such as the lanthipeptides nisin and haloduracin. Methylation occurred at the α-amino group of various amino acids as well as the ε-amino function of internal or C-terminal Lys units. The mononaridin MT MonM was shown to also catalyze N-terminal alkylation reactions with SAM analogs. The ability of MonM to catalyze allylation of the nisin N-terminus allowed fluorescent labeling of nisin for a cell imaging study.104
An α-N-methylating enzyme, termed PhomM, was also reported for phomopsins, RiPP natural products of ascomycete fungi (Section 3.17).16 The enzyme, obtained from E. coli expressions, generated mono- and dimethylated products, with only the monomethylated version thus far found in nature.
Divamide A (Fig. 83) was isolated and characterized from the marine tunicate Didemnum molle,381 and is structurally related to the class II lanthipeptide cinnamycin produced by Streptomyces cinnamoneous,382 featuring three methyllanthionines, a lysinoalanine, and a β-hydroxy-Asp residue (Fig. 83). Additionally, divamide A features a rare N-trimethylated Glu residue and possesses anti-HIV activity.381 The BGC was identified in the uncultivated symbiotic cyanobacterium Prochloron didemni by searching for candidate precursor-encoding sequences in the tunicate metagenome. The entire BGC was expressed heterologously in E. coli to yield an unmethylated precursor lacking the lysinoalanine residue, a feature subsequently introduced chemically by a previously established method.383 The presence of the lysinoalanine (see Section 5.10), was essential for the activity of DivMT, the MT responsible for catalyzing N-terminal trimethylation in vitro.381
Initial in vitro experiments using NosN and a MIA thioester of N-acetylcysteamine (SNAC), a surrogate for MIA bound as a thioester to the acyl carrier protein NosJ, confirmed that the enzyme is responsible for indole methylation at C4.342,385 When CD3-SAM was supplied, only two deuterium atoms were transferred to the substrate, as reported for class C368 and A rSAM MTs.174 Thus, like TbtI (Section 5.3.3), NosN also is not a methyl transferase but a methylase.385 The third methyl deuterium from CD3-SAM was incorporated into 5′-dA, a coproduct of the reaction.385 An additional similarity between NosN and TbtI is the essentiality of only those Cys residues that are involved in coordination of the [4Fe–4S] cluster, which indicated that methyl transfer is not mediated by a protein Cys residue,342 as it is for class A rSAM MTs.174
An additional intermediate believed to be 4-methylene-3-methylindolic acid-SNAC (MMIA-SNAC, see Fig. 84A) containing an electrophilic C4 methylene group has been detected in NosN reconstitution experiments.385 3,4-Dimethyl-2-indolic acid-SNAC (DMIA-SNAC, see Fig. 84A) was also observed at low intensity. Given that NosN was shown to only catalyze a single turnover at most with the MIA-SNAC substrate and that the resulting product MMIA-SNAC is slowly converted to DMIA-SNAC, it was proposed that MIA-NosJ might not be the natural enzyme substrate. Indeed, NosN was shown to catalyze formation of a macrocyclic product when a synthetic pentathiazolyl substrate mimic was used spanning residues 42–46 of the precursor peptide NosM including the thioester linkage between Cys8 and MIA (Fig. 84B).385 This result was later recapitulated using polyhistidine-tagged variants of the substrate NosM. Subsequent mutational analysis and labelling of the substrate confirmed that Glu6 of the pentathiazolyl substrate is responsible for trapping the exocyclic methylene intermediate generated by NosN to close the side-ring macrocycle through an ester bond.385 The use of additional substrate mimics and the capture of NosN reaction intermediates have provided further information regarding the timing of NosN macrocyclization during nosiheptide maturation, showing that NosN acts prior to pyridine ring-formation by NosO and most likely also prior to NosD/E-catalyzed dehydrations of Ser/Thr residues (Section 3.2).386,387
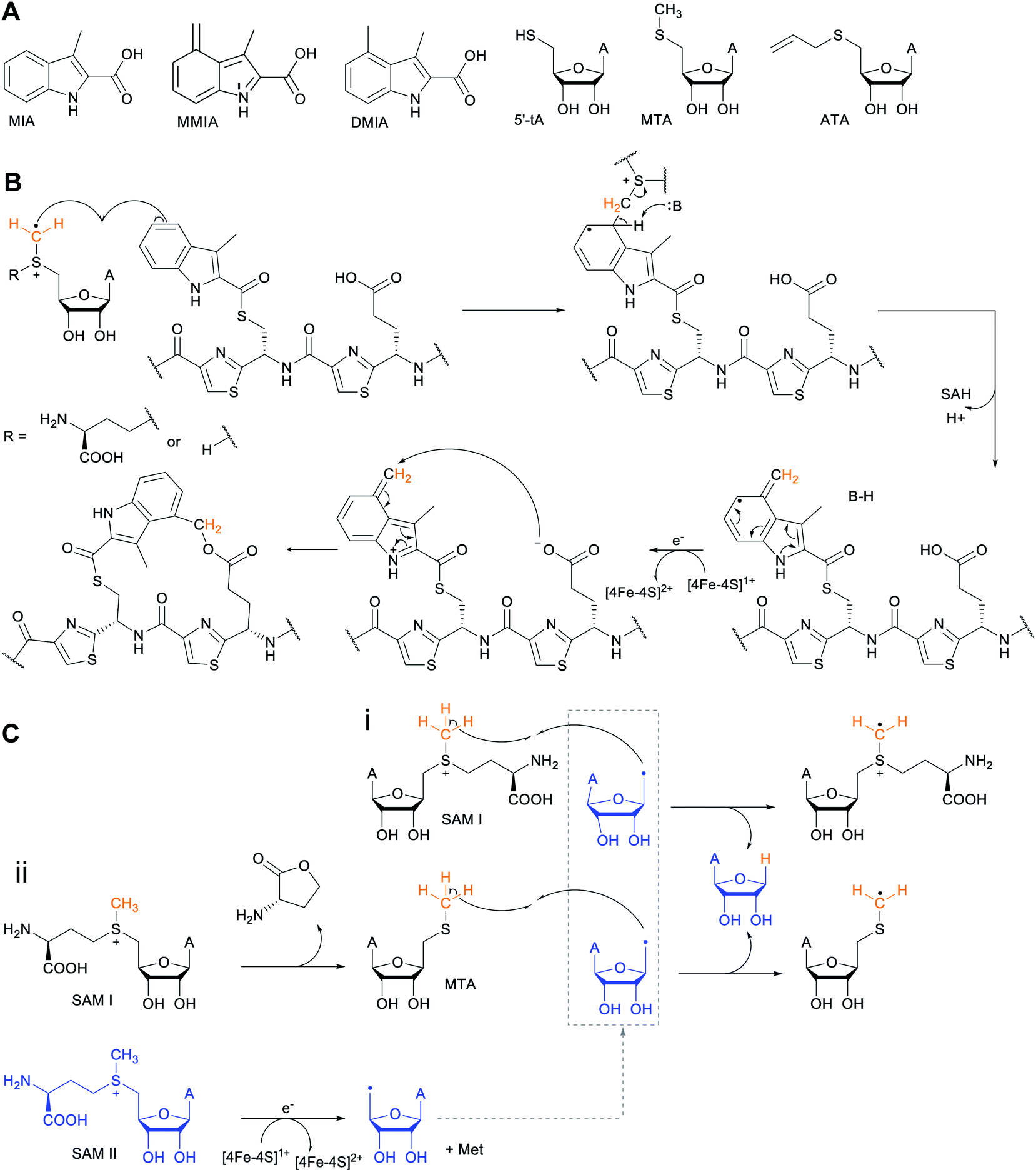 | ||
| Fig. 84 Proposed mechanisms of NosN catalysis. (A) Structures of various proposed or detected intermediates and products in the reaction of NosN with native substrates or substrate analogs. (B) Mechanism of methylene transfer and intramolecular trapping by a Glu residue. (C) Two mechanisms that differ in whether 5′-dA˙ abstracts a hydrogen from SAM (mechanism i) or from MTA (mechanism ii). | ||
The described observations permitted a working hypothesis for the NosN mechanism (Fig. 84B) that initially proceeds in an analogous manner to that described for TbtI (Section 5.3.3) with the [4Fe–4S]-mediated homolytic cleavage of SAM to produce Met and a 5′-dA˙ radical. The 5′-dA˙ abstracts a hydrogen from the methyl group of a second molecule of SAM to produce a methylene radical, which adds to C4 of MIA that has already been connected to Cys8 of the core peptide containing the thiazole groups. The resulting aryl radical intermediate is deprotonated at C4, triggering the elimination of SAH and the generation of a substrate radical intermediate bearing a C4 exocyclic methylene group. This species is oxidized by the [4Fe–4S] cluster to give the electrophilic MMIA species attached to Cys8, which is subsequently attacked by the Glu6 carboxyl group, resulting in ester bond formation and closure of the nosiheptide secondary heterocycle.385
An alternative mechanism (Fig. 84C) has been put forward in which 5′-methylthioadenosine (MTA, Fig. 84A) is the methyl donor, based on the detection of 5′-thioadenosine (5′-tA, Fig. 84A)388 and on recent in vitro NosN experiments in which a pantetheinylated SAM substrate (MIA-pant) was incubated with allyl-SAM.389 This approach permitted the trapping of 5′-dA˙ by the olefin moiety of a second allyl-SAM unit, consistent with the use of two molecules of SAM. Omission of MIA-pant from the reaction resulted in the production of 5′-allylthioadenosine (ATA, Fig. 84A), which is proposed to be generated in an analogous manner to 5′-tA from MTA, suggesting its involvement in the NosN mechanism. Consequently, a variation on the mechanism has been proposed in which binding of the MIA-containing substrate to the enzyme triggers the conversion of SAM to MTA (Fig. 84C). 5′-dA˙ generated by the reductive cleavage of a second SAM molecule then abstracts a hydrogen atom from MTA, generating a methylene radical that subsequently attacks the C4 position of the MIA moiety. 5′-tAH released as a coproduct was suggested to react with homoserine lactone to produce SAH, which has been identified as a NosN coproduct. This mechanism was supported by density functional theory calculations.388,390 However, the production of 5′-tA has been questioned and the recent characterization of two on-pathway SAM adduct intermediates casts doubt on the involvement of MTA as an intermediate in the NosN-catalyzed reaction.385,386 Further biochemical and structural investigations will be required to distinguish between the two mechanisms of methyl transfer.
5.3.7.1 Methylation of the C-terminal carboxylate. Several O-methyltransferases have been reported to methylate the C-terminal carboxylate of peptides. The first protein performing methylation of the C-terminus of a RiPP was reported in 2013 with the discovery of aeruginosamide B, a linear cyanobactin (Section 3.6) in which the N-terminus is prenylated and the C-terminus methylated (Fig. 85A).391 Its BGC encodes a didomain protein AgeMTPT with prenyltransferase and methyl transferase domains (Fig. 85B). The enzyme was subsequently reconstituted and demonstrated to prenylate thiazoline-containing peptides.392
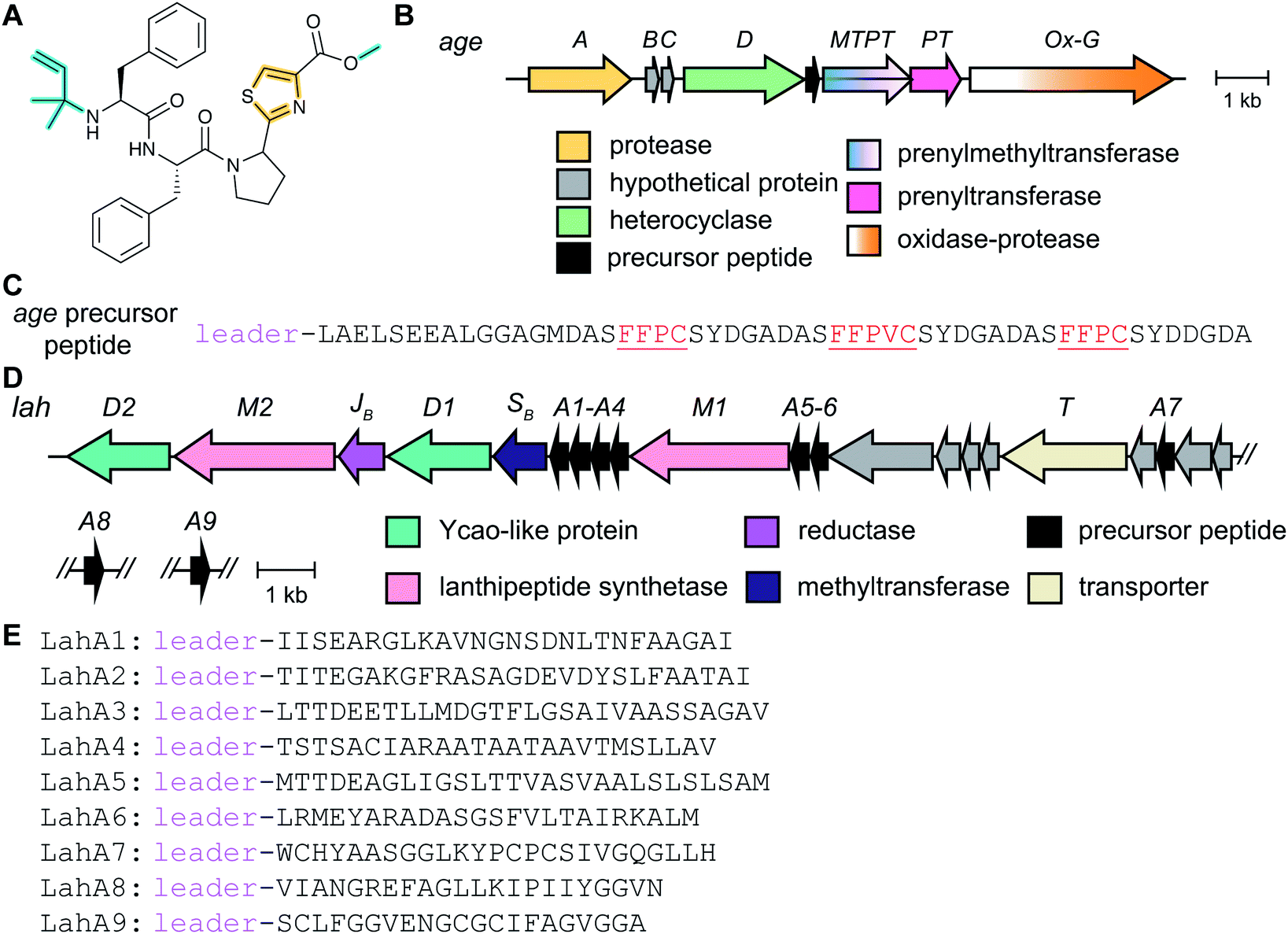 | ||
| Fig. 85 Biosynthesis of RiPPs that are methylated on the C-terminus. (A) Structure of aeruginosamide B that is N-prenylated and methylated on its C-terminal carboxylate shown in cyan. (B) BGC of aeruginosamide B. (C) Precursor peptide for aeruginosamide B with the core peptides in red. (D) RiPP BGC in Lachnospiraceae. (E) Sequences of the seven precursor peptides of the lah BGC, some of which are methylated on the C-terminus by LahSB. | ||
Lassomycin is an anti-mycobacterial lasso peptide (see Section 3.8) with an O-methylated C-terminus.393 Using the predicted MT within the lassomycin BGC as a BLAST query, a small number of lassomycin-like BGCs were identified in bacterial genomes,394 of which the stsp cluster, located in a Streptomyces sp. genome, served as the source for in vitro MT studies. These experiments showed that StspM attaches a methyl group to the carboxyl moiety of the C-terminal Pro residue. The enzyme converted the full-length open-chain precursor peptide with a predicted 16 amino acid core peptide as well as N-terminally truncated variants down to eight residues, with decreasing efficiency for shorter peptides. Further mutational studies showed that the enzyme tolerates exchange of the terminal Pro to Ala and of more extended amino acid substitutions close to the modification site.
An MT that displays substrate tolerance was also encoded in a BGC of the human microbiome member of the Lachnospiraceae group (LahSB; Fig. 85D). This enzyme was shown to methylate the C-terminus of a subset of precursor peptides (LahA2–5; Fig. 85E). Its activity was LP independent and a crystal structure was reported for the enzyme.307 The final structures of the RiPPs produced by this BGC are currently not known.
In contrast to C-terminal carboxylate methylation, the carboxylate side chain of D-Asp in bottromycin is methylated by BmbA (Fig. 37).143In vitro assays using the S. scabies ortholog of this protein indicated that this is the final step in bottromycin biosynthesis.
5.3.7.2 Cryptic methylation: MT-mediated β-amino acid incorporation. Previous reports identified a family of O-methyltransferases that are commonly found encoded in actinobacterial class I lanthipeptide BGCs.33,395 Indeed, members of this family, referred to as LanSA enzymes, represent the third-most abundant enzymes associated with such BGCs after the core biosynthetic enzymes LanB and LanC (Section 3.1.1).62 LanSA enzymes share low homology with protein L-isoaspartate (D-aspartate) O-methyltransferases (PIMTs). Consistent with a role in PTM of Asp, associated LanA substrates all possess a conserved Asp residue in the core peptide region. Recently, a representative enzyme encoded in the olv BGC from S. olivaceus was studied both in vivo and in vitro, revealing that methylation of this conserved Asp residue (Asp6) sets the stage for a remarkable transformation that results in the incorporation of a β-amino acid residue in the peptide chain (Fig. 86).70
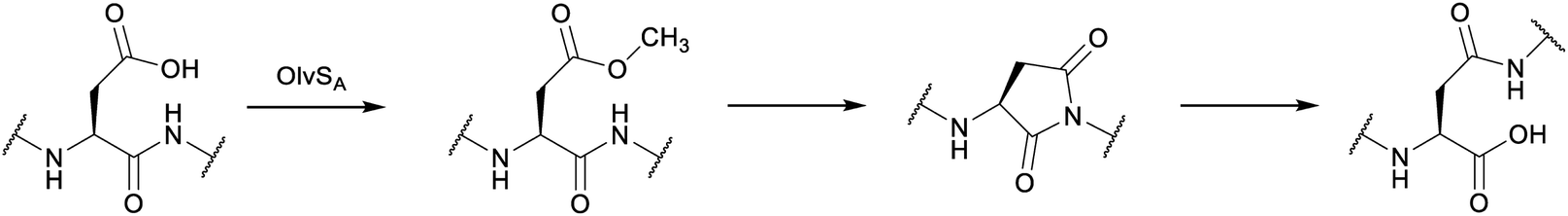 | ||
| Fig. 86 OlvSA-catalyzed L-Asp O-methyltransfer and formation of L-isoAsp via a succinimide intermediate. OlvSA orthologs are often encoded in class I lanthipeptide BGCs (Section 3.1.1) in Actinobacteria. | ||
Heterologous co-expression of His6–OlvA, which contains Thr, four Ser and four Cys residues, with the dehydratase OlvB and cyclase OlvC in the presence or absence of OlvSA, resulted in a mass consistent with a 4-fold dehydrated product. However, despite having identical masses, the 4-fold dehydrated products of the two experiments possessed different retention times during RP-HPLC analysis, suggesting structurally different products. Furthermore, a second minor product corresponding to a 5-fold dehydrated peptide was detected exclusively in the presence of OlvSA. This product disappeared on extended sample incubation to leave only the 4-fold dehydrated product, hinting that the 5-fold dehydrated peptide might represent an intermediate in the reaction. Thiol derivatization experiments provided strong support that all four Cys residues in the core peptide are involved in the formation of (methyl)lanthionine crosslinks, which prevented informative tandem MS analyses.
To elucidate the structure of OlvSA-generated products and the nature of the transformation catalyzed, NMR analyses were performed and solution structures were obtained.70 Critically, following reactions in the presence of OlvSA, strong NOE signals were observed indicative of a β-peptide linkage between the side chain carboxylate of Asp6 and the amino group of Gly7. This conversion of Asp to isoAsp introduces an additional methylene group into the peptide backbone, and an unusual carboxyl side chain, explaining the shift in retention time observed during RP-HPLC analysis. To probe the possible reaction mechanism of OlvSA, its activity was reconstituted in vitro using 4-fold cyclized His6–OlvA. In time course experiments, SAM-dependent methylation was observed. Subsequently, 5-fold dehydrated product consistent with the formation of succinimide was detected, concomitant with the appearance of a 4-fold dehydrated product (Fig. 86). In addition to the NMR structure of the final product with an isoAsp residue, complementary in vitro hydrazine trapping and 18O-labeling experiments provided further evidence of likely succinimide formation during the transformation. 18O-labeling experiments further indicated that multiple cycles of methylation occur via the succinimide intermediate, and that OlvSA is also able to accept isoAsp containing cyclic peptides. This latter observation suggests that the electrophilic succinimide might be the intended product, and further experimentation will be required to explore this possibility.
5.4 Arginine to ornithine conversion during landornamide biosynthesis
Landornamide A (Fig. 87B) represents the most recent addition to the proteusins, and is distinct from other, polytheonamide-type197,202 family members (Section 3.12) in that it is a bicyclic peptide rather than forming a beta-helix, and because it possesses ornithine residues as a novel PTM.204 Landornamide A is the product of the osp (renamed osp1) BGC encoded in Kamptonema (Oscillatoria) sp. The BGC (Fig. 87A) features genes encoding OspD, a previously characterized rSAM epimerase (Section 5.1.1) that installs two D-amino residues into the OspA precursor core peptide,196,329,331 a class II lanthionine synthetase (OspM), and OspR, an initially hypothetical protein. OspR does not resemble any previously characterized maturase enzymes, but weak homology to arginases suggested it might be the enzyme responsible for the hydrolysis of two L-Arg residues in the OspA core peptide to L-Orn.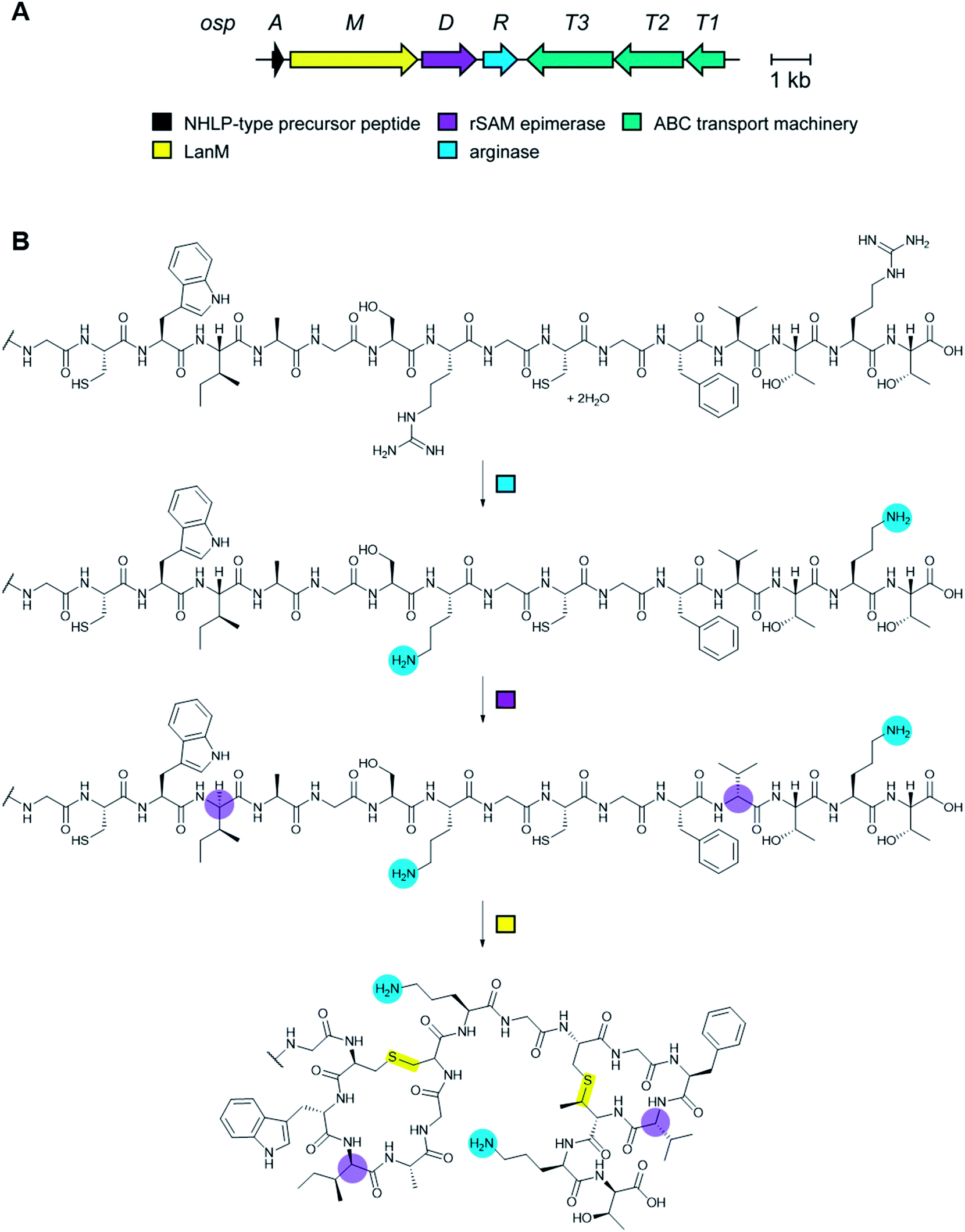 | ||
| Fig. 87 (A) BGC for landornamide. (B) Biosynthetic pathway towards landornamide. Since proteusins do not have class-defining PTMs (Table 1), the color-coding is based on the enzymes that install each modification. | ||
To access the final product of the pathway and to characterize the individual maturation steps, the ospAMDR genes were cloned and expressed in E. coli as a multi-gene unit retaining native intergenic regions, with ospA modified to introduce an N-terminal His6-tag. Subsequent purification, proteolytic digestion and LCMS analysis of an OspA core fragment revealed an overall loss of 120 Da relative to an unmodified peptide, corresponding to two dehydrations and the loss of two urea equivalents, which was consistent with the predicted activities of OspM and OspR, respectively. Expression of pathway variants containing single-point mutations within PTM enzyme loci were performed in order to probe the function and timing of each of these enzymes during biosynthesis. Mutation of the predicted OspR manganese-binding motif resulted in a product bearing two lanthionine bridges and two D-amino acid residues at the expected positions, but lacking Orn residues in place of Arg8 and Arg15, confirming the role of the enzyme in Arg-to-Orn conversion. In all instances in which the native ospR coding sequence was retained, complete conversion of both Arg8 and Arg15 to Orn was observed, indicating that osp PTM enzymes function in a promiscuous manner and can perform catalysis irrespective of other PTMs.
Based on a proposed mechanism for rat liver arginase derived from structural data,396 OspR catalysis likely begins with the initial nucleophilic attack on the guanidinium group of Arg by an active site metal-bridging hydroxide ion, resulting in the formation of a metastable tetrahedral intermediate (Fig. 88). Subsequent proton transfer, possibly mediated via Asp39, to the amine results in collapse of the tetrahedral intermediate to yield L-Orn. The subsequent addition of a water molecule to the manganese cluster facilitates the departure of urea.
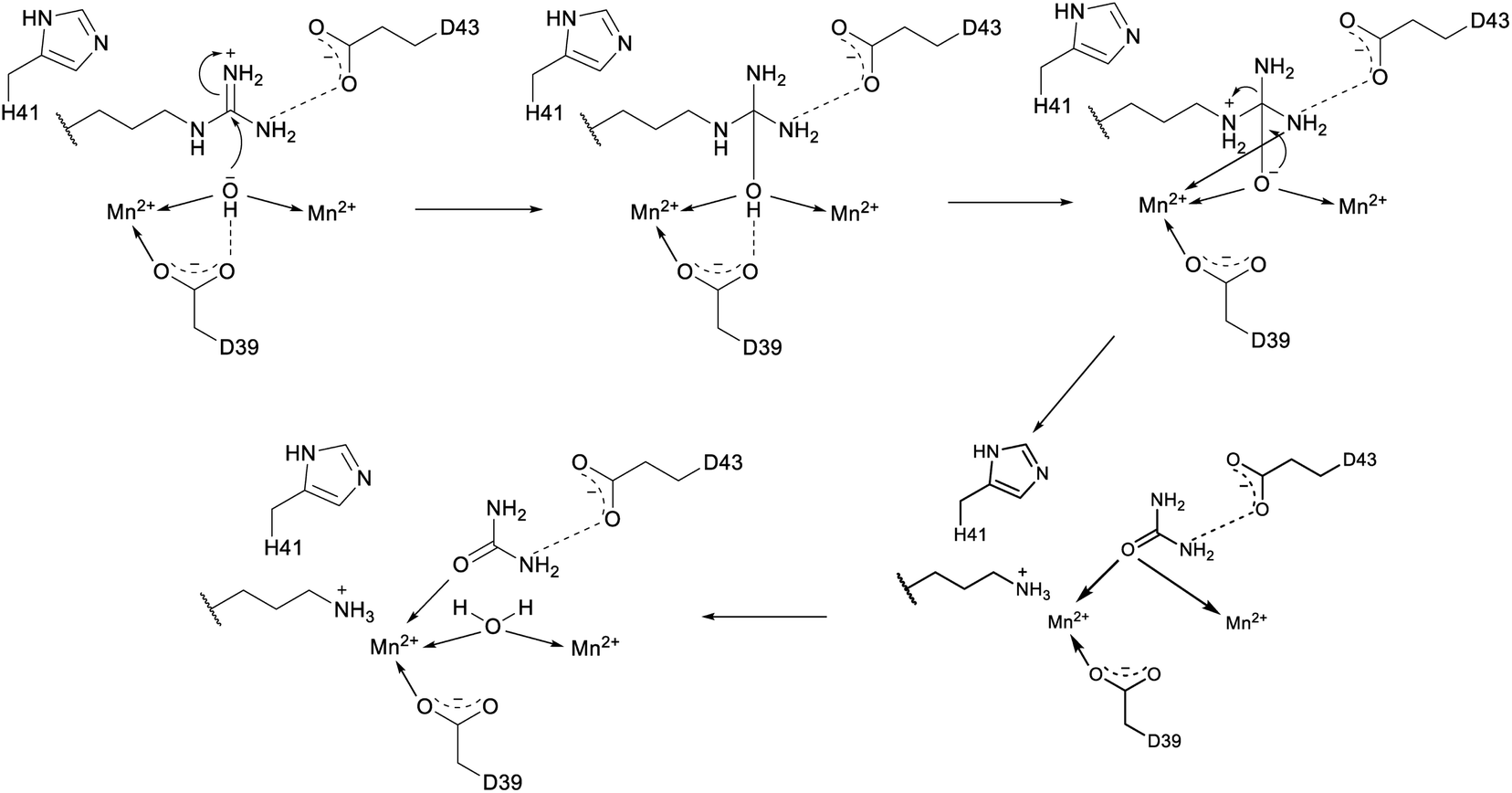 | ||
| Fig. 88 Proposed mechanism for the conversion of Arg to Orn by OspR during landornamide biosynthesis. | ||
5.5 Arginine to citrulline conversion during lasso peptide biosynthesis
Another recently characterized PTM involving Arg was found in the lasso peptide citrulassin. Arg9 in this compound is hydrolyzed to citrulline and the Arg involved is conserved in all 55 homologs of the precursor peptide.154 An obvious peptidylarginine deiminase is not encoded in the BGC, and therefore the origin of the citrulline is currently unknown.5.6 Phosphorylation during lasso peptide biosynthesis
During efforts to expand the lasso peptide family (Section 3.8) beyond members isolated and characterized from either proteobacteria or actinobacteria, a recent study focused on a family of BGCs found among firmicutes. Heterologous expression of a BGC from Paenibacillus dendritiformis led to the isolation of paeninodin, a lasso peptide bearing a phosphorylated C-terminal Ser residue.162 Indeed, a unique feature conserved among the BGCs of firmicutes studied is a gene encoding a putative HPr kinase (InterPro family: IPR011104), which are typically involved in sugar uptake processes. Deletion of the HPr-related kinase gene padeK from the expression plasmid abolished production of phosphorylated paeninodin, which was recovered by in trans expression from an additional vector carrying padeK. In vitro experiments confirmed that PadeK modifies the C-terminal Ser of the precursor peptide. A PadeK homolog from Thermobacillus composti (ThcoK) was shown to be exchangeable for PadeK both in vivo and in vitro and exhibited much higher phosphorylation activity.HPr kinases share a conserved active-site motif consisting of one His, one Lys, and two Asp residues (Fig. 89). Stabilized by hydrogen bonding with His123, the conjugate base of Asp179 deprotonates the Ser46 side chain of HPr for subsequent attack on the γ-phosphate group from ATP. Lys161 is responsible for ATP binding and Asp178 participates in Ca2+ coordination. Single and quadruple Ala substitutions of the corresponding residues, in addition to double Asp-to-Ala substitutions, in both PadeK and ThcoK abolished phosphorylation activity. These lasso peptide tailoring kinases have the ability to transfer multiple phosphate groups onto the precursor peptides, depending on the phosphate donor and the precursor sequence leading to polyphosphorylated lasso peptides.397 More recently, a second example of a lasso peptide with a phosphorylated tail was reported for pseudomycoidin.398 The authors reported that after phosphorylation, the phosphate group is diglycosylated, a unique PTM not previously reported. The BGC for this lasso peptide from Bacillus pseudomycoides resembles that of paeninodin and does not contain any obvious glycosyltransferases. Genetic experiments suggest that the nucleotidyltransferase PsmN may be responsible for glycosylation.
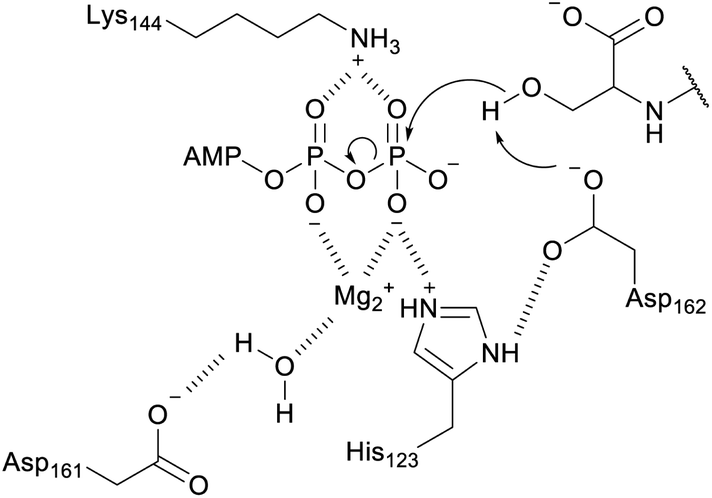 | ||
| Fig. 89 Proposed mechanism for phosphorylation of the lasso peptide paeninodin. | ||
A wide range of substrates varying in both length and amino acid composition were assayed to explore ThcoK substrate specificity. It was shown that even a variant comprising only the last five residues of the peptide, is modified albeit to a small extent. Substitution of the C-terminal Ser residue abolished phosphorylation activity, even when replaced with residues bearing a hydroxyl group side chain, and phosphorylation was also not detected following the substitution of core residues for Ser at several alternate locations. These findings indicate that ThcoK phosphorylation proceeds in a leader- and core-independent fashion and that the C-terminal Ser is essential for activity. Finally, both PadeK and ThcoK were unable to modify the mature paeninodin lasso peptide, suggesting that the lasso fold impedes access of the kinase to the C-terminal Ser and that phosphorylation likely precedes lasso fold formation.
