
The importance of the molecular weight of PEDOT hole transporting materials for efficient organic solar cells†
Qingyao
Fu
a,
Yuda
Li
 *a,
Xunchang
Wang
b,
Qi
Li
a,
Feng
Wang
a and
Renqiang
Yang
bc
*a,
Xunchang
Wang
b,
Qi
Li
a,
Feng
Wang
a and
Renqiang
Yang
bc
aKey Laboratory for Green Process of Ministry of Education, School of Chemical Engineering and Pharmacy, Wuhan Institute of Technology, Wuhan 430205, China. E-mail: psydli@wit.edu.cn
bCAS Key Laboratory of Bio-Based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
cKey Laboratory of Optoelectronic Chemical Materials and Devices (Ministry of Education), School of Chemical and Environmental Engineering, Jianghan University, Wuhan 430056, China
First published on 9th November 2020
Abstract
Poly(3,4-ethylenedioxythiophene) (PEDOT) conducting polymers have been extensively studied as hole transporting materials (HTMs) in organic electronic devices. However, almost none of the reports have revealed the molecular weight effect of PEDOT on the device performance. Herein, analysis of the molecular weight effect is enabled using a polymerization strategy on a representative model system of poly(styrenesulfonate) doped PEDOT (PEDOT:PSS) which produces conducting polymers with tunable PEDOT molecular weights. Experiments reveal that the power conversion efficiency (PCE) of PBDB-T:ITIC organic solar cells fabricated using these PEDOT:PSS HTMs first increases from nearly 0 to 1.32 and to 9.91% with increasing PEDOT molecular weight. This is primarily due to the enhancement of the doping degree of PEDOT and the phase seperation between PEDOT and PSS that produces enormous shifts in HTM conductivity from nearly insulating (7.70 × 10−7 S cm−1) to medially conductive (1.14 × 10−4 S cm−1) and to highly conductive (7.22 × 10−4 S cm−1). With the further increase of PEDOT molecular weight, the PCE rapidly reaches up to 10.36% and stays unchanged, which seems to be largely determined by the combined effect of enhanced conductivity and increased roughness of HTMs that produces an optimal balance between the hole collection and carrier recombination loss in devices. These findings highlight the importance of optimizing PEDOT molecular weight for PEDOT-based HTMs to help realize the full potential of organic optoelectronic devices.
1 Introduction
Bulk heterojunction organic solar cells (OSCs) have gained enormous attention over the past decades because of their great potential for solving energy and environment issues, such as fossil fuel exhaustion and environmental pollution.1–13 Recently, the power conversion efficiency (PCE) of single-junction OSCs has been dramatically enhanced to approach 18%, benefiting from the significant innovations in photoactive heterojunction materials and devices.14–23 In addition, the development of novel hole transporting materials (HTMs) is also a useful strategy to improve the device efficiency by promoting the collection of holes and suppressing the recombination of photogenerated excitons at the anode interface.24–27 Unfortunately, the progress in HTMs has been limited, with only a few materials available at present.24–27Poly(3,4-ethylenedioxythiophene) (PEDOT) conducting polymers have been proven to be an efficient HTM for organic electronic devices.28–32 Commonly, researchers select commodity PEDOT aqueous dispersion, i.e. poly(styrenesulfonate) doped PEDOT (PEDOT:PSS), as the benchmark HTM to fabricate organic electronic devices. However, PEDOT:PSS exhibits modest hole transporting capability which restricts the performance of devices.28–36 Over the past decades, huge strategies focused on tuning the PEDOT:PSS micro–nano structure have been proposed to overcome the above defect, such as (1) converting the PEDOT from weakly conductive benzoid conformation to highly conductive quinoid conformation, (2) enhancing the crystallinity and connectivity of PEDOT conducting networks, (3) enriching the high ionization potential component onto the surface of the PEDOT:PSS film, etc.31,32,35–38 Besides, selecting dopants to replace PSS to redesign the micro–nano structure of PEDOT conducting polymers is also regarded as a promising strategy for the development of high performance HTMs.39–46 For instance, Li et al.33 designed a highly homogeneous PEDOT HTM by using a branched dopant of MNSF, achieving excellent hole transporting performance in both organic light-emitting diodes and perovskite solar cells. And, they synthesized a novel PEDOT HTM with superior conductivity and work function by using a redox-active dopant of GSL, exhibiting obviously higher hole transporting performance in perovskite solar cell than PEDOT:PSS standard HTM (PCE 14.94% versus 12.6%).45
Apart from the above-mentioned micro–nano structural parameters, the molecular weight may also play a critical role in determining the hole transporting performance of conducting polymer-based HTMs because the properties of the conjugated polymer are partly determined by the molecular weight of the polymer.47–50 However, almost no studies have been devoted to understanding how the molecular weight of conjugated PEDOT influences the optical, morphological and electrical characteristics and thus the device performance of PEDOT-based HTMs. To the best of our knowledge, commonly, researchers synthesize PEDOT-based HTMs without optimization of PEDOT molecular weight (see Fig. 1). Therefore, it can be concluded that there is a great potential for optimizing the PEDOT molecular weight for conducting polymers to help realize the full potential of HTMs in organic optoelectronic devices (see Fig. 1).
 | ||
| Fig. 1 (a) Visible (Vis) absorption spectra of neutral PEDOTs of previously reported PEDOT-based HTMs. Inset: Photographs of neutral PDA:PEDOT:LS, PEDOT:GSL, PEDOT:LS, PEDOT:MNSF and PEDOT:PSS solutions (left to right). (b) A comparison of the PCEs with previously reported perovskite solar cells (PSCs) with PEDOT-based HTMs.33,34,44,45 | ||
In this article, we chose the PEDOT:PSS conducting polymer, one of the most famous HTMs, as a model system to study the PEDOT molecular weight effect on the photovoltaic performance of OSCs. For a start, five batches of PEDOT:PSSs with a broad range of PEDOT molecular weights (ultralow, low, middle, high and ultrahigh) were obtained through a simple synthetic strategy based on oxidative polymerization, taking into account the effect of the FeCl3 catalyst amount or the ethylene glycol (EG) emulsifier amount. Secondly, the product polymers were characterized regarding the radical concentration, doping degree and microstructure by using electron spin resonance (ESR), X-ray photoelectron spectroscopy (XPS) and atomic force microscopy (AFM) imaging techniques, respectively. Furthermore, we correlated two electrical properties (conductivity and work function) of PEDOT:PSSs with their PEDOT molecular weights. Eventually, the effects of PEDOT molecular weight on the photovoltaic performance of OSCs were discussed in detail based on a PBDB-T:ITIC heterojunction-based device and a deep insight into the different photovoltaic performances was provided by dark current density–voltage (J–V) measurements in terms of the hole extraction and electron blocking properties of HTMs.
