
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of small Ag–Sb–Te nanocrystals with composition control†
Annina
Moser
,
Olesya
Yarema
,
Maksym
Yarema
and
Vanessa
Wood
 *
*
Department of Information Technology and Electrical Engineering, ETH Zurich, Gloriastrasse 35, CH-8092 Zurich, Switzerland. E-mail: vwood@ethz.ch
First published on 14th October 2020
Abstract
Ternary telluride nanocrystals have gained increasing interest as materials for thermoelectric, optoelectronic, and phase-change memory applications. Synthetic approaches for colloidal multicomponent tellurides however remain sparse. Here, we report a convenient, amide-promoted synthesis for Ag–Sb–Te nanocrystals with small sizes and narrow size distributions (e.g., nanocrystal diameters of 3.5 ± 0.8 nm). We focus on achieving composition control for Ag–Sb–Te nanocrystals by adjusting the ratio of cationic precursors and find a broad solid solution range for AgxSb1−xTe1.5−x nanocrystals (x is from 0.3 and 0.6), which extends beyond that measured in Ag–Sb–Te thin films. The ability to produce size- and composition-controlled Ag–Sb–Te nanocrystals is a first step in achieving bottom-up assembled Ag–Sb–Te semiconductors for device applications.
Introduction
Tellurides are promising candidates for a broad range of applications, such as infrared detection,1 photoluminescence,2 photovoltaics,3 phase change memory,4 and thermoelectrics.5 Taking the example of thermoelectrics, several tellurides exhibit high figures-of-merit, zT (e.g., Bi2Te3,6 PbTe,7 GeTe,8 and AgSbTe29,10). Ternary Ag–Sb–Te is particularly interesting for thermoelectrics, owing to spontaneous cationic ordering in nanoscale domains11,12 and bond anharmonicity.13 This contributes to glass-like phonon scattering, decreasing the lattice thermal conductivity while still enabling good electrical conductivity, therefore increasing the thermoelectric figure of merit.The bulk Ag–Sb–Te material system has a single ternary phase with a nominal stoichiometry of AgSbTe2.14 This phase has a characteristic octahedral rock-salt-type arrangement of atoms, tolerating small non-stoichiometry (e.g., Sb-rich AgxSb1−xTe1.5−x with x up to 0.4114,15). Understanding the formation and compositional range of rock-salt-type Ag2Te–Sb2Te3 solid solution in the Ag–Sb–Te material system is important for designing thermoelectric devices due to composition-dependent effects such as nanoscale domain ordering, atomic vacancy concentration, and inclusions of secondary phase.11,16–18
In general, colloidal nanocrystals promise new opportunities for device engineering. In addition to allowing non-vacuum solution-based fabrication, colloidal nanocrystals can be thought of as building blocks, enabling the properties of the thin film to be tuned through judicious control of the size, composition, and surface chemistry of the nanocrystals.19 In the context of thermoelectrics, such bottom-up fabrication of semiconductors has been shown to increase interface scattering20,21 and thus improve thermoelectric performance. Thermoelectrics assembled from nanocrystals of Bi2Te3,22,23 PbS–Ag,21,24 CuFeSe225 and other materials,26,27 which are then sintered, show lower thermal conductivity compared to bulk materials. Achieving better control of the individual nanocrystal building blocks and nanocrystal surface (i.e., which often become the grain boundaries of sintered films) allow highly flexible design of thermoelectric devices.
Telluride nanocrystals remain notably less developed than other chalcogenide materials. This is likely associated with the lack on tellurium precursors and the air sensitivity of telluride nanomaterials. While many binary telluride nanocrystals have been successfully synthesized (i.e., CdTe,28 HgTe,29 GeTe,30etc.), there is only a handful of reports of size-uniform multicomponent telluride nanocrystals.2,31 In fact, synthesis of colloidally stable AgSbTe2 nanocrystals has only been reported once.32 The authors prepared ternary AgSbTe2 and quaternary Ag–Pb–Sb–Te nanocrystals via reverse micellar approach, performed at room temperature. However, this synthesis lasts more than 10 hours and results in large size dispersion of nanocrystals, ranging from 3 to 15 nm. In a later publication, good composition and size control for colloidally stable quaternary AgPbmSbTe2+m (m between 1 and 18) is achieved.33 While promising properties for application in thermoelectrics are demonstrated, the Pb-free composition (i.e., AgSbTe2) is not reported.
Here, we present an amide-promoted synthesis, which yields small AgxSb1−xTe1.5−x nanocrystals with tunable composition between Ag0.3Sb0.7Te1.2 and Ag0.6Sb0.4Te0.9 (i.e., x ranges from 0.3 to 0.6). The choice of synthetic approach is motivated by previous success in preparing various chalcogenide nanocrystals.2,34,35 Due to improved nucleation rates, amide-promoted synthesis typically provides small size nanocrystals and quantitative reaction yields at short reaction times.