Though phosphorylation was localized to the C-terminal Ser residue, the modification could occur on either the side chain or the C-terminus. To distinguish between these two possibilities, NMR analysis comparing synthetic and enzymatically phosphorylated PadeA 11-mer variants definitively demonstrated that phosphorylation occurs at the Ser side chain.162
5.7 Hydroxylation and halogenation
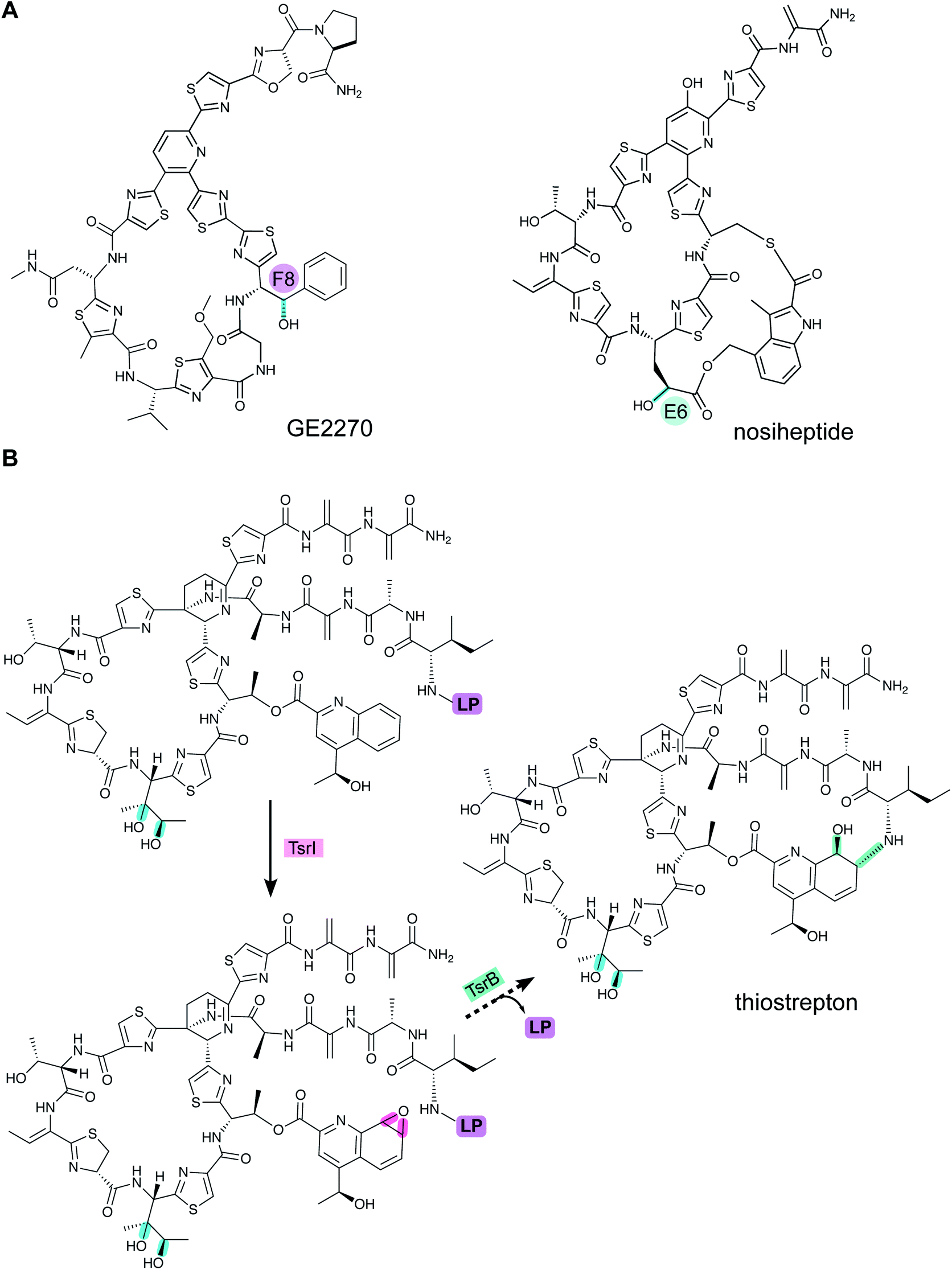 | ||
| Fig. 90 (A) Structures of GE2270 and nosiheptide with the oxidations by characterized oxidative tailoring enzymes indicated in cyan. (B) The cyclization of the secondary macrocycle of thiostrepton involves epoxidation of the quinaldic acid and subsequent epoxide ring opening by the N-terminal Ile that is generated by LP removal by TsrB. Cyclization is shown here to occur as the last step of biosynthesis, but this hypothesis has not been experimentally verified as of yet. Dihydroxylation of Ile10, catalyzed by TsrK (TsrR) is shown in cyan. | ||
To generate thiostrepton derivatives (Fig. 90B), Streptomyces laurentii ΔtsrM (also called ΔtsrT), which cannot form quinaldic acid,399 was fed with 7′ or 8′-fluorinated quinaldic acid analogs.400 Rather than identifying thiostrepton analogs, only a truncated product was found in which the macrocycle containing the quinaldic acid was lacking. These findings suggested that epoxidation of the quinaldic acid moiety is required prior to LP removal and subsequent cyclization by attack of the liberated amino group of Ile1 onto the epoxide (Fig. 90B). TsrI (also called TsrP) and TsrK (also called TsrR) are homologous to cytochrome P450 enzymes (InterPro family: IPR001128) and are predicted to perform the bishydroxylation of Ile10 and epoxidation of the quinaldic acid. Genetic deletion of tsrI yielded an intermediate that shares the same structure as the product identified with the fluorinated analogs, suggesting that the epoxidation reaction is performed by TsrI. Deletion of tsrK resulted in the loss of product formation, suggesting that dihydroxylation of Ile10 is perhaps an early checkpoint during thiostrepton biosynthesis.400
Nosiheptide contains two hydroxyl groups in the macrocyclic scaffold that are post-translationally installed (Fig. 90A). Genetic and biochemical characterization of nosB and nosC predicted to encode P450 monooxygenases revealed their role in nosiheptide maturation.401 Deletion of nosB from Streptomyces actuosus, resulted in production of a compound where the hydroxylation at the γ-carbon of Glu6 was missing. Subsequent in vitro expression of NosB and incubation with des-hydroxy nosiheptide yielded mature nosiheptide. Deletion of nosC resulted in an intermediate that lacked the hydroxyl on the pyridine ring, but also contained a bis-Dha tail. Recombinant NosC hydroxylated this intermediate on the pyridine ring.401
A cytochrome P450 enzyme is also believed to be responsible for the hydroxylation of a Pro residue in the biosynthesis of the lanthipeptide NAI-107,402,403 but this activity has not yet been reconstituted in vitro. P450-catalyzed hydroxylation of Phe has also been proposed in the biosynthesis of the thioamitide thioalbamide11 and gene deletions in the thiovarsolin BGC suggest that VarO, a heme oxygenase (InterPro family: IPR016084), is involved in dehydrogenation of an arginine side chain.14
5.7.3.1 Formation of the quinaldic acid of thiostrepton. Like nosiheptide, the thiopeptide thiostrepton features two macrocycles (Fig. 90A and B). One macrocycle is formed by the class-defining [4 + 2]-cycloaddition reaction forming a six-membered, nitrogen-containing heterocycle (Section 3.2). The second macrocycle is specific to the nosiheptide and thiostrepton sub-families of thiopeptides. In both cases, part of the second macrocycle is fashioned from L-Trp. For nosiheptide, MIA formation and macrocyclization involves two rSAM proteins discussed in Sections 5.2 and 5.3.6. For thiostrepton, the quinaldic acid moiety (Fig. 90B) is also believed to be derived from L-Trp by a different rSAM-catalyzed process. Genetic studies suggested that tsrM (also called tsrT), tsrV (also called tsrA), and tsrQ (also called tsrE) are required for quinaldic acid biosynthesis.399 TsrM was characterized as a rSAM methyltransferase to produce 2-methyl-L-Trp (Fig. 80, Section 5.3.1). To determine the roles of tsrV and tsrQ, the corresponding enzymes were produced in E. coli and the bacteria were provided with 2-methyl-DL-Trp.399 Production of a quinolone ketone was only observed when both enzymes were expressed. Purified TsrV catalyzed the pyridoxyl-5′-phosphate dependent transformation of 2-methyl-L-Trp to indolylpyruvate using a variety of ammonia acceptor co-substrates (Fig. 91). Furthermore, reconstitution of TsrQ with FAD, NADPH, O2, and a flavin reductase resulted in the conversion of indolylpyruvate to the corresponding quinolone ketone.399 At lower temperatures, selective hydroxylation at the 3′-position of indolylpyruvate was detected. The TsrQ product is highly reactive and can readily hydrate to the unstable 2,3-dihydroxy-pyrroline (Fig. 91). This intermediate can undergo ring expansion by breaking the N1–C2 bond, condensation of the amine onto the ketone, and subsequent dehydration-rearomatization to generate the quinoline ketone (Fig. 91).
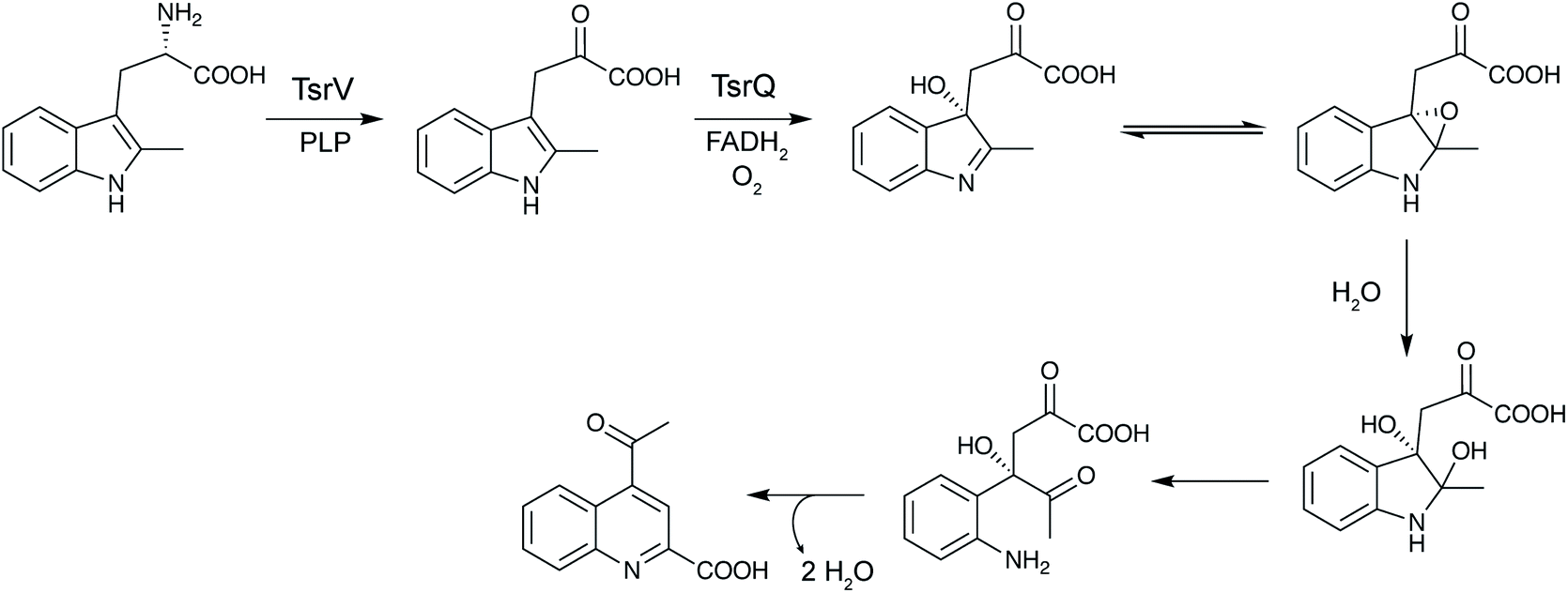 | ||
| Fig. 91 Biosynthesis of quinaldic acid from 2-methyl-L-Trp by TsrV (TsrA) and TsrQ (TsrE) during thiostrepton biosynthesis. For the mechanism of 2-methyl-L-Trp formation, see Fig. 80. For the structure of thiostrepton, see Fig. 90B. | ||
5.7.3.2 Oxime formation in azolemycins. Azolemycins are a family of LAPs (Section 3.4) produced by Streptomyces sp.404 The azolemycins contain unique oxime moieties installed on the N-terminal amine of Val1 with either the E or Z stereochemistry (Fig. 92). Subsequent experiments using Marfey's reagent permitted the configuration of all stereocenters in azolemycin A to be elucidated. In addition to genes encoding the canonical apparatus necessary for the introduction of the characteristic azoles in the biosynthesis of other LAPs (Section 3.4), the BGC encodes a SAM-dependent MT (AzmE), and a flavin-dependent monooxygenase (AzmF, InterPro family: IPR036661).404 Subsitution of the azmF gene with a selection marker in Streptomyces sp. lead to abolishment of azolemycin A–D production. Instead, compounds were isolated comprising an amino group in place of the oxime moiety or lacking the N-terminal Val residue altogether. These observations are consistent with a role for AzmF in oxime formation, a modification not previously documented in the final structure of a RiPP, but which has been seen previously as an intermediate in ustiloxin maturation where it is installed by the flavin monooxygenase UstF2 (Section 3.17).229 A survey of AzmF homologs in actinomycetes revealed that the majority are found in putative BGCs for non-ribosomal peptides and that only a single other example is found in a putative LAP pathway (encoded by Actinomadura oligospora).
 | ||
| Fig. 92 Structure of the LAP azolemycin A. Class-defining PTMs are yellow while secondary PTMs are cyan. The N-terminal oxime is believed to be formed by the flavin-dependent monooxygenase AzmF. | ||
5.7.3.3 2-Aminovinylcysteine formation by oxidative decarboxylation. AviCys and the related avionin structures are found in several RiPPs including class I and class II lanthipeptides, thioamitides (Fig. 2), lipolanthines (Fig. 11), lanthidins (Fig. 18A), and linaridins (Fig. 27). Recent studies have explored the biosynthetic LanD flavoenzymes (InterPro family: IPR003382) involved in the oxidative decarboxylation of the C-terminal Cys for various RiPPs. NAI-107 is a class I lanthipeptide containing a C-terminal AviCys that is installed by MibD, a flavin-dependent decarboxylase. Recombinant MibD displayed decarboxylation activity of the terminal Cys residue in both full length MibA as well as in the MibA core peptide. Thus, the enzyme does not require the LP nor do the thioether rings need to be installed first. However, MibD did not display the large substrate tolerance previously described in early studies of the lanthipeptide decarboxylase EpiD,405 and instead tolerates a more limited sequence motif. The oxidative decarboxylation of the linaridin cypemycin is catalyzed by CypD,406 whose crystal structure was solved (PDB: 6JDD). The enzyme is structurally similar to previously characterized lanthipeptide LanD proteins despite low sequence similarity. Investigation of AviCys formation for the lanthipeptide mutacin 1140 showed that a lanthionine was formed in the absence of the MutD decarboxylase,407 suggesting that oxidative decarboxylation is not required for ring formation. Co-expression of TvaA, the precursor peptide to thioviridamide, with the decarboxylase TvaF in E. coli led to the production of a C-terminal thioenol, consistent with observations for AviCys biosynthesis in lanthipeptides. TvaF can modify both the full length TvaA and a mutated core peptide, TvaAS8A(2–13), albeit at a significantly lower rate, indicating that the leader peptide is not strictly required but likely assists with substrate recognition by TvaF.129 The biosynthesis of the lanthidin cacaoidin has not yet been investigated but its BGC contains a LanD-like decarboxylase (Fig. 18B).54
For lipolanthines, the LanD decarboxylase MicD was shown to work in concert with the MicKC labionin synthetase, with the activity of both greatly affected by the presence of its partner.88
5.7.3.4 Chlorination of NAI-107 and ammosamide A. All known Trp halogenases (InterPro family: IPR006905) prior to 2013 used L-Trp as the substrate. Recently, two enzymes have been identified that act on Trp imbedded in a peptide. The lanthipeptide NAI-107 contains a 5-chloro-Trp residue that is generated by a FADH2-dependent halogenase, MibH. MibH collaborates with MibS, the cognate NADH-dependent flavin reductase.408 MibH exhibits unusual substrate specificity, because the enzyme was incapable of chlorinating free Trp, Trp in a linear peptide, and Trp in mutacin 1140, which has a similar set of rings as NAI-107. Therefore, MibH seemingly recognizes the overall structure of NAI-107, including the amino acid sequence of the C-terminal rings. Structural characterization of MibH showed a large substrate binding cleft afforded by subtle amino acid changes compared to previously characterized Trp halogenases explaining the selectivity for a macrocyclic substrate.408 Although the enzyme is very selective for its peptide substrate, it is tolerant to using other halides as brominated variants of NAI-107 were obtained by growing producing organisms from either Actinoallomurus or Microbispora in media supplemented with KBr.409
Ammosamide A (Fig. 16) belongs to a group of RiPPs known as the pearlins (see Section 2.15). Ammosamide A and its congeners are produced by marine Streptomyces sp. The BGC of the ammosamides contains a Trp halogenase, amm3 that is homologous to MibH. Genetic deletion of amm3 resulted in complete loss of product formation,51 suggesting that chlorination of the indole ring occurs early in the biosynthesis. Chemical complementation with 6-Cl-Trp did not restore ammosamide production, further supporting the hypothesis that the chlorination event occurs on a specific Trp in a peptidic substrate.
5.7.3.5 Thioether oxidation. The terminal methyllanthionine in the lanthipeptide actagardine is oxidized to a sulfoxide by a dedicated oxidase, GarO. The enzyme is homologous to luciferase-like monooxygenases. Co-expression of the substrate GarA, the lanthipeptide synthetase GarM (Section 3.1.2), and GarO in E. coli resulted in the production of the cyclic, sulfoxide-containing product.410 Purified GarO selectively oxidized the terminal thioether ring of cyclized GarA in the presence of FMN and NADH. GarO did not require the LP to catalyze thioether oxidation, and likely recognizes the terminal methyllanthionine in the macrocycle.
Thioether oxidation is also found in the fungal RiPP α-amanitin (Fig. 57, Section 3.18), but the enzyme involved in the oxidation the Cys–Trp crosslink to the sulfoxide is not yet identified. In contrast, the class B flavin monooxygenase UstF1 was shown in vitro to oxidize the thioether of an advanced intermediate to a sulfoxide (Fig. 55, Section 3.17).229
5.7.4.1 Asp hydroxylation in the lanthipeptide duramycin and lasso peptide canucin A. Duramycin, a lanthipeptide that binds phosphatidylethanolamine, contains an erythro-3-hydroxy-aspartic acid residue that is also found in the structurally similar divamides (Fig. 83). The hydroxy-Asp is installed by an α-ketoglutarate/iron(II)-dependent hydroxylase, DurX.411 Mutational analysis of the DurA precursor peptide determined that the glycine immediately C-terminal to the Asp residue is necessary for DurX activity. Additionally, DurX prefers cyclized substrate over linear peptide. Interestingly, the hydroxylation of the highly conserved Asp is required for the next PTM,412 the installation of a lysinoalanine crosslink that is characteristic for this group of lanthipeptides (see Section 5.10).
Asp hydroxylation was also reported for the lasso peptide canucin A. In vitro the α-ketoglutarate/iron(II)-dependent hydroxylase CanE oxidized the linear precursor peptide CanA prior to lasso formation in a reaction that was facilitated by the stand-alone RRE protein CanB1.164
5.7.4.2 β-Hydroxylation of Val and Asn in polytheonamides. β-Hydroxylation of polytheonamides occurs at Val and Asn residues (Fig. 47). Co-expression studies confirmed that PoyI, a putative α-ketoglutarate/iron(II)-dependent enzyme, catalyzes the selective hydroxylation of Val32, Asn38, and Val24.28 The β-hydroxylation events seem to be both regioselective and side chain specific. Some mutations in PoyA such as V31L and N38H were tolerated and afforded the hydroxylated products, whereas N38Q and V31H did not appear to be substrates for PoyI.
Likewise, two other characterized dikaritins, phomopsin A and asperipin-2a (Fig. 3), contain a similar pattern of aromatic hydroxylation. Phomopsin A contains β-hydroxy-3′,4′-dihydroxy-Phe, which is possibly formed by the homologous PhomQ, PhomYa, and PhomYb proteins encoded in the phomopsin BGC.16 In asperipin-2a, it has been hypothesized that AprY, a DUF3328 containing protein, is responsible for α-hydroxylation–dehydration to yield an imine intermediate that can hydrolyze to the ketone, which may serve as a substrate for the reductase AprR to yield the β-hydroxy residue.414 However, more study is required to validate this hypothesis.
5.8 Prenylation
Cyanobactin prenyltransferases (PTases) increase the structural diversity of this natural product family through the O-prenylation of Tyr, Thr and Ser residues in the forward or reverse prenyl orientation.326,415,416 Cyanobactins (Section 3.6) bearing reverse O-prenylated Tyr can undergo a further Claisen rearrangement to yield forward C-prenylated Tyr.326,415 Prenylation of Trp residues has also been documented, both at the N-1 (ref. 417 and 418) or C-3 (ref. 419–421) positions of the indole ring, in addition to N-prenylation of the amino terminus of some linear cyanobactins.391 Recently, the biosynthesis of the bis-prenylated linear peptide alkaloids muscoride A and B was described, which feature unusual prenyl groups that protect both amino- (reverse prenyl on α-nitrogen of Val) and carboxy- (forward prenyl on carboxy group of terminal methyloxazole) termini.422,423 These enzymes (InterPro family: IPR031037) therefore significantly expand the cyanobactin structural diversity and also represent promising tools for biocatalysis. Although the enzymology of cyanobactin prenylation has been reviewed previously,1 more recent structural, biochemical and molecular dynamics studies have shed further light on the basis of broad substrate tolerance of the TruF family of PTases and are summarized here.Co-crystal structures of the Tyr O-prenylating enzyme PagF from the prenylagaramide biosynthetic pathway from Oscillatoria agardhii in complex with linear (PDB: 5TU4 and 5TU5) and macrocyclic (PDB: 5TU6) peptide substrates have highlighted a number of key features that explain the high substrate promiscuity of TruF-type PTases, despite the absence of obvious recognition sequences.416 Structural data revealed that PagF (PDB: 5TTY) comprises an α/β PT barrel fold like those observed in bacterial ABBA enzymes and fungal indole PTases.424 However, in PagF, the barrel fold is truncated, resulting in a solvent-exposed channel that must be occluded by the peptide substrate to prevent nonproductive quenching of the dimethylallyl pyrophosphate-derived carbocation intermediate, a role performed by structural elements in characterized small-molecule PTases. Binding of peptide substrate in PagF create a deep, encapsulated hydrophobic cavity that effectively shields the allylic carbocation from solvent, as observed in cocrystal structures with a dimethylallyl S-thiolodiphosphate analog.416 A series of hydrophobic residues line the circumference of the active-site tunnel that further protect reactive intermediates.
In addition to solvent exclusion from the PagF active site, peptide binding also serves to orient the Tyr hydroxyl group for prenylation in the vicinity of the prenyl donor for direct electrophilic attack on the Tyr oxygen. Superposition of cocrystal structures with either cyclic or linear peptide substrates resulted in the almost identical placement of the isoprene-accepting Tyr residue.416 This observation suggested that PagF can modify Tyr residues in any substrate, provided the geometric and orientation constraints imposed by the active sites can be satisfied. A minimal N-Tyr-ψ-R (ψ = any aliphatic/aromatic residue) motif was hypothesized, consistent with molecular dynamics simulations, and was validated biochemically using non-physiological substrates that were monitored for O-prenylation in vitro.416
Though the vast majority of RiPP PTases catalyze the regioselective transfer of C5 dimethylallyl donors to their cognate peptide substrates, reconstitution of the activity of PirF from piricyclamide in Microcystis aeruginosa confirmed its Tyr O-geranylation activity.425,426In vitro reconstitution experiments elucidated that PirF specifically geranylates Tyr residues when presented with Ser, Thr, Trp, or Tyr derivatives as substrates.425 Experiments with further small molecule substrates revealed a broad substrate scope for PirF, which is able to geranylate L- and D-Tyr, in addition to a selection of small phenolic compounds, though larger Tyr-containing peptide substrates are favored to small amino acids. A Tyr–Tyr–Tyr tripeptide substrate was a particularly favored substrate and was used to explore isoprenoid specificity using C5–C20 donors showing selective utilization of the C10 donor GPP.425 To determine the basis for C10 donor utilization, the crystal structure of PirF was elucidated and uncovered a single amino acid mutation in the vicinity of the isoprene binding pocket that is distinct from C5-utilizing homologs such as PagF.426 In PagF, Phe222 represents one of the hydrophobic residues mentioned previously that serve to stabilize the allylic carbocation. In PirF, the equivalent residue is a Gly, which results in an enlargement of the active site pocket to accommodate GPP. Indeed, PagF-F222G and F222A variants showed robust geranyltransferase activity, concomitant with a 10 to 20-fold loss of C5 isoprene activity that rules out a simple gain of function mutation.426 Co-crystallization of the F222A variant with GPP and MgCl2 permitted the visualization of GPP extension into the active site space created by the F222A substitution.
Aside from cyanobactin biosynthesis, prenylation is also a critical step in the maturation of the RiPP ComX,1 a quorum sensing pheromone from Bacillus species that involves geranylation or farnesylation at the indolic C-3 position of a Trp residue in the core peptide.427 Recent mutational analyses have revealed the essential nature of an Asp residue that is necessary for preComX-binding by the prenyl transferase ComQ, in addition to residues that determine specificity for either C10 or C15 donor substrates.428 Reconstitution of ComQnatto, responsible for farnesylation of a Trp residue in the ComXnatto core peptide from B. subtilis subsp. natto, demonstrated broad peptide substrate tolerance for the enzyme.429 ComQnatto accepted a range of N- and or C-terminally truncated ComXnatto analogs, in addition to even a single Trp substrate, suggesting a leader sequence is not necessary for substrate recognition.
5.9 Additional modifications of N- and C-termini
The N- and C-termini of peptides are vulnerable to exopeptidase degradation. Therefore, many RiPP BGCs encode enzymes that modify these termini, either during the primary scaffold installation (e.g. via macrocyclization) or by secondary tailoring processes. Methylation (Section 5.3.7.1) and prenylation (Section 5.8) of N- and C-termini were discussed in previous sections as well as oxidation of the N-terminus to an oxime (Section 5.7.3.2) and oxidative decarboxylation of Cys at the C-terminus (Section 5.7.3.3). Several other strategies have emerged that are briefly discussed in this section.Dehydration of Ser/Thr in the first position of the core peptide results in Dha/Dhb residues, which upon proteolytic removal of the LP tautomerize to the corresponding α-keto amides (Fig. 93A). In some cases, such as the polytheonamides (Fig. 47; Section 3.12), this structure is further methylated by rSAM enzymes (Section 5.3.2), whereas for other compounds the ketone is reduced to the corresponding alcohol, either by a dehydrogenase in the BGC, or by an unknown enzyme (e.g.Fig. 2). In the case of the biosynthesis of the lanthipeptide epilancin 15X the reduction is catalyzed by the NADH-dependent oxidoreductase ElxO encoded in the BGC and has been reconstituted in vitro.312 Modification of C-termini has also been reported to involve dehydration of a Ser in the biosynthesis of the thiopeptide nosiheptide (Fig. 90A).430–432 Incubation of a biosynthetic intermediate containing a C-terminal Dha with recombinant NosA resulted in enamide hydrolysis to afford a C-terminal amide (Fig. 93B). A crystal structure of the enzyme (PDB: 4ZA1) provided insights into the mechanism of amide formation.431 NosA analogs are encoded in the BGCs of a number of other amidated thiopeptides such as nocathiacin,433 GE2270A,367 and berninamycin434 suggesting a common mechanism of C-terminal amide formation.
 | ||
| Fig. 93 (A) Blocking the N-terminus of peptides from aminopeptidase activity by hydrolysis of dehydroamino acids. (B) Blocking the C-terminus of peptides by amidation from a dehydro amino acid by NosA. | ||
The chemistry leading to amidation of thiostrepton (for structure, see Fig. 90B) is different. Genetic studies showed that its C-terminus first undergoes a cryptic methylation by an unknown enzyme, possibly to facilitate dehydration of the C-terminal Ser.435In vitro experiments showed that the resulting methyl ester is then hydrolyzed to the carboxylate by TsrB, and gene inactivation studies indicated that the Asn-synthetase-like amidotransferase TsrC subsequently converts the carboxylate to the corresponding carboxamide.
Another modification of the C-terminus is found in the thiopeptide micrococcin P1. Genetic studies suggest the short chain dehydrogenase TclP performs an oxidative decarboxylation of the C-terminal Thr to the corresponding 2-oxopropylamine. This ketone is subsequently reduced to the alcohol by TclS.90,436 Genetic evidence also implicates C-terminal oxidative decarboxylation in bottromycin biosynthesis by a cytochrome P450 enzyme.143
Other RiPPs are N-terminally acetylated such as the LAP goadsporin (Fig. 97), the lasso peptide albusnodin (Section 3.8), and many microviridins/graspetides (Section 3.15). For goadsporin, the GCN5-related N-acetyltransferase (GNAT) family member GodH catalyzes the acetylation.437 Similarly, the N-terminal acetylation of albusnodin is catalyzed by a GNAT protein.163 For microviridins, the protoype MdnE is also a GNAT acetyl transferase that acetylates the N-terminus.206 The gene encoding such an acetyltransferase is sometimes missing from microviridin BGCs,438 but at present it is unclear if this leads to a non-acetylated product or whether the acetyltransferase is encoded outside of the gene cluster.