2 Results and discussion
2.1 Synthesis and characterization of PEDOT:PSS series
PEDOT:PSS conducting polymers were synthesized using the oxidative polymerization method and the synthetic routes are outlined in Fig. 2a. After finding conditions that yield different molecular weight PEDOTs, the catalyst and emulsifier amounts were varied to synthesize five batches of PEDOT:PSSs. The UV-vis-NIR absorption spectra of as-synthesized PEDOT:PSS solutions are shown in Fig. 2c. It can be seen that all the samples exhibit obvious polaron and bipolaron absorptions between 700 and 1300 nm.52 This phenomenon confirms the successful preparation of PEDOT:PSS conducting polymers. Moreover, we found that these five samples exhibit similar absorbance in the visible light region (400–800 nm). | ||
| Fig. 2 (a) Synthetic route of the PEDOT:PSS series with varied PEDOT molecular weights. (b) Band diagram for neutral, polaron and bipolaron states in PEDOT conducting polymers.51 This figure is used for the interpretation of the different optical characteristics of PEDOTs in the neutral and polaronic/bipolaronic states. (c) UV-vis-NIR absorption spectra of as-synthesized PEDOT:PSS aqueous dispersions. (d) Vis-NIR absorption spectra of dilute PEDOT:PSS solutions after dedoping treatments. Inset: Photographs of dedoped PEDOT:PSS solutions with mass concentrations of about 0.3% (left to right: P-UL, P-L, P-M, P-H, P-UH). | ||
It is well known that the quantitative detection of PEDOT molecular weights is still a challenge. Considering that the π-conjugated polymers with higher molecular weights exhibit more red-shifted absorption spectra,41,53 we proposed to apply Vis-NIR absorption spectra to qualitatively analyse the relative magnitude of the molecular weight of the five batches of PEDOT. However, because the as-synthesized PEDOT is common in the doped state (polaron or bipolaron state), the characteristic highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) transition absorption cannot be detected directly (see Fig. 2b). Therefore, we conducted a dedoping experiment using a strong reducing agent ethanediamine on the as-synthesized five batches PEDOT to prepare the neutral PEDOTs that can be used for the detection of HOMO–LUMO transition absorption. As a result, the PEDOT:PSS synthesized using the APS oxidant exhibits an intense peak at about 583 nm after dedoping treatment (red curve in Fig. 2d) which can be assigned to the HOMO–LUMO transition absorption of neutral PEDOT.54 Moreover, it is worth noting that this characteristic absorption peak gradually shifts to the long wavelength region with the increase of FeCl3 amount (green, purple and pink curves) and to the short wavelength region due to the use of EG (black curve), respectively. Thus, it can be concluded that five batches of PEDOT:PSSs with varied PEDOT molecular weights were successfully prepared. Besides, we found that the dedoped sample with a different concentration exhibits the same absorption spectrum (see Fig. S1, ESI†). This result indicates that the aggregation behaviour has only a negligible effect on the absorption spectrum. In conclusion, according to these absorption spectra, we can divide the PEDOT molecular weights into five grades of ultralow, low, middle, high and ultrahigh in this study. For brevity, these five polymers are named as P-UL, P-L, P-M, P-H and P-UH, respectively.
2.2 Effect of molecular weight on the microstructure of the PEDOT:PSS series
Firstly, to detect the population of radicals on the PEDOT backbone, ESR measurements were performed and the spectra are shown in Fig. 3a. The population of radicals was calculated by double integrating of the ESR intensity and normalizing the integrated value to the sample weight. The changes in the double integrated ESR intensity with PEDOT molecular weight are displayed in Fig. 3c. As shown in Fig. 3a, all the samples exhibit an intense and broad single-line ESR signal (g = 2.0033). This is a typical observation for PEDOT radicals with large spin-delocalization within the entire PEDOT backbone.55,56 As shown in Fig. 3c, the double integrated ESR intensity first enhances monotonically with the increase of PEDOT molecular weight and reaches the maximum point (191.4) at the critical molecular weight of middle grade. With a further increase of the PEDOT molecular weight, the double integrated ESR intensity gradually decreases to 54.8 for P-UH. These results indicate that the PEDOT gradually changes from its non-spin paired polaron state (s = 1/2, ESR active) to the zero-spin bipolaron state (s = 0, ESR silent) with the increase of molecular weight. In other words, the oxidation level/doping degree of PEDOT enhances monotonically with the increase of molecular weight. | ||
| Fig. 3 (a) ESR and (b) XPS spectra of PEDOT:PSS powders. (c) Plots of double integrated ESR intensity and doping degree as a function of PEDOT molecular weight. | ||
Secondly, to get the doping degree of PEDOT, XPS measurements were performed and the S 2p spectra are shown in Fig. 3b and Fig. S2 (ESI†). The characteristic peaks of S 2p at around 166 and 163 eV were decomposed into three doublets: the S signal for the sulphonate group of PSS on the high binding energy side (∼168.0 and ∼166.9 eV), the S signal coming from oxidized PEDOT at the middle binding energy region (∼165.1 and ∼164.1 eV), and the S signal for neutral PEDOT on the low binding energy side (∼163.9 and ∼162.8 eV).57,58 The ratio of the contributions of different S signals of PEDOT provides a direct estimation of the doping degree.57,58 The changes in the doping degree with the PEDOT molecular weight are displayed in Fig. 3c. Clearly, the doping degree of PEDOT enhances monotonically from 0.081 to 0.209 with the increase of molecular weight, which is in good agreement with the ESR results.