Experimental
Materials
Antimony(III) chloride (SbCl3, 99.999%) and tellurium (Te, broken ingots, 99.999%) are purchased from STREM; silver trifluoroacetate (CF3COOAg, AgTFA, 98%), lithium bis(trimethylsilyl)amide (LiN[Si(CH3)3]2, 95%), oleylamine (techn., 80–90%) and ethanol (anh., >95%) from Acros Organics; oleic acid (techn., 90%), tri-n-octylphosphine (TOP, 97%), hexane (anh., 95%) and toluene (anh., 99.8%) from Sigma Aldrich.General remarks on synthesis
All syntheses are carried out in an air-free environment using standard Schlenk line technique. Oleylamine and oleic acid are purified from water residues by heating to 100 °C under vacuum for at least 1 h. The solvents are then transferred into the glovebox. All other chemicals are used as purchased. Injection mixtures and stock solutions are prepared in a N2-filled glovebox. Stock solutions of 0.1 M cation salts are prepared by dissolving respective amounts in oleylamine at 70 °C. In order to handle SbCl3 in oleylamine, the precursor solution needs to be heated to 40 °C. A stock solution of 1 M tellurium in TOP (i.e., TOP:Te) is prepared by dissolving elemental Te at 220 °C and subsequent filtering of the cold solution.Synthesis of AgSbTe2 and AgxSb1−xTe1.5−x nanocrystals
In a typical synthesis, 0.5 mL AgTFA in oleylamine (0.05 mmol) and 0.5 mL SbCl3 in oleylamine (0.05 mmol) are diluted in 5.5 mL dried oleylamine and transferred to a three-neck flask. After heating the solution under vacuum to 80 °C, the atmosphere is changed to N2. Next, a mixture of 2.5 mmol LiN[Si(CH3)3]2 dissolved in 1 mL TOP and 1 mL TOP:Te (1 mmol) is swiftly added to the flask. After 1 h at 80 °C the heating mantle is removed and 8 mL toluene and 8 mL oleic acid are added to the hot crude solution.After cooling down naturally, the solution is transferred into the glovebox, where another 12 mL toluene is added. Upon mixing with an equal amount of ethanol, the turbid mixture is centrifuged at 6000 rpm for 3 minutes. The precipitate is redispersed in 4 mL toluene.
Non-stoichiometric AgxSb1−xTe1.5−x (i.e., other than AgSbTe2) nanocrystals are achieved by changing the Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sb precursor ratio (0.1 mmol cation precursors in total), while the large excess of Te and LiN[Si(CH3)3]2 is maintained. In this work, a Ag:Sb precursor ratio from 2:8 to 7:3 is explored.
:Sb precursor ratio (0.1 mmol cation precursors in total), while the large excess of Te and LiN[Si(CH3)3]2 is maintained. In this work, a Ag:Sb precursor ratio from 2:8 to 7:3 is explored.
Characterization of nanocrystals
TEM images are acquired on a Hitachi HT7700 operating at 100 keV, while HRTEM and STEM/EDX images are taken on a FEI Talos operating at 200 keV. For transmission electron microscopy (TEM), the crude nanocrystal solution is drop-cast on a Cu-mesh TEM grid. Ethanol soaking allows a sufficient removal of organics. For the detailed imaging of the stoichiometric AgSbTe2 samples, the TEM samples are prepared with purified and filtered colloidal solutions. Size distributions are evaluated by measuring >100 particles per sample with ImageJ software.Energy-dispersive X-ray spectroscopy (EDX) data are measured with a FEI Quanta 200 FEG SEM microscope, operating at 30 keV. X-Ray diffraction (XRD) measurements are carried out on a Rigaku SmartLab 9 kW System with a rotating Cu anode and a HyPix-3000SL 2D solid-state detector. Rietveld refinement is performed with FullProf_Suite software. For EDX and XRD analysis, colloidal solutions are additionally purified with ethanol and centrifugation, redispersed in hexane and drop-cast onto the respective sample holders. Details of ICP-OES measurements are given in the ESI.†
Results and discussion