5.10 Lysinoalanine crosslink formation
Many RiPPs feature macrocycles that introduce conformational restraints that facilitate target binding and reduce proteolytic susceptibility. Section 3 discussed nature's strategies for macrocyclization that introduce the crosslinks that distinguish different RiPP classes. Sometimes, additional crosslinks are formed resulting in secondary macrocycles. Examples already discussed include the compound-specific second macrocycles found in the thiostrepton and nosiheptide groups (Fig. 90A and B) of thiopeptides. Another example is a group of class II lanthipeptides (Section 3.1.2) including cinnamycin, duramycin and the divamides that contain a lysinoalanine crosslink (Fig. 83). A small protein encoded in the BGCs of these compounds was recently shown to form the crosslink by catalyzing the addition of a Lys to a Dha.412 For duramycin the crystal structure of this protein (DurN) bound to its product or biosynthetic intermediates (PDB: 6C0G, 6C0H, 6C0Y) demonstrated that the enzyme does not seem to make any contacts with the substrate near the site of the chemical reaction. Instead the enzyme binds the substrate to hold it in a reactive conformation and a hydroxylated Asp in the substrate appears to act as the base that deprotonates the Lys nucleophile. Biochemical and structural studies showed that the hydroxylation of this Asp is critical for lysinoalanine formation.412 Thus, prior PTM of an Asp in the substrate, as discussed in Section 5.7.4.1, is required for the formation of the next PTM. Such a strict order of PTMs is not uncommon in RiPP biosynthesis, but the substrate-assisted nature of lysinoalanine formation is at present unique. Recently, substrate assistance was also proposed for Asp epimerization during bottromycin biosynthesis (Section 5.1.3).1445.11 RiPP glycosylation
The glycocins are RiPPs for which glycosylation is the class-defining PTM (Section 3.25). Surprisingly, very few other glycosylations have been reported of members of other RiPP classes, unlike the ubiquitous glycosylated natural products from non-RiPP biosynthetic pathways.439,440 Several glycosylated thiopeptides are known in series e441 such as glycothiohexide α, philipimycin, and nocathiacin I, but although the BGC of the latter has been determined,433 the glycosylation steps have not yet been investigated. Two examples of glycosylated lanthipeptides have been reported. The class III lanthipeptide NAI-112 (Section 3.1.3; Fig. 22) is glycosylated on the indole nitrogen of Trp.84 Like the glycocins listeriocytocin and enterocin 96,287 the class V lanthipeptide cacaoidin is a diglycosylated RiPP. However, unlike these glycocins that are diglucosylated on Ser, cacaoidin carries a 6-deoxygulopyranosyl-(rhamnopyranose) disaccharide on Tyr.54 Several glycosyl transferases as well as enzymes involved in rhamnose biosynthesis are encoded in the cacaoidin BGC (Fig. 18B). Finally, the lasso peptide pseudomycoidin is glycosylated on a C-terminal phosphorylated Ser residue (Section 5.6).3986 RiPP genome mining tools
6.1 BAGEL
BActeriocin GEnome mining tool, BAGEL (http://bagel4.molgenrug.nl/) was the first web-based tool that enabled users to screen genetic information to find RiPP BGCs (Table 2). Originally reported in 2006, BAGEL has undergone periodic updates.442–445 Next to RiPPs, BAGEL also predicts genetically encoded non-post translationally modified bacteriocins. BAGEL detects specific RiPP classes based on defined properties of the modification enzymes (for instance a LanB and a LanC enzyme for class I lanthipeptides, Section 3.1.1) and based on specific properties required for the core peptide (e.g. contains Cys and Thr/Ser). BAGEL requires prokaryotic nucleotide sequence (FASTA format) as input. The workflow of BAGEL4 (ref. 445) is as follows: first, the program performs a six-frame translation. Second, the hypothetical proteins are screened for the presence of motifs found in modification enzymes for the defined RiPP classes. The third step entails defining the so-called areas of interest in the genome, based on the hits of step 2. In the final step, these areas are analyzed in detail, i.e. first genes are called using GLIMMER446 and annotated based on Basic Local Alignment Search Tool (BLAST)447 with the UniRef90 database. Modification enzymes are annotated based on protein domains they entail. Subsequently, all possible small open reading frames (ORFs) are called up to a set maximum size. The core peptide is either detected based on homology to known core peptides or filtered based on set properties expected for core peptides of the given class. A new feature in BAGEL4 is the option to upload RNA expression data that can be visualized along with the BGC to inform whether the RiPP is expressed in the tested condition. Additionally, BAGEL4 offers an indication of the modifications taking place in the core peptide based on homology to known modified peptides. This data is downloaded from UnitProt directly and visualized based on homology, giving a fast overview of the similarity to known RiPPs and expected modifications.| Name | Input | Availability | RiPP classes supported | |
|---|---|---|---|---|
| Webtool | Download | |||
| antiSMASH | Genomic data | Yes | Yes | Lanthipeptides, rSAM-modified RiPPs, microviridins, head-to-tail cyclized peptides, bottromycins, sactipeptides, lasso peptides, LAPs, glycocins, cyanobactins, linaridins, thiopeptides |
| BAGEL4 | Genomic data | Yes | Yes | Lanthipeptides, lasso peptides, cyanobactins, microcins, thiopeptides, head-to-tail cyclized peptides, glycocins, sactipeptides, bottromycins, LAPs |
| NeuRiPP | Protein accession number | No | Yes | Class independent |
| RiPP-PRISM | Genomic data | Yes | Yes | Autoinducing peptides, bottromycins, ComX, cyanobactins, glycocins, head-to-tail cyclized peptides, lanthipeptides, lasso peptides, linaridins, LAPs, microviridins, prochlorosins, proteusins, sactipeptides, streptide, thiopeptides, trifolitoxin, thioviridamide, YM-216391 |
| RiPPER | Protein accession number | No | Yes | Class independent |
| RiPPMiner | Protein sequence | Yes | No | Lanthipeptides, bottromycins, cyanobactins, glycocins, lasso peptides, linaridins, LAPs, microcins, sactipeptides, thiopeptides, ComX, autoinducing peptides, head-to-tail cyclized peptides |
| RiPPQuest | MSMS data and genome | Yes | No | Lanthipeptides, LAPs, lasso peptides, linaridins, proteusins, cyanobactins, methanobactins |
| RODEO | Protein accession number | Yes | Yes | Lasso peptides, class I–IV lanthipeptides, ranthipeptides, sactipeptides, thiopeptides |
| IMG-ABC | Data in its database | Yes | No | Lanthipeptides, rSAM-modified RiPPs, microviridins, head-to-tail cyclized peptides, bottromycins, sactipeptides, lasso peptides, LAPs, glycocins, cyanobactins, linaridins, thiopeptides |
| RRE-Finder | Protein sequence | Yes (RODEO-integrated) | Yes | RRE-dependent RiPP classes |
| decRiPPter | Genomic data | No | Yes | Class-independent |
| DeepRiPP | Genomic data | Yes | Yes | Class-independent |
BAGEL is widely used as a quick and easy way to check the RiPP/bacteriocin potential in genomes. It is used to improve annotations of genomes and to discover new BGCs. For instance, BAGEL was used to discover almost 400 new potential head-to-tail cyclized peptides of which many were found in the genus of Bacillus.448 BAGEL was also used to characterize the RiPP expression potential of strains with plant-growth promoting properties449 and also of lactic acid bacteria with desired properties for starter cultures.450
6.2 RODEO
Rapid ORF Description & Evaluation Online, RODEO (http://rodeo.scs.illinois.edu/), a genome-mining tool reported in 2017, was the first to use machine learning to predict RiPP precursor peptides (Table 2).154 The workflow of RODEO is as follows: when a protein or a list of proteins are used as input queries, the algorithm retrieves the relevant genomic records from GenBank. RODEO then uses profile Hidden Markov Model (pHMM)-based analysis to annotate the function of genes flanking the query protein(s). To do so, RODEO utilizes the HMMER toolkit451 and compares proteins to Pfam, TIGRFAM, or customized HMM databases.452,453 Since RiPP precursor peptides are often small and hypervariable, they frequently are not predicted as protein-coding sequences. Therefore, RODEO performs a six-frame nucleotide translation of the intergenic regions in search for possible precursor peptides. These translated reads are then analyzed using a scoring function, specific to a chosen RiPP class, which combines heuristic scoring, motif analysis, and supervised machine learning. The output is an annotated BGC with a list of scored, predicted precursor peptides, provided in both graphical (html) and tabular (csv) formats. The current version, RODEO 2.0, supports precursor peptide prediction for lasso peptides, class I–IV lanthipeptides, thiopeptides, and sactipeptides with the latter sufficient to also identify ranthipeptides.43,62,132,154,454 The predictive capabilities of RODEO were incorporated into antiSMASH4.0 (and later versions), a secondary metabolite BGC prediction software that performs analysis on single genome inputs (Section 6.8).454 RODEO is available to the user in both webtool and command-line formats (recommended for large jobs, >1000 queries).RODEO's utility was first demonstrated by the broad-scale investigation of lasso peptides, revealing more than 1400 lasso peptide BGCs, a 10-fold expansion over what was previously appreciated.154 The predictive capabilities of RODEO were further bolstered by the isolation and characterization of new lasso peptides (Section 3.8), such as LP2006, citrulassin, and lagmycin,154 prioritized based on predicted structural novelty. RODEO has been used to perform in-depth mining of four other RiPP classes: the sactipeptides and ranthipeptides,43 the thiopeptides,132 and class I–IV lanthipeptides.62 These studies highlight the utility of RODEO in performing in-depth, high-throughput analyses of a specific RiPP class. RODEO-enabled analysis of multiple inputs in a single query also allows for the quick generation of large, reliable data sets that can be leveraged to glean new bioinformatic insights into a RiPP family. For instance, studies on the lasso peptide family revealed the “YxxP” motif in the LP region which was predicted to be important for recognition by the RRE (Section 4.1.1).154 These predictions were corroborated by biochemical characterization in several studies.158,160,161,292,293 Very little information exists regarding how RiPP biosynthetic complexes are formed and regulated, and data sets that are sufficiently large can be leveraged to predict protein–protein interaction surfaces that mediate RiPP biosynthesis.455 As new compounds of a RiPP class are described, the scoring modules can continually be improved and used to update the RiPP family of interest, as recently demonstrated for the lasso peptides158 and lanthipeptides.62 New scoring modules could also be developed to aid in the analysis of newer RiPP classes by adjusting the scoring metrics. Although RODEO was originally developed as a RiPP genome-mining tool, it could be utilized to analyze non-RiPP BGCs by using custom configuration files and pHMMs. Similar to other genome mining tools,456,457 the capabilities of RODEO are reliant to a certain extent on the user's knowledge of a RiPP class.
6.3 RiPPER
RiPP Precursor Peptide Enhanced Recognition (RiPPER; https://github.com/streptomyces/ripper) is a tool meant for the class-independent discovery of RiPPs.14 RiPPER leverages RODEO to fetch nucleotide data corresponding to a query protein(s) and its local genomic neighborhood. Unlike RODEO that performs translation on all six frames in the intergenic regions of the query,154 RiPPER employs a modified version of Prodigal (PROkaryotic Dynamic programming Gene-finding Algorithm) to identify candidate precursor peptides (20–120 amino acids).14 Prodigal is an algorithm that uses features such as ribosomal binding sites, GC content and codon bias to identify bacterial coding sequences.458 Since RiPP precursors tend to be in the same direction as that of the tailoring enzymes, Prodigal scores are enhanced if the identified precursor peptide is encoded as such. The top three high-scoring gene products within 8 kb from the gene encoding the query protein, along with additional genes scoring beyond a threshold Prodigal score, are listed in a text file with associated information such as conserved domains and Prodigal score, among others.14 Multiple peptides are retrieved per query, which may not all be plausible precursor peptides as shown by test analyses on lasso peptides, graspetides, and thiopeptides. Therefore, peptide similarity networks and BGC analysis must be done on multiple queries in parallel to identify possible precursors. This approach was employed to analyze 229 TfuA-containing BGCs in Actinobacteria (Section 3.5), revealing the diversity of thioamidated RiPPs. This dataset was leveraged to isolate thioamidated tripeptides, termed thiovarsolins, from Streptomyces varsoviensis via heterologous expression.14 Thiovarsolins A–D derive from one precursor peptide VarA, which features repeats in the core peptide. The BGC also encodes an ATP-grasp ligase and amidinotransferase that are conserved among homologous BGC but are not functional under the conditions expressed; therefore, thiovarsolins have been suggested to be shunt products.14 Unlike RODEO, antiSMASH, and RiPP-PRISM that are available as webtools, RiPPER is currently available only in the command-line format.6.4 NeuRiPP
NeuRiPP is another addition to the suite of tools for RiPP precursor peptide prediction (https://github.com/emzodls/neuripp). Aimed at tailoring enzyme-independent prediction of RiPP precursor peptides, NeuRiPP employs deep neural networks trained on experimentally validated RiPPs, and precursors predicted with a high degree of confidence by existing RiPP genome mining tools.459 In addition to identifying precursor peptides predicted by RODEO and RiPPER on the test set, NeuRiPP identifies additional precursor peptides, most of which hit HMMs developed for precursor peptides of known RiPP classes.459 NeuRiPP awaits validation as new compounds are yet to be isolated from NeuRiPP-predicted precursor peptides. Like RiPPER, NeuRiPP also lacks a web-based interface and thus is currently restricted to users comfortable with a command-line interface.6.5 RiPPMiner
RiPPMiner is an online resource (http://www.nii.ac.in/rippminer.html) that offers tools, based on a curated database,460 allowing users to assess if a protein sequence is RiPP-related and to which known class it belongs. Additionally, RiPPMiner attempts to predict the LP cleavage site for lanthipeptides, cyanobactins, lasso peptides, and thiopeptides. The curated database of RiPPs was generated from previously published databases and currently entails 513 records. This database was used for machine learning to generate a support vector machine able to discriminate RiPPs from other small proteins. To predict LP cleavage sites, a random-forest approach is employed. RiPPMiner also predicts cross-links within RiPP structures based on a support vector machine classification. Recently a genome search option was added that allows the user to upload genomic data (FASTA format) to mine for RiPP BGCs.6.6 RiPPQuest
RiPPQuest is an online program (https://gnps.ucsd.edu/) that matches tandem MS data to RiPP BGCs encoded by a particular organism.461 The search space is limited to the vicinity of the encoded RiPP biosynthesis genes identified in the genome. The generated database for the proteomic search includes an extensive number of peptide modifications to enable RiPP discovery. Although it is desirable to couple genetic potential with experimental validation, this also means that detection depends on finding the producing culture conditions and sample preparation. This platform can help to further automate RiPP discovery and enables researchers to go beyond genome-based identification in a more automated way.6.7 RiPP-PRISM
PRediction Informatics for Secondary Metabolomes (PRISM) was originally developed to enable identification of non-ribosomal peptide and polyketide BGCs and predict structures of the associated natural products.462 In a recent version, PRISM 3 (http://grid.adapsyn.com/prism/#!/prism), the functionality was expanded to include BGC identification and structure prediction for RiPPs, among other classes of natural products.456 RiPP-PRISM identifies RiPP BGCs in an input query using a set of rules that includes comparison of genes to a library of 154 Hidden Markov Models (HMMs) associated with 21 RiPP classes.463 The precursor peptides in the BGC, recognized using a combination of heuristic scoring and HMM analyses, are further subjected to motif analysis using a collection of 54 RiPP-associated motifs to identify potential LP/follower peptide cleavage sites. Tailoring reactions based on domains identified in the BGC are then virtually performed on potential sites in the predicted core peptide, to generate a combinatorial library of hypothetical structures for each identified RiPP BGC.456,463 One caveat, also common to many other RiPP genome mining tools,154,454,457 is that all predictions are based on homology to proteins associated with known RiPP classes. As a result, RiPP-PRISM cannot identify novel RiPP families and also cannot predict novel PTMs. Nevertheless, RiPP-PRISM is unique among other RiPP genome mining tools by facilitating comprehensive analysis of genomes, beginning with the identification of potential RiPP BGCs and providing a library of possible structures, thus enhancing the prospects of RiPP discovery.456,463 RiPP-PRISM was used to perform a global analysis on 65421 prokaryotic genomes in the database of the National Center for Biotechnology Information, which predicted 30261 RiPP BGCs that would encode 2231 potentially unique RiPP products.463 This study underscored the global prevalence of RiPP BGCs across prokaryotes, highlighting several bacterial phyla previously underappreciated as RiPP producers, such as Thermotogae, Fusobacteria, Dictoglomi, and others. Structural predictions generated by RiPP-PRISM were used in tandem with automated LC-MS/MS searches using the GNP (Genomes to Natural Products) platform to isolate and characterize a novel RiPP, aurantizolicin, from Streptomyces aurantiacus JA 450 extracts.463,464 Aurantizolicin belongs to a small family of RiPPs termed YM-216391 that are characterized by an azole-rich macrocycle.1,465,466 The compounds are structurally similar to head-to-tail cyclized cyanobactins, but are produced by actinobacteria rather than cyanobacteria. Only two representative BGCs in the genomes were identified during the study. This highlights the utility of RiPP-PRISM to be used in combination with other natural product discovery platforms in expediting the targeted discovery of RiPPs.
6.8 AntiSMASH
AntiSMASH (Antibiotics & Secondary Metabolite Analysis Shell) is an online program (https://antismash.secondarymetabolites.org/) that was originally published in 2011 (ref. 467) and has since undergone frequent updates and enhancements.454,468–470 In addition to its use for RiPP discovery, it also supports the mining of non-ribosomally encoded compounds. It supports the input of prokaryotic and eukaryotic DNA. As other tools, it integrated previously described homology models for the identification of core peptides. Recently, the RODEO program has been integrated into the pipeline (antiSMASH 4.0 and later). In the most recent version (antiSMASH 5.0), more non-RiPP classes have been added, the run times have been improved, and the output has been adapted to a format (JSON) that enables automated downstream processing of the generated data. The user-friendly interface combined with the many classes of compounds it can mine for have made this the most highly cited genome mining tool for natural product identification.6.9 IMG-ABC
![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif) ntegrated
ntegrated ![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) icrobial
icrobial ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) enomes-
enomes-![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) tlas of
tlas of ![[B with combining low line]](https://www.rsc.org/images/entities/char_0042_0332.gif) iosynthetic gene lusters, IMG-ABC (https://img.jgi.doe.gov/cgi-bin/abc/main.cgi), is a publicly available database of predicted and experimentally verified natural product BGCs and their associated products.289,471,472 IMG-ABC utilizes ClusterMine360,473 DoBiscuit,474 antiSMASH,454,470,475,476 PubChem Compound,477 and MeSH library478 to curate characterized natural products and their associated BGCs. It uses ClusterFinder479 and antiSMASH454,470,475,476 to predict and annotate putative BGCs in genomic and metagenomic data in IMG.480 An update performed in 2016 integrated a tool called ClusterScout with IMG-ABC, allowing for targeted identification of custom BGCs.472 The latest update, IMG-ABC v5,289 uses antiSMASH 5.0,470 expanding its predictive capabilities to additional BGC types including several classes of RiPPs. IMG-ABC v5 (as of September 2019) contains a total of 330884 verified and predicted BGCs in 42892 publicly available genomes and 4944 metagenome-derived scaffolds.289 Searches on IMG-ABC can be performed using attributes associated with the secondary metabolite or the BGC of interest. Alternatively, IMG-ABC can also be browsed for secondary metabolites and BGCs by taxonomy, ecosystem, natural product/BGC type, gene count, and Pfam information for functional assignment of neighboring genes. IMG-ABC provides the most comprehensive information on secondary metabolites and BGCs in genomic and metagenomic data in IMG, with links to external databases and tools within IMG allowing users to perform large-scale analyses with any desired level of detail.289,471,472
iosynthetic gene lusters, IMG-ABC (https://img.jgi.doe.gov/cgi-bin/abc/main.cgi), is a publicly available database of predicted and experimentally verified natural product BGCs and their associated products.289,471,472 IMG-ABC utilizes ClusterMine360,473 DoBiscuit,474 antiSMASH,454,470,475,476 PubChem Compound,477 and MeSH library478 to curate characterized natural products and their associated BGCs. It uses ClusterFinder479 and antiSMASH454,470,475,476 to predict and annotate putative BGCs in genomic and metagenomic data in IMG.480 An update performed in 2016 integrated a tool called ClusterScout with IMG-ABC, allowing for targeted identification of custom BGCs.472 The latest update, IMG-ABC v5,289 uses antiSMASH 5.0,470 expanding its predictive capabilities to additional BGC types including several classes of RiPPs. IMG-ABC v5 (as of September 2019) contains a total of 330884 verified and predicted BGCs in 42892 publicly available genomes and 4944 metagenome-derived scaffolds.289 Searches on IMG-ABC can be performed using attributes associated with the secondary metabolite or the BGC of interest. Alternatively, IMG-ABC can also be browsed for secondary metabolites and BGCs by taxonomy, ecosystem, natural product/BGC type, gene count, and Pfam information for functional assignment of neighboring genes. IMG-ABC provides the most comprehensive information on secondary metabolites and BGCs in genomic and metagenomic data in IMG, with links to external databases and tools within IMG allowing users to perform large-scale analyses with any desired level of detail.289,471,472
6.10 DeepRiPP
DeepRiPP (http://deepripp.magarveylab.ca/) is a recent addition to the genome mining tools specific for RiPPs, and similar to RODEO, the tool employs a machine learning based approach.481 DeepRiPP consists of three intergrated algorithms: NLPPrecursor to identify RiPP precursor peptides in a class-independent fashion, BARLEY, to aid in the cheminformatic identification of potentially novel compounds, and CLAMS, to match genomic information with comparative metabolomic data. DeepRiPP was used to analyze more than 10000 extracts from nearly 500 bacterial strains. These efforts led to the discovery of three novel RiPPs: deepstreptin (a lasso peptide, Section 3.8), and deepflavo and deepginsen (lanthipeptides, Section 3.1).
6.11 RRE-Finder
RRE-Finder (http://rodeo.scs.illinois.edu/), another recent addition to the RiPP genome mining toolkit, facilitates detection of RRE domains (Section 4.1.1) in known and potentially novel RRE-dependent RiPP classes.482 The tool is available to the users in two modes of operation: precision mode and exploratory mode. Precision mode utilizes 35 custom HMMs built using RRE domains from largely known RiPP classes but includes a few high-confidence RREs from predicted new RiPP classes. The intent is for precision mode to enable class-dependent discovery of RRE domain-containing proteins. Exploratory mode uses a truncated version of the HHPred483 workflow and a custom database of ∼2400 sequence-divergent RREs to facilitate discovery of novel RRE-dependent RiPP classes. Using RRE-Finder working in precision mode, ∼35000 RRE domain-containing proteins were identified in the UniProtKB protein database,484 revealing many novel, yet-to-be explored, RRE-dependent RiPP classes. RRE-Finder is available to the user as a downloadable command-line tool. The precision mode of RRE-Finder has been integrated into RODEO (Section 6.2) and will be integrated into the future release of antiSMASH (Section 6.8).
6.12 decRiPPter
decRiPPter (https://github.com/Alexamk/decRiPPter) is yet another recent addition to the growing list of class-independent RiPP genome mining tools.56 This algorithm also utilizes machine learning, specifically, a support vector approach, in combination with a pan-genomic analysis to identify genes associated with RiPP biosynthesis within operon-like contexts. Two unique features consider whether these genes are distinct from the core genome (i.e. part of the accessory genome) and whether they encode sequences that overlap with known biosynthetic proteins already in the antiSMASH and MIBiG databases. Together, these filters reduce retrieval of primary metabolic pathways and previously described RiPP classes. When visualizing the results from the genus Streptomyces using sequence similarity networking, 42 new candidate RiPP families were found with experimental data supporting the identification of a novel class V lanthipeptide (Section 2.17).7 Engineering, screening, and production of RiPP variants
The great expansion of knowledge regarding RiPP biosynthesis in the last two decades has catalyzed a large engineering effort. The manipulation of leader and core peptides as well as PTM enzymes through single and multiple point mutations has allowed the assessment of substrate tolerance of the biosynthetic enzymes. Initial demonstration of the versatility of the enzymes to accept core peptides attached to their cognate LP prompted the development of RiPP modular systems, where the PTM enzymes, the LP and the core peptide are considered as separate modules. The lanthipeptide nisin (Section 3.1.1) served as the first example of RiPP-inspired modular design in Lactococcus lactis,485 but many other examples have since been reported and are reviewed here. Since RiPP engineering was not covered in the previous RiPP community-wide review,1 this section of the current review also includes studies predating 2013. However, we will not cover the very large volume of site-directed mutagenesis studies on the core peptide, which has greatly aided understanding RiPP biosynthesis and structure–activity relationships.151,486,487 Instead, we focus on new technologies and novel strategies to make RiPP analogs.Manipulation of core peptides to challenge the PTM enzymes and to study their specificity, binding motifs, mechanism, and processivity has been common in RiPP research. More recently, different engineering approaches have been applied to access completely novel chemical entities, yielding different new functions. We here discuss efforts and successes in modifying rings of macrocyclic RiPPs, the incorporation of non-canonical amino acids (ncAAs), the development of various in vitro engineering systems, and the use of high throughput screening and display systems that have been of particular interest and fruitfulness in the last seven years.
7.1 RiPP variants and hybrids: novel combinations of known functional moieties
RiPP structural genes commonly encode a peptide consisting of minimally a leader and a core peptide. Research conducted in the last years demonstrated that these sequences often have motifs with concrete functions. Sequences have been identified that correspond to recognition elements for PTM enzymes, spacers to allow the correct positioning of the core peptide in the enzyme's active site, LP cleavage sequences, or structural motifs within the core peptide that specifically interact with the enzymes. With this continuously expanding repository of functional sequences, the combinatorial assembly of moieties with a well-defined function, originating from different RiPPs, unmodified peptides or synthetic compounds, together with selected PTM enzymes, offers the prospect of biosynthesizing a vast array of new-to-nature bioactive compounds (Fig. 94).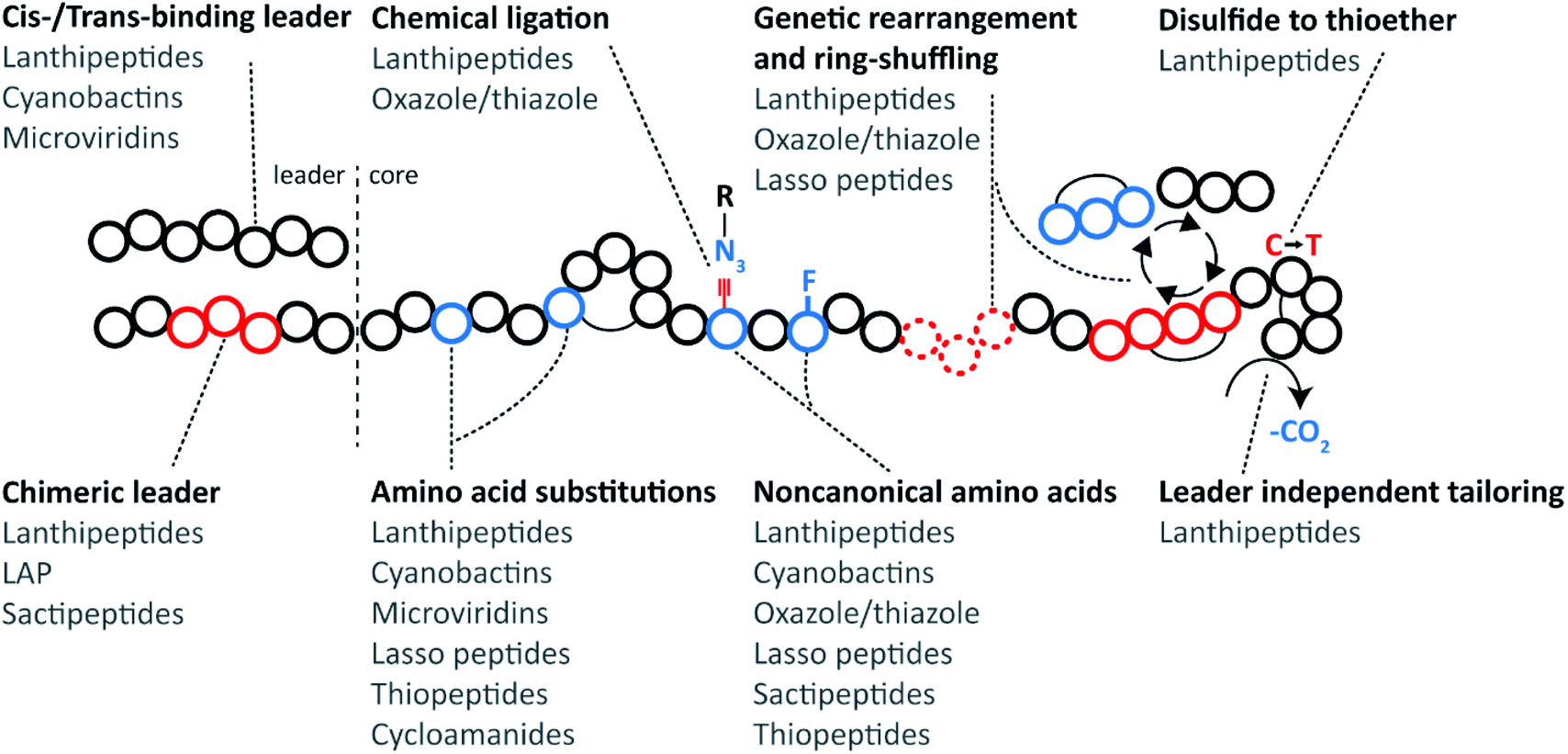 | ||
| Fig. 94 Overview of peptide modifications discussed in Section 7. Cis-/Trans-binding LP: leaderless substrates can be modified by modification enzymes that have their LP covalently bound (in cis) or added as a separate molecule (in trans) to the reaction (Section 7.2). Chemical ligation: a molecule is added through a reaction not related to cellular metabolism (e.g. click chemistry). Genetic rearrangement and ring-shuffling: peptide elements are added, removed, or recombined at the DNA level. Disulfide to thioether: replacement of cystine by lanthionine (Section 7.4.2). Chimeric leader: the LP is comprised of modules from two or more molecules, allowing for modification by PTM enzymes from more than one RiPP system (Section 7.2). Amino acid substitutions: one or more amino acids are simultaneously replaced by residues that do not naturally occur in those positions (Section 7.6). Noncanonical amino acids: one or more amino acids are replaced by non-proteinogenic residues through one of several methods (Section 7.4). Leader-independent tailoring: the peptide is modified by secondary PTM enzymes that recognize a section of the core peptide, instead of a LP. | ||
Precursor peptides containing multiple core sequences have some distinctive advantages in RiPP engineering. The most straightforward application is the use of multiple core peptides to increase yield: the recombinant yield of lyciumins (Section 2.9) in tobacco was improved by 5- to 10-fold by including 5 or 10 repeats of the same core peptide in a single precursor peptide.32 However, such increases in yield are not found with all pathways/substrates and need to be empirically determined.302 Another potential advantage is in engineering of novel RiPPs.488 Some core peptides have slightly different sequence selectivity and post-translational modification requirements,217 such that a desired modification that fails at one core peptide may work at another. The spacing between enzyme recognition sequences and core peptides may be more flexible in precursors containing multiple core peptides, enabling synthesis of hybrid products using enzymes from more than one biosynthetic pathway.489,490 Finally, another as-yet unexplored benefit might be the design or high-throughput synthesis of synergistically acting products. With careful design, it may be possible to generate these products in the desired ratio. For example, in peptides with two different core sequences, sometimes compounds are generated from each core with equal stoichiometry.302
7.2 RiPP analogs accessed by leader peptide engineering
The role of the LP in RiPP biosynthesis was discussed in Section 4.1. For many RiPP classes, the RRE66 mediates the interaction between the biosynthetic proteins and the peptide substrate. This LP does not necessarily need to be covalently attached to the core peptide to achieve enzyme activation. Complementation in cis (covalent linkage of the LP to its cognate PTM enzyme, e.g. the lanthipeptide synthetase LctM,491 cyanobactin heterocyclases PatD,489 LynD,115 and TruD,302,392 and microviridin cyclases MvdCD)215 or in trans (i.e. addition of the LP in vitro,492 or coexpression of the LP and the core peptide as separate molecules)493 results in core peptide modification. This observation simplifies core peptide design and reduces the precursor peptide size, which facilitates chemical synthesis of peptides or libraries for in vitro modification.494,495 For instance, the complete synthesis of the linear cyanobactin-like RiPP aeruginosamide (Fig. 85) was reconstructed in vitro using the heterocyclase TruD attached to its LP.392 This platform facilitated the characterization of the order of N- and C-alkylation by the enzyme AgeMTPT, which prenylates the N- and C-terminus (Sections 5.3.7.1 and 5.8).392 Similarly, MvdD and MvdC covalently fused to the LP or with added LP, allowed the reconstruction of the complete microviridin biosynthesis pathway (Section 3.15) in a LP-independent fashion.215 This strategy was used for in vitro screening of microviridin libraries for protease inhibition (Section 7.7.1).LP engineering has allowed the design of new-to-nature RiPP hybrids in E. coli that are derived from the combination of known pathways.496 In this strategy, recognition motifs of different LPs are fused to structural elements that chosen enzymes are able to modify in their native system. As an example, a LAP (Section 3.4) and class I lanthipeptide hybrid precursor peptide (Section 3.1.1) was designed (Fig. 95) that contained the RS for the cyclodehydratase HcaDF (Section 3.4), the key FNLD motif of the nisin LP for recognition by the dehydratase NisB and the cyclase NisC (Section 3.1.1), and a hybrid core peptide with pieces of their respective cognate core peptides. This design added a second function to the nisin LP as a spacer sequence between the HcaDF recognition motif and its substrate sequence GGRCG. Similarly, the C-terminal residues behind the FNLD motif and the GGRCG in the core peptide served as a spacer between the FNLD motif and the amino acids to be modified by NisBC. Upon co-expression of the designed peptide with HcaDF and NisBC, a peptide containing the desired thiazoline, dehydrobutyrine, lanthionine, and methyllanthionine was the main product (Fig. 95).496
 | ||
| Fig. 95 Concept of hybrid RiPP biosynthesis through leader and core peptide hybridization. By combining the key recognition motifs for leader-dependent PTM enzymes, a core peptide can be modified by enzymes from multiple systems. In this example, the HcaDF RS was combined with the NisBC RS to create a hybrid LP. The hybrid core peptide existed of sequences from LAPs and the class I lanthipeptide nisin.496 The product contained PTMs from LAP (blue) and lanthipeptide (red) biosynthesis. | ||
A second hybrid class was formed by the combination of HcaDF with the subtilosin rSAM enzyme AlbA (Section 3.9) to produce thiazoline-containing sactipeptides. A similar precursor peptide architecture was designed with the RS of HcaDF, the LP of subtilosin, and a hybrid core peptide consisting mainly of the subtilosin precursor with replacement of ACLVD by RCGGC. Upon co-expression of this chimeric peptide with HcaDF and AlbA, two thiazolines and two sactionines were present in the product.496 Since engineering enzyme–enzyme interactions (for a review, readers are referred to reference ref. 93) in such a way that the partially modified core peptide is acted on by different enzymes in the right order is an (almost) impossible task, point mutations that favor/hamper one enzyme over the other were used to manipulate the modification order.496
E. coli was also transformed with a combination of genes encoding HcaDF and the class II lanthipeptide synthetase ProcM (Section 3.1.2).497 In the previous two examples, the enzymes used contained RRE motifs (Section 4.1.1), but this is not the case for ProcM. A hybrid LP was constructed with the minimal binding motifs for the heterocyclase HcaDF and ProcM. Point mutations in the prochlorosin-derived core peptide introduced residues that provided a sequence amenable for HcaDF modification. Since the core peptides were natural substrates of ProcM, initially no thiazoline could be installed indicating outcompetition of HcaDF by ProcM for Cys modification. This problem was solved by reducing the ProcM expression level. Thus, a peptide containing both thiazoline and lanthionine was produced albeit as a mixture with a peptide without thiazoline. These and other experiments demonstrated that core peptide design and fine-tuning expression levels are pivotal to achieve the desired modification pattern.496
LP combinations to use different biosynthesis pathways concomitantly have also been used in flexible in vitro translation systems. This approach has allowed the reconstitution of thiopeptide biosynthesis with a set of enzymes from diverse RiPP enzymes in one pot (Section 7.3).490
The addition of tailoring enzymes from different BGCs to existing pathways has also been accomplished by combining dehydro amino acid reductases (Section 5.1.2) or C-terminal decarboxylases (Section 5.7.3.3) with the nisin biosynthetic machinery to form hybrids in vivo.498 These additional secondary post-translational tailoring modifications do not rely on LP recognition. Similarly, HcaDF–ProcM modified peptides were expressed together with either the dehydrogenase NpnJ (InterPro IPR013149) or the decarboxylase MibD (InterPro family: IPR003382) in E. coli.496 Further modification using other LP-independent enzymes or PTM enzymes activated by covalently attached cognate LPs (vide supra) might further enlarge the repertoire of available PTMs in the future.
A different approach to producing non-cognate RiPPs uses chimeric substrates composed of leader and core peptides from different members of a certain class. This approach was first demonstrated for chimeras of the LP of the class I lanthipeptide subtilin and core peptides made up of nisin and subtilin fragments.499,500 More recently, a set of more than 60 putative lanthipeptides was detected in silico, several of which were produced by attaching the bioinformatically identified non-cognate core peptides to the NisA LP and using the nisin biosynthetic machinery.501,502 Similarly, the tru biosynthetic enzymes served as a production platform in E. coli for the products of cyanobactin core peptides (Section 3.6) detected in silico that were inserted into peptides containing the TruE LP and RSs.503 A chimeric approach was also taken with the LP of the class II lanthipeptide prochlorosin 2.8 and the core peptide of the unrelated lanthipeptide lacticin 481. The class II lanthipeptide synthetase ProcM converted this chimeric substrate into a product with the correct lacticin 481 ring pattern.504 The same strategy has also been used for lasso peptides (Section 3.8). For instance, the fusilassin/fuscanodin biosynthetic machinery was used to make the non-cognate citrulassin and cellulonodin-1 in vitro.150,158 Notably, whereas fusilassin/fuscanodin contains a Trp1–Glu9 macrolactam, citrulassin possesses a Leu1–Asp8 macrolactam and a core peptide that is three residues shorter than that of fusilassin/fuscanodin, and cellulonodin and fusilassin/fuscanodin share identity in the core peptide at only 8 of 19 positions. These examples illustrate the power of using appropriate enzymatic systems to access natural products that may be difficult to obtain from the native organisms.