Thirdly, to obtain information on the structure of PEDOT:PSS blends, AFM measurements were performed. The AFM height and phase images are shown in Fig. 4a–j, respectively. As shown in the height images, the P-UL and P-L films exhibit a similar flat surface with low root mean square (RMS) values of around 1.00 nm. As the PEDOT molecular weight increases, the RMS value rises from 1.23 for P-M to 1.67 for P-H and to 1.90 nm for P-UH, gradually. Simultaneously, we observed a sharp transition in the morphology between the P-M and P-H films, that is, the P-UL, P-L and P-M films with relatively low PEDOT molecular weights exhibit blurred amorphous morphologies whereas the P-H and P-UH films with relatively high PEDOT molecular weights display clear crystalline morphologies. This phenomenon suggests that high PEDOT molecular weight may result in a strong aggregation behaviour, which is similar to the observations of the conjugated polymer of P3HT.59 Besides, it should be noted that the increased surface roughness may cause a negative effect that reduces the charge selectivity of PEDOT:PSS-based HTMs.28,60
 | ||
| Fig. 4 (a)–(e) 2 × 2 μm scaled AFM height and (f)–(j) 1 × 1 μm scaled AFM phase images of P-UL (a and f), P-L (b and g), P-M (c and h), P-H (d and i) and P-UH (e and j) films. | ||
In the AFM phase images, the P-UL and P-L films show a similar continuous microphase structure with a lot of large aggregates. As the PEDOT molecular weight increases to middle grade, the microphase structure turns out to be discontinuous and a lot of isolated PEDOT phases appear, indicating the presence of a critical molecular weight where enhanced phase separation starts to occur. This enhanced phase separation is considered to be the main reason for the roughness enhancement of the P-M film. Moreover, the enhanced phase separation will be beneficial for the electron transport of PEDOT:PSS conducting polymers.61 When the PEDOT molecular weight further increases to high grade, the phase separation becomes more significant and some continuous big PEDOT phases are formed, which supports the further increase in the surface roughness. Moreover, when the PEDOT molecular weight increases to ultrahigh grade, the phase morphology becomes inhomogeneous with varied aggregate sizes. This phenomenon indicates that the excessively high molecular weight is likely to increase the irregularity of PEDOT aggregates which may increase the surface roughness of the PEDOT:PSS film.
2.3 Electrical properties of PEDOT:PSS series
The conductivities of the five PEDOT:PSS conducting polymers were determined from a diode device with the ITO/PEDOT:PSS/Al architecture. The film thicknesses were measured to be 50, 50, 50, 30 and 40 nm for P-UL, P-L, P-M, P-H and P-UH, respectively. The current–voltage (I–V) curves and the calculated conductivities are provided in Fig. 5a and b, respectively. Apparently, the conductivity exhibits a positive correlation with the PEDOT molecular weight, increasing from 7.70 × 10−7 to 1.08 × 10−3 S cm−1 with the PEDOT molecular weight increasing from ultralow to ultrahigh grades. The main reason for the improved conductivity is the increased oxidation level of PEDOT (enhanced intrinsic intramolecular conductivity) and the enhanced phase separation between PEDOT and PSS (increased intermolecular conductivity).62,63 More importantly, we observed a successive jump in the conductivity from nearly insulating (7.70 × 10−7 S cm−1) to medially conductive (1.14 × 10−4 S cm−1) and to highly conductive (7.22 × 10−4 S cm−1) with PEDOT molecular weight increasing from ultralow to middle grades. On the contrary, as the PEDOT molecular weight further increases, the increase amplitude of conductivity obviously decreases (see Fig. 5b). These findings highlight the importance of optimizing PEDOT molecular weight in the design of highly conductive conducting polymers.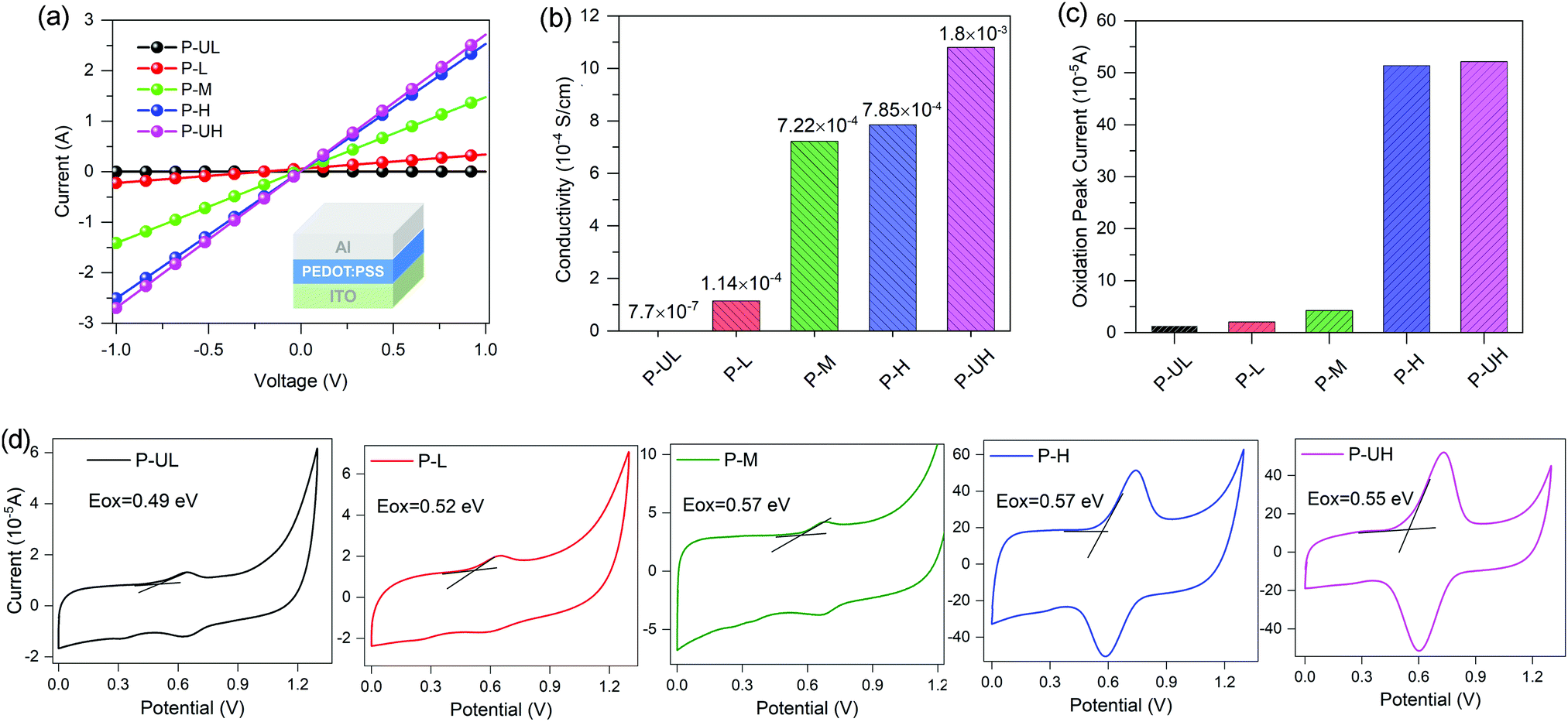 | ||
| Fig. 5 (a) I–V curves of the PEDOT:PSS-based diode devices. (b) Conductivities of different PEDOT:PSS films. (c) Redox peak current of different PEDOT:PSS films obtained from CV measurements. (d) CV curves of PEDOT:PSS films coated on ITO substrates. | ||