Multicomponent nanocrystals containing cations from different groups (e.g., I–III–VI or I–V–VI) pose a synthetic challenge due to the need to balance the reactivity of the constituent cations.19 In particular, Ag and Sb exhibit very different electronegativities, and Sb3+ is known to easily undergo disproportionation.36 Furthermore, while a number of synthetic protocols exist for ternary I–V–VI sulfides37–39 and selenides,35,40–42 synthesis of I–V–VI tellurides poses additional challenges due to a high reactivity of Te-precursors and decreased stability.32 Since amide-promoted syntheses have enabled size and composition control of various nanocrystals including silver chalcogenides,34 I–III–VI selenides43 and tellurides,2 I–V–VI selenides42 and GeTe,30 we explore an amide-promoted synthesis for I–V–VI tellurides.A schematic of the reaction is shown in Fig. 1a. The use of Ag acetate, AgCl or AgI salts, as well as of SbI3, resulted in formation of binary Ag2Te, so we select AgTFA34 and SbCl3 as elemental sources. The combination of highly acidic AgTFA and relatively stable SbCl3 compound allow formation of the desired ternary product. Lithium amide and tellurium in TOP are swiftly added to the cation salts dissolved in oleylamine. Due to the higher reactivity of Ag compared to Sb, this reaction results in the initial formation of Ag2−xTe seeds followed by the incorporation of Sb (Fig. 1b and c). The cation-exchange process appears slow and Ag–Sb–Te nanocrystals prepared at shorter times are Sb-deficient. This can be explained by the rearrangement of the anionic sublattice upon conversion from Ag2−xTe to AgSbTe2. A reaction time of 1 h is therefore used to ensure a completion of this cation-exchange process. A measured yield of reaction is over 90%, which further corroborates a near complete uptake of all cations. Long reaction times at low temperatures have been previously shown to not deteriorate the size distribution of chalcogenide nanocrystals, prepared via amide-promoted syntheses.34
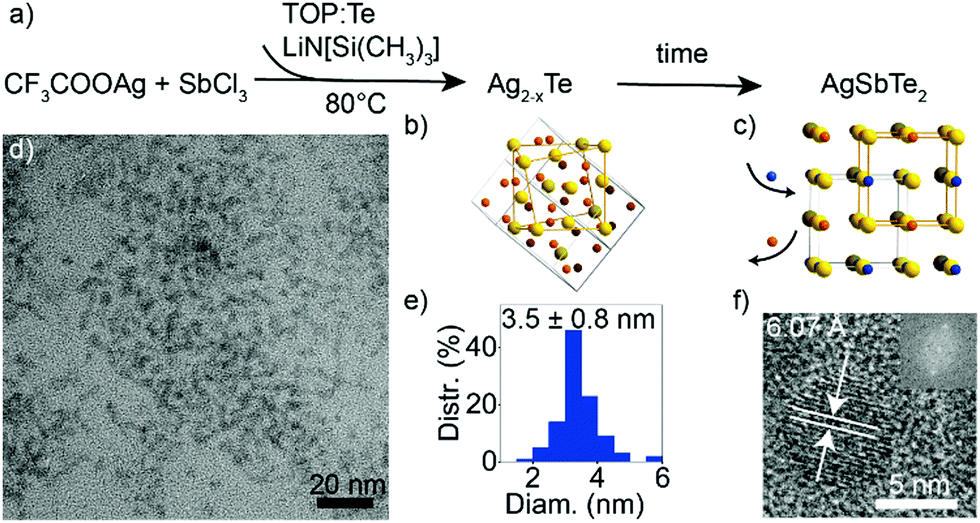 | ||
| Fig. 1 (a) Reaction schematic of amide-promoted synthesis of AgSbTe2 nanocrystals; (b) and (c) crystal structures of Ag2Te and AgSbTe2 (Ag atoms shown in orange, Sb in blue, and Te in yellow); (d) and (e) TEM image of stoichiometric AgSbTe2 nanocrystals and corresponding size histogram; (f) HR-TEM image of a single AgSbTe2 nanocrystal with its Fourier transform as inset. | ||
Synthesis of AgxSb1−xTe1.5−x nanocrystals requires an excess of lithium amide, which facilitates a fast conversion of the initial Ag and Sb precursors to intermediates with high enough and balanced reactivity. When halving the amount of amide, formation of binary Ag2Te byproduct is observed. The tellurium precursor (TOP:Te solution) is also added in excess (here, 10-fold with respect to cations). Lower concentrations of tellurium under the same reaction conditions lead to an incomplete synthesis, thus impeding composition control. Also, remaining SbCl3 reacts to Sb2O3 upon addition of oleic acid. We hypothesize that high excess of tellurium is necessary due to low reactivity and slow diffusion of the TOP:Te adduct at the studied reaction temperatures. Finally, a successful synthesis is observed only in a narrow temperature window around 80 °C. This temperature likely slows the reaction enough to prevent immediate formation of silver telluride compounds, while still being high enough to enable antimony to be incorporated in the structure.
The resulting AgSbTe2 nanocrystals have a size of 3.5 ± 0.8 nm (Fig. 1d and e). HR-TEM shows single crystalline particles with a characteristic interatomic distance of 6.07 Å (Fig. 1f and Fig. S1, S2, ESI†), which is close to the experimental bulk lattice constant (a = 6.0667 Å).44 STEM-EDX measurements of nanocrystal sub-monolayers confirm that indeed ternary nanocrystals have been formed (Fig. S3, ESI†).