7.3 In vitro translation and post-translational modification
The systems discussed in Section 7.2 highlight both the opportunities but also the difficulties of producing each PTM enzyme in the right amount, at the right time, and with all the necessary cofactors in vivo. This challenge can be avoided when the PTM enzymes are purified and fully characterized, so that all the necessary cofactors will be present during the reaction in vitro. In addition, tolerance of the PTM for non-cognate peptides can increase due to the option to use longer reaction times and artificially increased enzyme:substrate ratios. Coupling in vitro translation systems with the appropriate PTM enzymes can aid with expression timing and competition of different enzymes for the same amino acid to insert different PTMs, thereby improving the homogeneity of the final product. In vitro biosynthesis can also simplify the incorporation of non-proteinogenic amino acids.
The main limitation of this strategy is that the enzyme needs to be (heterologously) expressed and purified and a thorough knowledge of the cofactors, tolerance of reaction conditions, and (minimal) RSs is required in order to provide optimal conditions. In vitro translation technology has drastically evolved in the last decades, as the required cellular components are better understood and the addition of PTM enzymes can be achieved with less interference on the reaction mixture (for a review see ref. 505). The fact that no living cells are involved also solves potential problems that novel RiPPs can pose for heterologous expression systems when the novel products exhibit antimicrobial activity. A great advance is the development of flexizymes and flexible in vitro translation (FIT) systems, which allow not only RiPP biosynthesis but also enable genetic code reprogramming.506,507 Flexizymes are acylation ribozymes that can acylate tRNA using many acyl-donors (e.g. amino acids with non-proteinogenic side chains, N-alkylated, N-acylated, β-, and D-amino acids, and even non-amino acids) without the requirement for structural similarity as in the case of conventional amino acyl-tRNA synthetases. This flexibility is extended to the tRNAs they can accept via recognition of the 3′ end of the tRNA molecule. Compatibility with the ribosome and the translation machinery is the main limitation for this system.
The cyclodehydratase PatD involved in the biosynthesis of the cyanobactin patellamide (Section 3.6; Fig. 35) has been used in the FIT system to investigate leader and core peptide tolerance in a simplified manner.489 A structural gene was designed encoding the patellamide LP, RSs, core peptide, and C-terminal extension. When the core peptide was that of patellamide, the expected product containing four azolines was obtained. The FIT system was then used to determine the minimal size of the substrate for PatD.489 Thus, it was shown that the sequence between the LP and the core sequence is merely a spacer that is not required for PatD although it serves as the RS for the protease PatA. Indeed, replacing this sequence by a different protease cleavage sequence still allowed PatD modification.508 In addition, only the C-terminal six residues of the LP were required for recognition and modification. In trans supplementation with the LP suggests that it activates the enzyme as a trans-acting chaperone and not as an essential in cis element. In addition to interrogation of the LP and RSs, extensive mutagenesis of core peptides was performed with the FIT system.489 PatD modified even a 36-amino acid core peptide in a context-independent fashion, with a preference for Cys over Ser and Thr. This substrate tolerance is not shared in vitro or in vivo by similar cyclodehydratases from other LAP systems despite belonging to similar families.108,509
In addition, PatD has been used to create novel heterocycles using the FIT system. Threonine analogs with arylated or alkylated β-carbons were correctly processed into substituted azolines with PatD activity requiring the 2-S configuration (Fig. 96, panel II). In addition to its use to form substituted heterocycles, PatD accepted an amino group as a nucleophile in the β-position to yield imidazoline, as well as either amino or thiol groups at the γ-carbon. In the latter cases, a mass loss consistent with heterocyclization was observed, but the structure of the heterocycle was not determined (Fig. 96, panel IV).510
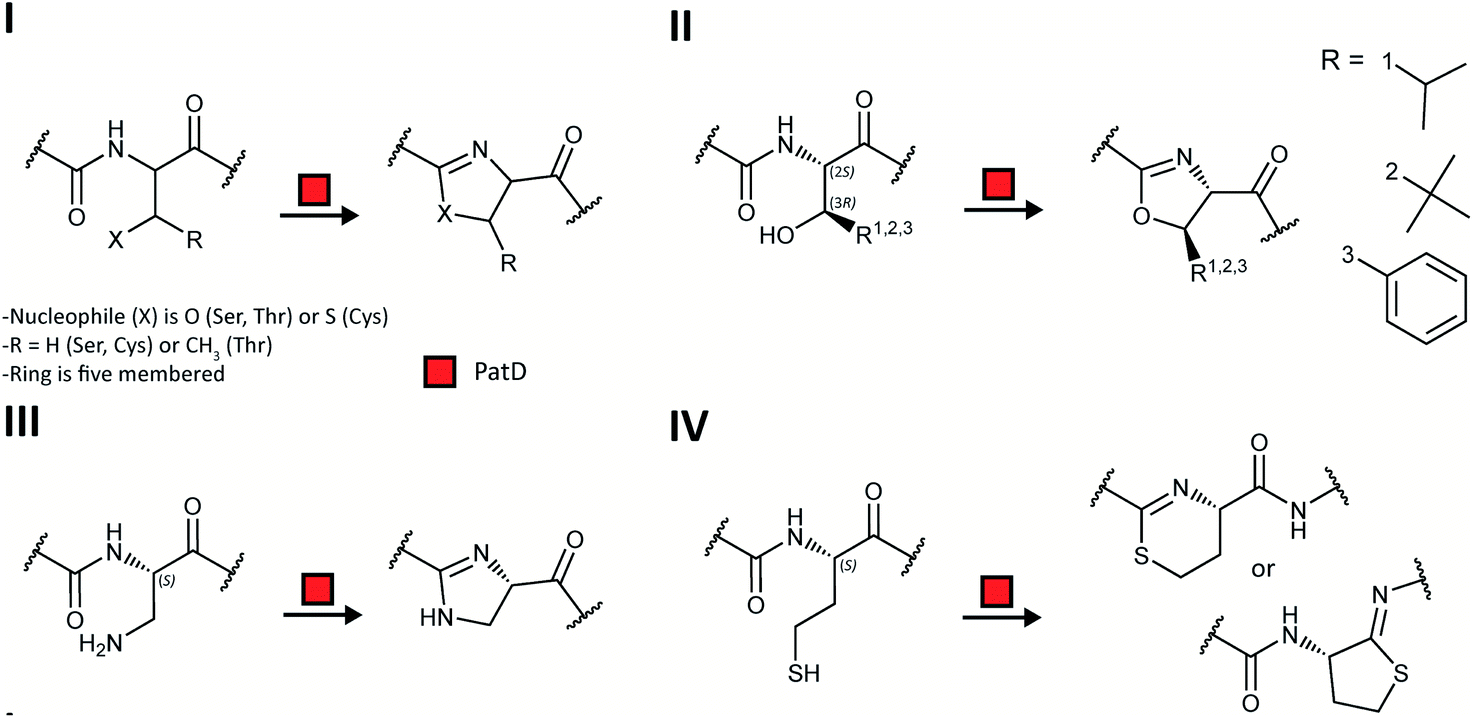 | ||
| Fig. 96 Employment of the FIT system in RiPP biosynthesis. Use of the FIT system for installation of ncAAs in cyclodehydratase substrates and in vitro modification by PatD. (I) The natural substrates (Ser, Thr and Cys residues) result in oxazolines (Ser/Thr) and thiazoline (Cys). (II) PatD installs substituted oxazolines from Thr analogs carrying alkyl or aryl groups on the β-carbon. (III) PatD is able to form an imidazoline. (IV) Substrates with either a thiol group or amino group (not shown) as the nucleophile at the γ-carbon are modified by PatD. The formation of a six or five membered ring could not be distinguished.510 | ||
The FIT system has also been used for the reconstitution of goadsporin biosynthesis, aiding in the determination of the order of the enzymatic modifications and the tolerance towards variations in the core peptide.414 Goadsporin is a LAP (Section 3.4) but also contains dehydro amino acids introduced by a split lanthipeptide dehydratase (Section 3.2; Fig. 97). Experimental difficulties necessitated using a PTM enzyme from a different BGC. Thus, the enzymes GodDEFH were expressed in E. coli, whereas the glutamate elimination enzyme GodG was replaced by LazF originating from the lactazole biosynthesis pathway (lactazole is a thiopeptide, Section 3.2).511 Sequential addition of PTM enzymes to the reaction showed that first GodD and GodE are required for full cyclodehydration of the core peptide. In the absence of the oxidase GodE, GodD installed a variable number of azolines, indicating that azoline oxidation is required for complete heterocyclization. Serine dehydration via GodF-catalyzed glutamylation required azoles to be installed, but only one residue was glutamylated. When the azole-containing peptide was incubated with GodF and LazF, both Ser residues were dehydrated to Dha. This reaction proceeded with N- to C-directionality, indicating that the first dehydration is essential for the second one to take place. After proteolytic removal of the LP, GodH (Section 5.9.1) acetylated the fully modified core peptide. Different deletions in the core peptide and mutations of a minimal core peptide identified an X-T/S/C-Y motif for cyclodehydration activity, where X is a small uncharged amino acid and Y a non-acidic amino acid.437
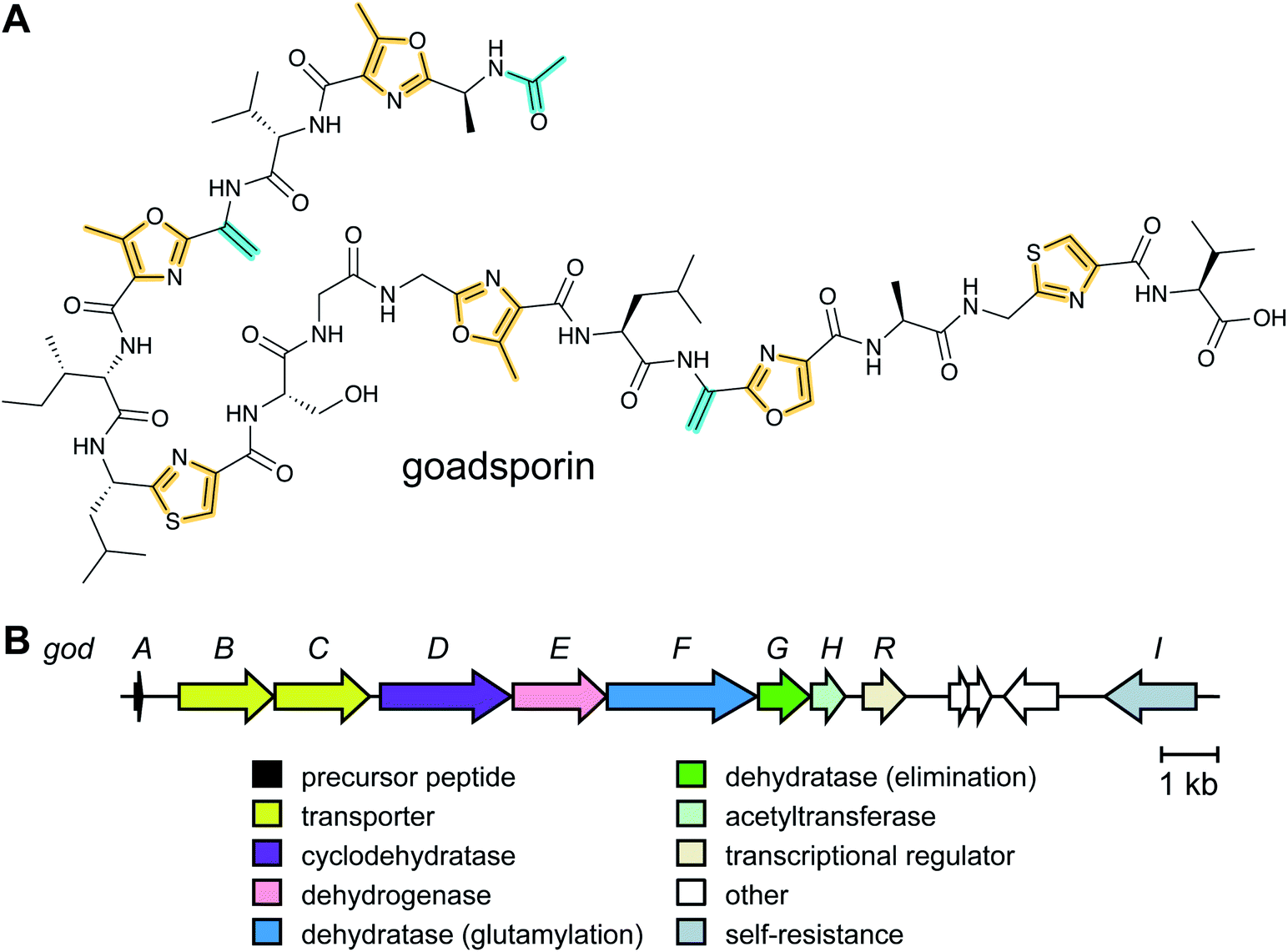 | ||
| Fig. 97 Biosynthesis of goadsporin. (A) Structure of goadsporin which contains features of LAPs (thiazoles and oxazoles; Section 3.4) and lanthipeptides (Dha; Section 3.1.1). (B) BGC for goadsporin containing LAP-type azole-installing machinery (GodDE) as well as a split dehydratase (GodFG; Section 3.2). GodH catalyzes the N-terminal acetylation.437 | ||
The analogous function of enzymes from different RiPP families was also exploited with the FIT system for the synthesis of thiopeptides (Fig. 98).490 Instead of reconstituting the canonical pathway, enzymes with an analogous function were borrowed from alternative soluble and well-established in vitro systems. In addition, the incorporation of the ncAA phenylselenocysteine (SecPh) by codon reprogramming allowed orthogonal chemical oxidation to yield Dha without needing the two proteins of a dehydratase (Section 3.2). Compared to in vitro thiopeptide biosynthesis accomplished previously using six canonical enzymes purified from heterologous hosts,71,89 using this engineered system minimizes the number of PTM enzymes required to three. In this strategy the cyanobactin cyclodehydratase LynD produces azolines that are oxidized to azoles by the thiopeptide dehydrogenase TbtE. Chemical oxidation of selenocysteine derivatives produces the dehydroalanines that serve as a substrate for pyridine formation catalyzed by TclM (InterPro family: IPR023809) from the thiocillin gene cluster (Section 3.2). FIT also was used to test different arrangements of the LP recognition motifs for LynD and TclM in order to obtain the correct PTMs.490
 | ||
| Fig. 98 Employment of the FIT system in thiopeptide biosynthesis. Use of FIT and enzymes from different pathways to reduce the complexity of thiocillin biosynthesis. (I) BGC for thiocillin. (II) The precursor peptide containing SecPh is produced using FIT, after which the combination of LynD (thiazoline formation) and TbtE (thiazoline dehydrogenase) installs triazoles. The SecPh residues are converted to Dha by H2O2, eliminating the need for dehydratases. In a final step, the macrocyclic ring is formed by TclM (Section 3.2). Using this pathway, three enzymes are required rather than the original six.71,490 | ||
More recently, the FIT system has been also combined with recombinantly produced lactazole biosynthetic enzymes, achieving in vitro biosynthesis of the thiopeptide lactazole A and its analogs.512 Stepwise addition of the enzymes to the precursor peptide resulted in either under- or overdehydrated thiopeptides while selective production of the native lactazole A occurred only when the full enzyme set was present from the beginning, implying orchestrated actions of the enzymes during cooperative maturation of the thiopeptide. Intensive mutagenesis of the precursor using this system has revealed remarkable substrate tolerance of the biosynthetic enzymes and demonstrated production of diverse designer lactazole analogs with 10 consecutive mutations, 14- to 62-membered macrocycles, and 18 amino acid-long tail regions, as well as hybrid thiopeptides containing multiple non-canonical amino acids (see Section 7.4.3).512
Very recently, a cell-free protein synthesis (CFPS) system was also used for the preparation of variants of the lanthipeptide nisin.513 The levels of purified dehydratase NisB and cyclase NisC (Section 3.1.1) were first optimized with the sequence of the precursor peptide encoding nisin Z. Then available genomes were searched for natural nisin analogs, and the 18 core peptide variants thus identified were fused to the nisin Z leader peptide. The CFPS system was then used to produce the nisin variants, with four peptides displaying antimicrobial activities, which were then produced in larger amounts using heterologous expression in E. coli. A library of an additonal 3000 variants made by CFPS was screened for activity against E. coli and two nisin analogs were identified with improved Gram-negative activity compared to nisin Z.
In vitro translation systems can also be engineered in an enzyme-free fashion to create RiPPs. A screening system for functional lanthipeptides (Section 3.1) was constructed in which 1011 genes were simultaneously screened by crosslinking the lanthipeptide to its coding mRNA during translation.514 The in vitro translation system substituted lysine with the structural homolog 4-selenalysine (SeLys), which in the presence of hydrogen peroxide was converted into Dha. This Dha was attacked non-enzymatically under alkaline conditions by a neighboring cysteine rendering a lanthionine, although the stereochemistry was not controlled. The chemical attachment of the coding mRNA to its lanthipeptide was achieved using a puromycin-tagged short oligonucleotide that partly annealed to the 3′ end of the mRNA. This approach induces formation of a covalent bond between peptide and mRNA by the ribosome, and therefore phenotype and genotype are linked. A library encoding a hypervariable xxK′xxCxxxx sequence (with K′ = SeLys) was synthesized. After transcription, puromycin-labeled mRNA was generated and translated. Upon purification of the translated library and reverse transcription, about 1013 molecules of peptide–mRNA conjugate were produced, providing 24-fold coverage of the library. This library was tested for binding to staphylococcal sortase A as a drug target using five selection rounds that provided 42 clones in which hydrophobic residues were the most abundant, especially Trp. Chemically synthesized positive hits, including all the possible stereochemical variants, were tested for affinity in vitro providing five candidates with micromolar affinity for sortase A.514
Collectively, these studies demonstrate the versatility of in vitro translation systems for analysis of full biosynthetic routes, albeit at small scale. Mixing biosynthetic routes, expanding the chemical diversity with ncAAs, and using large gene libraries that do not rely on transformation efficiencies of the heterologous host for full coverage, will drastically enlarge the analytical possibilities of the FIT system.
7.4 Engineering RiPPs containing non-canonical amino acids
The structural diversity of RiPPs brought about by PTM enzymes constitutes a great advantage over unmodified peptides in terms of bioactivity and stability. An even higher level of chemical diversity has been introduced using non-canonical amino acids (ncAAs) that can confer attractive properties. The presence of certain ncAAs can increase the stability of the peptide by reducing the accessibility to and recognition by proteases (e.g. by use of D-amino acid, β-amino acids, and N-alkylamino acids). Some ncAAs such as fluorinated amino acids can enhance the half-life and bioavailability of a peptide in vivo.515 Moreover, ncAAs open a wide spectrum of bio-orthogonal chemistry that affords conjugation opportunities in a context-independent manner. These ncAAs can be coupled to diverse probes by standard bio-orthogonal chemical reactions, such as copper-catalyzed alkyne–azide cycloadditions (CuAAC or click-chemistry), olefin metathesis, and metalloporphyrin-catalyzed alkylation.516 Click-based examples in peptide chemistry include the coupling of peptides to polyethylene glycols to increase their solubility and half-life, to fluorophores in order to monitor the distribution or mode of action of a compound, and to sugars or any other chemical moiety to alter the activity spectrum.517 ncAAs have been incorporated into RiPPs through three main routes, (1) by chemical synthesis of the linear precursor peptide containing the ncAAs in the desired locations for further in vitro modification, (2) by supplementation-based incorporation or (3) by codon reprogramming.LctM was also exploited to modify synthetic lacticin 481 precursor peptides with ortho-nitro-phenylglycine derivatives at position 1. This ncAA allowed photocleavage of the LP with good yields in a context-independent manner, thereby obviating the use of leader peptidases and allowing the activation of the fully modified (antimicrobial) RiPP in situ.518 A further step to improve this methodology is represented by a PTM enzyme activated with its cognate LP fused (Section 7.2), which avoids the necessity of removing the LP. Using this approach, lacticin 481 variants containing ncAAs were produced that outperformed the native compound.491
Similarly, the macrocyclase PatG (Section 3.19, InterPro family IPR034056) has been used in vitro with synthetic peptides containing ncAAs (e.g. diamino propionate, ornithine, and aminohexanoic acid) including the N-terminal residue that forms the lactam. Although the reaction is slow,519 diverse circular peptides could be produced. Furthermore, full reconstruction of the cyanobactin pathway has been accomplished.301 Trimming the essential RSs allowed a drastic reduction of the structural peptide to approximately one third of the original length. Sequential addition of the enzymes showed a specific order for the system where the cyclodehydratase ThcD, leader peptidase PatA, macrocyclase PatG and prenyltransferase TruF (InterPro family: IPR031037) worked step-wise to completely transform and decorate the core peptide. Moreover, tailoring enzymes were exchanged to alter the modification pattern. For instance, the prenyltransferase PagF prenylated tyrosine instead of serine and threonine that were modified by TruF.301 It is noteworthy that the full route involved PTM enzymes from three different cyanobactin BGCs, which still allowed conversion of 95% of the synthetic peptide into the final product. One-pot synthesis was demonstrated as well, highlighting the proteases PatA and PatG as critical points. When PatA is present in high amounts and cleaves the LP, heterocyclization is almost abolished because the recognition by the heterocyclase PatD disappears. A solution for this drawback was the use of a different protease and cleavage site (e.g. TEV site) instead of PatA and its canonical sequence. In this way the distance between the cyclodehydratase recognition sequence and the modifiable residues was maintained.508
The use of synthetic peptides allowed increasing the ring size up to 22 amino acids using PatG.301 In future studies, substrate tolerant macrocyclases such as butelase-1 or other asparaginyl endopeptidases from the cyclotide pathway (Section 3.19),239,240 prolyl oligopeptidases from the amatoxin cluster (Section 3.18),235 or PCY1 from the orbitide biosynthesis route (Section 3.19),248 might facilitate the overall process as a replacement for PatG, since their structure and recognition elements are now very well understood. These enzymes display faster kinetics than PatG, and were used to cyclize larger peptides such as head-to-tail cyclized bacteriocins.520
The increased interest in peptide cyclization has driven the in vitro characterization of macrocyclase requirements. The necessity for proline or thiazole as P1 residue for cyanobactin cyclases (e.g. PatG) has been challenged using triazole or vicinal cysteine disulfide bonds, which are readily introduced during peptide synthesis prior to enzymatic cyclization.521,522 In addition, several triazoles, polyethylene glycol, amino benzoic acid or amino n-alkyl acids (up to 8-amino octanoic acid) were inserted in the peptide backbone and still cyclized by PatG.523 Moreover, cyanobactin macrocyclases (Section 3.6) with increased substrate tolerance have been used to increase ring sizes, incorporate D-amino acids, and process peptides with Cys- and Ser-pseudoproline at the P1 position.241
Butelase-1 involved in cyclotide biosynthesis (Section 3.19) is currently the fastest cyclase and has proven to have broad substrate tolerance in terms of peptide size (up to 200 residues). This enzyme requires Asn at position P1 and His–Val at P1′ and P2′, respectively. Residues in P1′′ and P2′′ also affect cyclization with Pro at P1′′ unfavorable and Ile, Leu, Val, Cys the preferred residues at P2′′;239 Gly at P1′′ allows the use of virtually any amino acid at P2′′, thus enabling a broader designer freedom.524 Butelase promiscuity affords the cyclization of peptides containing D-amino acids even at the P1′′ and P2′′ positions or even all D-amino acid-containing peptides (except for the P1, P1′ and P2′ positions) and has been used for the preparation of conotoxins, θ-defensin or SFTI.524
Thiopeptides have been prepared using a semisynthetic route. In this strategy, a synthetic thiocillin core peptide containing thiazole rings was fused to its LP using native chemical ligation (Section 3.2).94 Trimming the LP using this approach facilitated peptide synthesis and identified key residues for correct processing by TclM.95 This strategy opens possibilities to test this enzyme for further efforts including incorporation of ncAAs or variants with different ring sizes.525
In summary, the approach using purified PTM enzymes and synthetic peptides, referred to as in vitro mutasynthesis,494 offers a powerful platform to explore at an analytical level the tolerance and the effect of the ncAAS. The main limitations are the availability of well-characterized enzymes that are active in vitro (i.e. that can be expressed and purified in an active soluble form), the reduced amounts of material that can be produced, and sometimes slow kinetics of the PTM enzymes.
The SPI method has been used to incorporate a variety of analogs into different RiPPs. Using a range of auxotrophic E. coli strains, Met, Pro, and Trp analogs were incorporated into the class II lanthipeptide lichenicidin (see Section 3.1.2 for general biosynthetic pathway).530 The most successful incorporation was that of the Met analogs homopropargylglycine, azidohomoalanine, norleucine, and ethionine. The first two analogs served as a basis for further bio-orthogonal CuAAC. All the Met variants were incorporated and rendered compounds with antimicrobial activity close to that of the wild-type lanthipeptide. CuAAC was performed and showed successful addition of a glucopyranoside and fluorescein as a proof of concept for bio-orthogonal conjugation.530
The use of SPI for RiPP production faces two major hurdles. First, the incorporation of the ncAA into host proteins (including PTM enzymes) can result in the production of inactive proteins, which causes host stress. Secondly, usually slower tRNA acylation kinetics are associated with mischarging tRNAs with ncAAs. In order to solve the first issue, control over the timing of enzyme(s) and structural gene expression using different promoters can provide a cleaner production system. This method has been tested using Trp analogs with fluorine, methyl, or hydroxyl groups at position 5 of the indole to access ncAA-containing variants of the class I lanthipeptide nisin (Section 3.1.1) in L. lactis. In this approach, a nisin inducible promoter was first induced to express the dehydratase, cyclase and transporter (NisB, NisC, NisT) prior to the zinc-controlled expression of nisin precursor peptide mutants containing Trp analogs at position 1, 4, 17 or 32. The slower tRNA acylation was compensated by including an additional copy of the tryptophanyl-tRNA-synthetase that was expressed at the same time as the nisA structural gene.531 With these experimental improvements, incorporation efficiency ranged between 69% and 97%, with production levels up to 50% when compared to typical nisin production. Yields varied greatly with the selected ncAA and the position of incorporation. Consistently, 5-methyl-Trp was incorporated with a reduced yield in all locations tested and mutants in position 4 and 32 favored the production of partly dehydrated nisin. Dehydration efficiency of Ser and Thr was affected in some of the variants, when they were positioned close to ncAAs.
In addition to lanthipeptides, the lasso peptide capistruin (Section 3.8) has been produced in a methionine auxotrophic E. coli strain using SPI. Ring, loop and tail amino acids were individually replaced by azidohomoalanine and homopropargylglycine albeit in low yields, particularly when the ring amino acid was mutated.529 However, all analog-carrying lasso peptides that were produced in quantifiable amounts were shown to have the lasso structure. This result shows compatibility of the capistruin modification machinery with the non-natural side chains present in the ncAA-containing peptides. This paper also describes achieving similar features by using the stop-codon suppression method (Section 7.4.3), which typically increases the production yields.
Amber codon suppression has been tested with diverse archaea-derived orthogonal amino acyl-tRNA synthetases and their respective tRNAs in both Gram-positive and Gram-negative hosts to produce several RiPP classes. The most broadly used orthogonal pairs are pyrrolysyl-tRNA synthetase (PylRS) and tRNAPyl from Methanosarcina barkeri or Methanosarcina mazei as well as O-methyltyrosine-tRNA synthethase and tRNAO-Me-Tyr from Methanococcus jannaschii, and the most commonly used host is E. coli. Expression of RiPP pathways in E. coli was achieved early on for cyanobactins534 and lanthipeptides,535 and hence the first examples of SCS were reported for these compounds.503,536
The class II lanthipeptide synthetases ProcM, NukM, HalM1 and HalM2 (Section 3.1.2), and the class I machinery of nisin (Section 3.1.1) were heterologously expressed with their respective substrates in E. coli and shown to be fully active. The relaxed substrate specificity of ProcM497 was used for the proof-of-concept in vivo incorporation of ncAAs in ProcA3.2. The simultaneous expression of ProcM, ProcA3.2 and an orthogonal tRNA/amino acyl-tRNA synthetase pair allowed the incorporation of the photo-crosslinking residue p-benzoyl-L-phenylalanine.536 Subsequently, similar class II lanthipeptide production systems have been engineered using the lacticin 481 and nukacin ISK-1 systems. Variants containing hydroxy acid derivatives of Lys, Phe, and Tyr in the first position of the core peptide resulted in an ester bond connecting LP and core peptide, thus allowing cleavage of the LP by simple hydrolysis.537 In addition, substitutions of Trp19 and Phe21 of lacticin 481, which were shown with synthetic peptides to lead to improved variants (Section 7.4.1), were produced using SCS technology.538 The range of lanthipeptide producing strains that can incorporate ncAAs has also been extended. Recently, the incorporation of Boc-Lys in the class I lanthipeptide nisin was achieved with the pyrrolysyl pair in E. coli for screening purposes and then transferred to L. lactis.539 The constitutive expression of pylRS greatly simplified the number and/or size of vectors required, thereby making the expression chassis more stable and with faster growth. Additionally, constitutive expression of amino acyl-tRNA synthetases increased the final yield.539
An improvement in ncAA incorporation in class I lanthipeptides in E. coli has been the supplementation with additional copies of genes encoding tRNAGlu,538,540 required by NisB to glutamylate the Ser/Thr residues prior to dehydration (Section 3.1.1).49 Thus, Ser5 could be replaced by keto-, azide or acetylene containing amino acids with yields up to 5 mg L−1 providing sufficient material for downstream orthogonal chemistry.540 With increased product homogeneity, remodeling nisin's ring structure was also accomplished. The typical lanthionine thioether crosslink was replaced using p-chloroacetamide phenylalanine that can react non-enzymatically with cysteine. Although the antimicrobial activity of nisin was lost, this study established proof-of-principle for ring chemistry alteration in vivo.540 Other improvements for the use of SCS with the nisin system involved fine-tuning the expression levels of the biosynthetic enzymes and the ncAA incorporation machinery, and the use of an expression host lacking endogenous amber stop codons (for more discussion of this strain, see below).538 Although successful, production yields decreased by 50–100-fold compared to production of nisin in E. coli without ncAA incorporation.
The class II lanthipeptide cinnamycin (see Sections 3.1.2 and 5.10) has been produced with ncAAs using amber suppression with the pylRS pair in the Gram-positive heterologous host Streptomyces albus.541 In this case, the complete cinnamycin biosynthetic cluster (similar in structure to duramycin and the divamides, Fig. 83), including tailoring enzymes was expressed together with a TAG-encoding core peptide. Notably, Asp hydroxylation by CinX was not complete and therefore CinX was eliminated from the production system to gain homogeneity in the final product and increase the production yield 5-fold. Considering that non-hydroxylated cinnamycin is less bioactive than the fully modified peptide, this observation could indicate that the immunity determinants are not fully functional in the heterologous host. Thus, full modification of the core peptide in heterologous hosts might not be advisable if a high titer is desired. Strategies that may increase the yield include production of the fully modified peptide with the LP still attached for in vitro activation.535,536,542–544
In addition to lanthipeptides, other RiPP families have been produced in vivo using codon reprogramming. The complete cyanobactin pathway (Section 3.6) leading to the cyanobactin trunkamide was reconstituted in E. coli as the first example of a multistep RiPP assembly platform with ncAA incorporation.503 Cyanobactin structural genes often contain two core peptides. Thus, mutating the second core peptide to introduce the amber codon, while keeping the first cognate core peptide intact, provided an internal control of production. This work demonstrated broad substrate tolerance of the cyanobactin modification machinery. Also, it showed that highly mutated core sequences linked to the LP and flanked by RSs were modified, provided the last amino acid in the core peptide is a cysteine that is subsequently heterocyclized.503 A thorough adjustment of the plasmid system and expression timing of the different enzymes was required for the production of compounds with the right modification pattern.503,545 Tuning of expression was also required for the amino acyl-tRNA synthetase for ncAA incorporation. In this system, constitutive expression of the amino acyl-tRNA synthetase rendered the highest yield of ncAA-containing cyanobactins.
A systematically optimized production system for ncAA-containing thiopeptides (Section 3.2) has been constructed in B. cereus.546 Several DNA-regulating sequences (promoter, terminator and Shine–Dalgarno sequence) were assayed to control expression of tRNAPyl and a codon optimized tRNAPyl synthetase using a TAG-containing GFP reporter. Thus, increased fluorescence was paired to optimal promoter combinations for each member of the orthogonal pair. The production of thiocillin variants containing an amber codon at different positions together with a chromosomally integrated tclABCDI cluster under the control of native regulatory systems, allowed the expression of ncAA-containing thiocillin variants albeit with reduced production yields and antimicrobial activity. Interestingly, an alkyne functionalized Lys derivative was used to orthogonally incorporate a biotin attached to a photo-reactive diazirine. This thiocillin derivative was used as a photocrosslinking probe to determine the natural target of thiocillin, showing binding to protein L11 of the 50S ribosome.546
A sactipeptide production system containing the radical SAM enzyme AlbA responsible for the sulfur-to-α-carbon (sactionine) linkages in subtilosin A (Section 3.9) has been heterologously expressed with an O-methyl tyrosine-tRNA synthetase pair547 in a recombinant TAG-codon deprived E. coli strain.548 The use of such strains can, in principle, reduce the synthesis of large aberrant proteins encoded in the chromosome (vide infra). Notably, the heterologous expression system could better tolerate mutations in the Phe residues involved in sactionines than the native system in B. subtilis, although the stereochemistry of the variants has not yet been determined. Similar E. coli expression systems for the lasso peptides microcin J25 and capistruin (Section 3.8; Fig. 38) have been developed.529,549 Whereas the amber codon in the lasso tail reduced capistruin production, mutations of microcin J25 ring, loop and lock residues to Phe derivatives allowed production of the engineered compounds. The production yield was more dependent on the chemical structure of the ncAA than on the position, as has been observed also with other RiPP classes. The presence of ncAAs decreased the antimicrobial activity of the variants to a variable degree depending on the position and the structure of the ncAA. The ability to incorporate p-bromophenylalanine into microcin J25 was critical to the solution of the crystal structure of the RNA polymerase-microcin J25 complex.550
Engineering approaches using peptides and proteins containing ncAAs obtained by SCS offers opportunities for improved protein production. In this sense, the ability of macrocyclases to stabilize peptides and proteins and the combination with click-chemistry represents a robust and potent route to improve potential peptidic pharmaceuticals. As an example, the remarkable promiscuity of butelase-1 as a macrocyclase (Section 3.19) and its tolerance to ncAAs (see Section 7.4.1) has been exploited on dihydrofolate reductase (DHFR).551 Expression in E. coli of DHFR containing ncAA that display alkyne groups and a C-terminal extension with the Asn–His–Val recognition sequence for butelase-1 allowed the production of a cyclic DHFR variant. Next, CuAAC was used to conjugate either biotin or a cell-penetrating peptide to the cyclic DHFR for enzyme inmobilization or intracellular delivery, respectively. The cyclized enzyme survival time inside cells was longer than that of linear counterparts.