The work functions (WFs) of the five PEDOT:PSS conducting polymers were evaluated by cyclic voltammetry (CV) measurements. The redox curves of the five PEDOT:PSS films and the internal standard of ferrocene/ferrocenium are shown in Fig. 5d and Fig. S3 (ESI†), respectively. Unlike the conductivity, the WF in this study is not a monotonic function of PEDOT molecular weight and the WFs of the PEDOT:PSS series lie from ∼4.89 to ∼4.97 eV. This small variation in the WFs of PEDOT:PSSs is expected to yield similar energy-level offsets between the PEDOT:PSS HTMs and the polymer donor material. Furthermore, the redox peak current of different PEDOT:PSS films increases with the increase of PEDOT molecular weight (see Fig. 5c), which is in good agreement with the conductivity studies.
2.4 Photovoltaic performance and charge transport characteristics of PEDOT:PSS series-based OSCs
To investigate the PEDOT molecular weight effect of PEDOT:PSS HTMs on the OSC performance, conventional OSCs with a device structure of ITO/PEDOT:PSS/PBDB-T:ITIC/PDINO/Al were fabricated, where PBDB-T was used as the donor, ITIC, a non-fullerene compound firstly reported by Zhan's group,4,8 was used as the acceptor, and PDINO was used as the electron transporting material. The J–V curves of the PEDOT:PSSs-based OSCs and the related photovoltaic characteristics are given in Fig. 6 and Table 1, respectively. It can be observed that the device open-circuit voltage (Voc) exhibits negligible dependence on the PEDOT molecular weight and the values lie from 0.90 to 0.93 V. This phenomenon may be ascribed to the small and irregular variation in WFs of PEDOT:PSS HTMs as a function of PEDOT molecular weight which results in the similar energy-level offsets between the HTMs and the PBDB-T. On the contrary, the device short-circuit current (Jsc) enhances monotonically from 0.054 to 15.17 mA cm−2 with the increase of PEDOT molecular weight, which coincides well with the conductivity studies. Moreover, the device fill factor (FF) increases from 13.4 to 74.8% in the molecular weight range of ultralow to high grades and then decreases to 74.3% at the ultrahigh grade. Taken together, the device PCE first improves with the increase of PEDOT molecular weight and then reaches up to a saturation value of 10.36% at the critical molecular weight of high grade. Of note is that the P-UL-based device exhibits a PCE of ∼0% with a Jsc of ∼0% which may be attributed to the nearly insulating nature of the P-UL HTM (7.70 × 10−7 S cm−1) that severely restricts the extraction of photogenerated carriers. With increasing PEDOT molecular weight from ultralow to middle grade, we observed a successive jump in the PCE from ∼0 to 1.32 and to 9.91% synchronized with a successive jump in the Jsc from ∼0 to 8.26 and to 14.71 mA cm−2 which may be due to the enormous shifts in the HTM conductivity from nearly insulating (7.70 × 10−7 S cm−1) to medially conductive (1.14 × 10−4 S cm−1) and to highly conductive (7.22 × 10−4 S cm−1).64,65 Moreover, we found that the PCE may eventually reach saturation point (10.36%) with further increase of PEDOT molecular weight, which suggests the existence of a critical molecular weight where there is an optimal balance between hole collection and carrier recombination loss.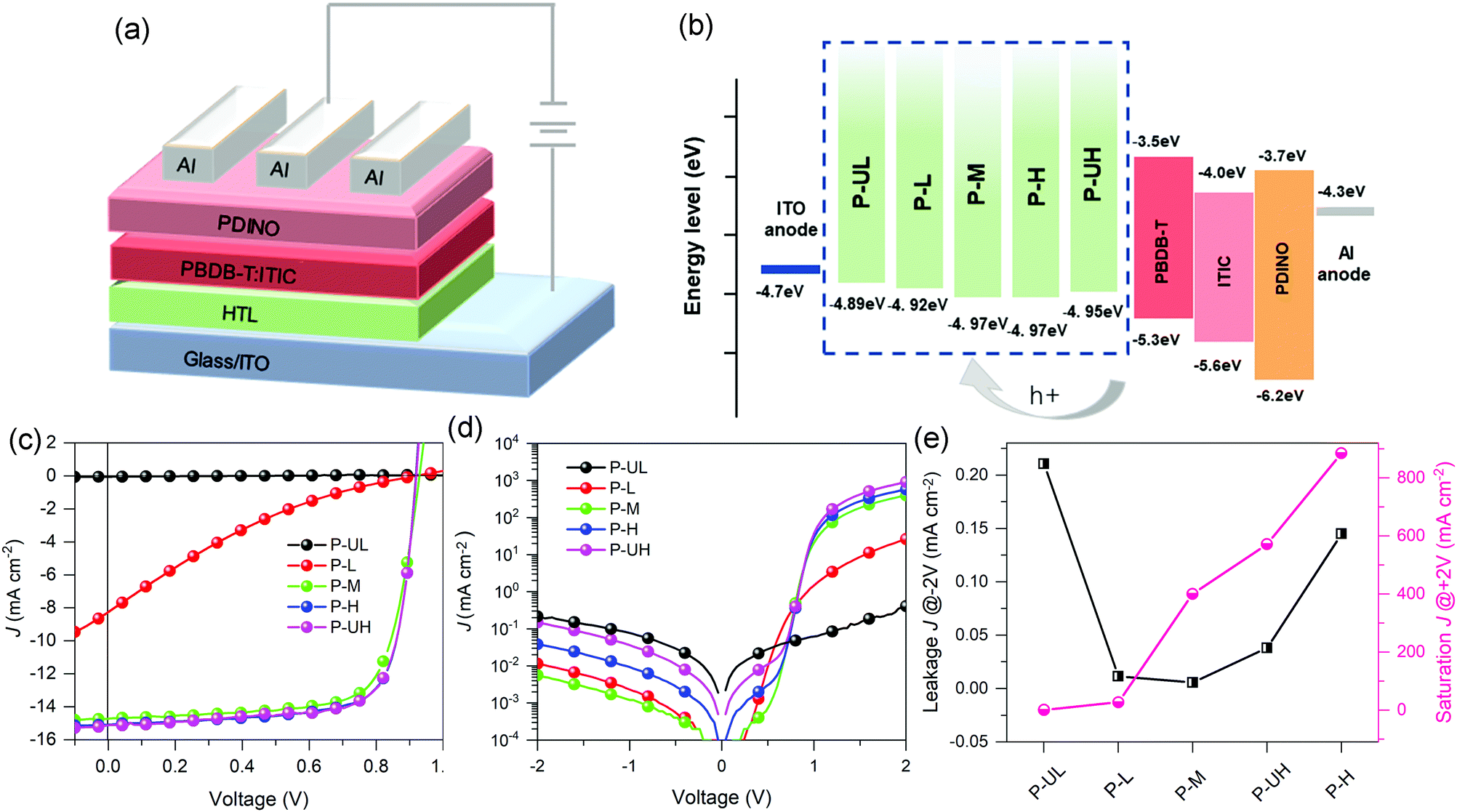 | ||
| Fig. 6 (a) Device configuration and (b) energy level alignment of OSCs. (c) J–V curves of OSCs. (d) Dark J–V curves of OSCs. (e) Plots of leakage and saturation current densities of OSCs against PEDOT molecular weight. | ||
| PEDOT:PSS HTL | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| P-UL | 0.90 | 0.054 | 13.4 | 0.0065 |
| P-L | 0.92 | 8.26 | 17.5 | 1.32 |
| P-M | 0.93 | 14.71 | 72.5 | 9.91 |
| P-H | 0.92 | 15.07 | 74.8 | 10.36 |
| P-UH | 0.92 | 15.17 | 74.3 | 10.36 |
To gain deep insight into the impact of PEDOT molecular weight of HTMs on the carrier transport and recombination of devices, dark J–V characteristics of the PEDOT:PSSs-based OSCs were measured and the curves are depicted in Fig. 6d. The leakage current density at −2 V and saturation current density at 2 V as a function of PEDOT molecular weight are summarized in Fig. 6e. It is known from the literature that a lower device leakage current represents a more favorable electron-blocking property for HTM while a higher device saturation current implies a stronger hole extraction capability for HTM.28,29,66 The leakage current curves indicate that the electron-blocking capability of PEDOT:PSS HTMs first enhances with the increase of PEDOT molecular weight and reaches the maxima at the middle grade molecular weight, and then diminishes gradually with further increase in molecular weight. The decrease in electron-blocking capability of PEDOT:PSS HTMs may be attributed to the elevation in their film surface roughness.28,66 The saturation current curves suggest that the hole extraction capability of PEDOT HTMs is positively correlated with PEDOT molecular weight, following the same variation trend as HTM conductivity. By combining the leakage and saturation current analysis, we believe that there exists a critical PEDOT molecular weight that produces optimal balance between carrier recombination and charge transport and thus resulting in the maxima device performance, highlighting the importance of optimizing PEDOT molecular weight for conducting polymers to help realize the full potential of HTMs in organic optoelectronic devices.
3 Conclusions
In conclusion, we have synthesized five batches of PEDOT:PSS HTMs with varied PEDOT molecular weights to explore the fundamental relationship between the PEDOT molecular weight, the HTM surface and electrical characteristics and the OSC performance. Experimental results reveal that both the surface roughness and conductivity of PEDOT:PSS films increase monotonically with the increase of PEDOT molecular weight, which are supported by the analysis of the PEDOT:PSS microstructures. The CV-derived WFs of the PEDOT:PSS films lie from ∼4.89 to ∼4.97 eV and display negligible dependence on the PEDOT molecular weight. When fabricated into PBDB-T:ITIC OSCs using the PEDOT:PSS series, we found that the device performance is highly PEDOT molecular weight dependent. With PEDOT molecular weight increasing from ultralow to middle grades, we observed a successive jump in the device PCE from ∼0 to 1.32% and to 9.91% accompanied by a significant enhancement in device Jsc and FF, which can be explained by the increasing conductivity. When the PEDOT molecular weight further increases, the device PCE reaches a saturation point of 10.36% which is thought to be largely determined by the combined effect of enhanced conductivity and increased roughness of HTMs that produces an optimal balance between the hole collection and carrier recombination loss in devices, as revealed by the dark J–V analysis. These findings highlight the important role of the PEDOT molecular weight of the conducting polymers for organic electronic devices. We hope that our PEDOT molecular weight–HTM properties–device performance relationship will serve as the basis to synthesize PEDOT conducting polymers and eventually help realize the full potential of HTMs in organic optoelectronic devices.4 Experimental section
4.1 Materials
3,4-Ethylenedioxythiophene (EDOT) monomer was purchased from Bayer AG. PSS (Mw 300 kg mol−1) dopant was purchased from Alfa Aesar. Ammonium persulfate (APS) oxidant, FeCl3 catalyst, EG emulsifier, ethanediamine reducing agent and organic solvents including methanol, chlorobenzene, acetone and isopropanol were purchased from Sinopharm Chemical and used without further purification. PBDB-T, ITIC and PDINO were purchase from Solarmer Materials.PEDOT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PSS (mass ratio 1:6) was synthesized by using oxidative polymerization following the literature procedure.41,67 The polymerizations were conducted at room temperature for 96 h. The catalyst and emulsifier amounts were tuned to vary the PEDOT molecular weights. The mole ratio of EDOT, APS, FeCl3 and EG are as follows: 1:1.5:0:0, 1:1.5:0.001:0, 1:1.5:0.044:0, 1:1.5:0.15:0, 1:1.5:0.02:20. And, the above reaction conditions will be abbreviated as APS, APS + 0.1% FeCl3, APS + 4.4% FeCl3, APS + 15% FeCl3 and APS + 2% FeCl3 + EG, respectively.
:PSS (mass ratio 1:6) was synthesized by using oxidative polymerization following the literature procedure.41,67 The polymerizations were conducted at room temperature for 96 h. The catalyst and emulsifier amounts were tuned to vary the PEDOT molecular weights. The mole ratio of EDOT, APS, FeCl3 and EG are as follows: 1:1.5:0:0, 1:1.5:0.001:0, 1:1.5:0.044:0, 1:1.5:0.15:0, 1:1.5:0.02:20. And, the above reaction conditions will be abbreviated as APS, APS + 0.1% FeCl3, APS + 4.4% FeCl3, APS + 15% FeCl3 and APS + 2% FeCl3 + EG, respectively.
4.2 PEDOT:PSS characterization
Ultraviolet-visible-near infrared (UV-vis-NIR) absorption spectra were obtained on a Hitachi U-4100 spectrophotometer. Raman spectra were measured using a DXR Raman Microscope. ESR spectra were obtained on a Bruker EMXplus ESR spectrometer using the following conditions: microwave frequency of 9.845 GHz, gain of 1 dB, modulation frequency of 100.00 kHz, power of 0.20 mV, power attenuator of 30.0 dB, modulation amplitude of 3.00 G, conversion time of 17.12 ms and sweep width of 185.1 G. XPS spectra were recorded on a Thermo Fisher VG Scienta R400 instrument. AFM images were obtained on an Agilent 5400 AFM instrument. CV measurements were recorded on a CHI660D electrochemical workstation using a PEDOT:PSS-coated ITO working electrode, an Ag/AgCl reference electrode and a platinum wire counter electrode. Tetrabutylammonium hexafluorophosphate in dichloromethane (0.1 M) was applied as the supporting electrolyte. Conductivity measurements were conducted based on a conductive device with the structure of ITO/PEDOT:PSS/Al and the conductivity value (σ) was calculated from the I/V curves by using the formula of σ = dI/AV, where d is the thickness of PEDOT:PSS film, I is the current, A is the area of the device (0.1 cm2) and V is the voltage. The conductive devices were fabricated with the following procedures. Firstly, PEDOT:PSS aqueous dispersions were spin-coated onto the clean ITO substrates at 4000 rpm for 40 s and annealed at 160 °C for 20 min. Then, the samples were transferred into a N2-filled glovebox. Finally, an Al electrode with the thickness of 100 nm was deposited at 5 × 10−4 bar in a thermal evaporator. Film thickness measurements were recorded on an Alpha step 500 surface profilometer.4.3 Solar cell fabrication and characterization
The patterned indium tin oxide (ITO) glass substrates were ultrasonically cleaned stepwise in detergent water, deionized water, acetone and isopropanol for 8 min each. Then, the substrates were blow-dried by N2 and treated by plasma for 13 min. After that, PEDOT:PSS aqueous dispersions were spin-coated onto the substrates at 4000 rpm for 40 s and annealed at 160 °C for 20 min. And then, the samples were transferred into the N2-filled glovebox. Subsequently, the PBDB-T:ITIC blend (weight ratio 1:1, 10 mg mL−1 in chlorobenzene with 1.0 vol% DIO) was spun at 2000 rpm for 30 s followed by an annealing treatment at 110 °C for 10 min to obtain an active layer. Next, PDINO electron transporting layer was spun at 3000 rpm for 30 s. Finally, the Al electrode was deposited at 5 × 10−4 bar in a thermal evaporator and the effective device area was 0.1 cm2. J–V measurements were tested in a N2-filled glovebox using a Keithley 2400 source meter under AM 1.5G (100 mW cm−2) illumination. Dark current measurements were implemented using the same instrument in the dark.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51773220, 51573205 and 22008184), the Education Ministry of China (Program for NCET-12-0714), the Educational Commission of Hubei Province of China (D20181505), the Scientific Research Foundation of Wuhan Institute of Technology (K201915), the Xiaozhang Foundation of Wuhan Institute of Technology (XZJJ2020007), the Open Project of Key Laboratory of Green Chemical Process of Ministry of Education (GCP20190209), and the Opening Project of Key Laboratory of Optoelectronic Chemical Materials and Devices of Ministry of Education Jianghan University (JDGD-202003).References
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS.
- D. Qian, Z. Zheng, H. Yao, W. Tress, T. R. Hopper, S. Chen, S. Li, J. Liu, S. Chen, J. Zhang, X.-K. Liu, B. Gao, L. Ouyang, Y. Jin, G. Pozina, I. A. Buyanova, W. Chen, O. Inganas, V. Coropceanu, J.-L. Bredas, H. Yan, J. Hou, F. Zhang, A. A. Bakulin and F. Gao, Nat. Mater., 2018, 17, 703–709 CrossRef CAS.
- H. Jiang, X. Li, H. Wang, Z. Ren, N. Zheng, X. Wang, Y. Li, W. Chen and R. Yang, Adv. Sci., 2020, 7, 1903455 CrossRef CAS.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS.
- C. Yang, S. Zhang, J. Ren, M. Gao, P. Bi, L. Ye and J. Hou, Energy Environ. Sci., 2020, 13, 2864–2869 RSC.
- C. Zhu, J. Yuan, F. Cai, L. Meng, H. Zhang, H. Chen, J. Li, B. Qiu, H. Peng, S. Chen, Y. Hu, C. Yang, F. Gao, Y. Zou and Y. Li, Energy Environ. Sci., 2020, 13, 2459–2466 RSC.
- J. Wu, J. Lee, Y.-C. Chin, H. Yao, H. Cha, J. Luke, J. Hou, J.-S. Kim and J. R. Durrant, Energy Environ. Sci., 2020, 13, 2422–2430 RSC.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS.
- J. Kim, D. Ouyang, H. Lu, F. Ye, Y. Guo, N. Zhao and W. C. H. Choy, Adv. Energy Mater., 2020, 10, 1903919 CrossRef CAS.
- S. Liu, W. Su, X. Zou, X. Du, J. Cao, N. Wang, X. Shen, X. Geng, Z. Tang, A. Yartsev, M. Zhang, W. Gruber, T. Unruh, N. Li, D. Yu, C. J. Brabec and E. Wang, J. Mater. Chem. A, 2020, 8, 5995–6003 RSC.
- X. Wang, J. Han, H. Jiang, Z. Liu, Y. Li, C. Yang, D. Yu, X. Bao and R. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 44501–44512 CrossRef CAS.
- Y. Tong, Z. Xiao, X. Du, C. Zuo, Y. Li, M. Lv, Y. Yuan, C. Yi, F. Hao, Y. Hua, T. Lei, Q. Lin, K. Sun, D. Zhao, C. Duan, X. Shao, W. Li, H.-L. Yip, Z. Xiao, B. Zhang, Q. Bian, Y. Cheng, S. Liu, M. Cheng, Z. Jin, S. Yang and L. Ding, Sci. China: Chem., 2020, 63, 758 CrossRef CAS.
- L. K. Reb, M. Böhmer, B. Predeschly, S. Grott, C. L. Weindl, G. I. Ivandekic, R. Guo, C. Dreißigacker, R. Gernhäuser, A. Meyer and P. Müller-Buschbaum, Joule, 2020, 4, 1880–1892 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma, C. An, C. He, Z. Wei, F. Gao and J. Hou, Adv. Mater., 2020, 32, 1908205 CrossRef CAS.
- X. Wang, Z. Du, K. Dou, H. Jiang, C. Gao, L. Han and R. Yang, Adv. Energy Mater., 2019, 9, 1802530 CrossRef.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- X. Wang, K. Dou, B. Shahid, Z. Liu, Y. Li, M. Sun, N. Zheng, X. Bao and R. Yang, Chem. Mater., 2019, 31, 6163–6173 CrossRef CAS.
- Z. Zhou, J. Duan, L. Ye, G. Wang, B. Zhao, S. Tan, P. Shen, H. S. Ryu, H. Y. Woo and Y. Sun, J. Mater. Chem. A, 2020, 8, 9684–9692 RSC.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094 CrossRef CAS.
- L. Sun, W. Zeng, C. Xie, L. Hu, X. Dong, F. Qin, W. Wang, T. Liu, X. Jiang, Y. Jiang and Y. Zhou, Adv. Mater., 2020, 32, 1907840 CrossRef CAS.
- V. Coropceanu, X.-K. Chen, T. Wang, Z. Zheng and J.-L. Bredas, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- N. Gasparini, A. Salleo, I. Mcculloch and D. Baran, Nat. Rev. Mater., 2019, 4, 229–242 CrossRef.
- N. Y. Doumon, F. V. Houard, J. Dong, P. Christodoulis, M. V. Dryzhov, G. Portale and L. J. A. Koster, J. Mater. Chem. A, 2019, 7, 5104–5111 CAS.
- J. Urieta-Mora, I. Garcia-Benito, A. Molina-Ontoria and N. Martin, Chem. Soc. Rev., 2018, 47, 8541–8571 RSC.
- H. Xu, F. Yuan, D. Zhou, X. Liao, L. Chen and Y. Chen, J. Mater. Chem. A, 2020, 8, 11478–11492 RSC.
- Z. Yu, W. Feng, W. Lu, B. Li, H. Yao, K. Zeng and J. Ouyang, J. Mater. Chem. A, 2019, 7, 11160–11169 RSC.
- J. Wang, Z. Zheng, D. Zhang, J. Zhang, J. Zhou, J. Liu, S. Xie, Y. Zhao, Y. Zhang, Z. Wei, J. Hou, Z. Tang and H. Zhou, Adv. Mater., 2019, 31, 1806921 CrossRef.
- Y. Lin, B. Adilbekova, Y. Firdaus, E. Yengel, H. Faber, M. Sajjad, X. Zheng, E. Yarali, A. Seitkhan, O. M. Bakr, A. El-Labban, U. Schwingenschlögl, V. Tung, I. McCulloch, F. Laquai and T. D. Anthopoulos, Adv. Mater., 2019, 31, 1902965 CrossRef CAS.
- B. Liu, Y. Wang, P. Chen, X. Zhang, H. Sun, Y. Tang, Q. Liao, J. Huang, H. Wang, H. Meng and X. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 33505–33514 CrossRef CAS.
- M. Zeng, X. Wang, R. Ma, W. Zhu, Y. Li, Z. Chen, J. Zhou, W. Li, T. Liu, Z. He, H. Yan, F. Huang and Y. Cao, Adv. Energy Mater., 2020, 10, 2000743 CrossRef CAS.
- S. Ahn, S.-H. Jeong, T.-H. Han and T.-W. Lee, Adv. Opt. Mater., 2017, 5, 1600512 CrossRef.
- B. Xu and J. Hou, Adv. Energy Mater., 2018, 8, 1800022 CrossRef.
- Y. Li, M. Liu, Y. Li, K. Yuan, L. Xu, W. Yu, R. Chen, X. Qiu and H.-L. Yip, Adv. Energy Mater., 2017, 7, 1601499 CrossRef.
- Y. Li, T. Liu, X. Qiu, Y. Zhou and Y. Li, ACS Sustainable Chem. Eng., 2019, 7, 961–968 CrossRef CAS.
- H. Cho, S.-H. Jeong, M. H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350, 1222 CrossRef CAS.
- H. Kim, K.-G. Lim and T.-W. Lee, Energy Environ. Sci., 2016, 9, 12–30 RSC.
- J. P. Thomas, L. Zhao, D. McGillivray and K. T. Leung, J. Mater. Chem. A, 2014, 2, 2383–2389 RSC.
- J. P. Thomas and K. T. Leung, J. Mater. Chem. A, 2016, 4, 17537–17542 RSC.
- S. Carli, M. D. Lauro, M. Bianchi, M. Murgia, A. D. Salvo, M. Prato, L. Fadiga and F. Biscarini, ACS Appl. Mater. Interfaces, 2020, 12, 29807–29817 CAS.
- Y. Li and N. Hong, J. Mater. Chem. A, 2015, 3, 21537–21544 RSC.
- Y. Li, H. Lou, F. Wang, Y. Pang and X. Qiu, ChemistrySelect, 2019, 4, 11406–11412 CrossRef CAS.
- A. I. Hofmann, D. Katsigiannopoulos, M. Mumtaz, I. Petsagkourakis, G. Pecastaings, G. Fleury, C. Schatz, E. Pavlopoulou, C. Brochon, G. Hadziioannou and E. Cloutet, Macromolecules, 2017, 50, 1959–1969 CrossRef CAS.
- B. Guo, Q. Yin, J. Zhou, W. Li, K. Zhang and Y. Li, ACS Sustainable Chem. Eng., 2019, 7, 8206–8214 CrossRef CAS.
- J. Huang, C. Wang, Z. Liu, X. Qiu, J. Yang and J. Chang, J. Mater. Chem. C, 2018, 6, 2311–2318 RSC.
- Y. Wu, J. Wang, X. Qiu, R. Yang, H. Lou, X. Bao and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 12377–12383 CrossRef CAS.
- W. Yu, X. Xie, Y. Li, Y. Li, R. Chen, X. Qiu and W. Huang, J. Mater. Chem. C, 2016, 4, 8077–8085 RSC.
- Z. Wang, H. Jiang, X. Liu, J. Liang, L. Zhang, L. Qing, Q. Wang, W. Zhang, Y. Cao and J. Chen, J. Mater. Chem. A, 2020, 8, 7765–7774 RSC.
- W. Wang, Q. Wu, R. Sun, J. Guo, Y. Wu, M. Shi, W. Yang, H. Li and J. Min, Joule, 2020, 4, 1070–1086 CrossRef CAS.
- H. Kang, M. A. Uddin, C. Lee, K.-H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS.
- N. Zhou, A. S. Dudnik, T. I. N. G. Li, E. F. Manley, T. J. Aldrich, P. Guo, H.-C. Liao, Z. Chen, L. X. Chen, R. P. H. Chang, A. Facchetti, M. O. de la Cruz and T. J. Marks, J. Am. Chem. Soc., 2016, 138, 1240–1251 CrossRef CAS.
- S. Ghosh, V. Gueskine, M. Berggren and I. V. Zozoulenko, J. Phys. Chem. C, 2019, 123, 15467–15476 CrossRef CAS.
- N. Massonnet, A. Carella, O. Jaudouin, P. Rannou, G. Laval, C. Celle and J.-P. Simonato, J. Mater. Chem. C, 2014, 2, 1278–1283 RSC.
- I. Zozoulenko, A. Singh, S. K. Singh, V. Gueskine, X. Crispin and M. Berggren, ACS Appl. Polym. Mater., 2019, 1, 83–94 CrossRef CAS.
- J.-H. Huang and C.-W. Chu, Electrochim. Acta, 2011, 56, 7228–7234 CrossRef CAS.
- Y. Li, L. Li, Y. Wu and Y. Li, J. Phys. Chem. C, 2017, 121, 8579–8588 CrossRef CAS.
- R. Jalili, J. M. Razal, P. C. Innis and G. G. Wallace, Adv. Funct. Mater., 2011, 21, 3363–3370 CrossRef CAS.
- X. Hu, G. Chen, X. Wang and H. Wang, J. Mater. Chem. A, 2015, 3, 20896–20902 RSC.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS.
- F. Liu, D. Chen, C. Wang, K. Luo, W. Gu, A. L. Briseno, J. W. P. Hsu and T. P. Russell, ACS Appl. Mater. Interfaces, 2014, 6, 19876–19887 CrossRef CAS.
- N. Wijeyasinghe, F. Eisner, L. Tsetseris, Y.-H. Lin, A. Seitkhan, J. Li, F. Yan, O. Solomeshch, N. Tessler, P. Patsalas and T. D. Anthopoulos, Adv. Funct. Mater., 2018, 28, 1802055 CrossRef.
- H. Shi, C. Liu, Q. Jiang and J. Xu, Adv. Electron. Mater., 2015, 1, 1500017 CrossRef.
- A. de Izarra, S. Park, J. Lee, Y. Lansac and Y. H. Jang, J. Am. Chem. Soc., 2018, 140, 5375–5384 CrossRef CAS.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- W. Li, X. Zhang, X. Zhang, J. Yao and C. Zhan, ACS Appl. Mater. Interfaces, 2017, 9, 1446–1452 CrossRef CAS.
- B. Xu and J. Hou, Adv. Energy Mater., 2018, 8, 1800022 CrossRef.
- Q. Yang, S. Yu, P. Fu, W. Yu, Y. Liu, X. Liu, Z. Feng, X. Guo and C. Li, Adv. Funct. Mater., 2020, 30, 1910205 CrossRef CAS.
- J. Friedrich and K. Werner, DE Pat., 4229192, 1994 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04372a |
| This journal is © The Royal Society of Chemistry 2020 |