While the crude solution is filterable, purification is a crucial step to obtain Ag–Sb–Te nanocrystals in volatile solvents. Although AgSbTe2 is known to be notoriously unstable,32 we found that it is possible to preserve colloidal stability for 1–2 days by adding oleic acid and toluene to the hot crude solution. During nanocrystal growth the solution is dark purple from an excess of tellurium (Fig. S4, ESI†) and with the addition of oleic acid, the solution turns brown and warms up. After addition of the oleic acid and toluene, the purification procedure (i.e., addition of ethanol, centrifugation, and redispersion in toluene) should be performed without delay or else leaching of Ag is observed. Oleic acid replaces the oleylamine at the surface of the nanocrystals and the toluene decreases the viscosity of the solution to ensure good mixing. In particular, surface Sb-atoms are stabilized well with oleic acid since they form a hard Lewis acid–base pair. Attempting to remove the oleylamine without addition of oleic acid leads to nanocrystals that are no longer colloidally stable.
Long reaction times and a large excess of tellurium precursor enables incorporation of all cations. Therefore, by changing the ratio of the cation precursors, AgTFA and SbCl3, it is possible to control the composition of nanocrystals over a wide range (Fig. 2a and Table S1, ESI†). EDX analysis reveals that the nanocrystal cation ratio generally follows the trend in cation precursor ratio. ICP-OES measurements confirm the composition range, as well as absence of Li cations (Fig. S5, ESI†). Towards Sb-rich AgxSb1−xTe1.5−x nanocrystals, there is an increase in Te-content, which likely balances a higher amount of positive charges (Table S1, ESI†). For cation precursor ratios Ag:Sb between 3:7 and 6:4, the resulting products are AgxSb1−xTe1.5−x nanocrystals of similar shape and size (Fig. 2b–g and Fig. S6, ESI†). If the content of Ag precursor exceeds this range, only a negligible amount of Sb is incorporated. Meanwhile, platelet formation is observed for large Sb precursor amounts, which is typical for hexagonal Sb2Te318 (Fig. S7 and Table S1, ESI†).
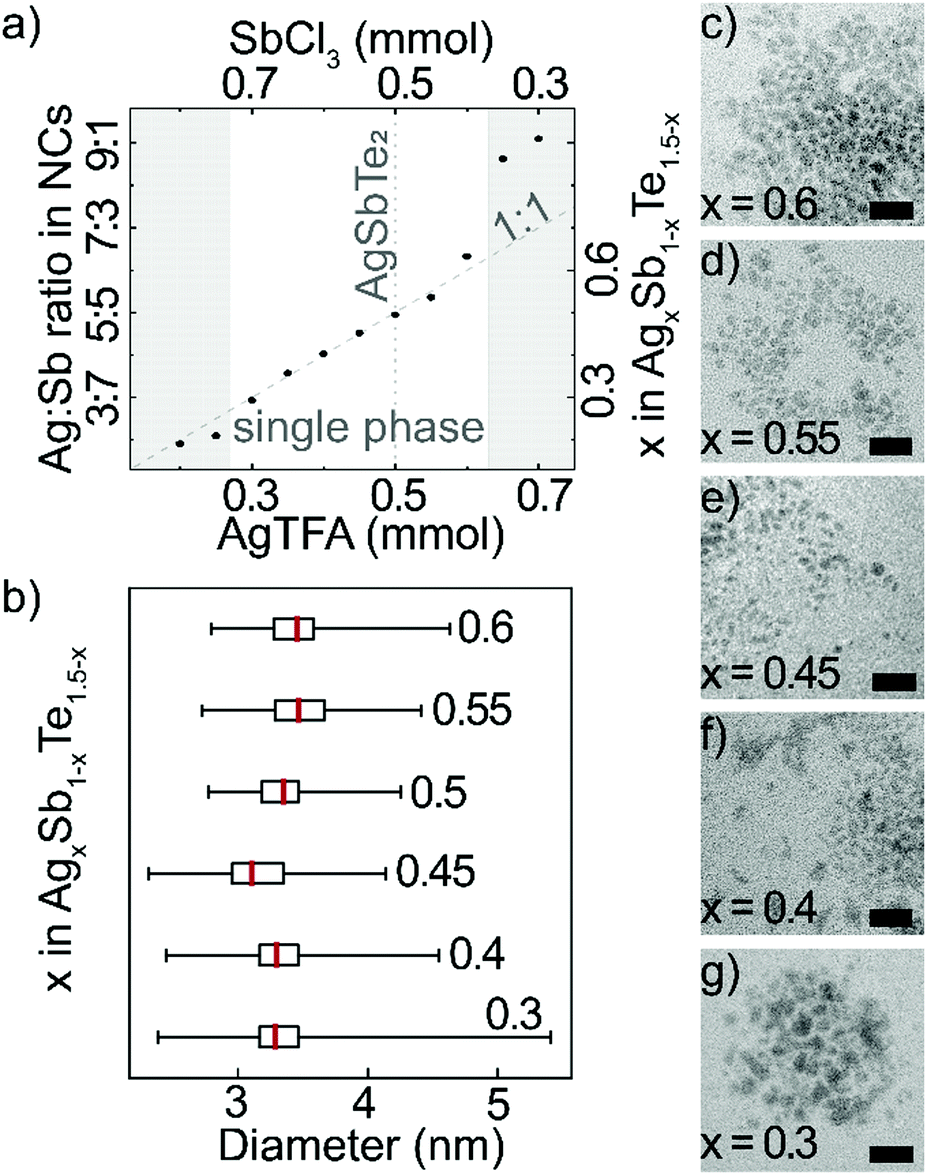 | ||
| Fig. 2 (a) Composition of AgxSb1−xTe1.5−x nanocrystals as a function of cation precursor ratio; (b) size distributions and (c)–(g) TEM images of AgxSb1−xTe1.5−x with variable x. Scale bars in (c)–(g) are 20 nm. | ||
Through HR-TEM (Fig. 1f and Fig. S1, S2, ESI†) and XRD (Fig. 3a), we are able to identify single crystalline, cubic phase nanocrystals, which indicate that nanocrystalline AgxSb1−xTe1.5−x material exhibit a broad solid solution. The diffraction patterns across this range correspond to the rock-salt phase of bulk AgSbTe2. Further analysis of the crystal structure is given in the ESI† (Fig. S8 and Table S2). With increasing Sb-content in AgxSb1−xTe1.5−x nanocrystals, the XRD peaks shift to higher angles (Fig. 3b). This corresponds to a smaller crystal lattice constant with increasing Sb-content, which is consistent with Sb3+ being a smaller cation (rionic,Sb = 0.85 Å) than Ag+ (rionic,Ag = 1.15 Å). At the same time, the small shift of XRD peaks suggests a high degree of covalent bonding in rock-salt AgxSb1−xTe1.5−x nanocrystals (Ag and Sb have similar covalent radii of 1.45 and 1.39 Å respectively) and thus only small ionic bonding character. The tellurium sublattice of AgSbTe2 and all solid solution AgxSb1−xTe1.5−x materials is also notably smaller than for binary Ag2Te or Sb2Te3 materials (Fig. S9 and S10, ESI†),44–46 additionally proving a ternary composition of obtained nanocrystals.
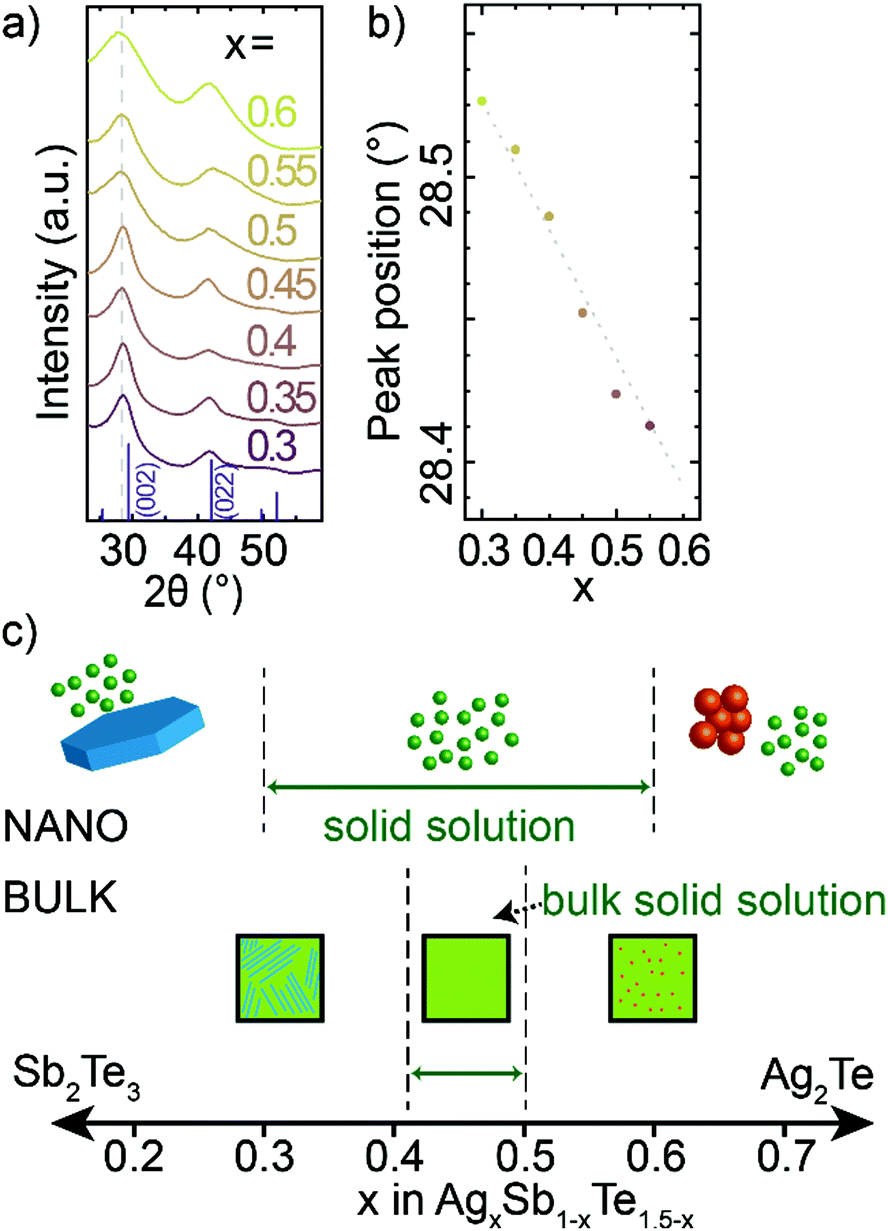 | ||
| Fig. 3 (a) XRD patterns of composition series of AgxSb1−xTe1.5−x nanocrystals and a bulk AgSbTe2; (b) positions of first peak as a function of composition; (c) proposed composition range of ternary Ag–Sb–Te phase for bulk14–16,50 and for nanocrystalline AgxSb1−xTe1.5−x materials. | ||
This composition study highlights that it is possible to make AgxSb1−xTe1.5−x alloys with x between 0.3 and 0.6 at the nanoscale, which represents a larger range for solid solutions than in bulk where solid solutions have only been reported only between Ag0.41Sb0.59Te1.09 and AgSbTe2 (Fig. 3c).14–16,47 This larger accessible composition range in nanocrystals compared to the bulk can be associated with the soft crystal boundaries (i.e., the small physical dimension of nanocrystals) or with the cation-exchange formation mechanism (i.e., facilitating metastable crystal structures).48,49
Conclusion and outlook