Like SPI, the SCS method has specific limitations. The orthogonal tRNA that recognizes the stop codon is competing with release factor-1 (RF1). The function of RF1 is to recognize the amber stop codon and, by binding to it, to facilitate the termination of translation. When this happens, the protein is truncated at the site at which the ncAA was intended to be incorporated.552 Several methods have been described where the competition between ncAA incorporation and RF1 binding was improved towards the former e.g. by engineering of the orthogonal tRNA553 or directed evolution of its tRNA-synthetase.552 In one study, the authors replaced all 321 amber stop codons in an E. coli expression strain with ochre stop codons, allowing complete deletion of RF1 from the genome, whilst keeping the resulting C321.Δ1 strain viable.548 In essence, the UAG codon was completely repurposed. C321.Δ1 showed improved applicability for incorporation of ncAAs, without reducing fitness and has been used for ncAA incorporation in RiPPs.538,547 A second problem is that near-cognate tRNAs, with anti-codons strongly resembling that of the amber stop codon, can mismatch and incorporate their canonical amino acid instead of the ncAA.554 This problem especially arises when RF1 is deleted.555 As these near-cognate tRNAs are essential, the problem can hardly be circumvented in living cells.
Both of the aforementioned SCS limitations are addressed in cell-free CFPS systems. CFPS encompasses a range of protein biosynthesis methods that employ biological machinery without using intact, living cells. Hence, essential cellular mechanisms that hamper ncAA incorporation can be removed without consequence. A typical CFPS setup involves a cell lysate, containing the necessary protein synthesis machinery, supplemented with a range of compounds or enzymes to support or modify the production effort. Using this method, the aforementioned problem of near-cognate tRNAs competing with orthogonal tRNAs can be addressed. For instance, near cognate tRNAs for the amber stop codon in the synthesis mixture have been inactivated by adding antisense oligonucleotides, which led to a 5-fold decrease in near cognate suppression without impairing normal protein expression.555
While the previously described advancements have not been widely implemented in the CFPS production of ncAA-containing RiPPs, this approach has recently received more attention. C321.Δ1 has been used as a basis for the CFPS system, allowing for cell-free synthesis without RF1.556 When taking the recent advancements into account, a logical next step is the incorporation of ncAAs in RiPPs, using one of the improved SCS methods by CFPS. Recently, the FIT system (Section 7.3; Fig. 96), in which multiple sense codons originally encoding proteinogenic amino acids can be simultaneously reprogrammed to designated ncAAs, has allowed in vitro production of ncAA-containing RiPPs. The combination of FIT-mediated genetic code reprogramming with RiPP biosynthetic enzymes has been demonstrated for in vitro production, not only of peptides with novel azoline derivatives as previously described510 (Fig. 96), but also of hybrid thiopeptides with a variety of ncAAs such as N-methylated amino acids, D-amino acids, β-amino acids, and hydroxy acids.512
In summary, proof-of-concept has been achieved for ncAA incorporation into diverse RiPP classes and heterologous hosts. Mutasynthesis for analytical approaches, when the PTM enzymes are available and the ncAAs are compatible (Section 7.4.1), using peptide synthesis can provide a suitable starting point for screening. In vivo technologies, in particular stop-codon suppression, are being optimized for higher production yields and increased homogeneity in the final product. Special attention should be paid to requirements such as cofactors or maturation conditions540,547 that each enzyme in the biosynthesis route might have, as well as compartmentalization and expression timing of the different enzymes in complex pathways.557 Optimization of growth conditions, promoters, Shine–Dalgarno sequences and cluster arrangement can influence the success or failure in RiPP engineering. Thus far, constitutive expression of the tRNA synthetase and inducible expression of precursor peptides, tRNA and biosynthetic enzymes have been the most successful strategies. Last, design of the biosynthesis of non-cognate peptides must also consider sensitivity of the heterologous host to the new RiPP analog, which can be detrimental for high yield production.
7.5 RiPP mutasynthesis and engineering strategies based on growth conditions
An alternative means of accessing RiPP analogs is feeding non-natural building blocks or applying alterations of the growth medium and fermentation conditions. Growing microbisporicin-producing streptomycetes in the presence of NaBr was sufficient to change the typical chlorinated tryptophan in this class I lanthipeptide (Section 3.1.1) into the brominated form, which was more active than the parent compound.409 Considering that microbisporicin is undergoing clinical evaluation, this is an important achievement. Similarly, reconstruction of the quinaldic acid pathway for the thiopeptide thiostrepton in Streptomyces laurentii (Sections 3.2 and 5.7.3.1) showed that indolic ring rearrangement involves the formation of an intermediate ketone 4-acetylquinoline-2-carboxylic acid (Fig. 90) that can be externally supplied. The stereospecific oxidoreductase acting on this ketone can use a 6-fluorinated variant that serves as a substrate for the downstream processing enzymes, resulting in a fluorinated thiostrepton analog that was 4-fold more active than the original compound.558 Similarly, fluorinated Trp was used for the fluoroindolic acid ring closure of nosiheptide by NosL (Section 5.2).344 Halogenated indole supplementation provides a certain flexibility in the ring closure of some thiopeptides, and may avoid side effects caused by forced feeding using Trp-derived ncAAs. Notably, studies with the cyanobactin pathway showed that changes in the growth media and conditions can be crucial to increase yields (up to 30000-fold) due to alteration of the metabolic routes in the heterologous host.325
Expression of BGCs in the cyanobacterium Nostoc punctiforme was shown to be greatly upregulated by high density fermentation conditions involving high concentrations of bicarbonate.321 These conditions resulted in expression of four RiPP gene clusters including the mvd BGC, which led to the discovery of a new microviridin (Section 3.15) that is not acylated at its N-terminus but instead is extended with different numbers of amino acids originating from the leader peptide.321 In addition to finding conditions that allowed isolation of previously uncharacterized compounds, these findings also suggest that leader peptide removal may be a stepwise aminopeptidase process similar to that discussed in Section 4.4.4.
7.6 Accessing bioactive scaffolds by rational design using RiPP biosynthetic enzymes
As discussed in Section 3.14, rSAM enzymes convert α-amino acid dipeptide sequences into β-peptide structures (Fig. 50). In addition to the extended backbone as a new feature, the introduced carbonyl group is also of interest as a handle for further manipulation. Ketoamides are known pharmacophores of non-ribosomal and synthetic peptides that inhibit Ser and Cys proteases, with the ketone functional group important in mechanism-based inhibition. Examples of therapeutic relevance are the drugs boceprevir and telaprevir, peptidomimetics used for hepatitis C treatment that were developed from synthetic peptide leads. Exploring the promiscuity of the splicease, six amino acids around the splice site of its substrate were modified to mimic the boceprevir lead sequence, resulting in successful conversion.41 The keto group could also be modified via hydrazone formation to access conjugates, exemplified by a fluorescent moiety that was attached to the spliced core peptide. Thus, diverse applications of spliceases might be envisaged.Similarly, the dehydrogenase NpnJA that reduces Dha to D-Ala residues during lanthipeptide biosynthesis (Section 5.1.2) modified a variety of non-lanthipeptide substrates. The enzyme was used to generate an analog of the heptapeptide dermorphin, a μ-opioid receptor agonist produced by South American frogs.334 These results highlight the substrate tolerant nature of this enzyme and its potential for bioengineering.
Integrin binders can inhibit angiogenesis during tumor development, thereby reducing tumor growth, which makes them interesting molecules for drug discovery and development. In proof-of-principle studies, the lasso peptide microcin J25 (Section 3.8) was used as scaffold for epitope grafting employing the RGD-integrin-binding motif.559,560 The integrin binding motif was also inserted in ProcA2.8 at three different positions providing bicyclic compounds with nanomolar binding affinity upon reaction with the class II lanthipeptide synthetase ProcM (Section 3.1.2). Cyclization improved the inhibition capacity by one order of magnitude compared to the linear counterpart and provided proteolytic resistance against trypsin.561
7.7 RiPP library generation and peptide display methods
An NNT codon library coding for 15 possible amino acids was used to simultaneously replace three positions in the lasso peptide microcin J25 (Section 3.8), providing 3375 different clones.562 For screening purposes, an E. coli host expressing the structural gene library, immunity protein and PTM enzymes under the control of three different promoters was used. In this construction, the gene library expression was controlled by IPTG, the enzymes were constitutively expressed and immunity was induced by arabinose. Thus, the library was replicated on three plates, one containing IPTG and glucose, the second IPTG and arabinose, and the third with no inducer. Clones that were unable to grow with IPTG and glucose were used in a second screen using supernatants of cultures grown with IPTG and arabinose for antimicrobial activity against Salmonella serotype Newport and E. coli. Twelve new improved variants of microcin J25, against one or both strains, with up to three simultaneous mutations, were isolated.562 In another approach, the lasso peptide astexin-1 has been proposed as a suitable candidate for library screening (including surface exposed libraries, vide infra) owing to the demonstration of high flexibility in the C-terminal region of its precursor peptide. Fusions of the precursor with GFP or leucine zipper protein A1 were still modified, indicating that a bulky C-terminal protein domain does not abolish production.563
Macrocyclic peptide libraries have been developed using the fungal machinery of the cyclic hexapeptide cycloamanide in E. coli.233 The POPB catalyzing the cyclization reaction (Section 3.18; InterPro family: IPR002470) was used with a set of substrate variants with different lengths fused to the maltose binding protein (MBP), and peptides up to 16 amino acids were cyclized in vitro. An NNK library randomizing three positions in the core peptide maintained key residues for cyclization, including the important C-terminal Pro in the LP. Random selection of transformants identified a large diversity in the sequences. For faster production screening, groups of eight colonies were grown together. After in vitro removal of MBP and cyclization, MS analyses confirmed that most peptides were cyclized, except for those with charged residues or Tyr at position 1.
An extended screening method for circular RiPPs including heterocyclization and prenylation, was developed with the cyanobactin pathway in E. coli (Section 3.6).503 Trunkamide was taken as a scaffold and a Pro was maintained in position 3 and a Cys as the last amino acid of the core peptide since a heterocycle is mandatory at this position. Single, double, triple and quadrupole mutant libraries were synthesized in which any codon was introduced. Therefore, stop codons were detected as well as frameshift mutations. Manual replication of the clones in growth media, extraction and mass-spectrometry coupled to DNA sequencing, demonstrated that sequences unrelated to trunkamide were processed, although some amino acid residues were never present at certain positions and some others, such as basic residues, were unfavorable. This study provided an extensive evaluation of the substrate tolerance towards heterocyclization and prenylation.488
In vivo thiopeptide expression (Section 3.2) has been optimized (30-fold yield improvement) using modified Streptomyces coelicolor, in which the BGCs coding natural products had been previously deleted.564 In this strain, a vector containing the entire cluster with the structural gene inactivated was complemented with an E. coli-Streptomyces shuttle vector harboring a library of structural gene variants. Saturation mutagenesis with NNK degenerate primers allowed the creation of seven libraries, six with randomized positions in the macrocycle and one outside of it. Screening of the library for antimicrobial activity was done by assays in solid media. In addition to obtaining valuable information about the impact of mutations on PTM installation, a variant with improved activity was isolated with this methodology. Similarly, a site-saturation library was generated varying each position in the precursor peptide that is incorporated into the macrocycle of the thiopeptide thiocillin. Expression in B. cereus and antimicrobial screening resulted in eight variants with improved activity.565
A microviridin library and screening system was developed to isolate specific protease inhibitors (see Section 7.2).566 Simultaneous mutation of five residues was achieved based on previous single point mutant analyses. Thus, three amino acids were used for positions 1, 2, 3 and 15, and ten possibilities were created for position 5. A previously developed heterologous production platform in E. coli was applied.214 In this platform, essential RiPP-regulatory sequences were identified and used, as well as a minimal LP sequence. Several clones produced a compound with low inhibitory concentration against at least one of the proteases tested. Position 5 was pivotal in protease selectivity, thus saturation mutagenesis on this position was performed and the products screened in a colorimetric assay for protease specificity. Subsequently, a p-nitroanilide fused to peptides specifically targeted by one protease were mixed with culture supernatants of the producer cells to assay directly for protease inhibition. Eventually, variants F5L, F5V and F5K were obtained as subtilisin-specific inhibitors with no activity on chymotrypsin or thrombin.
An alternative method that does not require peptide purification from E. coli fermentations, and can test partly modified microviridins has also been developed. A LP covalently bound on the ATP-grasp ligases MvdD and MvdC (Section 3.15), together with the leader independent N-acetyl transferase MvdB were used for one-pot in vitro synthesis of microviridins.215 Two core peptide libraries containing any amino acid at position 5 of either microviridin K or microviridin B were synthesized and processed with the three enzymes, and partially or fully modified peptides were tested. In addition to determining unfavorable amino acids at this position that prevented correct cyclization, the libraries were evaluated for subtilisin and trypsin inhibition with the colorimetric assay previously mentioned.566 Protease-specific inhibitors with potencies in the low micromolar or nanomolar range were identified, highlighting the effect of each ring and acetylation on activity.215
Several studies have performed extensive library screening of lanthipeptides (Section 3.1) with in situ activation of candidate peptides by LP removal coupled to antimicrobial activity detection. In one set of studies, the class I lanthipeptide nisin inspired the architecture of the variants.567 In one approach, the NisA LP was fixed and the cognate core peptide was divided in different sections corresponding to functional domains: a lipid II binding domain, a hinge region, and a membrane insertion/pore forming domain. Each of these sections was diversified with modules originating from twelve lanthipeptides, both class I and class II, for which the structure and mechanism of action was (partly) known, connected by designed linkers that confer flexibility resembling the hinge region of nisin. Although the N-terminus was limited to conserved sequences from nisin and gallidermin to keep a proper cleavage site for the protease NisP (InterPro family: IPR008357)311 and to facilitate NisBC modification (Section 3.1.1),501 the remaining regions were designed with larger variability.567 A library encoding all possible combinations of these modules was assembled by controlled pore glass synthesis combined with a mix-and-split approach (ca. 6000 possible genes). The library was cloned and maintained in E. coli and used to transform L. lactis expressing NisBCT but not the protease NisP. External NisP addition resulted in activation by LP removal and the library was screened using a miniaturized Fleming's assay that was termed nano-Fleming. In this screening platform, the producer cells were resuspended together with the sensor strain in an alginate solution and encapsulated by dropping the alginate cell suspension in a CaCl2 solution. By manipulating dropping speed and vibration, the particle size of the alginate beads was set to an approximate diameter of 0.5 mm and volume of 65 nL. These dimensions ensured an average of 0.3 producer and 150 sensor cells per alginate bead, effectively converting every bead into a typical petri dish for antimicrobial activity testing. The beads remained isolated from the environment by incubation in a hydrophobic phase, preventing crosstalk between the beads. As the producer and sensor strains were labeled with red and green fluorescence markers, respectively, beads that produced an active compound after NisP cleavage, could be selected based on their fluorescence emission. A particle sorting device efficiently selected beads where the green fluorescence was low, indicating that an active lanthipeptide was produced. These beads were used for further isolation of the producer strain and sequencing for the identification of the lanthipeptide gene sequence. This system allowed the screening of 3.2 × 105 nL reactors (ensuring full coverage of the library) from which 839 clones were isolated. A secondary assay using the 205 unique peptide sequences retrieved showed that even active peptides produced in low yield were identified during particle sorting indicating high sensitivity of the method. Purification of the lanthipeptide and MS characterization showed that even peptides with every position occupied by a module from a different lanthipeptide were present in the positive hits. Although the production yield and heterogeneity in dehydration patterns are areas for improvement, this strategy allowed the identification of novel hybrid lanthipeptides with low MIC values against several Gram-positive pathogens, including strains resistant against nisin.567 The screening platform shows great versatility and potential for further development with other RiPP families or indicator strains for targeted antimicrobial discovery.
A second lanthipeptide screening platform was recently reported using class II lanthipeptide synthetases in E. coli (Section 3.1.2).568 Spatial separation of the leader peptidase and the precursor modification machinery was used as in the previous system to ensure high production of the modified precursor before activation and antimicrobial activity screening. In this strategy, the leader peptidase LicP from the lichenicidin gene cluster313,569 was fused to the OmpA secretion signal leading to accumulation of the protease in the periplasm of E. coli. The PTM enzyme and a library of precursor peptides were co-expressed in the cytoplasm. Last, the producer organism was engineered to induce autolysis involving phage λ SRRz under the control of a temperature-regulated promoter. Thus, increasing the incubation temperature induced cell lysis, removing the physical barrier between the leader peptidase in the periplasm and the fully modified core peptides in the cytoplasm. Lysis thus results in core peptide release and diffusion around the producer colony causing inhibition of the sensor strain if the mutant is active. This in-colony activation system has been used with the Halβ peptide from the two-component lanthipeptide haloduracin, where the LP was modified to include a LicP cleavage sequence. An NNK saturation library was used for each residue that is not essential for ring formation, and after induction, lysis and addition of Halα for full activity, halo sizes and modification extent of the compounds were analyzed. This approach allowed the creation of an activity-based heat-map for each position and each amino acid. Although the controls indicated that lysis and/or compartmentalization are not completely tight, and the activity can be affected by activation rate, diffusion, host protease activity and production level (as with most heterologous systems discussed in Section 7), the system constitutes a versatile platform for screening.568
In addition to manipulating their native bioactivities, RiPPs have also been used as a scaffold for completely new functions. Lanthionine rings introduce stable thioether bonds that encompass a variable number of amino acids. Pioneering work was performed using NisBC from the BGC of the class I lanthipeptide nisin (Section 3.1.1) to stabilize the peptide hormone angiotensin, replacing the natural disulfide bridge by an engineered lanthionine.570 This structural change rendered a more potent and stable molecule with promising clinical applications.571 Class II lanthipeptide synthetases (Section 3.1.2) have also been tested for such purpose. ProcM displayed high tolerance to sequence variability in the ring sequences of one of its substrates, ProcA2.8, as well as the linker region connecting the two rings naturally present in this molecule. Several point mutations, deletions and insertions in the first ring were tolerated, although the second ring was not amenable for insertions.561
In addition to grafting protein-binding motifs on a RiPP scaffold (Section 7.6), de novo development of bioactive peptides that can disrupt protein–protein interactions could have interesting medical applications. Reverse two-hybrid systems provide a robust screening platform for such inhibitors.572 NWY codons were used for randomizing the positions within the two rings of the class II lanthipeptide prochlorosin 2.8 (Section 3.1.2). This codon usage results in eight different amino acids at each position and avoids both stop codons as well as Ser, Thr and Cys that might interfere with the canonical ring formation and create aberrant rings. A variant library of 109 members was used to transform E. coli, resulting in a reduction of the library size to 106 due to limited transformation efficiency. This library was used with a previously established reverse two-hybrid bacterial system involving the p6-UEV interaction. P6 is an HIV protein that interacts with the TSG101 human protein at the ubiquitin E2 variant domain (UEV). If this interaction is blocked, viral egress can no longer take place. Briefly, in the reverse two-hybrid system, p6 was translationally fused to P22 and UEV to 422, forming a P22–422 transcriptional repressor complex when p6 and UEV interact. The repressor prevents transcription of HIS3, kanamycin resistance gene and lacZ leading to a strong growth defect. When the p6-UEV interaction is blocked by a member of the RiPP library the repressor is released and transcription is enabled.573 Using this system, faster growing colonies were selected from the library and false positives were removed. Additional controls involved testing initial hits in a different reverse two-hybrid system to rule out non-specific inhibition.572 Out of 80 initial hits, one passed the three selection rounds and was purified. Notably, its sequence was unrelated to known inhibitors of the p6-UEV interaction. Thus, this large randomization effort gave access to a novel binding inhibition motif, highlighting the advantages of an unbiased screening process for the identification of new therapeutic targets. After LP removal the protein–protein inhibitory activity in vitro was potentiated to low micromolar concentrations and binding to UEV was dependent on lanthionine ring formation. Since the size of this compound compromised its cell permeability, it was fused to a Tat-peptide for transport inside eukaryotic cells where viral assembly was inhibited at low concentrations.572 This system can be further optimized by reducing plasmid size to increase transformation efficiency.
A bacterial surface display system for lanthionine-containing peptides modified by the class I lanthipeptide biosynthetic machinery of nisin (Section 3.1.1) was developed using L. lactis as an expression host.574 The nisin precursor peptide was fused to a sortase recognition motif and co-expressed with NisBC. Sortases catalyze peptide bond formation between an LPXTG sequence in a secreted peptide and a short peptide on the peptidoglycan. The display system produced nisin anchored to the cell wall as demonstrated by adding trypsin to the culture, thus releasing active nisin that inhibited a sensor strain in co-culture. Next, a library of a randomized 3-mer sequence using the NNS codon flanked by Ser and Cys was successfully dehydrated by NisB and cyclized by NisC. The library of 8000 possible sequences was screened using magnetic beads coated with streptavidin yielding a potent binder with the sequence SMNVC that was correctly cyclized.574 More recently, the system was used to screen NisB variants that are capable of dehydrating precursor peptides that are not good substrates for wild-type NisB.575
Another RiPP display system targeting neuropilin-1 and neuropilin-2 (involved in angiogenesis and possible anticancer targets576) has been developed in E. coli.577 This system was inspired by cyclotides (Section 3.19), macrocyclic peptides from plants (for a review of peptide grafting using circular plant peptides, see ref. 578). Since surface display requires a covalent bond between the RiPP and the surface protein, macrocyclization of the N- and C-termini is not feasible. Thus, the lactam forming step was removed from the expression system relying on cystine-knot formation for cyclization. Cyclotides have shown great sequence tolerance as long as the knotted disulfide bonds are correctly positioned. Kalata B1 was fused to the circularly permutated OmpX outer membrane protein.579 Loop 6 in kalata B1 was randomized to generate a library of 6 × 109 possible sequences that was tested for neuropilin-1 receptor binding through successive rounds of cell sorting. The best candidate was used to construct a second library randomizing additional positions in loop 5. The best three hits bound the neuropilin-1 receptor with a KD of 40–60 nM, which was 200-fold better than the best hit after randomizing only loop 6. Chemical synthesis of acyclic and cyclic derivatives of the hits showed that the cyclic peptides displayed increased protease resistance against most of the proteases tested and more potent inhibition of cell migration during in vitro testing.577
More recently, phage display of class II lanthipeptides (Section 3.1.2) has been achieved using the ProcM enzyme in E. coli as a host to screen for two novel biological activities.580 N- and C-terminal fusions or the prochlorosin precursor ProcA3.3 to the pIII protein of the M13 phage were tested. The unusual and less commonly used C-terminal fusion provided improved expression and cyclization due to longer residence time in the cytoplasm, where ProcM was expressed. Conversely, the N-terminal fusions did not result in modification. Thus, the expression systems used consisted of the pIII coat protein followed by a linker and the leader and core peptides. A C-terminal His-tag was added for convenience. Whereas most of the expression systems used in RiPP engineering use high-copy number vectors and express the PTM enzymes from strong inducible promoters, low constitutive expression provided the best results for this system.580 Three libraries based on the topology of prochlorosins 1.1, 2.8 and 2.11 were synthesized. In these constructs, the Cys and Ser/Thr residues were kept at their original position and the other residues in the core peptides were varied resulting in 109 sequences for each library. The phages pooled after expression of the substrate libraries with ProcM were evolved for streptavidin or urokinase-type plasminogen activator (uPA) binding during three rounds of selection. The streptavidin selection provided 82 hits corresponding to two unrelated sequences. One was dehydrated but linear, whereas the other contained one essential lanthionine ring. Screening on the more relevant biological target uPA, involved in cancer development and many other pathologies,581 provided a total of 368 clones from the three libraries that shared common motifs.582 Purification of selected variants demonstrated that the peptides were modified and bound to uPA, inhibiting its catalytic activity. Analysis of the identified hits also showed some heterogeneity in the modification state of the displayed peptides, which can hamper the identification of the best binder.
A second lanthipeptide phage display system using the modification machinery of class I lanthipeptides (Section 3.1.1) and N-terminal fusions of the precursor peptide to protein III was also reported.583 Since phage assembly takes place in the periplasm and the Sec-mediated transport cannot translocate structured proteins easily, Tat-mediated transport was chosen. The fully modified nisin precursor peptide containing a Tat export signal was detected in the periplasm of E. coli upon modification by NisBC expressed in the cytoplasm. Thus, for phage display a gene was constructed encoding the Tat signal peptide followed by a His-tagged nisin LP that was attached to a library of nisin core peptide variants that was fused to a truncated pIII. The library randomized the five residues in rings A and B of nisin with an NNK-codon (3.2 × 106 possible clones). Co-expression with NisBC and helper plasmid encoding wild-type pIII resulted in phage displaying the modified nisin variants. Expression of the library was followed by digestion with externally added leader peptidase NisP to remove the LP (Section 4.4.2), thus uncapping the N-terminal rings A and B of the nisin analogs that bind lipid II. The phages were incubated with biotinylated lipid II and binders were recovered for E. coli infection and genetic identification of the successful clones. Four clones were enriched in the selection. One of them was expressed yielding a correctly modified peptide. Surprisingly, this variant lacked antimicrobial activity due to binding to lipid II in a different way than nisin.583
The first RiPP-inspired yeast display system was developed using the cyclotide scaffold (Section 3.19) to produce libraries of cystine-knot peptide (knottins)584 that could bind to the integrin ανβ3. The sequence of the cystine-knot peptide AgRP was fused to the yeast surface agglutinin Aga2. Aga2 is attached to Aga1 by disulfide bonds and the latter is covalently anchored to the yeast surface. The RGD integrin-binding sequence was fixed at positions 3–5 in AgRP loop 4, whereas the surrounding six amino acids were randomized providing a library of 5 × 106 clones. After three selection rounds using cell sorting several clones were isolated. Usually a Gly was located in front of the RGD sequence followed by hydrophilic/charged amino acids. These peptides bound integrin with nanomolar affinity. Next, loops 1–3 of the best integrin-binder were randomized for new yeast display selection, in order to increase the affinity. Although the affinity was not increased, selectivity for ανβ3 over other isoforms was achieved.584
The first successful yeast display of RiPPs has been recently reported.583 C-terminal fusions of a class II lanthipeptide-based library to Aga2 were explored using the precursor peptide of lacticin 481, LctA, expressed with the synthetase LctM in S. cerevisiae. In these conditions LctA was fully modified and the rings correctly formed. Similar results were obtained using the class II lanthipeptide system of HalA2 and HalM2 indicating that the N-terminal Aga2 domain does not hamper enzymatic modification, similar to the findings in the case of MBP or the pIII coat protein for phage display (see above).580 In order to allow PTM and correct surface display, the design in the yeast platform required appropriate compartmentalization signals for the enzymes and the combinatorial peptides. A strep-tag added on LctA/HalA2 allowed the detection of the peptide on the surface and tandem MS analysis demonstrated their correct modification. ανβ3 integrin binders were selected with a library in which the 12 amino acids of the lacticin 481 ring C were mutated using an NDT-codon library (3 × 106 possibilities). This library ensured that the epitope RGD would be present. Two rounds of fluorescence activated cell sorting provided a fast and sensitive screening method for integrin binders. Nearly 95% of the isolated interactions contained the RGD motif. Expression of the positive hits and structural analysis showed they had the right ring pattern and could bind integrin in the nanomolar range.583
8 Outlook
Just seven years after the first comprehensive review of RiPP biosynthesis, the field has exploded in terms of discovery of new compound classes, discovery of new compounds within each class, new genome mining tools specifically designed for RiPPs, and a multitude of engineering tools that take full advantage of the genetically encoded precursor peptides. Moreover, the enzymology of RiPP biosynthesis has continued to reveal unprecented biochemical reactions and substrate recognition is now well established for many systems.With the new advances have also come new questions and challenges. While individual enzymes are increasingly understood at the molecular level, the mechanisms of communication between biosynthetic enzymes and the structures of enzyme complexes are still mostly unresolved.93 Similarly, although the interaction of enzymes with their leader peptides, follower peptides, and recognition sequences is now understood for a subset of RiPP biosynthetic pathways, the interactions with the core peptides are much less well established, especially for enzymes that operate iteratively on substrates that are changing in structure with each successive reaction.
RiPPs are currently also still underdeveloped in terms of commercial usage, but the advances in discovery and engineering should aid in future applications. A particularly interesting observation is that RiPPs appear to be highly abundant in microbiomes, and several compounds have been linked to human disease and disease prevention.585,586 At present the RiPP field is defined largely based upon the chemistry of microbial metabolism, with a few exceptions in animals and plants.587 In the longer term, it will be worth exploring commonalities with human and animal hormones and signaling peptides. These peptides seem to occupy the domain of primary metabolism from the anthropocentric perspective, but in comparison to bacteria, higher animals occupy a small branch on the evolutionary tree, and their products are also quite specialized to a relatively narrow taxonomic branch in comparison to the much larger web of life.
Other areas that will likely see new developments are structure prediction based on the make-up of the BGC, improved understanding of the mode of action of RiPPs, and further exploration of the many exciting engineering approaches described in this review. Thus, the future holds tremendous potential.
9 Author contributions
This review was written by Manuel Montalbán-López (University of Granada, Section 7), Thomas Scott and Jörn Piel (ETH, parts of Sections 3 and 5), Jakob Viel, Auke J. van Heel, and Oscar Kuipers (University of Groningen, Sections 6 and 7), Sangeetha Ramesh and Douglas Mitchell (University of Illinois at Urbana-Champaign, parts of Sections 3 and 6), and Imran Rahman and Wilfred van der Donk (University of Illinois at Urbana-Champaign, Sections 2 and 4 and parts of Sections 3 and 5). All authors proofread the entire review and provided suggestions for improvement on all sections. The resulting draft was sent to all other co-authors who attended the First International Conference on RiPPs (https://www.rippsconference.org/) in 2019 and those scientists who provided feedback for additions, corrections, and clarifications were added as authors.10 Conflicts of interest
C. Hill has an equity interest in Artugen Therapeutics, J. Koehnke has intellectual property interest in GyreOx, A. J. link is a consultant for Lassogen, and D. A. Mitchell is a cofounder and owns stock options in Lassogen, Inc. J. K. Weng is a co-founder, a member of the Scientific Advisory Board, and a shareholder of DoubleRainbow Biosciences, which develops biotechnologies related to natural products.11 Acknowledgments
RiPP research in the laboratories of the corresponding authors is supported by grants from the National Institutes of Health (GM123998 to DAM, AI144967 to WAV and DAM, GM058822 to WAV, and 5T32-GM070421 to IRR), the EU FW7 programme SynPeptide (to MML, OPK and JP), the EU Horizon 2020 programme Rafts4Biotech (to AJvH), the Swiss National Science Foundation (407240_1167051 to JP), the Dutch Research Council (NWO-ALWOP-214 to JV), a European Molecular Biology Organization Long-Term Fellowship (ALTF 344-2018 to TAS), and the Howard Hughes Medical Institute (to WAV).12 References
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- Y. Li and S. Rebuffat, J. Biol. Chem., 2020, 295, 34–54 CrossRef CAS.
- M. A. Funk and W. A. van der Donk, Acc. Chem. Res., 2017, 50, 1577–1586 CrossRef CAS.
- X. Yang and W. A. van der Donk, Chem.–Eur. J., 2013, 19, 7662–7677 CrossRef CAS.
- A. Plat, A. Kuipers, R. Rink and G. N. Moll, Curr. Protein Pept. Sci., 2013, 14, 85–96 CrossRef CAS.
- E. W. Schmidt and M. S. Donia, Methods Enzymol., 2009, 458, 575–596 CAS.
- M. Demerec, E. A. Adelberg, A. J. Clark and P. E. Hartman, Genetics, 1966, 54, 61–76 CAS.
- Y. Hayakawa, K. Sasaki, H. Adachi, K. Furihata, K. Nagai and K. Shin-ya, J. Antibiot., 2006, 59, 1–5 CrossRef CAS.
- M. Izumikawa, I. Kozone, J. Hashimoto, N. Kagaya, M. Takagi, H. Koiwai, M. Komatsu, M. Fujie, N. Satoh, H. Ikeda and K. Shin-ya, J. Antibiot., 2015, 68, 533–536 CrossRef CAS.
- M. Izawa, T. Kawasaki and Y. Hayakawa, Appl. Environ. Microbiol., 2013, 79, 7110–7113 CrossRef CAS.
- L. Frattaruolo, R. Lacret, A. R. Cappello and A. W. Truman, ACS Chem. Biol., 2017, 12, 2815–2822 CrossRef CAS.
- L. Kjaerulff, A. Sikandar, N. Zaburannyi, S. Adam, J. Herrmann, J. Koehnke and R. Müller, ACS Chem. Biol., 2017, 12, 2837–2841 CrossRef CAS.
- T. Kawahara, M. Izumikawa, I. Kozone, J. Hashimoto, N. Kagaya, H. Koiwai, M. Komatsu, M. Fujie, N. Sato, H. Ikeda and K. Shin-Ya, J. Nat. Prod., 2018, 81, 264–269 CrossRef CAS.
- J. Santos-Aberturas, G. Chandra, L. Frattaruolo, R. Lacret, T. H. Pham, N. M. Vior, T. H. Eyles and A. W. Truman, Nucleic Acids Res., 2019, 47, 4624–4637 CrossRef CAS.
- M. Umemura, N. Nagano, H. Koike, J. Kawano, T. Ishii, Y. Miyamura, M. Kikuchi, K. Tamano, J. Yu, K. Shin-ya and M. Machida, Fungal Genet. Biol., 2014, 68, 23–30 CrossRef CAS.
- W. Ding, W. Q. Liu, Y. Jia, Y. Li, W. A. van der Donk and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3521–3526 CrossRef CAS.
- N. Nagano, M. Umemura, M. Izumikawa, J. Kawano, T. Ishii, M. Kikuchi, K. Tomii, T. Kumagai, A. Yoshimi, M. Machida, K. Abe, K. Shin-Ya and K. Asai, Fungal Genet. Biol., 2016, 86, 58–70 CrossRef CAS.
- Y. Ye, T. Ozaki, M. Umemura, C. Liu, A. Minami and H. Oikawa, Org. Biomol. Chem., 2019, 17, 39–43 RSC.
- M. Noike, T. Matsui, K. Ooya, I. Sasaki, S. Ohtaki, Y. Hamano, C. Maruyama, J. Ishikawa, Y. Satoh, H. Ito, H. Morita and T. Dairi, Nat. Chem. Biol., 2015, 11, 71–76 CrossRef CAS.
- D. H. Haft, BMC Genomics, 2011, 12, 21 CrossRef CAS.
- R. S. Ayikpoe and J. A. Latham, J. Am. Chem. Soc., 2019, 141, 13582–13591 CrossRef CAS.
- L. Peña-Ortiz, A. P. Graça, H. Guo, D. Braga, T. G. Köllner, L. Regestein, C. Beemelmanns and G. Lackner, Chem. Sci., 2020, 11, 5142–5156 RSC.
- M. Ibrahim, A. Guillot, F. Wessner, F. Algaron, C. Besset, P. Courtin, R. Gardan and V. Monnet, J. Bacteriol., 2007, 189, 8844–8854 CrossRef CAS.
- L. B. Bushin, K. A. Clark, I. Pelczer and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2018, 140, 17674–17684 CrossRef CAS.
- N. S. van der Velden, N. Kälin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS.
- S. Ramm, B. Krawczyk, A. Mühlenweg, A. Poch, E. Mösker and R. D. Süssmuth, Angew. Chem., Int. Ed., 2017, 56, 9994–9997 CrossRef CAS.
- K. Viehrig, F. Surup, C. Volz, J. Herrmann, A. Abou Fayad, S. Adam, J. Köhnke, D. Trauner and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 7407–7410 CrossRef CAS.
- M. F. Freeman, M. J. Helf, A. Bhushan, B. I. Morinaka and J. Piel, Nat. Chem., 2017, 9, 387–395 CrossRef CAS.
- A. Benjdia, A. Guillot, P. Ruffie, J. Leprince and O. Berteau, Nat. Chem., 2017, 9, 698–707 CrossRef CAS.
- S. Yahara, C. Shigeyama, T. Ura, K. Wakamatsu, T. Yasuhara and T. Nohara, Chem. Pharm. Bull., 1993, 41, 703–709 CrossRef CAS.
- J. Hattori, K. A. Boutilier, M. M. van Lookeren Campagne and B. L. Miki, Mol. Gen. Genet., 1998, 259, 424–428 CrossRef CAS.
- R. D. Kersten and J.-K. Weng, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10961–E10969 CrossRef CAS.
- Q. Zhang, J. R. Doroghazi, X. Zhao, M. C. Walker and W. A. van der Donk, Appl. Environ. Microbiol., 2015, 81, 4339–4350 CrossRef CAS.
- V. Wiebach, A. Mainz, M.-A. J. Siegert, N. A. Jungmann, G. Lesquame, S. Tirat, A. Dreux-Zigha, J. Aszodi, D. L. Beller and R. D. Süssmuth, Nat. Chem. Biol., 2018, 14, 652 CrossRef CAS.
- D. D. Luu, A. Joe, Y. Chen, K. Parys, O. Bahar, R. Pruitt, L. J. G. Chan, C. J. Petzold, K. Long, C. Adamchak, V. Stewart, Y. Belkhadir and P. C. Ronald, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8525–8534 CrossRef CAS.
- R. N. Pruitt, A. Joe, W. Zhang, W. Feng, V. Stewart, B. Schwessinger, J. R. Dinneny and P. C. Ronald, New Phytol., 2017, 215, 725–736 CrossRef CAS.
- P. Ronald and A. Joe, Ann. Bot., 2018, 121, 17–23 CrossRef CAS.
- Y. Amano, H. Tsubouchi, H. Shinohara, M. Ogawa and Y. Matsubayashi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18333–18338 CrossRef CAS.
- T. Wei, M. Chern, F. Liu and P. C. Ronald, Mol. Plant Pathol., 2016, 17, 1493–1498 CrossRef CAS.
- H. Baba and T. Ishibashi, Adv. Exp. Med. Biol., 2019, 1190, 165–179 CrossRef CAS.
- B. I. Morinaka, E. Lakis, M. Verest, M. J. Helf, T. Scalvenzi, A. L. Vagstad, J. Sims, S. Sunagawa, M. Gugger and J. Piel, Science, 2018, 359, 779–782 CrossRef CAS.
- D. H. Haft and M. K. Basu, J. Bacteriol., 2011, 193, 2745–2755 CrossRef CAS.
- G. A. Hudson, B. J. Burkhart, A. J. DiCaprio, C. J. Schwalen, B. Kille, T. V. Pogorelov and D. A. Mitchell, J. Am. Chem. Soc., 2019, 141, 8228–8238 CAS.
- A. Caruso, L. B. Bushin, K. A. Clark, R. J. Martinie and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 990–997 CrossRef CAS.
- A. Caruso, R. J. Martinie, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 16610–16614 CrossRef CAS.
- K. A. Clark, L. B. Bushin and M. R. Seyedsayamdost, J. Am. Chem. Soc., 2019, 141, 10610–10615 CrossRef CAS.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. Guo, A. Makriyannis, L. Linares-Otoya, N. Bohringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schaberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS.
- T. A. J. Grell, P. J. Goldman and C. L. Drennan, J. Biol. Chem., 2015, 290, 3964–3971 CrossRef CAS.
- M. A. Ortega, Y. Hao, Q. Zhang, M. C. Walker, W. A. van der Donk and S. K. Nair, Nature, 2015, 517, 509–512 CrossRef CAS.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS.
- P. A. Jordan and B. S. Moore, Cell Chem. Biol., 2016, 23, 1504–1514 CrossRef CAS.
- T. P. Wyche, A. C. Ruzzini, L. Schwab, C. R. Currie and J. Clardy, J. Am. Chem. Soc., 2017, 139, 12899–12902 CrossRef CAS.
- S. H. Reisberg, Y. Gao, A. S. Walker, E. J. N. Helfrich, J. Clardy and P. S. Baran, Science, 2020, 367, 458–463 CrossRef CAS.
- F. J. Ortíz-López, D. Carretero-Molina, M. Sánchez-Hidalgo, J. Martin, I. Gonzalez, F. Roman-Hurtado, M. de la Cruz, S. Garcia-Fernandez, F. Reyes, J. P. Deisinger, A. Muller, T. Schneider and O. Genilloud, Angew. Chem., Int. Ed. Engl., 2020, 59, 12654–12658 CrossRef.
- F. Román-Hurtado, M. Sánchez-Hidalgo, J. Martín, F. J. Ortíz-López and O. Genilloud, bioRxiv, 2020 DOI:10.1101/2020.05.20.105809.
- A. M. Kloosterman, P. Cimermancic, S. S. Elsayed, C. Du, M. Hadjithomas, M. S. Donia, M. A. Fischbach, G. P. van Wezel and M. H. Medema, bioRxiv, 2020 DOI:10.1101/2020.05.19.104752.
- M. Tao, M. Xu, F. Zhang, Z. Cheng, G. Bashiri, J. Wang, J. Hong, Y. Wang, L. Xu, X. Chen, S.-X. Huang, S. Lin and Z. Deng, Angew. Chem., Int. Ed. DOI:10.1002/anie.202008035.
- J. J. Hug, J. Dastbaz, S. Adam, O. Revermann, J. Koehnke, D. Krug and R. Müller, ACS Chem. Biol., 2020, 15, 2221–2231 CrossRef CAS.
- W. Tang and W. A. van der Donk, Nat. Chem. Biol., 2013, 9, 157–159 CrossRef CAS.
- W. Tang, G. Jimenez-Oses, K. N. Houk and W. A. van der Donk, Nat. Chem., 2015, 7, 57–64 CrossRef CAS.
- C. T. Lohans, J. L. Li and J. C. Vederas, J. Am. Chem. Soc., 2014, 136, 13150–13153 CrossRef CAS.
- M. C. Walker, S. M. Eslami, K. J. Hetrick, S. E. Ackenhusen, D. A. Mitchell and W. A. van der Donk, BMC Genomics, 2020, 21, 387 CrossRef CAS.
- K. I. Mohr, C. Volz, R. Jansen, V. Wray, J. Hoffmann, S. Bernecker, J. Wink, K. Gerth, M. Stadler and R. Müller, Angew. Chem., Int. Ed. Engl., 2015, 54, 11254–11258 CrossRef CAS.
- T. Caetano, W. van der Donk and S. Mendo, Microbiol. Res., 2020, 235, 126441 CrossRef CAS.
- N. Garg, L. M. Salazar-Ocampo and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7258–7263 CrossRef CAS.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS.
- I. R. Bothwell, D. P. Cogan, T. Kim, C. J. Reinhardt, W. A. van der Donk and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17245–17250 CrossRef CAS.
- L. M. Repka, K. J. Hetrick, S. H. Chee and W. A. van der Donk, J. Am. Chem. Soc., 2018, 140, 4200–4203 CrossRef CAS.
- M. A. Ortega, Y. Hao, M. C. Walker, S. Donadio, M. Sosio, S. K. Nair and W. A. van der Donk, Cell Chem. Biol., 2016, 23, 370–380 CrossRef.
- J. Z. Acedo, I. R. Bothwell, L. An, A. Trouth, C. Frazier and W. A. van der Donk, J. Am. Chem. Soc., 2019, 141, 16790–16801 CrossRef CAS.
- G. A. Hudson, Z. Zhang, J. I. Tietz, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 16012–16015 CrossRef CAS.
- X. Yang and W. A. van der Donk, ACS Chem. Biol., 2015, 10, 1234–1238 CrossRef CAS.
- A. Abts, M. Montalbán-Lopez, O. P. Kuipers, S. H. Smits and L. Schmitt, Biochemistry, 2013, 52, 5387–5395 CrossRef CAS.
- J. Reiners, A. Abts, R. Clemens, S. H. Smits and L. Schmitt, Sci. Rep., 2017, 7, 42163 CrossRef CAS.
- S. H. Dong, W. Tang, T. Lukk, Y. Yu, S. K. Nair and W. A. van der Donk, eLife, 2015, 4, e07607 CrossRef.
- Y. Habibi, K. A. Uggowitzer, H. Issak and C. J. Thibodeaux, J. Am. Chem. Soc., 2019, 141, 14661–14672 CrossRef CAS.
- I. R. Rahman, J. Z. Acedo, X. R. Liu, L. Zhu, J. Arrington, M. L. Gross and W. A. van der Donk, ACS Chem. Biol., 2020, 15, 1473–1486 CrossRef CAS.
- C. J. Thibodeaux, T. Ha and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 17513–17529 CrossRef CAS.
- C. J. Thibodeaux, J. Wagoner, Y. Yu and W. A. van der Donk, J. Am. Chem. Soc., 2016, 138, 6436–6444 CrossRef CAS.
- W. Tang, G. N. Thibodeaux and W. A. van der Donk, ACS Chem. Biol., 2016, 11, 2438–2446 CrossRef CAS.
- Y. Yu, Q. Zhang and W. A. van der Donk, Protein Sci., 2013, 22, 1478–1489 CrossRef CAS.
- S. C. Bobeica, L. Zhu, J. Z. Acedo, W. Tang and W. A. van der Donk, Chem. Sci., 2020 10.1039/d0sc01651a.
- N. A. Jungmann, B. Krawczyk, M. Tietzmann, P. Ensle and R. D. Süssmuth, J. Am. Chem. Soc., 2014, 136, 15222–15228 CrossRef CAS.
- M. Iorio, O. Sasso, S. I. Maffioli, R. Bertorelli, P. Monciardini, M. Sosio, F. Bonezzi, M. Summa, C. Brunati, R. Bordoni, G. Corti, G. Tarozzo, D. Piomelli, A. Reggiani and S. Donadio, ACS Chem. Biol., 2014, 9, 398–404 CrossRef CAS.
- H. Ren, C. Shi, I. R. Bothwell, W. A. van der Donk and H. Zhao, ACS Chem. Biol., 2020, 15, 1642–1649 CrossRef CAS.
- J. D. Hegemann and W. A. van der Donk, J. Am. Chem. Soc., 2018, 140, 5743–5754 CrossRef CAS.
- J. D. Hegemann, L. Shi, M. L. Gross and W. A. van der Donk, ACS Chem. Biol., 2019, 14, 1583–1592 CrossRef CAS.
- V. Wiebach, A. Mainz, R. Schnegotzki, M.-A. J. Siegert, M. Hügelland, N. Pliszka and R. Süssmuth, Angew. Chem., Int. Ed. Engl. DOI:10.1002/anie.202003804.
- Z. Zhang, G. A. Hudson, N. Mahanta, J. I. Tietz, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2016, 138, 15511–15514 CrossRef CAS.
- K. D. Bewley, P. R. Bennallack, M. A. Burlingame, R. A. Robison, J. S. Griffitts and S. M. Miller, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12450–12455 CrossRef CAS.
- T. Ozaki, Y. Kurokawa, S. Hayashi, N. Oku, S. Asamizu, Y. Igarashi and H. Onaka, ChemBioChem, 2016, 17, 218–223 CrossRef CAS.
- Y. Du, Y. Qiu, X. Meng, J. Feng, J. Tao and W. Liu, J. Am. Chem. Soc., 2020, 142, 8454–8463 CrossRef CAS.
- A. Sikandar and J. Koehnke, Nat. Prod. Rep., 2019, 36, 1576–1588 RSC.
- W. J. Wever, J. W. Bogart, J. A. Baccile, A. N. Chan, F. C. Schroeder and A. A. Bowers, J. Am. Chem. Soc., 2015, 137, 3494–3497 CrossRef CAS.
- W. J. Wever, J. W. Bogart and A. A. Bowers, J. Am. Chem. Soc., 2016, 138, 13461–13464 CrossRef CAS.
- J. W. Bogart and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 1842–1846 CrossRef CAS.
- D. P. Cogan, G. A. Hudson, Z. Zhang, T. V. Pogorelov, W. A. van der Donk, D. A. Mitchell and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12928–12933 CrossRef CAS.
- J. W. Bogart, N. J. Kramer, A. Turlik, R. M. Bleich, D. S. Catlin, F. C. Schroeder, S. K. Nair, R. T. Williamson, K. N. Houk and A. A. Bowers, J. Am. Chem. Soc., 2020, 142, 13170 CrossRef CAS.
- S. Ma and Q. Zhang, Nat. Prod. Rep., 2020 10.1039/c9np00074g.
- J. Claesen and M. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297–16302 CrossRef CAS.
- J. Claesen and M. J. Bibb, J. Bacteriol., 2011, 193, 2510–2516 CrossRef CAS.
- M. E. Rateb, Y. Zhai, E. Ehrner, C. M. Rath, X. Wang, J. Tabudravu, R. Ebel, M. Bibb, K. Kyeremeh, P. C. Dorrestein, K. Hong, M. Jaspars and H. Deng, Org. Biomol. Chem., 2015, 13, 9585–9592 RSC.
- Z. Shang, J. M. Winter, C. A. Kauffman, I. Yang and W. Fenical, ACS Chem. Biol., 2019, 14, 415–425 CrossRef CAS.
- F. Wang, W. Wanqing, J. Zhao, T. Mo, X. Wang, X. Huang, S. Ma, S. Wang, Z. Deng, W. Ding, Y. Liang and Q. Zhang, CCS Chem., 2020, 2, 1049–1057 CrossRef.
- T. Mo, W.-Q. Liu, W. Ji, J. Zhao, T. Chen, W. Ding, S. Yu and Q. Zhang, ACS Chem. Biol., 2017, 12, 1484–1488 CrossRef CAS.
- Y.-M. Li, J. C. Milne, L. L. Madison, R. Kolter and C. T. Walsh, Science, 1996, 274, 1188–1193 CrossRef CAS.
- C. L. Cox, J. R. Doroghazi and D. A. Mitchell, BMC Genomics, 2015, 16, 778 CrossRef.
- C. D. Deane, B. J. Burkhart, P. M. Blair, J. I. Tietz, A. Lin and D. A. Mitchell, ACS Chem. Biol., 2016, 11, 2232–2243 CrossRef CAS.
- Y. Igarashi, Y. Kan, K. Fujii, T. Fujita, K. Harada, H. Naoki, H. Tabata, H. Onaka and T. Furumai, J. Antibiot., 2001, 54, 1045–1053 CrossRef CAS.
- B. J. Burkhart, C. J. Schwalen, G. Mann, J. H. Naismith and D. A. Mitchell, Chem. Rev., 2017, 117, 5389–5456 CrossRef CAS.
- J. O. Melby, K. L. Dunbar, N. Q. Trinh and D. A. Mitchell, J. Am. Chem. Soc., 2012, 134, 5309–5316 CrossRef CAS.
- K. L. Dunbar, J. O. Melby and D. A. Mitchell, Nat. Chem. Biol., 2012, 8, 569–575 CrossRef CAS.
- K. L. Dunbar and D. A. Mitchell, J. Am. Chem. Soc., 2013, 135, 8692–8701 CrossRef CAS.
- J. Koehnke, A. F. Bent, D. Zollman, K. Smith, W. E. Houssen, X. Zhu, G. Mann, T. Lebl, R. Scharff, S. Shirran, C. H. Botting, M. Jaspars, U. Schwarz-Linek and J. H. Naismith, Angew. Chem., Int. Ed., 2013, 52, 13991–13996 CrossRef CAS.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. E. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS.
- Y. Ge, C. M. Czekster, O. K. Miller, C. H. Botting, U. Schwarz-Linek and J. H. Naismith, Biochemistry, 2019, 58, 2125–2132 CrossRef CAS.
- K. L. Dunbar, J. R. Chekan, C. L. Cox, B. J. Burkhart, S. K. Nair and D. A. Mitchell, Nat. Chem. Biol., 2014, 10, 823–829 CrossRef CAS.
- N. Mahanta, A. Liu, S. Dong, S. K. Nair and D. A. Mitchell, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3030–3035 CrossRef CAS.
- S.-H. Dong, A. Liu, N. Mahanta, D. A. Mitchell and S. K. Nair, ACS Cent. Sci., 2019, 5, 842–851 CAS.
- L. Franz, S. Adam, J. Santos-Aberturas, A. W. Truman and J. Koehnke, J. Am. Chem. Soc., 2017, 139, 18158–18161 CrossRef CAS.
- C. J. Schwalen, G. A. Hudson, S. Kosol, N. Mahanta, G. L. Challis and D. A. Mitchell, J. Am. Chem. Soc., 2017, 139, 18154–18157 CrossRef CAS.
- D. Ghilarov, C. E. M. Stevenson, D. Y. Travin, J. Piskunova, M. Serebryakova, A. Maxwell, D. M. Lawson and K. Severinov, Mol. Cell, 2019, 73, 749–762.e5 CrossRef CAS.
- D. A. Mitchell, S. W. Lee, M. A. Pence, A. L. Markley, J. D. Limm, V. Nizet and J. E. Dixon, J. Biol. Chem., 2009, 284, 13004–13012 CrossRef CAS.
- K. L. Dunbar, J. I. Tietz, C. L. Cox, B. J. Burkhart and D. A. Mitchell, J. Am. Chem. Soc., 2015, 137, 7672–7677 CrossRef CAS.
- J. O. Melby, X. Li and D. A. Mitchell, Biochemistry, 2014, 53, 413–422 CrossRef CAS.
- Y. Hayakawa, K. Sasaki, H. Adachi, K. Furihata, K. Nagai and K. Shin-ya, J. Antibiot., 2006, 59, 1–5 CrossRef CAS.
- M. Izawa, S. Nagamine, H. Aoki and Y. Hayakawa, J. Gen. Appl. Microbiol., 2018, 64, 50–53 CrossRef CAS.
- M. Izawa, T. Kawasaki and Y. Hayakawa, Appl. Environ. Microbiol., 2013, 79, 7110–7113 CrossRef CAS.
- J. Lu, J. Li, Y. Wu, X. Fang, J. Zhu and H. Wang, Org. Lett., 2019, 21, 4676–4679 CrossRef CAS.
- D. D. Nayak, N. Mahanta, D. A. Mitchell and W. W. Metcalf, eLife, 2017, 6, e29218 CrossRef.
- N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 142–163 CrossRef CAS.
- C. J. Schwalen, G. A. Hudson, B. Kille and D. A. Mitchell, J. Am. Chem. Soc., 2018, 140, 9494–9501 CrossRef CAS.
- N. Leikoski, L. Liu, J. Jokela, M. Wahlsten, M. Gugger, A. Calteau, P. Permi, C. A. Kerfeld, K. Sivonen and D. P. Fewer, Chem. Biol., 2013, 20, 1033–1043 CrossRef CAS.
- J. Martins and V. Vasconcelos, Mar. Drugs, 2015, 13, 6910–6946 CrossRef CAS.
- W. Gu, S. H. Dong, S. Sarkar, S. K. Nair and E. W. Schmidt, Methods Enzymol., 2018, 604, 113–163 CAS.
- V. Agarwal, E. Pierce, J. McIntosh, E. W. Schmidt and S. K. Nair, Chem. Biol., 2012, 19, 1411–1422 CrossRef CAS.
- W. E. Houssen, J. Koehnke, D. Zollman, J. Vendome, A. Raab, M. C. M. Smith, J. H. Naismith and M. Jaspars, ChemBioChem, 2012, 13, 2683–2689 CrossRef CAS.
- L. Huo, S. Rachid, M. Stadler, S. C. Wenzel and R. Müller, Chem. Biol., 2012, 19, 1278–1287 CrossRef CAS.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 3522 RSC.
- Y. Hou, Ma. D. B. Tianero, J. C. Kwan, T. P. Wyche, C. R. Michel, G. A. Ellis, E. Vazquez-Rivera, D. R. Braun, W. E. Rose, E. W. Schmidt and T. S. Bugni, Org. Lett., 2012, 14, 5050–5053 CrossRef CAS.
- W. J. K. Crone, F. J. Leeper and A. W. Truman, Chem. Sci., 2012, 3, 3516 RSC.
- G. Mann, L. Huo, S. Adam, B. Nardone, J. Vendome, N. J. Westwood, R. Müller and J. Koehnke, ChemBioChem, 2016, 17, 2286–2292 CrossRef CAS.
- W. J. K. Crone, N. M. Vior, J. Santos-Aberturas, L. G. Schmitz, F. J. Leeper and A. W. Truman, Angew. Chem., Int. Ed., 2016, 55, 9639–9643 CrossRef CAS.
- A. Sikandar, L. Franz, S. Adam, J. Santos-Aberturas, L. Horbal, A. Luzhetskyy, A. W. Truman, O. V. Kalinina and J. Koehnke, Nat. Chem. Biol., 2020, 16, 1013–1018 CrossRef.
- A. Sikandar, L. Franz, O. Melse, I. Antes and J. Koehnke, J. Am. Chem. Soc., 2019, 141, 9748–9752 CrossRef CAS.
- D. Y. Travin, M. Metelev, M. Serebryakova, E. S. Komarova, I. A. Osterman, D. Ghilarov and K. Severinov, J. Am. Chem. Soc., 2018, 140, 5625–5633 CrossRef CAS.
- J. D. Hegemann, M. Zimmermann, S. Zhu, H. Steuber, K. Harms, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2014, 53, 2230–2234 CrossRef CAS.
- J. D. Hegemann, C. D. Fage, S. Zhu, K. Harms, F. S. Di Leva, E. Novellino, L. Marinelli and M. A. Marahiel, Mol. BioSyst., 2016, 12, 1106–1109 RSC.
- C. D. Allen, M. Y. Chen, A. Y. Trick, D. T. Le, A. L. Ferguson and A. J. Link, ACS Chem. Biol., 2016, 11, 3043–3051 CrossRef CAS.
- J. D. Koos and A. J. Link, J. Am. Chem. Soc., 2019, 141, 928–935 CrossRef CAS.
- M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS.
- Y. Li, S. Zirah and S. Rebuffat, Lasso Peptides. Bacterial Strategies to Make and Maintain Bioactive Entangled Scaffolds, Springer-Verlag, New-York, 2015 Search PubMed.
- J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS.
- M. O. Maksimov and A. J. Link, J. Am. Chem. Soc., 2013, 135, 12038–12047 CrossRef CAS.
- K.-P. Yan, Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, ChemBioChem, 2012, 13, 1046–1052 CrossRef CAS.
- S. Zhu, C. D. Fage, J. D. Hegemann, A. Mielcarek, D. Yan, U. Linne and M. A. Marahiel, Sci. Rep., 2016, 6, 35604 CrossRef CAS.
- A. J. DiCaprio, A. Firouzbakht, G. A. Hudson and D. A. Mitchell, J. Am. Chem. Soc., 2019, 141, 290–297 CrossRef CAS.
- W. L. Cheung, M. Y. Chen, M. O. Maksimov and A. J. Link, ACS Cent. Sci., 2016, 2, 702–709 CrossRef CAS.
- T. Sumida, S. Dubiley, B. Wilcox, K. Severinov and S. Tagami, ACS Chem. Biol., 2019, 14, 1619–1627 CrossRef CAS.
- J. R. Chekan, C. Ongpipattanakul and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 24049–24055 CrossRef CAS.
- S. Zhu, J. D. Hegemann, C. D. Fage, M. Zimmermann, X. Xie, U. Linne and M. A. Marahiel, J. Biol. Chem., 2016, 291, 13662–13678 CrossRef CAS.
- C. Zong, W. L. Cheung-Lee, H. E. Elashal, M. Raj and A. J. Link, Chem. Commun., 2018, 54, 1339–1342 RSC.
- C. Zhang and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 890–894 CrossRef CAS.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 210–222 CrossRef CAS.
- M. Zimmermann, J. D. Hegemann, X. Xie and M. A. Marahiel, Chem. Biol., 2013, 20, 558–569 CrossRef CAS.
- Y. Li, R. Ducasse, S. Zirah, A. Blond, C. Goulard, E. Lescop, C. Giraud, A. Hartke, E. Guittet, J. L. Pernodet and S. Rebuffat, ACS Chem. Biol., 2015, 10, 2641–2649 CrossRef CAS.
- M. Zimmermann, J. D. Hegemann, X. Xie and M. A. Marahiel, Chem. Sci., 2014, 5, 4032–4043 RSC.
- J. Mevaere, C. Goulard, O. Schneider, O. N. Sekurova, H. Ma, S. Zirah, C. Afonso, S. Rebuffat, S. B. Zotchev and Y. Li, Sci. Rep., 2018, 8, 8232 CrossRef.
- M. Metelev, J. I. Tietz, J. O. Melby, P. M. Blair, L. Zhu, I. Livnat, K. Severinov and D. A. Mitchell, Chem. Biol., 2015, 22, 241–250 CrossRef CAS.
- S. Kodani, H. Hemmi, Y. Miyake, I. Kaweewan and H. Nakagawa, J. Ind. Microbiol. Biotechnol., 2018, 45, 983–992 CrossRef CAS.
- L. Flühe, T. A. Knappe, M. J. Gattner, A. Schäfer, O. Burghaus, U. Linne and M. A. Marahiel, Nat. Chem. Biol., 2012, 8, 350–357 CrossRef.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS.
- M. R. Bauerle, E. L. Schwalm and S. J. Booker, J. Biol. Chem., 2015, 290, 3995–4002 CrossRef CAS.
- A. P. Mehta, S. H. Abdelwahed, N. Mahanta, D. Fedoseyenko, B. Philmus, L. E. Cooper, Y. Liu, I. Jhulki, S. E. Ealick and T. P. Begley, J. Biol. Chem., 2015, 290, 3980–3986 CrossRef CAS.
- L. Flühe, O. Burghaus, B. M. Wieckowski, T. W. Giessen, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2013, 135, 959–962 CrossRef.
- O. Berteau, A. Guillot, A. Benjdia and S. Rabot, J. Biol. Chem., 2006, 281, 22464–22470 CrossRef CAS.
- T. A. J. Grell, W. M. Kincannon, N. A. Bruender, E. J. Blaesi, C. Krebs, V. Bandarian and C. L. Drennan, J. Biol. Chem., 2018, 293, 17349–17361 CrossRef CAS.
- A. Benjdia, A. Guillot, B. Lefranc, H. Vaudry, J. Leprince and O. Berteau, Chem. Commun., 2016, 52, 6249–6252 RSC.
- N. A. Bruender and V. Bandarian, Biochemistry, 2016, 55, 4131–4134 CrossRef CAS.
- W. M. Kincannon, N. A. Bruender and V. Bandarian, Biochemistry, 2018, 57, 4816–4823 CrossRef CAS.
- B. M. Wieckowski, J. D. Hegemann, A. Mielcarek, L. Boss, O. Burghaus and M. A. Marahiel, FEBS Lett., 2015, 589, 1802–1806 CrossRef CAS.
- T. L. Grove, P. M. Himes, S. Hwang, H. Yumerefendi, J. B. Bonanno, B. Kuhlman, S. C. Almo and A. A. Bowers, J. Am. Chem. Soc., 2017, 139, 11734–11744 CrossRef CAS.
- N. A. Bruender, J. Wilcoxen, R. D. Britt and V. Bandarian, Biochemistry, 2016, 55, 2122–2134 CrossRef CAS.
- T. Nakai, H. Ito, K. Kobayashi, Y. Takahashi, H. Hori, M. Tsubaki, K. Tanizawa and T. Okajima, J. Biol. Chem., 2015, 290, 11144–11166 CrossRef CAS.
- T. W. Precord, N. Mahanta and D. A. Mitchell, ACS Chem. Biol., 2019, 14, 1981–1989 CrossRef CAS.
- T. L. Grove, P. M. Himes, S. Hwang, H. Yumerefendi, J. B. Bonanno, B. Kuhlman, S. C. Almo and A. A. Bowers, J. Am. Chem. Soc., 2017, 139, 11734–11744 CrossRef CAS.
- K. R. Schramma, L. B. Bushin and M. R. Seyedsayamdost, Nat. Chem., 2015, 7, 431–437 CrossRef CAS.
- M. Ibrahim, A. Guillot, F. Wessner, F. Algaron, C. Besset, P. Courtin, R. Gardan and V. Monnet, J. Bacteriol., 2007, 189, 8844–8854 CrossRef CAS.
- B. Fleuchot, C. Gitton, A. Guillot, J. Vidic, P. Nicolas, C. Besset, L. Fontaine, P. Hols, N. Leblond-Bourget, V. Monnet and R. Gardan, Mol. Microbiol., 2011, 80, 1102–1119 CrossRef CAS.
- N. A. Isley, Y. Endo, Z.-C. Wu, B. C. Covington, L. B. Bushin, M. R. Seyedsayamdost and D. L. Boger, J. Am. Chem. Soc., 2019, 141, 17361–17369 CrossRef CAS.
- K. R. Schramma and M. R. Seyedsayamdost, ACS Chem. Biol., 2017, 12, 922–927 CrossRef CAS.
- K. M. Davis, K. R. Schramma, W. A. Hansen, J. P. Bacik, S. D. Khare, M. R. Seyedsayamdost and N. Ando, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10420–10425 CrossRef CAS.
- K. R. Schramma, C. C. Forneris, A. Caruso and M. R. Seyedsayamdost, Biochemistry, 2018, 57, 461–468 CrossRef CAS.
- D. H. Haft, M. K. Basu and D. A. Mitchell, BMC Biol., 2010, 8, 70 CrossRef.
- B. I. Morinaka, A. L. Vagstad, M. J. Helf, M. Gugger, C. Kegler, M. F. Freeman, H. B. Bode and J. Piel, Angew. Chem., Int. Ed. Engl., 2014, 53, 8503–8507 CrossRef CAS.
- M. F. Freeman, C. Gurgui, M. J. Helf, B. I. Morinaka, A. R. Uria, N. J. Oldham, H. G. Sahl, S. Matsunaga and J. Piel, Science, 2012, 338, 387–390 CrossRef CAS.
- T. Hamada, S. Matsunaga, G. Yano and N. Fusetani, J. Am. Chem. Soc., 2005, 127, 110–118 CrossRef CAS.
- T. Hamada, S. Matsunaga, M. Fujiwara, K. Fujita, H. Hirota, R. Schmucki, P. Guntert and N. Fusetani, J. Am. Chem. Soc., 2010, 132, 12941–12945 CrossRef CAS.
- A. Renevey and S. Riniker, Eur. Biophys. J., 2017, 46, 363–374 CrossRef CAS.
- Q. Zhang, W. A. van der Donk and W. Liu, Acc. Chem. Res., 2012, 45, 555–564 CrossRef CAS.
- A. Bhushan, P. J. Egli, E. E. Peters, M. F. Freeman and J. Piel, Nat. Chem., 2019, 11, 931–939 CrossRef CAS.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9508–9518 CrossRef CAS.
- N. Bosch, B. Mariana, U. Greczmiel, B. Morinaka, M. Gugger, A. Oxenius, A. L. Vagstad and J. Piel, Angew. Chem., Int. Ed. Engl., 2020, 59, 11763–11768 CrossRef.
- B. G. Butcher, Y. P. Lin and J. D. Helmann, J. Bacteriol., 2007, 189, 8616–8625 CrossRef CAS.
- N. Ziemert, K. Ishida, A. Liaimer, C. Hertweck and E. Dittmann, Angew. Chem., Int. Ed., 2008, 47, 7756–7759 CrossRef CAS.
- B. Philmus, G. Christiansen, W. Y. Yoshida and T. K. Hemscheidt, ChemBioChem, 2008, 9, 3066–3073 CrossRef CAS.
- H. Lee, Y. Park and S. Kim, Biochemistry, 2017, 56, 4927–4930 CrossRef CAS.
- H. Roh, Y. Han, H. Lee and S. Kim, ChemBioChem, 2019, 20, 1051–1059 CrossRef CAS.
- C. Lee, H. Lee, J.-U. Park and S. Kim, Biochemistry, 2020, 59, 285–289 CrossRef CAS.
- H. Lee, M. Choi, J.-U. Park, H. Roh and S. Kim, J. Am. Chem. Soc., 2020, 142, 3013–3023 CrossRef CAS.
- K. Li, H. L. Condurso, G. Li, Y. Ding and S. D. Bruner, Nat. Chem. Biol., 2016, 12, 973–979 CrossRef CAS.
- B. Philmus, J. P. Guerrette and T. K. Hemscheidt, ACS Chem. Biol., 2009, 4, 429–434 CrossRef CAS.
- A. R. Weiz, K. Ishida, K. Makower, N. Ziemert, C. Hertweck and E. Dittmann, Chem. Biol., 2011, 18, 1413–1421 CrossRef CAS.
- E. Reyna-González, B. Schmid, D. Petras, R. D. Süssmuth and E. Dittmann, Angew. Chem., Int. Ed., 2016, 55, 9398–9401 CrossRef.
- M. N. Ahmed, E. Reyna-González, B. Schmid, V. Wiebach, R. D. Süssmuth, E. Dittmann and D. P. Fewer, ACS Chem. Biol., 2017, 12, 1538–1546 CrossRef CAS.
- Y. Zhang, K. Li, G. Yang, J. L. McBride, S. D. Bruner and Y. Ding, Nat. Commun., 2018, 9, 1780 CrossRef.
- M. R. Quijano, C. Zach, F. S. Miller, A. R. Lee, A. S. Imani, M. Künzler and M. F. Freeman, J. Am. Chem. Soc., 2019, 141, 9637–9644 CrossRef CAS.