In this communication, we present a synthesis for small colloidal AgSbTe2 and AgxSb1−xTe1.5−x nanocrystals. A careful selection of cation and anion precursors, along with appropriate reaction temperature and time, allows the formation of ternary nanocrystals. Composition control is realized by varying the cation precursor ratio, revealing a significantly larger solid solution for AgxSb1−xTe1.5−x nanocrystals compared to bulk. This novel synthesis is an important step forward towards better understanding and further improvement of bottom-up thermoelectric, optoelectronic, and memory devices, which are built from telluride materials. Precise composition tuning for non-stoichiometric compounds may exhibit superior properties e.g., due to ordered network of atomic defects.43 The small size of nanocrystals may kinetically hinder phase separation even during the fabrication of thin films and pellets, thereby preserving the non-equilibrium Ag:Sb ratios in such devices. In the future, this amide-promoted synthesis could be up-scaled51 to achieve products on the gram-scale for device fabrication, as well as extended to prepare other nanoscale I–V–VI tellurides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the Swiss National Science foundation via a research grant (No. 175889), the NCCR “QSIT – Quantum Science and Technology”, and an Ambizione Fellowship (No. 161249). TEM and EDX measurements were performed at the Scientific Center for Optical and Electron Microscopy (ScopeM) of the Swiss Federal Institute of Technology. ICP measurements were performed at the Optical Materials Engineering Laboratory (OMEL) of the Swiss Federal Institute of Technology.References
- E. Lhuillier, S. Keuleyan, H. Liu and P. Guyot-Sionnest, Chem. Mater., 2013, 25, 1272–1282 CrossRef CAS.
- O. Yarema, M. Yarema, W. M. M. Lin and V. Wood, Chem. Commun., 2016, 52, 10878–10881 RSC.
- J. Britt and C. Ferekides, Appl. Phys. Lett., 1993, 62, 2851–2852 CrossRef CAS.
- L. Perniola, V. Sousa, A. Fantini, E. Arbaoui, A. Bastard, M. Armand, A. Fargeix, C. Jahan, J. F. Nodin, A. Persico, D. Blachier, A. Toffoli, S. Loubriat, E. Gourvest, G. Betti Beneventi, H. Feldis, S. Maitrejean, S. Lhostis, A. Roule, O. Cueto, G. Reimbold, L. Poupinet, T. Billon, B. De Salvo, D. Bensahel, P. Mazoyer, R. Annunziata, P. Zuliani and F. Boulanger, IEEE Electron Device Lett., 2010, 31, 488–490 CAS.
- H. Goldsmid, Materials, 2014, 7, 2577–2592 CrossRef CAS.
- D. Champier, Energy Convers. Manage, 2017, 140, 167–181 CrossRef.
- K. Kishimoto and T. Koyanagi, J. Appl. Phys., 2002, 92, 2544–2549 CrossRef CAS.
- E. Hazan, N. Madar, M. Parag, V. Casian, O. Ben-Yehuda and Y. Gelbstein, Adv. Electron. Mater., 2015, 1, 1500228 CrossRef.
- J. Ma, O. Delaire, A. F. May, C. E. Carlton, M. A. McGuire, L. H. VanBebber, D. L. Abernathy, G. Ehlers, T. Hong, A. Huq, W. Tian, V. M. Keppens, Y. Shao-Horn and B. C. Sales, Nat. Nanotechnol., 2013, 8, 445–451 CrossRef CAS.
- B. Du, H. Li, J. Xu, X. Tang and C. Uher, Chem. Mater., 2010, 22, 5521–5527 CrossRef CAS.
- J. Ma, O. Delaire, A. F. May, C. E. Carlton, M. A. McGuire, L. H. VanBebber, D. L. Abernathy, G. Ehlers, T. Hong, A. Huq, W. Tian, V. M. Keppens, Y. Shao-Horn and B. C. Sales, Nat. Nanotechnol., 2013, 8, 445–451 CrossRef CAS.
- W. Szczypka and A. Koleżyński, J. Alloys Compd., 2018, 732, 293–299 CrossRef CAS.
- D. T. Morelli, V. Jovovic and J. P. Heremans, Phys. Rev. Lett., 2008, 101, 035901 CrossRef CAS.
- R.-M. Marin, G. Brun and J.-C. Tedenac, J. Mater. Sci., 1985, 20, 730–735 CrossRef CAS.
- J. P. McHugh, W. A. Tiller, S. E. Haszko and J. H. Wernick, J. Appl. Phys., 1961, 32, 1785 CrossRef CAS.
- M. D. Nielsen, C. M. Jaworski and J. P. Heremans, AIP Adv., 2015, 5, 053602 CrossRef.
- J. D. Sugar and D. L. Medlin, J. Alloys Compd., 2009, 478, 75–82 CrossRef CAS.
- S. N. Zhang, T. J. Zhu, S. H. Yang, C. Yu and X. B. Zhao, Acta Mater., 2010, 58, 4160–4169 CrossRef CAS.
- O. Yarema, M. Yarema and V. Wood, Chem. Mater., 2018, 30, 1446–1461 CrossRef CAS.
- S. Ortega, M. Ibáñez, Y. Liu, Y. Zhang, M. V. Kovalenko, D. Cadavid and A. Cabot, Chem. Soc. Rev., 2017, 46, 3510–3528 RSC.
- M. Ibáñez, Z. Luo, A. Genç, L. Piveteau, S. Ortega, D. Cadavid, O. Dobrozhan, Y. Liu, M. Nachtegaal, M. Zebarjadi, J. Arbiol, M. V. Kovalenko and A. Cabot, Nat. Commun., 2016, 7, 10766 CrossRef.
- M. Scheele, N. Oeschler, K. Meier, A. Kornowski, C. Klinke and H. Weller, Adv. Funct. Mater., 2009, 19, 3476–3483 CrossRef CAS.
- Y. Liu, Y. Zhang, K. H. Lim, M. Ibáñez, S. Ortega, M. Li, J. David, S. Martí-Sánchez, K. M. Ng, J. Arbiol, M. V. Kovalenko, D. Cadavid and A. Cabot, ACS Nano, 2018, 12, 7174–7184 CrossRef CAS.
- M. Ibáñez, A. Genç, R. Hasler, Y. Liu, O. Dobrozhan, O. Nazarenko, M. De La Mata, J. Arbiol, A. Cabot and M. V. Kovalenko, ACS Nano, 2019, 13, 6572–6580 CrossRef.
- L. Vaure, Y. Liu, D. Cadavid, F. Agnese, D. Aldakov, S. Pouget, A. Cabot, P. Reiss and P. Chenevier, ChemNanoMat, 2018, 4, 982–991 CrossRef CAS.
- H. Yang, L. A. Jauregui, G. Zhang, Y. P. Chen and Y. Wu, Nano Lett., 2012, 12, 540–545 CrossRef CAS.
- M. Ibañez, R. Hasler, A. Genc, Y. Liu, B. Kuster, M. Schuster, O. Dobrozhan, D. Cadavid, J. Arbiol, A. Cabot and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 8025–8029 CrossRef.
- V. Kloper, R. Osovsky, J. Kolny-Olesiak, A. Sashchiuk and E. Lifshitz, J. Phys. Chem. C, 2007, 111, 10336–10341 CrossRef CAS.
- S. E. Keuleyan, P. Guyot-Sionnest, C. Delerue and G. Allan, ACS Nano, 2014, 8, 8676–8682 CrossRef CAS.
- O. Yarema, A. Perevedentsev, V. Ovuka, P. Baade, S. Volk, V. Wood and M. Yarema, Chem. Mater., 2018, 30, 6134–6143 CrossRef CAS.
- M.-A. Langevin, T. Pons, A. M. Ritcey and C. Nì Allen, Nanoscale Res. Lett., 2015, 10, 255 CrossRef.
- A. J. Karkamkar and M. G. Kanatzidis, J. Am. Chem. Soc., 2006, 128, 6002–6003 CrossRef CAS.
- I. U. Arachchige, J. Wu, V. P. Dravid and M. G. Kanatzidis, Adv. Mater., 2008, 20, 3638–3642 CrossRef CAS.
- M. Yarema, S. Pichler, M. Sytnyk, R. Seyrkammer, R. T. Lechner, G. Fritz-Popovski, D. Jarzab, K. Szendrei, R. Resel, O. Korovyanko, M. A. Loi, O. Paris, G. Hesser and W. Heiss, ACS Nano, 2011, 5, 3758–3765 CrossRef CAS.
- O. Yarema, M. Yarema, A. Moser, O. Enger and V. Wood, Chem. Mater., 2020, 32, 2078–2085 CrossRef CAS.
- R. Reiche, J. P. Holgado, F. Yubero, J. P. Espinos and A. R. Gonzalez-Elipe, Surf. Interface Anal., 2003, 35, 256–262 CrossRef CAS.
- B. Zhou, M. Li, Y. Wu, C. Yang, W.-H. Zhang and C. Li, Chem. – Eur. J., 2015, 21, 11143–11151 CrossRef CAS.
- N. Suriyawong, B. Aragaw, J.-B. Shi and M.-W. Lee, J. Colloid Interface Sci., 2016, 473, 60–65 CrossRef CAS.
- L. Hu, R. J. Patterson, Z. Zhang, Y. Hu, D. Li, Z. Chen, L. Yuan, Z. L. Teh, Y. Gao, G. J. Conibeer and S. Huang, J. Mater. Chem. C, 2018, 6, 731 RSC.
- D. B. Agocs, T. Danna and A. L. Prieto, ACS Appl. Energy Mater., 2019, 2, 1903–1910 CrossRef CAS.
- Y. Liu, D. Cadavid, M. Ibáñez, J. De Roo, S. Ortega, O. Dobrozhan, M. V. Kovalenko and A. Cabot, J. Mater. Chem. C, 2016, 4, 4756–4762 RSC.
- A. Das, B. Hsu, A. Shamirian, Z. Yang and P. T. Snee, Chem. Mater., 2017, 29, 4597–4602 CrossRef CAS.
- O. Yarema, M. Yarema, D. Bozyigit, W. M. M. Lin and V. Wood, ACS Nano, 2015, 9, 11134–11142 CrossRef CAS.
- E. Quarez, K.-F. Hsu, R. Pcionek, N. Frangis, E. K. Polychroniadis and M. G. Kanatzidis, J. Am. Chem. Soc., 2005, 127, 9177–9190 CrossRef CAS.
- J. Schneider and H. Schulz, Z. Kristallogr. – Cryst. Mater., 2014, 203, 1–16 Search PubMed.
- T. L. Anderson and H. B. Krause, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1307–1310 CrossRef CAS.
- O. Cojocaru-Mirédin, L. Abdellaoui, M. Nagli, S. Zhang, Y. Yu, C. Scheu, D. Raabe, M. Wuttig and Y. Amouyal, ACS Appl. Mater. Interfaces, 2017, 9, 14779–14790 CrossRef.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS.
- H. Li, M. Zanella, A. Genovese, M. Povia, A. Falqui, C. Giannini and L. Manna, Nano Lett., 2011, 11, 4964–4970 CrossRef CAS.
- B. Du, J. Xu, W. Zhang and X. Tang, J. Electron. Mater., 2011, 40, 1249–1253 CrossRef CAS.
- M. Yarema, O. Yarema, W. M. M. Lin, S. Volk, N. Yazdani, D. Bozyigit and V. Wood, Chem. Mater., 2017, 29, 796–803 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S10 and Tables S1, S2. See DOI: 10.1039/d0tc00880j |
| This journal is © The Royal Society of Chemistry 2020 |