- H. Song, J. R. Fahrig-Kamarauskaitè, E. Matabaro, H. Kaspar, S. L. Shirran, C. Zach, A. Pace, B.-A. Stefanov, J. H. Naismith and M. Künzler, ACS Chem. Biol., 2020, 15, 1901–1912 CrossRef CAS.
- H. Song, N. S. van der Velden, S. L. Shiran, P. Bleiziffer, C. Zach, R. Sieber, A. S. Imani, F. Krausbeck, M. Aebi, M. F. Freeman, S. Riniker, M. Künzler and J. H. Naismith, Sci. Adv., 2018, 4, eaat2720 CrossRef CAS.
- O. Sterner, W. Etzel, A. Mayer and H. Anke, Nat. Prod. Lett., 1997, 10, 33–38 CrossRef CAS.
- A. Mayer, H. Anke and O. Sterner, Nat. Prod. Lett., 1997, 10, 25–32 CrossRef CAS.
- C. Ongpipattanakul and S. K. Nair, ACS Chem. Biol., 2018, 13, 2989–2999 CrossRef CAS.
- M. Umemura, H. Koike, N. Nagano, T. Ishii, J. Kawano, N. Yamane, I. Kozone, K. Horimoto, K. Shin-ya, K. Asai, J. Yu, J. W. Bennett and M. Machida, PLoS One, 2013, 8, e84028 CrossRef.
- A. Yoshimi, M. Umemura, N. Nagano, H. Koike, M. Machida and K. Abe, AMB Express, 2016, 6, 9 CrossRef.
- M. Umemura, N. Nagano, H. Koike, J. Kawano, T. Ishii, Y. Miyamura, M. Kikuchi, K. Tamano, J. Yu, K. Shin-ya and M. Machida, Fungal Genet. Biol., 2014, 68, 23–30 CrossRef CAS.
- T. Tsukui, N. Nagano, M. Umemura, T. Kumagai, G. Terai, M. Machida and K. Asai, Bioinformatics, 2015, 31, 981–985 CrossRef CAS.
- G. A. Vignolle, R. L. Mach, A. R. Mach-Aigner and C. Derntl, BMC Genomics, 2020, 21, 258 CrossRef CAS.
- Y. Ye, A. Minami, Y. Igarashi, M. Izumikawa, M. Umemura, N. Nagano, M. Machida, T. Kawahara, K. Shin-ya, K. Gomi and H. Oikawa, Angew. Chem., Int. Ed., 2016, 55, 8072–8075 CrossRef CAS.
- D. C. Gournelis, G. G. Laskaris and R. Verpoorte, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Falk, G. W. Kirby, R. E. Moore and C. H. Tamm, Springer-Verlag/Wein, New York, 1998, vol. 75, pp. 1–179 Search PubMed.
- R. D. Johnson, G. A. Lane, A. Koulman, M. Cao, K. Fraser, D. J. Fleetwood, C. R. Voisey, J. M. Dyer, J. Pratt, M. Christensen, W. R. Simpson, G. T. Bryan and L. J. Johnson, Fungal Genet. Biol., 2015, 85, 14–24 CrossRef CAS.
- H. Luo, S. Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610–1617 CrossRef CAS.
- R. M. Sgambelluri, M. O. Smith and J. D. Walton, ACS Synth. Biol., 2018, 7, 145–152 CrossRef CAS.
- C. M. Czekster and J. H. Naismith, Biochemistry, 2017, 56, 2086–2095 CrossRef CAS.
- C. M. Czekster, H. Ludewig, S. A. McMahon and J. H. Naismith, Nat. Commun., 2017, 8, 1045 CrossRef.
- C. Gabrielsen, D. A. Brede, I. F. Nes and D. B. Diep, Appl. Environ. Microbiol., 2014, 80, 6854–6862 CrossRef.
- R. H. Perez, T. Zendo and K. Sonomoto, Front. Microbiol., 2018, 9, 2085 CrossRef.
- J. Koehnke, A. F. Bent, W. E. Houssen, G. Mann, M. Jaspars and J. H. Naismith, Curr. Opin. Struct. Biol., 2014, 29, 112–121 CrossRef CAS.
- G. K. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS.
- K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Nat. Commun., 2015, 6, 10199 CrossRef CAS.
- C. N. Alexandru-Crivac, C. Umeobika, N. Leikoski, J. Jokela, K. A. Rickaby, A. M. Grilo, P. Sjo, A. T. Plowright, M. Idress, E. Siebs, A. Nneoyi-Egbe, M. Wahlsten, K. Sivonen, M. Jaspars, L. Trembleau, D. P. Fewer and W. E. Houssen, Chem. Commun., 2017, 53, 10656–10659 RSC.
- K. Bernath-Levin, C. Nelson, A. G. Elliott, A. S. Jayasena, A. H. Millar, D. J. Craik and J. S. Mylne, Chem. Biol., 2015, 22, 571–582 CrossRef CAS.
- C. J. Barber, P. T. Pujara, D. W. Reed, S. Chiwocha, H. Zhang and P. S. Covello, J. Biol. Chem., 2013, 288, 12500–12510 CrossRef CAS.
- X. Hemu, A. El Sahili, S. Hu, K. Wong, Y. Chen, Y. H. Wong, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C.-F. Liu, J. Lescar and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11737–11746 CAS.
- A. M. James, J. Haywood, J. Leroux, K. Ignasiak, A. G. Elliott, J. W. Schmidberger, M. F. Fisher, S. G. Nonis, R. Fenske, C. S. Bond and J. S. Mylne, Plant J., 2019, 98, 988–999 CAS.
- J. Lee, J. McIntosh, B. J. Hathaway and E. W. Schmidt, J. Am. Chem. Soc., 2009, 131, 2122–2124 CrossRef CAS.
- K. S. Harris, R. F. Guarino, R. S. Dissanayake, P. Quimbar, O. C. McCorkelle, S. Poon, Q. Kaas, T. Durek, E. K. Gilding, M. A. Jackson, D. J. Craik, N. L. van der Weerden, R. F. Anders and M. A. Anderson, Sci. Rep., 2019, 9, 10820 CrossRef.
- J. R. Chekan, P. Estrada, P. S. Covello and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6551–6556 CrossRef CAS.
- H. Ludewig, C. M. Czekster, E. Oueis, E. S. Munday, M. Arshad, S. A. Synowsky, A. F. Bent and J. H. Naismith, ACS Chem. Biol., 2018, 13, 801–811 CrossRef CAS.
- C. Gabrielsen, D. A. Brede, Z. Salehian, I. F. Nes and D. B. Diep, J. Bacteriol., 2014, 196, 911–919 CrossRef.
- R. H. Perez, H. Sugino, N. Ishibashi, T. Zendo, P. Wilaipun, V. Leelawatcharamas, J. Nakayama and K. Sonomoto, Microbiology, 2017, 163, 431–441 CrossRef CAS.
- H. Chen, C. C. Tseng, B. K. Hubbard and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14901–14906 CrossRef CAS.
- J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS.
- I. Barr, J. A. Latham, A. T. Iavarone, T. Chantarojsiri, J. D. Hwang and J. P. Klinman, J. Biol. Chem., 2016, 291, 8877–8884 CrossRef CAS.
- J. A. Latham, A. T. Iavarone, I. Barr, P. V. Juthani and J. P. Klinman, J. Biol. Chem., 2015, 290, 12908–12918 CrossRef CAS.
- A. M. Martins, J. A. Latham, P. J. Martel, I. Barr, A. T. Iavarone and J. P. Klinman, J. Biol. Chem., 2019, 294, 15025–15036 CrossRef CAS.
- Q. Wei, T. Ran, C. Ma, J. He, D. Xu and W. Wang, J. Biol. Chem., 2016, 291, 15575–15587 CrossRef CAS.
- E. M. Koehn, J. A. Latham, T. Armand, R. L. Evans 3rd, X. Tu, C. M. Wilmot, A. T. Iavarone and J. P. Klinman, J. Am. Chem. Soc., 2019, 141, 4398–4405 CrossRef CAS.
- X. Tu, J. A. Latham, V. J. Klema, R. L. Evans 3rd, C. Li, J. P. Klinman and C. M. Wilmot, J. Biol. Inorg. Chem., 2017, 22, 1089–1097 CrossRef CAS.
- F. Bonnot, A. T. Iavarone and J. P. Klinman, Biochemistry, 2013, 52, 4667–4675 CrossRef CAS.
- D. H. Haft, P. G. Pierce, S. J. Mayclin, A. Sullivan, A. S. Gardberg, J. Abendroth, D. W. Begley, I. Q. Phan, B. L. Staker, P. J. Myler, V. M. Marathias, D. D. Lorimer and T. E. Edwards, Sci. Rep., 2017, 7, 41074 CrossRef CAS.
- B. Khaliullin, P. Aggarwal, M. Bubas, G. R. Eaton, S. S. Eaton and J. A. Latham, FEBS Lett., 2016, 590, 2538–2548 CrossRef CAS.
- N. A. Bruender and V. Bandarian, Biochemistry, 2016, 55, 2813–2816 CrossRef CAS.
- B. Khaliullin, R. Ayikpoe, M. Tuttle and J. A. Latham, J. Biol. Chem., 2017, 292, 13022–13033 CrossRef CAS.
- R. Ayikpoe, J. Salazar, B. Majestic and J. A. Latham, Biochemistry, 2018, 57, 5379–5383 CrossRef CAS.
- L. Peña-Ortiz, A. P. Graça, H. Guo, D. Braga, T. G. Köllner, L. Regestein, C. Beemelmanns and G. Lackner, bioRxiv, 2019, 821413 Search PubMed.
- G. Krishnamoorthy, P. Kaiser, L. Lozza, K. Hahnke, H.-J. Mollenkopf and S. H. E. Kaufmann, mBio, 2019, 10, e00190 CrossRef CAS.
- A. A. Dubey and V. Jain, Biochem. Biophys. Res. Commun., 2019, 516, 1073–1077 CrossRef CAS.
- M. Jin, S. A. Wright, S. V. Beer and J. Clardy, Angew. Chem., Int. Ed., 2003, 42, 2902–2905 CrossRef CAS.
- M. Jin, L. Liu, S. A. Wright, S. V. Beer and J. Clardy, Angew. Chem., Int. Ed., 2003, 42, 2898–2901 CrossRef CAS.
- S. V. Ghodge, K. A. Biernat, S. J. Bassett, M. R. Redinbo and A. A. Bowers, J. Am. Chem. Soc., 2016, 138, 5487–5490 CrossRef CAS.
- S. R. Fleming, P. M. Himes, S. V. Ghodge, Y. Goto, H. Suga and A. A. Bowers, J. Am. Chem. Soc., 2020, 142, 5024–5028 CrossRef CAS.
- G. E. Kenney and A. C. Rosenzweig, BMC Biol., 2013, 11, 17 CrossRef CAS.
- G. E. Kenney, L. M. K. Dassama, M. E. Pandelia, A. S. Gizzi, R. J. Martinie, P. Gao, C. J. DeHart, L. F. Schachner, O. S. Skinner, S. Y. Ro, X. Zhu, M. Sadek, P. M. Thomas, S. C. Almo, J. M. Bollinger Jr, C. Krebs, N. L. Kelleher and A. C. Rosenzweig, Science, 2018, 359, 1411–1416 CrossRef CAS.
- N. Nagano, C. A. Orengo and J. M. Thornton, J. Mol. Biol., 2002, 321, 741–765 CrossRef CAS.
- Z. Zhang and W. A. van der Donk, J. Am. Chem. Soc., 2019, 141, 19625–19633 CrossRef CAS.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS.
- A. Miyanaga, J. E. Janso, L. McDonald, M. He, H. Liu, L. Barbieri, A. S. Eustaquio, E. N. Fielding, G. T. Carter, P. R. Jensen, X. Feng, M. Leighton, F. E. Koehn and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 13311–13313 CrossRef CAS.
- G. E. Norris and M. L. Patchett, Curr. Opin. Struct. Biol., 2016, 40, 112–119 CrossRef CAS.
- H. Wang, T. J. Oman, R. Zhang, C. V. Garcia De Gonzalo, Q. Zhang and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 84–87 CrossRef CAS.
- C. V. Garcia De Gonzalo, L. Zhu, T. J. Oman and W. A. van der Donk, ACS Chem. Biol., 2014, 9, 796–801 CrossRef CAS.
- S. Biswas, C. V. Garcia De Gonzalo, L. M. Repka and W. A. van der Donk, ACS Chem. Biol., 2017, 12, 2965–2969 CrossRef CAS.
- Z. Amso, S. W. Bisset, S. H. Yang, P. W. R. Harris, T. H. Wright, C. D. Navo, M. L. Patchett, G. E. Norris and M. A. Brimble, Chem. Sci., 2018, 9, 1686–1691 RSC.
- S. W. Bisset, S. H. Yang, Z. Amso, P. W. R. Harris, M. L. Patchett, M. A. Brimble and G. E. Norris, ACS Chem. Biol., 2018, 13, 1270–1278 CrossRef CAS.
- C. Wu, S. Biswas, C. V. Garcia De Gonzalo and W. A. van der Donk, ACS Infect. Dis., 2019, 5, 454–459 CrossRef CAS.
- R. Nagar and A. Rao, Methods in molecular biology, Clifton, N.J., 2019, vol. 1954, pp. 279–296 Search PubMed.
- H. Ren, S. Biswas, S. Ho, W. A. van der Donk and H. Zhao, ACS Chem. Biol., 2018, 13, 2966–2972 CrossRef CAS.
- A. Kaunietis, A. Buivydas, D. J. Citavicius and O. P. Kuipers, Nat. Commun., 2019, 10, 1115 CrossRef.
- K. Palaniappan, I. A. Chen, K. Chu, A. Ratner, R. Seshadri, N. C. Kyrpides, N. N. Ivanova and N. J. Mouncey, Nucleic Acids Res., 2020, 48, D422–D430 CAS.
- R. L. Evans 3rd, J. A. Latham, Y. Xia, J. P. Klinman and C. M. Wilmot, Biochemistry, 2017, 56, 2735–2746 CrossRef.
- T. Y. Tsai, C. Y. Yang, H. L. Shih, A. H. Wang and S. H. Chou, Proteins, 2009, 76, 1042–1048 CrossRef CAS.
- H. Martin-Gómez, U. Linne, F. Albericio, J. Tulla-Puche and J. D. Hegemann, J. Nat. Prod., 2018, 81, 2050–2056 CrossRef.
- J. D. Hegemann, C. J. Schwalen, D. A. Mitchell and W. A. van der Donk, Chem. Commun., 2018, 54, 9007–9010 RSC.
- D. Ghilarov, C. E. M. Stevenson, D. Y. Travin, J. Piskunova, M. Serebryakova, A. Maxwell, D. M. Lawson and K. Severinov, Mol. Cell, 2019, 73, 749–762.e5 CrossRef CAS.
- S. W. Fuchs, G. Lackner, B. I. Morinaka, Y. Morishita, T. Asai, S. Riniker and J. Piel, Angew. Chem., Int. Ed. Engl., 2016, 55, 12330–12333 CrossRef CAS.
- K. M. Davis, K. R. Schramma, W. A. Hansen, J. P. Bacik, S. D. Khare, M. R. Seyedsayamdost and N. Ando, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10420–10425 CrossRef CAS.
- I. Zukher, M. Pavlov, D. Tsibulskaya, A. Kulikovsky, T. Zyubko, D. Bikmetov, M. Serebryakova, S. K. Nair, M. Ehrenberg, S. Dubiley and K. Severinov, mBio, 2019, 10, e00768 CrossRef CAS.
- S. H. Dong, A. Kulikovsky, I. Zukher, P. Estrada, S. Dubiley, K. Severinov and S. K. Nair, Chem. Sci., 2019, 10, 2391–2395 RSC.
- D. Tsibulskaya, O. Mokina, A. Kulikovsky, J. Piskunova, K. Severinov, M. Serebryakova and S. Dubiley, J. Am. Chem. Soc., 2017, 139, 16178–16187 CrossRef CAS.
- D. Sardar, E. Pierce, J. A. McIntosh and E. W. Schmidt, ACS Synth. Biol., 2015, 4, 167–176 CrossRef CAS.
- D. Sardar, Z. Lin and E. W. Schmidt, Chem. Biol., 2015, 22, 907–916 CrossRef CAS.
- W. Gu, D. Sardar, E. Pierce and E. W. Schmidt, J. Am. Chem. Soc., 2018, 140, 16213–16221 CrossRef CAS.
- D. D. Nayak, A. Liu, N. Agrawal, R. Rodriguez-Carerro, S. H. Dong, D. A. Mitchell, S. K. Nair and W. W. Metcalf, PLoS Biol., 2020, 18, e3000507 CrossRef CAS.
- B. Li, D. Sher, L. Kelly, Y. Shi, K. Huang, P. J. Knerr, I. Joewono, D. Rusch, S. W. Chisholm and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10430–10435 CrossRef CAS.
- D. Y. Lin, S. Huang and J. Chen, Nature, 2015, 523, 425–430 CrossRef CAS.
- V. Kieuvongngam, P. D. B. Olinares, A. Palillo, M. L. Oldham, B. T. Chait and J. Chen, eLife, 2020, 9, e51492 CrossRef CAS.
- L. Huo, X. Zhao, J. Z. Acedo, P. Estrada, S. K. Nair and W. A. van der Donk, ChemBioChem, 2020, 21, 190–199 CrossRef CAS.
- S. C. Bobeica, S. H. Dong, L. Huo, N. Mazo, M. I. McLaughlin, G. Jimenéz-Osés, S. K. Nair and W. A. van der Donk, eLife, 2019, 8, e42305 CrossRef.
- M. J. Helf, M. F. Freeman and J. Piel, J. Ind. Microbiol. Biotechnol., 2019, 46, 551–563 CrossRef CAS.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS.
- M. Montalbán-López, J. Deng, A. J. van Heel and O. P. Kuipers, Front. Microbiol., 2018, 9, 160 CrossRef.
- M. A. Ortega, J. E. Velasquez, N. Garg, Q. Zhang, R. E. Joyce, S. K. Nair and W. A. van der Donk, ACS Chem. Biol., 2014, 9, 1718–1725 CrossRef CAS.
- W. Tang, S.-H. Dong, L. M. Repka, C. He, S. K. Nair and W. A. van der Donk, Chem. Sci., 2015, 6, 6270–6279 RSC.
- W. Tang, S. C. Bobeica, L. Wang and W. A. van der Donk, J. Ind. Microbiol. Biotechnol., 2019, 46, 537–549 CrossRef CAS.
- Y. Xu, X. Li, R. Li, S. Li, H. Ni, H. Wang, H. Xu, W. Zhou, P. E. Saris, W. Yang, M. Qiao and Z. Rao, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1499–1505 CrossRef CAS.
- M. L. Kuhn, P. Prachi, G. Minasov, L. Shuvalova, J. Ruan, I. Dubrovska, J. Winsor, M. Giraldi, M. Biagini, S. Liberatori, S. Savino, F. Bagnoli, W. F. Anderson and G. Grandi, FASEB J., 2014, 28, 1780–1793 CrossRef CAS.
- M. Lagedroste, S. H. J. Smits and L. Schmitt, Biochemistry, 2017, 56, 4005–4014 CrossRef.
- K. Schardon, M. Hohl, L. Graff, J. Pfannstiel, W. Schulze, A. Stintzi and A. Schaller, Science, 2016, 354, 1594–1597 CrossRef CAS.
- D. Ghilarov, M. Serebryakova, C. E. M. Stevenson, S. J. Hearnshaw, D. S. Volkov, A. Maxwell, D. M. Lawson and K. Severinov, Structure, 2017, 25, 1549–1561.e5 CrossRef CAS.
- S. Chen, B. Xu, E. Chen, J. Wang, J. Lu, S. Donadio, H. Ge and H. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2533–2538 CrossRef CAS.
- D. Dehm, J. Krumbholz, M. Baunach, V. Wiebach, K. Hinrichs, A. Guljamow, T. Tabuchi, H. Jenke-Kodama, R. D. Süssmuth and E. Dittmann, ACS Chem. Biol., 2019, 14, 1271–1279 CrossRef CAS.
- G. H. Völler, B. Krawczyk, P. Ensle and R. D. Süssmuth, J. Am. Chem. Soc., 2013, 135, 7426–7429 CrossRef.
- Q. Zheng, S. Wang, P. Duan, R. Liao, D. Chen and W. Liu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14318–14323 CrossRef CAS.
- J. A. McIntosh, M. S. Donia and E. W. Schmidt, Nat. Prod. Rep., 2009, 26, 537–559 RSC.
- M. D. Tianero, E. Pierce, S. Raghuraman, D. Sardar, J. A. McIntosh, J. R. Heemstra, Z. Schonrock, B. C. Covington, J. A. Maschek, J. E. Cox, B. O. Bachmann, B. M. Olivera, D. E. Ruffner and E. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1772–1777 CrossRef CAS.
- J. A. McIntosh, Z. Lin, M. D. Tianero and E. W. Schmidt, ACS Chem. Biol., 2013, 8, 877–883 CrossRef CAS.
- D. Sardar, M. D. Tianero and E. W. Schmidt, Methods Enzymol., 2016, 575, 1–20 CAS.
- A. Cubillos-Ruiz, J. W. Berta-Thompson, J. W. Becker, W. A. van der Donk and S. W. Chisholm, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5424–E5433 CrossRef CAS.
- B. I. Morinaka, M. Verest, M. F. Freeman, M. Gugger and J. Piel, Angew. Chem., Int. Ed., 2017, 56, 762–766 CrossRef CAS.
- A. Parent, A. Benjdia, A. Guillot, X. Kubiak, C. Balty, B. Lefranc, J. Leprince and O. Berteau, J. Am. Chem. Soc., 2018, 140, 2469–2477 CrossRef CAS.
- A. L. Vagstad, T. Kuranaga, S. Puntener, V. R. Pattabiraman, J. W. Bode and J. Piel, Angew. Chem., Int. Ed., 2019, 58, 2246–2250 CrossRef CAS.
- P. D. Cotter, P. M. O'Connor, L. A. Draper, E. M. Lawton, L. H. Deegan, C. Hill and R. P. Ross, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18584–18589 CrossRef CAS.
- X. Yang and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 12426–12429 CrossRef CAS.
- L. Huo and W. A. van der Donk, J. Am. Chem. Soc., 2016, 138, 5254–5257 CrossRef CAS.
- G. Helynck, C. Dubertret, J. F. Mayaux and J. Leboul, J. Antibiot., 1993, 46, 1756–1757 CrossRef CAS.
- D. Fréchet, J. D. Guitton, F. Herman, D. Faucher, G. Helynck, B. Monegier du Sorbier, J. P. Ridoux, E. James-Surcouf and M. Vuilhorgne, Biochemistry, 1994, 33, 42–50 CrossRef.
- K. Yano, S. Toki, S. Nakanishi, K. Ochiai, K. Ando, M. Yoshida, Y. Matsuda and M. Yamasaki, Bioorg. Med. Chem., 1996, 4, 115–120 CrossRef CAS.
- M. Tsunakawa, S. L. Hu, Y. Hoshino, D. J. Detlefson, S. E. Hill, T. Furumai, R. J. White, M. Nishio, K. Kawano, S. Yamamoto, Y. Fukagawa and T. Oki, J. Antibiot., 1995, 48, 433–434 CrossRef CAS.
- Z. Feng, Y. Ogasawara, S. Nomura and T. Dairi, ChemBioChem, 2018, 19, 2045–2048 CrossRef CAS.
- G. Kreil, J. Biol. Chem., 1994, 269, 10967–10970 CAS.
- E. D. Badding, T. L. Grove, L. K. Gadsby, J. W. LaMattina, A. K. Boal and S. J. Booker, J. Am. Chem. Soc., 2017, 139, 5896–5905 CrossRef CAS.
- W. Ding, W. Ji, Y. Wu, R. Wu, W. Q. Liu, T. Mo, J. Zhao, X. Ma, W. Zhang, P. Xu, Z. Deng, B. Tang, Y. Yu and Q. Zhang, Nat. Commun., 2017, 8, 437 CrossRef.
- Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218–226 RSC.
- Q. Zhang, Y. Li, D. Chen, Y. Yu, L. Duan, B. Shen and W. Liu, Nat. Chem. Biol., 2011, 7, 154–160 CrossRef CAS.
- Y. Nicolet, L. Zeppieri, P. Amara and J. C. Fontecilla-Camps, Angew. Chem., Int. Ed., 2014, 53, 11840–11844 CrossRef CAS.
- D. M. Bhandari, H. Xu, Y. Nicolet, J. C. Fontecilla-Camps and T. P. Begley, Biochemistry, 2015, 54, 4767–4769 CrossRef CAS.
- X. Ji, Y. Li, W. Ding and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 9021–9024 CrossRef CAS.
- D. M. Bhandari, D. Fedoseyenko and T. P. Begley, J. Am. Chem. Soc., 2016, 138, 16184–16187 CrossRef CAS.
- X. Ji, Y. Li, Y. Jia, W. Ding and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 3334–3337 CrossRef CAS.
- X. Ji, W.-Q. Liu, S. Yuan, Y. Yin, W. Ding and Q. Zhang, Chem. Commun., 2016, 52, 10555–10558 RSC.
- X. Ji, Y. Li, L. Xie, H. Lu, W. Ding and Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 11845–11848 CrossRef CAS.
- G. Sicoli, J.-M. Mouesca, L. Zeppieri, P. Amara, L. Martin, A.-L. Barra, J. C. Fontecilla-Camps, S. Gambarelli and Y. Nicolet, Science, 2016, 351, 1320–1323 CrossRef CAS.
- D. M. Bhandari, D. Fedoseyenko and T. P. Begley, J. Am. Chem. Soc., 2018, 140, 542–545 CrossRef CAS.
- R. I. Sayler, T. A. Stich, S. Joshi, N. Cooper, J. T. Shaw, T. P. Begley, D. J. Tantillo and R. D. Britt, ACS Cent. Sci., 2019, 5, 1777–1785 CrossRef CAS.
- J. M. Kuchenreuther, W. K. Myers, T. A. Stich, S. J. George, Y. Nejatyjahromy, J. R. Swartz and R. D. Britt, Science, 2013, 342, 472–475 CrossRef CAS.
- P. Amara, J. M. Mouesca, M. Bella, L. Martin, C. Saragaglia, S. Gambarelli and Y. Nicolet, J. Am. Chem. Soc., 2018, 140, 16661–16668 CrossRef CAS.
- C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153–164 RSC.
- W. L. Kelly, L. Pan and C. X. Li, J. Am. Chem. Soc., 2009, 131, 4327–4334 CrossRef CAS.
- S. Pierre, A. Guillot, A. Benjdia, C. Sandstrom, P. Langella and O. Berteau, Nat. Chem. Biol., 2012, 8, 957–959 CrossRef CAS.
- A. Benjdia, S. Pierre, C. Gherasim, A. Guillot, M. Carmona, P. Amara, R. Banerjee and O. Berteau, Nat. Commun., 2015, 6, 8377 CrossRef CAS.
- A. J. Blaszczyk, A. Silakov, B. Zhang, S. J. Maiocco, N. D. Lanz, W. L. Kelly, S. J. Elliott, C. Krebs and S. J. Booker, J. Am. Chem. Soc., 2016, 138, 3416–3426 CrossRef CAS.
- P. Zhou, D. O'Hagan, U. Mocek, Z. Zeng, L. D. Yuen, T. Frenzel, C. J. Unkefer, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1989, 111, 7274–7276 CrossRef CAS.
- A. J. Blaszczyk, B. Wang, A. Silakov, J. V. Ho and S. J. Booker, J. Biol. Chem., 2017, 292, 15456–15467 CrossRef CAS.
- A. Parent, A. Guillot, A. Benjdia, G. Chartier, J. Leprince and O. Berteau, J. Am. Chem. Soc., 2016, 138, 15515–15518 CrossRef CAS.
- J. P. Gomez-Escribano, L. Song, M. J. Bibb and G. L. Challis, Chem. Sci., 2012, 3, 3522–3525 RSC.
- N. Mahanta, Z. Zhang, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, J. Am. Chem. Soc., 2017, 139, 4310–4313 CrossRef CAS.
- A. Tocchetti, S. Maffioli, M. Iorio, S. Alt, E. Mazzei, C. Brunati, M. Sosio and S. Donadio, Chem. Biol., 2013, 20, 1067–1077 CrossRef CAS.
- Z. Zhang, N. Mahanta, G. A. Hudson, D. A. Mitchell and W. A. van der Donk, J. Am. Chem. Soc., 2017, 139, 18623–18631 CrossRef CAS.
- M. J. Helf, A. Jud and J. Piel, ChemBioChem, 2017, 18, 444–450 CrossRef CAS.
- K. Hotta, R. M. Keegan, S. Ranganathan, M. Fang, J. Bibby, M. D. Winn, M. Sato, M. Lian, K. Watanabe, D. J. Rigden and C. Y. Kim, Angew. Chem., Int. Ed. Engl., 2014, 53, 824–828 CrossRef CAS.
- A. H. Al-Mestarihi, G. Villamizar, J. Fernandez, O. E. Zolova, F. Lombo and S. Garneau-Tsodikova, J. Am. Chem. Soc., 2014, 136, 17350–17354 CrossRef CAS.
- K. J. Molohon, J. O. Melby, J. Lee, B. S. Evans, K. L. Dunbar, S. B. Bumpus, N. L. Kelleher and D. A. Mitchell, ACS Chem. Biol., 2011, 6, 1307–1313 CrossRef CAS.
- R. Scholz, K. J. Molohon, J. Nachtigall, J. Vater, A. L. Markley, R. D. Sussmuth, D. A. Mitchell and R. Borriss, J. Bacteriol., 2011, 193, 215–224 CrossRef CAS.
- B. Kalyon, S. E. Helaly, R. Scholz, J. Nachtigall, J. Vater, R. Borriss and R. D. Süssmuth, Org. Lett., 2011, 13, 2996–2999 CrossRef CAS.
- K. Komiyama, K. Otoguro, T. Segawa, K. Shiomi, H. Yang, Y. Takahashi, M. Hayashi, T. Otani and S. Omura, J. Antibiot., 1993, 46, 1666–1671 CrossRef CAS.
- J. Claesen and M. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16297–16302 CrossRef CAS.
- J. Lee, Y. Hao, P. M. Blair, J. O. Melby, V. Agarwal, B. J. Burkhart, S. K. Nair and D. A. Mitchell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12954–12959 CrossRef CAS.
- N. A. Piwowarska, S. Banala, H. S. Overkleeft and R. D. Sussmuth, Chem. Commun., 2013, 49, 10703–10705 RSC.
- A. Sharma, P. M. Blair and D. A. Mitchell, Org. Lett., 2013, 15, 5076–5079 CrossRef CAS.
- Q. Zhang and W. A. van der Donk, FEBS Lett., 2012, 586, 3391–3397 CrossRef CAS.
- T. E. Smith, C. D. Pond, E. Pierce, Z. P. Harmer, J. Kwan, M. M. Zachariah, M. K. Harper, T. P. Wyche, T. K. Matainaho, T. S. Bugni, L. R. Barrows, C. M. Ireland and E. W. Schmidt, Nat. Chem. Biol., 2018, 14, 179–185 CrossRef CAS.
- D. A. Widdick, H. M. Dodd, P. Barraille, J. White, T. H. Stein, K. F. Chater, M. J. Gasson and M. J. Bibb, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4316–4321 CrossRef CAS.
- A. Ökesli, L. E. Cooper, E. J. Fogle and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 13753–13760 CrossRef.
- Y. Yu, L. Duan, Q. Zhang, R. Liao, Y. Ding, H. Pan, E. Wendt-Pienkowski, G. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855–864 CrossRef CAS.
- J. W. LaMattina, B. Wang, E. D. Badding, L. K. Gadsby, T. L. Grove and S. J. Booker, J. Am. Chem. Soc., 2017, 139, 17438–17445 CrossRef CAS.
- B. Wang, J. W. LaMattina, S. L. Marshall and S. J. Booker, J. Am. Chem. Soc., 2019, 141, 5788–5797 CrossRef CAS.
- Y. Qiu, Y. Du, S. Wang, S. Zhou, Y. Guo and W. Liu, Org. Lett., 2019, 21, 1502–1505 CrossRef CAS.
- W. Ding, Y. Li, J. Zhao, X. Ji, T. Mo, H. Qianzhu, T. Tu, Z. Deng, Y. Yu, F. Chen and Q. Zhang, Angew. Chem., Int. Ed. Engl., 2017, 56, 3857–3861 CrossRef CAS.
- X. Ji, D. Mandalapu, J. Cheng, W. Ding and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6601–6604 CrossRef CAS.
- W. Ding, Y. Wu, X. Ji, H. Qianzhu, F. Chen, Z. Deng, Y. Yu and Q. Zhang, Chem. Commun., 2017, 53, 5235–5238 RSC.
- N. Leikoski, L. W. Liu, J. Jokela, M. Wahlsten, M. Gugger, A. Calteau, P. Permi, C. A. Kerfeld, K. Sivonen and D. P. Fewer, Chem. Biol., 2013, 20, 1033–1043 CrossRef CAS.
- D. Sardar, Y. Hao, Z. Lin, M. Morita, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2017, 139, 2884–2887 CrossRef CAS.
- E. Gavrish, C. S. Sit, S. Cao, O. Kandror, A. Spoering, A. Peoples, L. Ling, A. Fetterman, D. Hughes, A. Bissell, H. Torrey, T. Akopian, A. Mueller, S. Epstein, A. Goldberg, J. Clardy and K. Lewis, Chem. Biol., 2014, 21, 509–518 CrossRef CAS.
- Y. Su, M. Han, X. Meng, Y. Feng, S. Luo, C. Yu, G. Zheng and S. Zhu, Appl. Microbiol. Biotechnol., 2019, 103, 2649–2664 CrossRef CAS.
- A. J. Marsh, O. O'Sullivan, R. P. Ross, P. D. Cotter and C. Hill, BMC Genomics, 2010, 11, 679 CrossRef CAS.
- Z. F. Kanyo, L. R. Scolnick, D. E. Ash and D. W. Christianson, Nature, 1996, 383, 554–557 CrossRef CAS.
- S. Zhu, C. D. Fage, J. D. Hegemann, D. Yan and M. A. Marahiel, FEBS Lett., 2016, 590, 3323–3334 CrossRef CAS.
- T. Zyubko, M. Serebryakova, J. Andreeva, M. Metelev, G. Lippens, S. Dubiley and K. Severinov, Chem. Sci., 2019, 10, 9699–9707 RSC.
- Z. Lin, J. Ji, S. Zhou, F. Zhang, J. Wu, Y. Guo and W. Liu, J. Am. Chem. Soc., 2017, 139, 12105–12108 CrossRef CAS.
- Q. Zheng, S. Wang, R. Liao and W. Liu, ACS Chem. Biol., 2016, 11, 2673–2678 CrossRef CAS.
- W. Liu, Y. Xue, M. Ma, S. Wang, N. Liu and Y. Chen, ChemBioChem, 2013, 14, 1544–1547 CrossRef CAS.
- S. I. Maffioli, M. Iorio, M. Sosio, P. Monciardini, E. Gaspari and S. Donadio, J. Nat. Prod., 2014, 77, 79–84 CrossRef CAS.
- M. Sosio, G. Gallo, R. Pozzi, S. Serina, P. Monciardini, A. Bera, E. Stegmann and T. Weber, Genome Announcements, 2014, 2, e01198 CrossRef.
- N. Liu, L. Song, M. Liu, F. Shang, Z. Anderson, D. J. Fox, G. L. Challis and Y. Huang, Chem. Sci., 2016, 7, 482–488 RSC.
- T. Kupke, C. Kempter, G. Jung and F. Götz, J. Biol. Chem., 1995, 270, 11282–11289 CrossRef CAS.
- T. Mo, H. Yuan, F. Wang, S. Ma, J. Wang, T. Li, G. Liu, S. Yu, X. Tan, W. Ding and Q. Zhang, FEBS Lett., 2019, 593, 573–580 CrossRef CAS.
- J. Escano, A. Ravichandran, B. Salamat and L. Smith, Appl. Environ. Microbiol., 2017, 83, e00668 CrossRef CAS.
- M. A. Ortega, D. P. Cogan, S. Mukherjee, N. Garg, B. Li, G. N. Thibodeaux, S. I. Maffioli, S. Donadio, M. Sosio, J. Escano, L. Smith, S. K. Nair and W. A. van der Donk, ACS Chem. Biol., 2017, 12, 548–557 CrossRef CAS.
- J. C. Cruz, M. Iorio, P. Monciardini, M. Simone, C. Brunati, E. Gaspari, S. I. Maffioli, E. Wellington, M. Sosio and S. Donadio, J. Nat. Prod., 2015, 78, 2642–2647 CrossRef CAS.
- Y. Shi, A. Bueno and W. A. van der Donk, Chem. Commun., 2012, 48, 10966–10968 RSC.
- L. Huo, A. Ökesli, M. Zhao and W. A. van der Donk, Appl. Environ. Microbiol., 2017, 83, e02698 Search PubMed.
- L. An, D. P. Cogan, C. D. Navo, G. Jiménez-Osés, S. K. Nair and W. A. van der Donk, Nat. Chem. Biol., 2018, 14, 928–933 CrossRef CAS.
- Y. J. Park, G. E. Kenney, L. F. Schachner, N. L. Kelleher and A. C. Rosenzweig, Biochemistry, 2018, 57, 3515–3523 CrossRef CAS.
- Y. Ye, T. Ozaki, M. Umemura, C. Liu, A. Minami and H. Oikawa, Org. Biomol. Chem., 2019, 17, 39–43 RSC.
- J. A. McIntosh, M. S. Donia, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2011, 133, 13698–13705 CrossRef CAS.
- Y. Hao, E. Pierce, D. Roe, M. Morita, J. A. McIntosh, V. Agarwal, T. E. Cheatham, E. W. Schmidt and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14037–14042 CrossRef CAS.
- L. Dalponte, A. Parajuli, E. Younger, A. Mattila, J. Jokela, M. Wahlsten, N. Leikoski, K. Sivonen, S. A. Jarmusch, W. E. Houssen and D. P. Fewer, Biochemistry, 2018, 57, 6860–6867 CrossRef CAS.
- K. Iwasaki, A. Iwasaki, S. Sumimoto, T. Sano, Y. Hitomi, O. Ohno and K. Suenaga, Tetrahedron Lett., 2018, 59, 3806–3809 CrossRef CAS.
- M. Okada, T. Sugita, K. Akita, Y. Nakashima, T. Tian, C. Li, T. Mori and I. Abe, Org. Biomol. Chem., 2016, 14, 9639–9644 RSC.
- T. Sano and K. Kaya, Tetrahedron Lett., 1996, 37, 6873–6876 CrossRef CAS.
- A. Parajuli, D. H. Kwak, L. Dalponte, N. Leikoski, T. Galica, U. Umeobika, L. Trembleau, A. Bent, K. Sivonen, M. Wahlsten, H. Wang, E. Rizzi, G. De Bellis, J. Naismith, M. Jaspars, X. Liu, W. Houssen and D. P. Fewer, Angew. Chem., Int. Ed., 2016, 55, 3596–3599 CrossRef CAS.
- A. Nagatsu, H. Kajitani and J. Sakakibara, Tetrahedron Lett., 1995, 36, 4097–4100 CrossRef CAS.
- A. Mattila, R. M. Andsten, M. Jumppanen, M. Assante, J. Jokela, M. Wahlsten, K. M. Mikula, C. Sigindere, D. H. Kwak, M. Gugger, H. Koskela, K. Sivonen, X. Liu, J. Yli-Kauhaluoma, H. Iwai and D. P. Fewer, ACS Chem. Biol., 2019, 14, 2683–2690 CrossRef CAS.
- U. Metzger, C. Schall, G. Zocher, I. Unsold, E. Stec, S. M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS.
- M. Morita, Y. Hao, J. K. Jokela, D. Sardar, Z. Lin, K. Sivonen, S. K. Nair and E. W. Schmidt, J. Am. Chem. Soc., 2018, 140, 6044–6048 CrossRef CAS.
- P. Estrada, M. Morita, Y. Hao, E. W. Schmidt and S. K. Nair, J. Am. Chem. Soc., 2018, 140, 8124–8127 CrossRef CAS.
- M. Okada, I. Sato, S. J. Cho, H. Iwata, T. Nishio, D. Dubnau and Y. Sakagami, Nat. Chem. Biol., 2005, 1, 23–24 CrossRef CAS.
- K. Hirooka, S. Shioda and M. Okada, Biosci., Biotechnol., Biochem., 2020, 84, 347–357 CrossRef CAS.
- T. Sugita, M. Okada, Y. Nakashima, T. Tian and I. Abe, Chembiochem, 2018, 19, 1396–1399 CrossRef CAS.
- W. Liu, M. Ma, Y. Xue, N. Liu, S. Wang and Y. Chen, ChemBioChem, 2013, 14, 573–576 CrossRef CAS.
- S. Liu, H. Guo, T. Zhang, L. Han, P. Yao, Y. Zhang, N. Rong, Y. Yu, W. Lan, C. Wang, J. Ding, R. Wang, W. Liu and C. Cao, Sci. Rep., 2015, 5, 12744 CrossRef CAS.
- Y. Wang, S. Liu, P. Yao, Y. Yu, Y. Zhang, W. Lan, C. Wang, J. Ding, W. Liu and C. Cao, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 1033–1037 CrossRef CAS.
- Y. Ding, Y. Yu, H. Pan, H. Guo, Y. Li and W. Liu, Mol. BioSyst., 2010, 6, 1180–1185 RSC.
- S. J. Malcolmson, T. S. Young, J. G. Ruby, P. Skewes-Cox and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8483–8488 CrossRef CAS.
- R. Liao and W. Liu, J. Am. Chem. Soc., 2011, 133, 2852–2855 CrossRef CAS.
- P. R. Bennallack, K. D. Bewley, M. A. Burlingame, R. A. Robison, S. M. Miller and J. S. Griffitts, J. Bacteriol., 2016, 198, 2431–2438 CrossRef CAS.
- T. Ozaki, K. Yamashita, Y. Goto, M. Shimomura, S. Hayashi, S. Asamizu, Y. Sugai, H. Ikeda, H. Suga and H. Onaka, Nat. Commun., 2017, 8, 14207 CrossRef CAS.
- K. Unno, I. Kaweewan, H. Nakagawa and S. Kodani, Appl. Microbiol. Biotechnol., 2020, 104, 5293–5302 CrossRef CAS.
- D. M. Liang, J. H. Liu, H. Wu, B. B. Wang, H. J. Zhu and J. J. Qiao, Chem. Soc. Rev., 2015, 44, 8350–8374 RSC.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 CrossRef CAS.
- A. de Jong, S. A. F. T. van Hijum, J. J. E. Bijlsma, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2006, 34, W273–W279 CrossRef CAS.
- A. de Jong, A. J. van Heel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2010, 38, W647–W651 CrossRef CAS.
- A. J. van Heel, A. de Jong, M. Montalbán-López, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2013, 41, W448–W453 CrossRef.
- A. J. van Heel, A. de Jong, C. Song, J. H. Viel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2018, 46, W278–W281 CrossRef CAS.
- A. L. Delcher, K. A. Bratke, E. C. Powers and S. L. Salzberg, Bioinformatics, 2007, 23, 673–679 CrossRef CAS.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS.
- A. J. van Heel, M. Montalban-Lopez, Q. Oliveau and O. P. Kuipers, Microb. Genomes, 2017, 3, e000134 Search PubMed.
- H. Wu, Q. Gu, Y. Xie, Z. Lou, P. Xue, L. Fang, C. Yu, D. Jia, G. Huang, B. Zhu, A. Schneider, J. Blom, P. Lasch, R. Borriss and X. Gao, Environ. Microbiol., 2019, 21, 3505–3526 CrossRef CAS.
- B. Markusková, A. Lichvariková, T. Szemes, J. Koreňová, T. Kuchta and H. Drahovská, FEMS Microbiol. Lett., 2018, 365, fny257 Search PubMed.
- R. D. Finn, J. Clements, W. Arndt, B. L. Miller, T. J. Wheeler, F. Schreiber, A. Bateman and S. R. Eddy, Nucleic Acids Res., 2015, 43, W30–W38 CrossRef CAS.
- R. D. Finn, P. Coggill, R. Y. Eberhardt, S. R. Eddy, J. Mistry, A. L. Mitchell, S. C. Potter, M. Punta, M. Qureshi, A. Sangrador-Vegas, G. A. Salazar, J. Tate and A. Bateman, Nucleic Acids Res., 2016, 44, D279–D285 CrossRef CAS.
- D. H. Haft, J. D. Selengut, R. A. Richter, D. Harkins, M. K. Basu and E. Beck, Nucleic Acids Res., 2013, 41, D387–D395 CrossRef CAS.
- K. Blin, T. Wolf, M. G. Chevrette, X. Lu, C. J. Schwalen, S. A. Kautsar, H. G. Suarez Duran, E. L. C. de los Santos, H. U. Kim, M. Nave, J. S. Dickschat, D. A. Mitchell, E. Shelest, R. Breitling, E. Takano, S. Y. Lee, T. Weber and M. H. Medema, Nucleic Acids Res., 2017, 45, W36–W41 CrossRef CAS.
- S. Ovchinnikov, H. Kamisetty and D. Baker, eLife, 2014, 3, e02030 CrossRef.
- M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef CAS.
- A. J. van Heel, A. de Jong, C. Song, J. H. Viel, J. Kok and O. P. Kuipers, Nucleic Acids Res., 2018, 46, W278–W281 CrossRef CAS.
- D. Hyatt, G.-L. Chen, P. F. LoCascio, M. L. Land, F. W. Larimer and L. J. Hauser, BMC Bioinf., 2010, 11, 119 CrossRef.
- E. L. C. de los Santos, Sci. Rep., 2019, 9, 13406 CrossRef.
- P. Agrawal, S. Khater, M. Gupta, N. Sain and D. Mohanty, Nucleic Acids Res., 2017, 45, W80–W88 CrossRef CAS.
- H. Mohimani, R. D. Kersten, W.-T. Liu, M. Wang, S. O. Purvine, S. Wu, H. M. Brewer, L. Pasa-Tolic, N. Bandeira, B. S. Moore, P. A. Pevzner and P. C. Dorrestein, ACS Chem. Biol., 2014, 9, 1545–1551 CrossRef CAS.
- M. A. Skinnider, C. A. Dejong, P. N. Rees, C. W. Johnston, H. Li, A. L. Webster, M. A. Wyatt and N. A. Magarvey, Nucleic Acids Res., 2015, 43, 9645–9662 CAS.
- M. A. Skinnider, C. W. Johnston, R. E. Edgar, C. A. Dejong, N. J. Merwin, P. N. Rees and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E6343–E6351 CrossRef CAS.
- C. W. Johnston, M. A. Skinnider, M. A. Wyatt, X. Li, M. R. M. Ranieri, L. Yang, D. L. Zechel, B. Ma and N. A. Magarvey, Nat. Commun., 2015, 6, 8421 CrossRef CAS.
- Z.-F. Pei, M.-J. Yang, L. Li, X.-H. Jian, Y. Yin, D. Li, H.-X. Pan, Y. Lu, W. Jiang and G.-L. Tang, Org. Biomol. Chem., 2018, 16, 9373–9376 RSC.
- I. Kaweewan, H. Komaki, H. Hemmi, K. Hoshino, T. Hosaka, G. Isokawa, T. Oyoshi and S. Kodani, J. Antibiot., 2019, 72, 1–7 CrossRef CAS.
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS.
- K. Blin, M. H. Medema, D. Kazempour, M. A. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS.
- M. Hadjithomas, I.-M. A. Chen, K. Chu, A. Ratner, K. Palaniappan, E. Szeto, J. Huang, T. B. K. Reddy, P. Cimermančič, M. A. Fischbach, N. N. Ivanova, V. M. Markowitz, N. C. Kyrpides and A. Pati, mBio, 2015, 6, e00932 CrossRef CAS.
- M. Hadjithomas, I.-M. A. Chen, K. Chu, J. Huang, A. Ratner, K. Palaniappan, E. Andersen, V. Markowitz, N. C. Kyrpides and N. N. Ivanova, Nucleic Acids Res., 2017, 45, D560–D565 CrossRef CAS.
- K. R. Conway and C. N. Boddy, Nucleic Acids Res., 2012, 41, D402–D407 CrossRef.
- N. Ichikawa, M. Sasagawa, M. Yamamoto, H. Komaki, Y. Yoshida, S. Yamazaki and N. Fujita, Nucleic Acids Res., 2013, 41, D408–D414 CrossRef CAS.
- K. Blin, M. H. Medema, D. Kazempour, M. A. Fischbach, R. Breitling, E. Takano and T. Weber, Nucleic Acids Res., 2013, 41, W204–W212 CrossRef.
- K. Blin, M. H. Medema, R. Kottmann, S. Y. Lee and T. Weber, Nucleic Acids Res., 2016, 45, D555–D559 CrossRef.
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang and S. H. Bryant, Nucleic Acids Res., 2009, 37, W623–W633 CrossRef CAS.
- F. B. Rogers, Bull. Med. Libr. Assoc., 1963, 51, 114–116 CAS.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS.
- V. M. Markowitz, I. M. Chen, K. Chu, E. Szeto, K. Palaniappan, M. Pillay, A. Ratner, J. Huang, I. Pagani, S. Tringe, M. Huntemann, K. Billis, N. Varghese, K. Tennessen, K. Mavromatis, A. Pati, N. N. Ivanova and N. C. Kyrpides, Nucleic Acids Res., 2014, 42, D568–D573 CrossRef CAS.
- N. J. Merwin, W. K. Mousa, C. A. Dejong, M. A. Skinnider, M. J. Cannon, H. Li, K. Dial, M. Gunabalasingam, C. Johnston and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 371 CrossRef CAS.
- A. M. Kloosterman, K. E. Shelton, G. P. van Wezel, M. H. Medema and D. A. Mitchell, mSystems, 2020, 5, e00267-20 CrossRef.
- J. Soding, A. Biegert and A. N. Lupas, Nucleic Acids Res., 2005, 33, W244–W248 CrossRef.
- C. UniProt, Nucleic Acids Res., 2015, 43, D204–D212 CrossRef.
- J. Lubelski, R. Rink, R. Khusainov, G. N. Moll and O. P. Kuipers, Cell. Mol. Life Sci., 2008, 65, 455–476 CrossRef CAS.
- O. P. Kuipers, G. Bierbaum, B. Ottenwälder, H. M. Dodd, N. Horn, J. Metzger, T. Kupke, V. Gnau, R. Bongers, P. van den Bogaard, H. Kosters, H. S. Rollema, W. M. de Vos, R. J. Siezen, G. Jung, F. Götz, H. G. Sahl and M. J. Gasson, Antonie van Leeuwenhoek, 1996, 69, 161–169 CrossRef CAS.
- J. Cortés, A. N. Appleyard and M. J. Dawson, Methods Enzymol., 2009, 458, 559–574 Search PubMed.
- D. E. Ruffner, E. W. Schmidt and J. R. Heemstra, ACS Synth. Biol., 2015, 4, 482–492 CrossRef CAS.
- Y. Goto, Y. Ito, Y. Kato, S. Tsunoda and H. Suga, Chem. Biol., 2014, 21, 766–774 CrossRef CAS.
- S. R. Fleming, T. E. Bartges, A. A. Vinogradov, C. L. Kirkpatrick, Y. Goto, H. Suga, L. M. Hicks and A. A. Bowers, J. Am. Chem. Soc., 2019, 141, 758–762 CrossRef CAS.
- T. J. Oman, P. J. Knerr, N. A. Bindman, J. E. Vélasquez and W. A. van der Donk, J. Am. Chem. Soc., 2012, 134, 6952–6955 CrossRef CAS.
- M. R. Levengood, G. C. Patton and W. A. van der Donk, J. Am. Chem. Soc., 2007, 129, 10314–10315 CrossRef CAS.
- R. Khusainov and O. P. Kuipers, ChemBioChem, 2012, 2433–2438 CrossRef CAS.
- M. R. Levengood, P. J. Knerr, T. J. Oman and W. A. van der Donk, J. Am. Chem. Soc., 2009, 131, 12024–12025 CrossRef CAS.
- P. J. Knerr, T. J. Oman, C. V. Garcia De Gonzalo, T. J. Lupoli, S. Walker and W. A. van der Donk, ACS Chem. Biol., 2012, 7, 1791–1795 CrossRef CAS.
- B. J. Burkhart, N. Kakkar, G. A. Hudson, W. A. van der Donk and D. A. Mitchell, ACS Cent. Sci., 2017, 3, 629–638 CrossRef CAS.
- S. Mukherjee and W. A. van der Donk, J. Am. Chem. Soc., 2014, 136, 10450–10459 CrossRef CAS.
- A. J. van Heel, D. Mu, M. Montalban-Lopez, D. Hendriks and O. P. Kuipers, ACS Synth. Biol., 2013, 2, 397–404 CrossRef CAS.
- O. P. Kuipers, H. S. Rollema, W. M. de Vos and R. J. Siezen, FEBS Lett., 1993, 330, 23–27 CrossRef CAS.
- A. Chakicherla and J. N. Hansen, J. Biol. Chem., 1995, 270, 23533–23539 CrossRef CAS.
- A. J. van Heel, T. G. Kloosterman, M. Montalban-Lopez, J. Deng, A. Plat, B. Baudu, D. Hendriks, G. N. Moll and O. P. Kuipers, ACS Synth. Biol., 2016, 5, 1146–1154 CrossRef CAS.
- J. A. Majchrzykiewicz, J. Lubelski, G. N. Moll, A. Kuipers, J. J. Bijlsma, O. P. Kuipers and R. Rink, Antimicrob. Agents Chemother., 2010, 54, 1498–1505 CrossRef CAS.
- M. D. Tianero, M. S. Donia, T. S. Young, P. G. Schultz and E. W. Schmidt, J. Am. Chem. Soc., 2012, 134, 418–425 CrossRef CAS.
- Q. Zhang, Y. Yu, J. E. Velásquez and W. A. van der Donk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18361–18366 CrossRef CAS.
- J. L. Dopp, D. D. Tamiev and N. F. Reuel, Biotechnol. Adv., 2019, 37, 246–258 CrossRef.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS.
- Y. Goto and H. Suga, Curr. Opin. Chem. Biol., 2018, 46, 82–90 CrossRef CAS.
- W. E. Houssen, A. F. Bent, A. R. McEwan, N. Pieiller, J. Tabudravu, J. Koehnke, G. Mann, R. I. Adaba, L. Thomas, U. W. Hawas, H. Liu, U. Schwarz-Linek, M. C. Smith, J. H. Naismith and M. Jaspars, Angew. Chem., Int. Ed. Engl., 2014, 53, 14171–14174 CrossRef CAS.
- C. D. Deane, J. O. Melby, K. J. Molohon, A. R. Susarrey and D. A. Mitchell, ACS Chem. Biol., 2013, 8, 1998–2008 CrossRef CAS.
- Y. Goto and H. Suga, ChemBioChem, 2020, 21, 84–87 CrossRef CAS.
- S. Hayashi, T. Ozaki, S. Asamizu, H. Ikeda, S. Omura, N. Oku, Y. Igarashi, H. Tomoda and H. Onaka, Chem. Biol., 2014, 21, 679–688 CrossRef CAS.
- A. A. Vinogradov, M. Shimomura, Y. Goto, T. Ozaki, S. Asamizu, Y. Sugai, H. Suga and H. Onaka, Nat. Commun., 2020, 11, 2272 CrossRef CAS.
- R. Liu, Y. Zhang, G. Zhai, S. Fu, Y. Xia, B. Hu, X. Cai, Y. Zhang, Y. Li, Z. Deng and T. Liu, Adv. Sci., 2020 DOI:10.1002/advs.202001616.
- F. T. Hofmann, J. W. Szostak and F. P. Seebeck, J. Am. Chem. Soc., 2012, 134, 8038–8041 CrossRef CAS.
- B. C. Buer and E. N. Marsh, Protein Sci., 2012, 21, 453–462 CrossRef CAS.
- R. V. Maaskant and G. Roelfes, ChemBioChem, 2019, 20, 57–61 CrossRef CAS.
- H. Li, R. Aneja and I. Chaiken, Molecules, 2013, 18, 9797–9817 CrossRef CAS.
- N. Bindman, R. Merkx, R. Koehler, N. Herrman and W. A. van der Donk, Chem. Commun., 2010, 46, 8935–8937 RSC.
- J. A. McIntosh, C. R. Robertson, V. Agarwal, S. K. Nair, G. W. Bulaj and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 15499–15501 CrossRef CAS.
- X. Hemu, Y. Qiu, G. K. Nguyen and J. P. Tam, J. Am. Chem. Soc., 2016, 138, 6968–6971 CrossRef CAS.
- E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., Int. Ed., 2016, 55, 5842–5845 CrossRef CAS.
- E. Oueis, H. Stevenson, M. Jaspars, N. J. Westwood and J. H. Naismith, Chem. Commun., 2017, 53, 12274–12277 RSC.
- E. Oueis, B. Nardone, M. Jaspars, N. J. Westwood and J. H. Naismith, ChemistryOpen, 2017, 6, 11–14 CrossRef CAS.
- G. K. Nguyen, X. Hemu, J. P. Quek and J. P. Tam, Angew. Chem., Int. Ed. Engl., 2016, 55, 12802–12806 CrossRef CAS.
- A. A. Bowers, M. G. Acker, T. S. Young and C. T. Walsh, J. Am. Chem. Soc., 2012, 134, 10313–10316 CrossRef CAS.
- J. T. Wong, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6303–6306 CrossRef CAS.
- J. C. van Hest and D. A. Tirrell, FEBS Lett., 1998, 428, 68–70 CrossRef CAS.
- N. Budisa, C. Minks, F. J. Medrano, J. Lutz, R. Huber and L. Moroder, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 455–459 CrossRef CAS.
- R. S. Al Toma, A. Kuthning, M. P. Exner, A. Denisiuk, J. Ziegler, N. Budisa and R. D. Sussmuth, ChemBioChem, 2015, 16, 503–509 CrossRef CAS.
- F. Oldach, R. Al Toma, A. Kuthning, T. Caetano, S. Mendo, N. Budisa and R. D. Süssmuth, Angew. Chem., Int. Ed., 2012, 51, 415–418 CrossRef CAS.
- L. Zhou, J. Shao, Q. Li, A. J. van Heel, M. P. de Vries, J. Broos and O. P. Kuipers, Amino Acids, 2016, 48, 1309–1318 CrossRef CAS.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS.
- J. W. Chin, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11020–11024 CrossRef CAS.
- E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS.
- J. Nagao, Y. Harada, K. Shioya, Y. Aso, T. Zendo, J. Nakayama and K. Sonomoto, Biochem. Biophys. Res. Commun., 2005, 336, 507–513 CrossRef CAS.
- Y. Shi, X. Yang, N. Garg and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 2338–2341 CrossRef CAS.
- N. A. Bindman, S. C. Bobeica, W. R. Liu and W. A. van der Donk, J. Am. Chem. Soc., 2015, 137, 6975–6978 CrossRef CAS.
- N. Kakkar, J. G. Perez, W. R. Liu, M. C. Jewett and W. A. van der Donk, ACS Chem. Biol., 2018, 13, 951–957 CrossRef CAS.
- M. Bartholomae, T. Baumann, J. H. Nickling, D. Peterhoff, R. Wagner, N. Budisa and O. P. Kuipers, Front. Microbiol., 2018, 9, 657 CrossRef.
- C. Zambaldo, X. Luo, A. P. Mehta and P. G. Schultz, J. Am. Chem. Soc., 2017, 139, 11646–11649 CrossRef CAS.
- M. Lopatniuk, M. Myronovskyi and A. Luzhetskyy, ACS Chem. Biol., 2017, 12, 2362–2370 CrossRef CAS.
- Y. Lin, K. Teng, L. Huan and J. Zhong, Microbiol. Res., 2011, 166, 146–154 CrossRef CAS.
- G. Valsesia, G. Medaglia, M. Held, W. Minas and S. Panke, Appl. Environ. Microbiol., 2007, 73, 1635–1645 CrossRef CAS.
- M. Singh, S. Chaudhary and D. Sareen, Mol. Microbiol., 2020, 113, 326–337 CrossRef CAS.
- M. S. Donia, D. E. Ruffner, S. Cao and E. W. Schmidt, ChemBioChem, 2011, 12, 1230–1236 CrossRef CAS.
- X. Luo, C. Zambaldo, T. Liu, Y. Zhang, W. Xuan, C. Wang, S. A. Reed, P. Y. Yang, R. E. Wang, T. Javahishvili, P. G. Schultz and T. S. Young, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3615–3620 CrossRef CAS.
- P. M. Himes, S. E. Allen, S. Hwang and A. A. Bowers, ACS Chem. Biol., 2016, 11, 1737–1744 CrossRef CAS.
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H. R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr, J. A. Mosberg, N. Rohland, P. G. Schultz, J. M. Jacobson, J. Rinehart, G. M. Church and F. J. Isaacs, Science, 2013, 342, 357–360 CrossRef CAS.
- F. J. Piscotta, J. M. Tharp, W. R. Liu and A. J. Link, Chem. Commun., 2015, 51, 409–412 RSC.
- N. R. Braffman, F. J. Piscotta, J. Hauver, E. A. Campbell, A. J. Link and S. A. Darst, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1273–1278 CrossRef CAS.
- X. Bi, J. Yin, X. Hemu, C. Rao, J. P. Tam and C.-F. Liu, Bioconjugate Chem., 2018, 29, 2170–2175 CrossRef CAS.
- A. E. Owens, K. T. Grasso, C. A. Ziegler and R. Fasan, ChemBioChem, 2017, 18, 1109–1116 CrossRef CAS.
- J. Guo, C. E. Melancon 3rd, H. S. Lee, D. Groff and P. G. Schultz, Angew. Chem., Int. Ed. Engl., 2009, 48, 9148–9151 CrossRef CAS.
- S. Venkat, H. Chen, Q. Gan and C. Fan, Front. Pharmacol., 2019, 10, 248 CrossRef CAS.
- Q. Gan and C. Fan, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3047–3052 CrossRef CAS.
- R. W. Martin, B. J. Des Soye, Y. C. Kwon, J. Kay, R. G. Davis, P. M. Thomas, N. I. Majewska, C. X. Chen, R. D. Marcum, M. G. Weiss, A. E. Stoddart, M. Amiram, A. K. Ranji Charna, J. R. Patel, F. J. Isaacs, N. L. Kelleher, S. H. Hong and M. C. Jewett, Nat. Commun., 2018, 9, 1203 CrossRef.
- M. H. Medema, R. Breitling, R. Bovenberg and E. Takano, Nat. Rev. Microbiol., 2011, 9, 131–137 CrossRef CAS.
- L. Duan, S. Wang, R. Liao and W. Liu, Chem. Biol., 2012, 19, 443–448 CrossRef CAS.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS.
- J. D. Hegemann, M. De Simone, M. Zimmermann, T. A. Knappe, X. Xie, F. S. Di Leva, L. Marinelli, E. Novellino, S. Zahler, H. Kessler and M. A. Marahiel, J. Med. Chem., 2014, 57, 5829–5834 CrossRef CAS.
- J. D. Hegemann, S. C. Bobeica, M. C. Walker, I. R. Bothwell and W. A. van der Donk, ACS Synth. Biol., 2019, 8, 1204–1214 CrossRef CAS.
- S. J. Pan and A. J. Link, J. Am. Chem. Soc., 2011, 133, 5016–5023 CrossRef CAS.
- C. Zong, M. O. Maksimov and A. J. Link, ACS Chem. Biol., 2016, 11, 61–68 CrossRef CAS.
- T. S. Young, P. C. Dorrestein and C. T. Walsh, Chem. Biol., 2012, 19, 1600–1610 CrossRef CAS.
- H. L. Tran, K. W. Lexa, O. Julien, T. S. Young, C. T. Walsh, M. P. Jacobson and J. A. Wells, J. Am. Chem. Soc., 2017, 139, 2541–2544 CrossRef CAS.
- A. R. Weiz, K. Ishida, F. Quitterer, S. Meyer, J.-C. Kehr, K. M. Müller, M. Groll, C. Hertweck and E. Dittmann, Angew. Chem., Int. Ed., 2014, 53, 3735–3738 CrossRef CAS.
- S. Schmitt, M. Montalbán-López, D. Peterhoff, J. Deng, R. Wagner, M. Held, O. P. Kuipers and S. Panke, Nat. Chem. Biol., 2019, 15, 437–443 CrossRef CAS.
- T. Si, Q. Tian, Y. Min, L. Zhang, J. V. Sweedler, W. A. van der Donk and H. Zhao, J. Am. Chem. Soc., 2018, 140, 11884–11888 CrossRef CAS.
- T. Caetano, J. M. Krawczyk, E. Mosker, R. D. Süssmuth and S. Mendo, Chem. Biol., 2011, 18, 90–100 CrossRef CAS.
- L. D. Kluskens, S. A. Nelemans, R. Rink, L. de Vries, A. Meter-Arkema, Y. Wang, T. Walther, A. Kuipers, G. N. Moll and M. Haas, J. Pharmacol. Exp. Ther., 2009, 328, 849–854 CrossRef CAS.
- A. Kuipers, G. N. Moll, E. Wagner and R. Franklin, Peptides, 2019, 112, 78–84 CrossRef CAS.
- X. Yang, K. R. Lennard, C. He, M. C. Walker, A. T. Ball, C. Doigneaux, A. Tavassoli and W. A. van der Donk, Nat. Chem. Biol., 2018, 14, 375–380 CrossRef CAS.
- A. Tavassoli, Q. Lu, J. Gam, H. Pan, S. J. Benkovic and S. N. Cohen, ACS Chem. Biol., 2008, 3, 757–764 CrossRef CAS.
- T. Bosma, A. Kuipers, E. Bulten, L. de Vries, R. Rink and G. N. Moll, Appl. Environ. Microbiol., 2011, 77, 6794–6801 CrossRef CAS.
- X. Zhao, R. Cebrián, Y. Fu, R. Rink, T. Bosma, G. N. Moll and O. P. Kuipers, ACS Synth. Biol., 2020, 9, 1468–1478 CrossRef CAS.
- B. Hu, P. Guo, I. Bar-Joseph, Y. Imanishi, M. J. Jarzynka, O. Bogler, T. Mikkelsen, T. Hirose, R. Nishikawa and S. Y. Cheng, Oncogene, 2007, 26, 5577–5586 CrossRef CAS.
- J. A. Getz, O. Cheneval, D. J. Craik and P. S. Daugherty, ACS Chem. Biol., 2013, 8, 1147–1154 CrossRef CAS.
- C. K. Wang and D. J. Craik, Nat. Chem. Biol., 2018, 14, 417–427 CrossRef CAS.
- J. J. Rice and P. S. Daugherty, Protein Eng., Des. Sel., 2008, 21, 435–442 CrossRef CAS.
- J. H. Urban, M. A. Moosmeier, T. Aumüller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Nat. Commun., 2017, 8, 1500 CrossRef.
- N. Mahmood, C. Mihalcioiu and S. A. Rabbani, Front. Oncol., 2018, 8, 24 CrossRef.
- S. Chen, I. Rentero Rebollo, S. A. Buth, J. Morales-Sanfrutos, J. Touati, P. G. Leiman and C. Heinis, J. Am. Chem. Soc., 2013, 135, 6562–6569 CrossRef CAS.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, ACS Cent. Sci., 2018, 4, 458–467 CrossRef CAS.
- A. P. Silverman, A. M. Levin, J. L. Lahti and J. R. Cochran, J. Mol. Biol., 2009, 385, 1064–1075 CrossRef CAS.
- S. G. Kim, S. Becattini, T. U. Moody, P. V. Shliaha, E. R. Littmann, R. Seok, M. Gjonbalaj, V. Eaton, E. Fontana, L. Amoretti, R. Wright, S. Caballero, Z.-M. X. Wang, H.-J. Jung, S. M. Morjaria, I. M. Leiner, W. Qin, R. J. J. F. Ramos, J. R. Cross, S. Narushima, K. Honda, J. U. Peled, R. C. Hendrickson, Y. Taur, M. R. M. van den Brink and E. G. Pamer, Nature, 2019, 572, 665–669 CrossRef CAS.
- Y. Duan, C. Llorente, S. Lang, K. Brandl, H. Chu, L. Jiang, R. C. White, T. H. Clarke, K. Nguyen, M. Torralba, Y. Shao, J. Liu, A. Hernandez-Morales, L. Lessor, I. R. Rahman, Y. Miyamoto, M. Ly, B. Gao, W. Sun, R. Kiesel, F. Hutmacher, S. Lee, M. Ventura-Cots, F. Bosques-Padilla, E. C. Verna, J. G. Abraldes, R. S. Brown Jr, V. Vargas, J. Altamirano, J. Caballeria, D. L. Shawcross, S. B. Ho, A. Louvet, M. R. Lucey, P. Mathurin, G. Garcia-Tsao, R. Bataller, X. M. Tu, L. Eckmann, W. A. van der Donk, R. Young, T. D. Lawley, P. Starkel, D. Pride, D. E. Fouts and B. Schnabl, Nature, 2019, 575, 505–511 CrossRef CAS.
- S. Luo and S.-H. Dong, Molecules, 2019, 24, 1541 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |