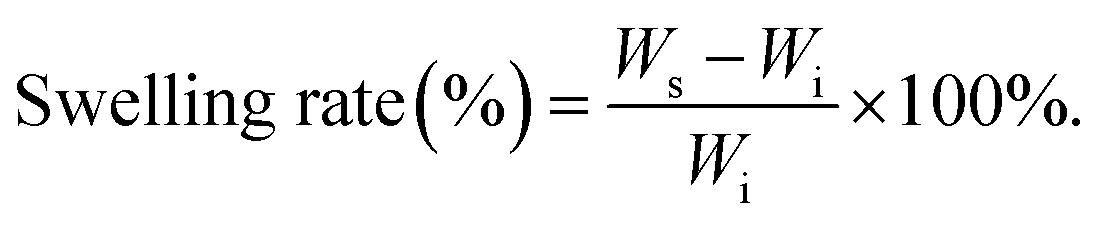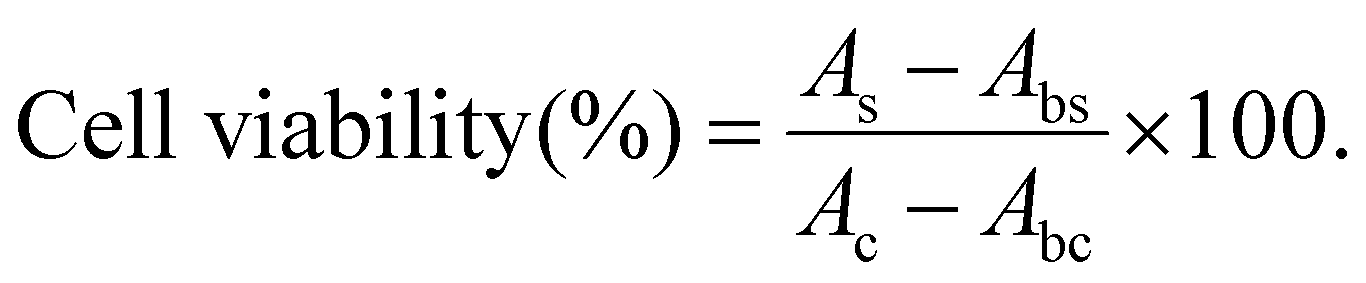A high-strength double network polydopamine nanocomposite hydrogel for adhesion under seawater†
Received
25th February 2020
, Accepted 29th June 2020
First published on 2nd July 2020
Abstract
Mussel-inspired catechol-based strategy has been widely used in the development of underwater adhesives. Nonetheless, the properties of the adhesives were still severely limited under harsh environments. A facile approach was proposed herein to prepare a double network hydrogel adhesive with low swelling rate and high strength in seawater, where the first network was polyacrylamide (PAM) and the second network was alginate (Alg). Meanwhile, polydopamine (PDA) nanoparticles, which were formed through self-polymerization as adhesion anchoring sites, distributed evenly throughout the double network hydrogel and effectively enhanced the adhesion capability of the hydrogel. The properties of the resulting hydrogel have been fully characterized. The optimal adhesion strength of the hydrogel adhesive in seawater was as high as 146.84 ± 7.78 kPa. Furthermore, the hydrogel also has excellent ability to promote the growth of zooxanthellae. Our studies provide useful insights into the rational design of underwater adhesives with high performances even beyond nature.
1 Introduction
Wet adhesives have drawn much attention in a wide range of fields such as biomedical materials for surgical adhesives, industrial applications for underwater repair, and various functional coatings for surface modification.1 Conventional adhesives are frequently frustrated in underwater conditions, especially in physiological and seawater environments, because the interfacial water molecules block adhesive-substrate interactions.2 When the adhesive materials are applied to submerged substrates, surface-bound waters will weaken the interfacial adhesion and destroy the integrity of the adhesive, creating a barrier to adhesive materials preventing them from making beneficial contacts.3 Although various strategies have been designed to fabricate various underwater adhesives for wet or even underwater conditions, including biomimetic adhesives,4,5 polymeric adhesives,6–9 and protein adhesives,10,11 most of the current underwater adhesives are prepolymers which display a flow behavior of viscoelastic fluid to achieve effective dispersion of adhesives. On the one hand, this kind of prepolymerized adhesive usually cures into a bonding joint without flexibility after reacting with water molecules, limiting the practical applications in resisting underwater mechanical movement. On the other hand, poor biocompatibility and weak ability to load active factors of the prepolymerized adhesives limit their application in the biomedical field. Hydrogels, as a water-swollen three-dimensional network based on hydrophilic polymer chains, have special rheological properties, good ductility and strong toughness.12 The hydrogel can be used typically as a viscoelastic solid adhesive for underwater adhesion to compensate the defects of viscoelastic fluid adhesive. However, traditional hydrogel adhesives are liable to lose their adhesion ability and mechanical properties in a wet environment in a shorter period, because of their strong swelling hydration.13 In general, in view of the complexity of surface modification,14,15 the development of a tough underwater hydrogel adhesive completely based on a chemical reaction with strong and durable adhesion under harsh conditions (e.g., physiological environment and seawater) has remained a challenge in adhesion science and materials engineering.
Nature always inspires human beings in many ways. Mussels living in the ocean secrete mussel foot proteins (mfps) in the form of a dense coacervate with two oppositely charged functional groups, which are impressive for their glue power on diverse submerged substrates in the ocean.16 These mfps are mainly composed of 20–30 mol% 3,4-dihydroxyphenylalanine (DOPA) and ∼20 mol% cationic lysine. The synergistic effect of these two functionalities make low interfacial energy of the coacervate to realize robust underwater adhesion.17 Therefore, in order to address the above challenges, it is very attractive to prepare catechol-based hydrogel adhesives inspired by mussel's excellent anti-wet adhesion performance. Quite often, derivatives of catechol, including dopamine (DA) and polydopamine (PDA), are used to simulate mfps.18,19 PDA is a nanocluster hybrid formed by oxidation of DA. The molecular structure of PDA is similar to that of mfps and PDA can easily adhere to almost all types of surfaces.20,21 In addition, the reversible non-covalent interactions within PDA, including π–π stacking and hydrogen bonds, can effectively dissipate energy during hydrogel deformation and prevent crack propagation by broken bonds during mechanical failure to enhance the toughness and stiffness of hydrogels.22 More noteworthy is that phenolic hydroxyl groups on the PDA chains enable superior cell viability and tissue adhesion properties.23
It has been elucidated that catechol-based hydrogel adhesives can strongly adhere to any rough surface based on the combination of noncovalent and/or covalent interactions with the substrate.24,25 However, a single network design of catechol-based hydrogel adhesives reported previously usually showed weak mechanical strength and poor deformability after long-term application in a wet environment,26 and most of them are metal ion-crosslinked hydrogels with potential toxicity.27 Therefore, double network hydrogels, as a kind of double-reinforced polymer network material, can provide a possibility for long-term underwater applications of adhesives to meet the requirements of significant mechanical properties and highly reversible deformation properties.
Recently, an ionically and covalently crosslinked polyacrylamide-alginate (PAM–Alg) double network hydrogel with highly stretchable capability and exceptional toughness was reported.28–30 PAM and Alg also provide a strengthening mechanism and have a cooperative effect. The high viscosity of sodium alginate (Alg-Na) and the gelling ability of alginate networks with divalent/trivalent cations make the PAM–Alg hydrogel particularly suitable for high salinity environments such as seawater.31,32 Furthermore, it has been proved that some double network hydrogels, one of the networks of which was catechol-alginate polymer chain, can achieve strong adhesion effect underwater. The main focus of this kind of hydrogel was covalent attachment of catechol groups to side-groups of alginate.33,34 However, the decrease of adhesion ability caused by the inevitable oxidation of catechol groups and the complexity of preparation of these hydrogels caused by grafting catechol groups imposed a series of limitations on mussel-inspired adhesives.35 Consequently, it is more preferred to construct a catechol-based double-network hydrogel adhesive with strong and durable adhesion under seawater using a simple method.
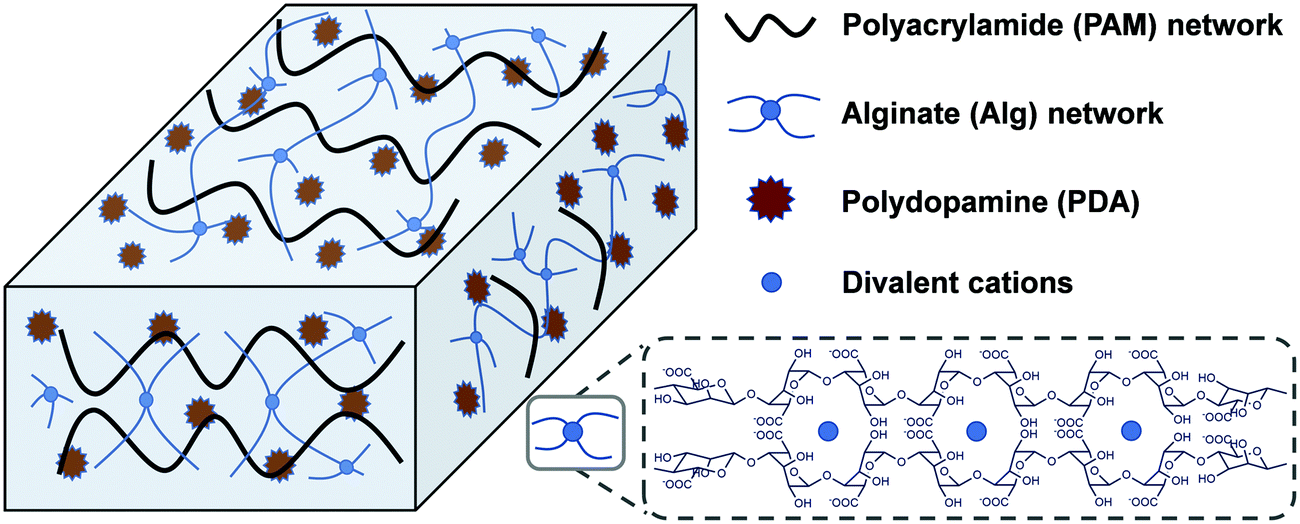
Herein, a mussel and algae inspired synergy strategy was proposed to fabricate a low swellable, high adhesion and strength nanocomposite hydrogel under seawater. This hydrogel consisted of two networks which are crosslinked physically and chemically. One is a covalently crosslinked PAM network, and another is cation (Ca2+ or Mg2+)–chelated calcium alginate (Alg-Ca) or magnesium alginate (Alg-Mg) network. The self-polymerized PDA nanoparticles distributed uniformly throughout the hydrogel system were first formed by rapid oxidation of DA in an alkalescent environment. In the process of formation of PDA, the free-radical polymerization of acrylamide (Am) monomer was triggered to form a PAM network with addition of a crosslinking agent, the formed PAM network can further prevent excessive oxidation of DA to maintain enough free phenolic hydroxyl groups. The existence of a finally formed alginate network (Alg-Ca or Alg-Mg) contributed to low swelling rate of the composite hydrogel, and thus inhibited the destruction of the hydrogel by seawater hydration (Scheme 1). These multiple interactions among phenolic hydroxyl groups of PDA, hydroxyl groups of Alg, and amino groups in PAM, as well as reversible non-covalent interactions in PDA, enable hydrogels to transfer loads effectively during mechanical deformation. In general, the PAM–Alg–PDA hydrogel combined the advantages of a double network and PDA together, and therefore it can achieve high adhesion capability, excellent mechanical properties and remarkable cell compatibility in seawater. The hydrogel adhesive exhibited the potential for wet environment applications in the biomedical field, such as tissue adhesives.
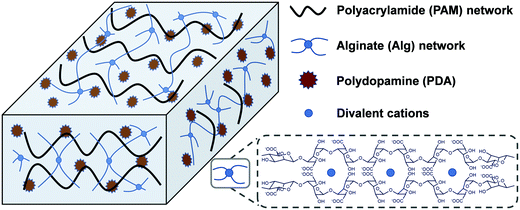 |
| | Scheme 1 Schematic structure of PAM–Alg–PDA hydrogel adhesive. | |
2 Experimental
2.1 Materials
Sodium alginate (Alg-Na, 200–500 mPa s), dopamine hydrochloride (DA), triethanolamine (TEA), acrylamide (Am), ammonium persulfate (APS), N,N′-methylene bis-acrylamide (MBAm) and all inorganic salts used in the preparation of artificial seawater were purchased from Aladdin Industrial Inc. (Shanghai, China). Zooxanthellae and F/2 Medium were purchased from Guangyu Biological Co., Ltd (Shanghai, China). A CCK-8 cell viability assay kit was purchased from Kaiji Biotechnology co., Ltd (Jiangsu, China). All chemicals and solvents were used without further purification.
2.2 Preparation of hydrogel adhesives
Alg-Na, Am and MBAm with different mass fractions (Table S1, ESI†) were fully dissolved in TEA buffer solution (0.2 M, pH = 9.8), then DA was added with stirring for 20 min to make DA self-oxidize to PDA (Fig. S1, ESI†). APS solution and the above solution were uniformly mixed to form a single network PAM–PDA hydrogel. The unreactive monomer was removed by rinsing with deionized water, and then the hydrogel was completely immersed in artificial seawater (Table S2, ESI†) to further form double network PAM–Alg–PDA hydrogel adhesive, which can work in seawater.
2.3 Structural and morphological characterization of hydrogels
The structural analysis of hydrogel adhesives was performed using a Fourier transform infrared (FTIR) spectrometer (Nicolet 5700, Thermo Fisher Scientific, America). The samples were recorded in the wavelength range of 4000–500 cm−1 using KBr. The microstructure and surface morphology of the freeze-dried hydrogels was observed using a field emission scanning electron microscope (Ultra Plus, Carl Zeiss, Germany).
2.4 Adhesion test
The prepared PAM–Alg–PDA hydrogel sample [cylinder, diameter (D) = 35 mm, height (H) = 1.5 mm] was directly adhered between the center of the polypropylene cap and other different substrates (e.g., high density polyethylene (HDPE), polytetrafluoroethylene (PTFE), rubber, steel, shell, and glass) as an intuitive display of the adhesion effect under seawater. The adhesion properties of the hydrogels were investigated by glass lap shear tests according to ASTM F2255-05 (2015) on the tensile testing machine (FLR-303, Flora Automatic Technology, China) at the testing speed of 10 mm min−1. All glass substrates were pretreated by rinsing with ethanol and deionized water and then dried before the test. The lap shear joint [length (L) × width (W) = 25 × 25 mm] was completely fabricated by adding 100 μL pre-gel adhesive solution between two substrates in air (dry environment groups) and in seawater (in seawater groups), respectively. Addition of a weight (100 g) held the substrates together for 30 min, the prepared joint with an iron clip was placed in air and artificial seawater for 6 h and 24 h, respectively. The samples of the seawater group were taken out from seawater and dried in air for 6 h. Shear adhesive strength was defined as the ratio of maximum tensile force at joint failure to the joint overlap area. The measurements of each sample were repeated at least five times (N = 5).
2.5 Swelling behavior
The prepared hydrogel sample [cube, L = W = H = 10 mm] was immersed in artificial seawater and weighed at specific time to investigate the swelling performance via the traditional swelling method at room temperature. The swelling rate (SR) was defined as follows (N = 5):| | 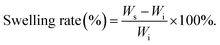 | (1) |
where Ws and Wi are the weight of the swelling samples at different time points and the original samples, respectively.
2.6 Rheological measurements
The dynamic rheological properties of pre-formed hydrogel samples [cylinder, D = 25 mm, H = 1.0 mm] were characterized at 25 °C using a strain-controlled rheometer equipped with 25 mm parallel plates (MCR 302, Anton Paar, Austria). The dynamic frequency sweeps were performed in the linear viscoelastic region of materials at 1.0% strain amplitude.
2.7 Mechanical test
The tensile samples [Cuboid, L = 45 mm, W = 1–3 mm, H = 40 mm] with a spacing of the clamps of 10 mm and compressive samples [cylinder, D = 25 mm, H = 20 mm] were prepared to perform the tensile test (FLR-303, Flora Automatic Technology, China) and compressive test (Instron 5940, Instron, America) at the speed of 5 mm min−1. The nominal stress is the applied force divided by the cross-sectional area of the undeformed sample. The strain is the length of a deformed sample divided by the initial length. The elastic modulus of tensile samples is obtained by calculating the slope of the stress–strain curve in the linear region. Compressive strength of the sample was determined by the ratio of the force at 90% strain (before immersion groups) or the maximum force required to crush the sample (after immersion groups) to the contact area (N = 5).
2.8 Cytocompatibility evaluation
Zooxanthellae cells were used to evaluate the cell compatibility of hydrogels through using CCK-8. Briefly, suspended zooxanthellae cells collected from symbiotic corals were cultured on F/2 medium (23 °C, 12 h light/12 h dark). The samples (S = 0.32 cm2, H = 1 mm) were purified by deionized water, 75% ethanol and F/2 medium, and then laid on the bottom of a 96-well plate. A count of 2 × 104 cells per well was seeded into a 96-well plate and incubated with a hydrogel.
The cell viability was assessed after 2 days, 3 days, 4 days, 5 days, and 6 days of culture. 10 μL CCK-8 solution was added to each well and incubated at 23 °C under light for 6 h. Later, 100 μL of the supernatant solution were transferred to another 96-well plate. The absorbance of the solution was measured using a Microplate Reader (Multiskan FC, Thermo, America) at 450 nm. The cell viability was calculated using the following equation (N = 5):
| | 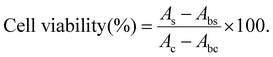 | (2) |
where
As,
Ac,
Abs and
Abc represent the absorbance of the sample groups, control groups (without hydrogels), blank groups for the sample (without cells), and blank groups for the control (without cells and hydrogels), respectively.
2.9 Statistical analysis
Statistical analysis of data was performed by one-way analysis of variance (ANOVA) using IBM SPSS 23.0 software. Post hoc comparisons were performed using Tukey's honest significant difference (HSD) test to compare means of multiple groups. Results were expressed as mean ± standard deviation, with p values < 0.05 indicating significance.
3 Results and discussion
3.1 Structural and morphological analysis of PAM–Alg–PDA hydrogel
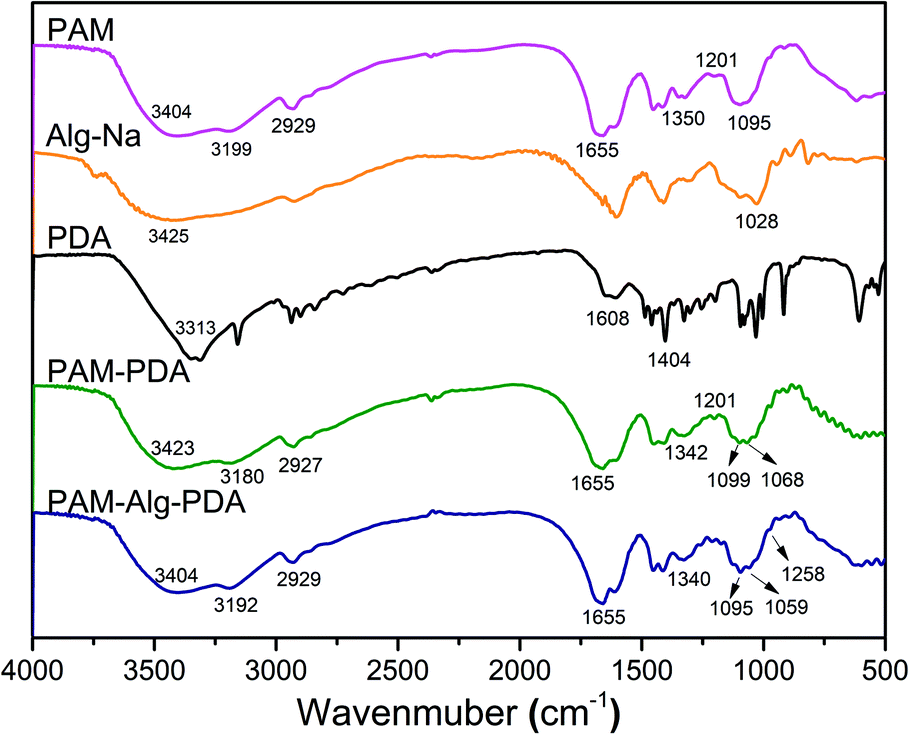
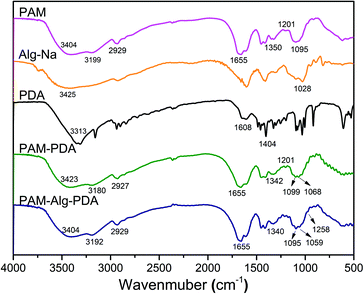
The FTIR spectra of hydrogel materials and individual components are shown in Fig. 1. The strong absorption band of Alg-Na at 1028 cm−1 refers to the –CO– stretching vibration peak of an acetal structure, which is the characteristic peak of a β-1,4 glycosidic bond.36 The peaks of PDA near 1500 cm−1 correspond to aromatic rings, and the bands near 3400 cm−1 refer to the overlap of water adsorbed in PDA polymers and the hydroxyl groups of PDA.37 The PAM hydrogel has the following peaks: 3500–3100 cm−1 (stretching vibration of N–H), 2929 cm−1 (antisymmetric stretching vibration of –CH2), 1655 cm−1 (stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, amide I), 1618 cm−1 (in-plane deformation of primary amine –NH, amide II), 1450 cm−1 (shear vibration of –CH2 in plane), 1419 cm−1 (stretching vibration of primary amide –CN), 1350 cm−1 (deformation of –CH) and 1095 cm−1 (in-plane swing of –NH2). The relative intensity of the absorption peak at 1201 cm−1 (stretching vibration of aliphatic amines C–N) of the PAM–PDA hydrogel was increased in comparison to the PAM hydrogel and PDA, indicating that C–NH2 of PAM reacted with the phenolic hydroxyl groups of PDA.38 In addition, a hydrogen bond causes the band to shift toward low frequency for stretching vibration, while a hydrogen bond causes the band to shift toward high frequency for bending vibration.36 Most of the characteristic stretching vibration absorption peaks of PAM–PDA hydrogels shifted toward low frequency in comparison to a single component, indicating that there was a certain combination between the components caused by non-covalent forces, such as hydrogen bonds. By analyzing the shift of characteristic absorption peaks, it can be found that Alg-Na weakened the partial non-covalent interaction between PDA and PAM. Moreover, compared with PAM–PDA hydrogels, the absorption band of PAM–Alg–PDA hydrogel showed a new peak at 1258 cm−1 (stretching vibration of C–N), which can be assigned to the multiple interactions among C–NH2 of PAM and phenolic hydroxyl groups of PDA and/or hydroxyl groups of Alg.38
O, amide I), 1618 cm−1 (in-plane deformation of primary amine –NH, amide II), 1450 cm−1 (shear vibration of –CH2 in plane), 1419 cm−1 (stretching vibration of primary amide –CN), 1350 cm−1 (deformation of –CH) and 1095 cm−1 (in-plane swing of –NH2). The relative intensity of the absorption peak at 1201 cm−1 (stretching vibration of aliphatic amines C–N) of the PAM–PDA hydrogel was increased in comparison to the PAM hydrogel and PDA, indicating that C–NH2 of PAM reacted with the phenolic hydroxyl groups of PDA.38 In addition, a hydrogen bond causes the band to shift toward low frequency for stretching vibration, while a hydrogen bond causes the band to shift toward high frequency for bending vibration.36 Most of the characteristic stretching vibration absorption peaks of PAM–PDA hydrogels shifted toward low frequency in comparison to a single component, indicating that there was a certain combination between the components caused by non-covalent forces, such as hydrogen bonds. By analyzing the shift of characteristic absorption peaks, it can be found that Alg-Na weakened the partial non-covalent interaction between PDA and PAM. Moreover, compared with PAM–PDA hydrogels, the absorption band of PAM–Alg–PDA hydrogel showed a new peak at 1258 cm−1 (stretching vibration of C–N), which can be assigned to the multiple interactions among C–NH2 of PAM and phenolic hydroxyl groups of PDA and/or hydroxyl groups of Alg.38
 |
| | Fig. 1 The FTIR spectra of hydrogel materials and individual components. | |
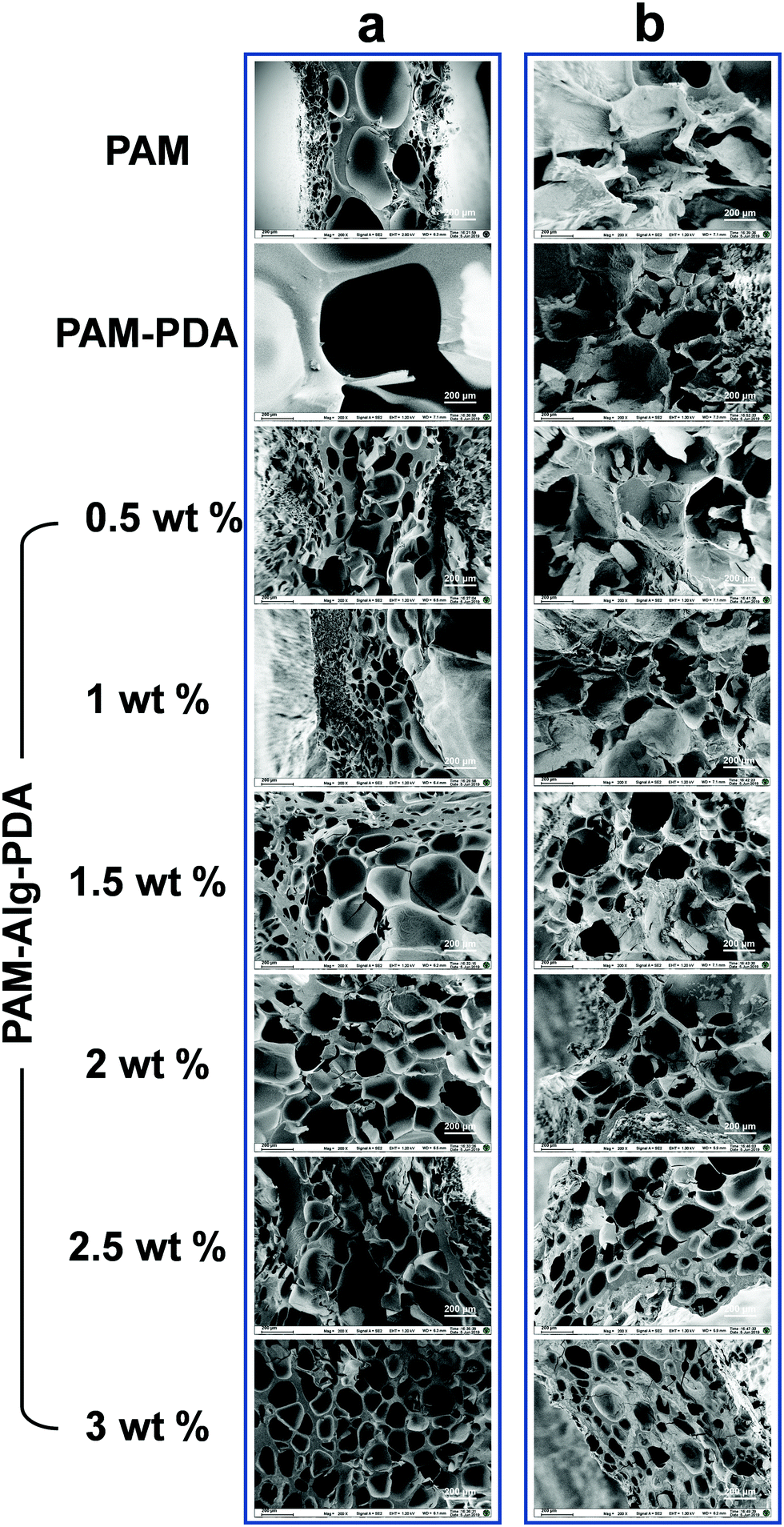
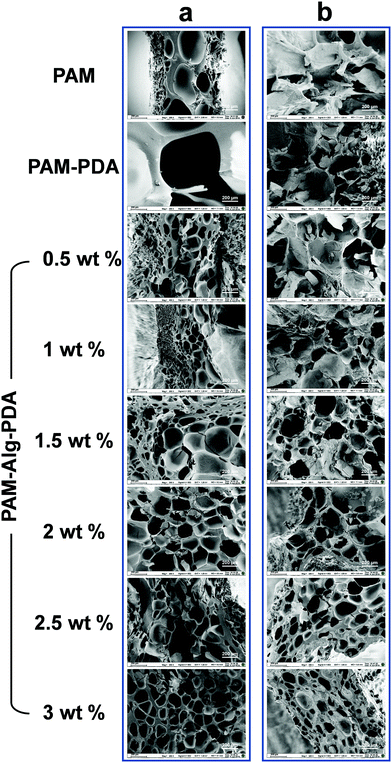
The internal morphology of the freeze-dried hydrogels was observed under scanning electron microscope (Fig. 2). The inner surface of PAM and PAM–PDA single-network hydrogels before immersion in seawater was relatively smooth, and their structure with extremely large pores was easily collapsed. Alg-Na, as a high molecular weight polymer, was responsible for supporting the structure of the hydrogel. Therefore, with an increase of Alg-Na concentration, the interconnected three-dimensional porous network structure tends to be regular and compact. After seawater immersion, there was a certain degree of erosion inside all samples, which was affected by the complex ionic components in seawater and the hydration for a long period.39 The structure of PAM and PAM–PDA hydrogels became compact with pores basically lost. Thanks to the secondary network constructed by Alg-Na with divalent cations in seawater, the PAM–Alg–PDA hydrogel still has a porous network structure. Compared with the particularly large or loose pore structure, such interconnected pores can provide excellent gas penetration paths, which are structurally beneficial for the enhancement of underwater adhesion ability and cell proliferation.
 |
| | Fig. 2 SEM morphology images of cross sections of PAM–Alg–PDA hydrogel with different concentrations of Alg-Na, PAM–PDA hydrogel and PAM hydrogel (a) before immersion in artificial seawater and (b) after immersion in artificial seawater for 3 days. | |
3.2 Adhesion ability of PAM–Alg–PDA hydrogel
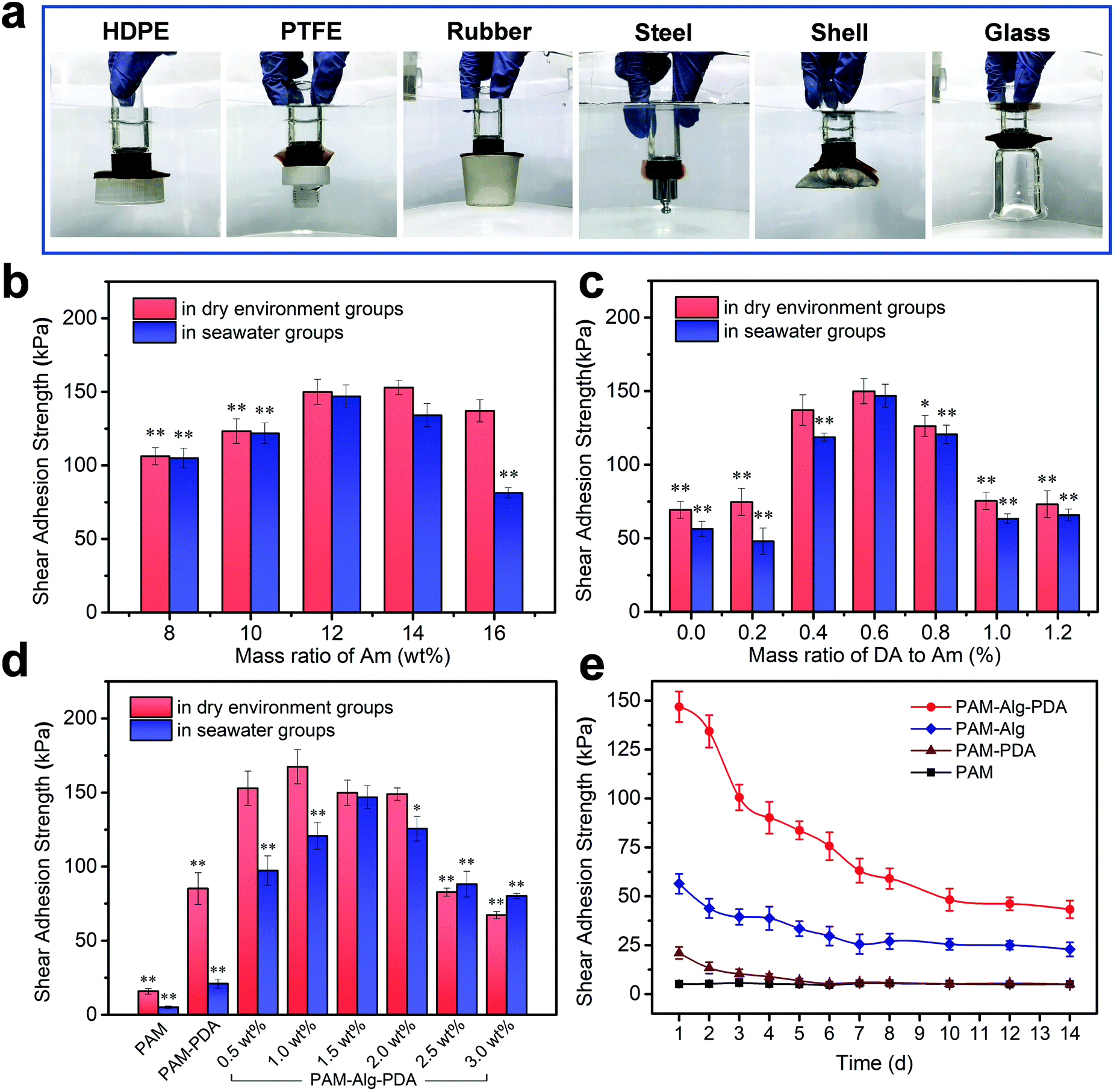
The PDA nanohybrid was formed by self-polymerization in alkalescent TEA buffer solution. Over the polymerization time, the size of PDA nanoparticles also increased, indicating the different degree of oxidation (Fig. S1, ESI†). After about 15 min of polymerization, the PDA polymerization rate gradually slowed, accompanied by stable adhesion ability (Fig. S2, ESI†). Besides, the content of DA and MBAm in the PAM–PDA hydrogel was systematically optimized in terms of dry adhesion performance (Fig. S3, ESI†). The PAM hydrogel showed little adhesion force, while PAM–Alg–PDA hydrogels have excellent adhesion properties because they simulated the principle of underwater adhesion of mussels. They can successfully adhere to solid substrates with different surface energies including organic (HDPE, PTFE, rubber) and inorganic (steel, shell, glass) surfaces in seawater (Fig. 3a). The excellent adhesive force was mainly ascribed to the physical interactions between the free catechol groups of PAM–Alg–PDA hydrogels and the solid substrate. In addition, the “crosslinked network” formed via autoxidation of PDA, the mechanism of which has not been fully elucidated, also plays an important role in adhesion.40 The hybrid structure of PDA can interact with various contact surfaces to form cation-π interactions or π–π stacking, so that the PAM–Alg–PDA hydrogel can adhere to various substrate surfaces.41 Due to the non-covalent chemical adhesion of the hydrogel, it can repeatedly adhere to glass, ceramic and steel substrates in a dry environment. Reversible surface adhesion tests on the glass showed that the interfacial toughness of the hydrogel decreased by 56% after 10 cycles (Fig. S4, ESI†). Unfortunately, the lack of strategies to remove the surface hydration layer makes it difficult for hydrogel to achieve repeatable adhesion in seawater (Fig. S4 and S5, ESI†).
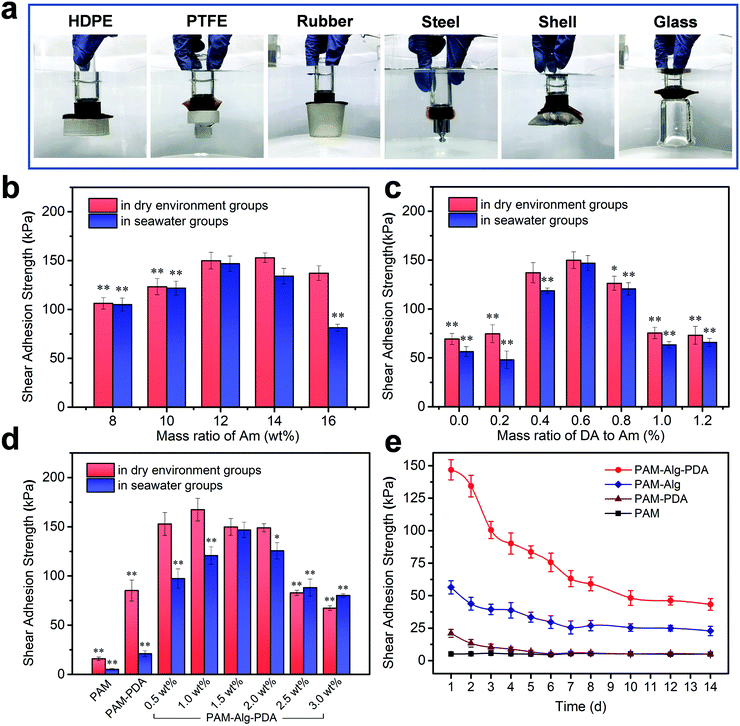 |
| | Fig. 3 Adhesion ability of PAM–Alg–PDA hydrogel. (a) PAM12–Alg1.5–PDA0.6 hydrogel adhered to various materials in seawater. HDPE and PTFE represent high density polyethylene and polytetrafluoroethylene, respectively; the shear adhesion strength of PAM–Alg–PDA hydrogel with different concentrations of Am (b), DA (c), and Alg-Na (d) on glass in a dry environment and artificial seawater, respectively (*p < 0.05, **p < 0.01, vs. the maximum adhesion strength in the same test environment); (e) the shear adhesion of hydrogels on glass after immersion in artificial seawater for different times. | |
Moreover, glass was chosen as the major challenging substrate for quantified shear adhesion tests in here due to its lower surface roughness and weaker interfacial interactions than aluminum.39,42 Concentration effects of three different network components on the adhesion strength of the hydrogel was evaluated. For PAM, as the sparsely cross-linked first network (mMBAm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mAm = 0.8%), the swelling and hydration caused by its high concentration will destroy the adhesion ability in seawater (Fig. 3b). The concentration of PDA was an important parameter to affect the adhesion. Low concentration PDA can effectively enhance the adhesion of double network hydrogel. Nevertheless, PDA can compete with Am for free radicals triggered by APS in the polymerization reactions, so excessive PDA concentration will seriously affect the gelling effect and adhesion (Fig. 3c). The Alg-Na content can also have a major impact upon adhesion (Fig. 3d). The adhesion force in a dry environment and in seawater increased first and then decreased with higher Alg-Na concentration. It is well known that an effective underwater adhesion requires both maximum interfacial contact area as well as strong mechanical strength of the material to resist water molecule or other destructive forces.43,44 Consequently, the balance between them is crucial in underwater adhesion. On the one hand, shifting the balance too much toward bond forming results in a bond that lacks internal strength,45 as shown by the samples of Alg-Na content less than 1.5 wt%. It can be seen from SEM that these samples have a large pore structure to absorb more water molecules, so the lack of mechanical strength makes them suffer more negative effects under long-term hydration, even if the high viscosity of Alg-Na can contribute to excellent adhesion in a dry environment. On the other hand, erring in the other direction will diminish interfacial adhesion,9 as shown by the samples of Alg-Na content greater than 1.5 wt%. Although the chelation of excessive Alg-Na with divalent cations in seawater provides sufficient mechanical strength to the hydrogel, and the dense pore structure weakens the hydration effect. An excessive content of Alg-Na may weaken the adhesion site effect of PDA and influence the penetration of divalent cations, which may cause poor underwater adhesion. Besides, ANOVA with the Tukey's HSD test indicated partially statistical differences between mean adhesion of hydrogels at varying content of Am and DA and Alg-Na, respectively (Tables S3–S5, ESI†). In general, the surface adhesion and mechanical strength of PAM12–Alg1.5–PDA0.6 hydrogel reached the optimal balance, and its adhesion strength was determined to be 146.84 ± 7.78 kPa in seawater after 24 h. The value of adhesion strength of an optimized hydrogel was relatively high in the current literature reports on the underwater adhesion property of hydrogel materials (Table S6, ESI†). Nonetheless, it must be noted that despite the data presented, the adhesion strength may vary largely because of differences in measuring conditions. Furthermore, the long-term adhesion on glass of the hydrogel adhesive in seawater was also measured (Fig. 3e). It can be seen that the adhesion strength of hydrogels decreased over immersing time. The lap-shear adhesion strength of PAM12–Alg1.5–PDA0.6 hydrogel was decreased to 43.28 ± 4.45 kPa after immersion in seawater for 14 days. Even so, its adhesion strength was still far higher than that of PAM, PAM–PDA and PAM–Alg hydrogels. It has been widely accepted that the catechol structure of DA can be easily oxidized into quinone or semi-quinone structure, thereby weakening the adhesion performance.46 Distinct from DA covalently grafted on the side-groups of Alg-Na or other hydrogels, PDA nanoparticles were introduced in a double network hydrogel through polymerization in situ before hydrogel formation. This strategy is expected to provide long-term adhesion for PAM–Alg–PDA hydrogels by effectively reducing the negative effect caused by the slow oxidation of catechol groups on the adhesion capability.
:mAm = 0.8%), the swelling and hydration caused by its high concentration will destroy the adhesion ability in seawater (Fig. 3b). The concentration of PDA was an important parameter to affect the adhesion. Low concentration PDA can effectively enhance the adhesion of double network hydrogel. Nevertheless, PDA can compete with Am for free radicals triggered by APS in the polymerization reactions, so excessive PDA concentration will seriously affect the gelling effect and adhesion (Fig. 3c). The Alg-Na content can also have a major impact upon adhesion (Fig. 3d). The adhesion force in a dry environment and in seawater increased first and then decreased with higher Alg-Na concentration. It is well known that an effective underwater adhesion requires both maximum interfacial contact area as well as strong mechanical strength of the material to resist water molecule or other destructive forces.43,44 Consequently, the balance between them is crucial in underwater adhesion. On the one hand, shifting the balance too much toward bond forming results in a bond that lacks internal strength,45 as shown by the samples of Alg-Na content less than 1.5 wt%. It can be seen from SEM that these samples have a large pore structure to absorb more water molecules, so the lack of mechanical strength makes them suffer more negative effects under long-term hydration, even if the high viscosity of Alg-Na can contribute to excellent adhesion in a dry environment. On the other hand, erring in the other direction will diminish interfacial adhesion,9 as shown by the samples of Alg-Na content greater than 1.5 wt%. Although the chelation of excessive Alg-Na with divalent cations in seawater provides sufficient mechanical strength to the hydrogel, and the dense pore structure weakens the hydration effect. An excessive content of Alg-Na may weaken the adhesion site effect of PDA and influence the penetration of divalent cations, which may cause poor underwater adhesion. Besides, ANOVA with the Tukey's HSD test indicated partially statistical differences between mean adhesion of hydrogels at varying content of Am and DA and Alg-Na, respectively (Tables S3–S5, ESI†). In general, the surface adhesion and mechanical strength of PAM12–Alg1.5–PDA0.6 hydrogel reached the optimal balance, and its adhesion strength was determined to be 146.84 ± 7.78 kPa in seawater after 24 h. The value of adhesion strength of an optimized hydrogel was relatively high in the current literature reports on the underwater adhesion property of hydrogel materials (Table S6, ESI†). Nonetheless, it must be noted that despite the data presented, the adhesion strength may vary largely because of differences in measuring conditions. Furthermore, the long-term adhesion on glass of the hydrogel adhesive in seawater was also measured (Fig. 3e). It can be seen that the adhesion strength of hydrogels decreased over immersing time. The lap-shear adhesion strength of PAM12–Alg1.5–PDA0.6 hydrogel was decreased to 43.28 ± 4.45 kPa after immersion in seawater for 14 days. Even so, its adhesion strength was still far higher than that of PAM, PAM–PDA and PAM–Alg hydrogels. It has been widely accepted that the catechol structure of DA can be easily oxidized into quinone or semi-quinone structure, thereby weakening the adhesion performance.46 Distinct from DA covalently grafted on the side-groups of Alg-Na or other hydrogels, PDA nanoparticles were introduced in a double network hydrogel through polymerization in situ before hydrogel formation. This strategy is expected to provide long-term adhesion for PAM–Alg–PDA hydrogels by effectively reducing the negative effect caused by the slow oxidation of catechol groups on the adhesion capability.
3.3 Swelling behavior of PAM–Alg–PDA hydrogels
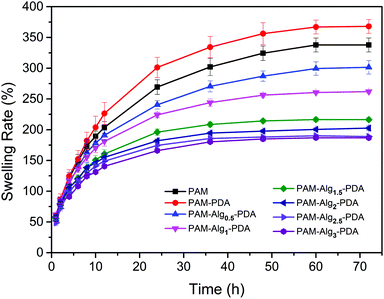
Swelling rate is one of the vital properties of hydrogels. In consideration of the destruction of the interface between adhesives and substrates due to hydration, the hydrogel with lower swelling rate has more advantages when it adheres to substrates in seawater.47 As shown in Fig. 4, the equilibrium swelling of all hydrogels was achieved after 3 days immersion in seawater. The PAM–PDA hydrogel exhibited the highest swelling rate in all samples, approaching 337.77 ± 11.38%, followed by the PAM hydrogel. The reason for this phenomenon is that the PAM–PDA hydrogel had a more internal porous structure than that of the PAM hydrogel, and PDA contains a large number of hydrophilic groups. However, the swelling rate of the prepared PAM–Alg–PDA hydrogels only ranged from 186.85 ± 2.23% to 301.45 ± 11.01% at 3 days, and it was decreased with higher Alg-Na concentration. The construction of a double network in PAM–Alg–PDA hydrogels made the internal structure of the hydrogel more compact, which can be observed in SEM. Consequently, the chelation of Alg-Na with divalent cations could effectively reduce the swelling rate of a composite hydrogel in a seawater environment, thereby reducing the damage of seawater to the adhesive surface and bulk mechanical strength.
 |
| | Fig. 4 The swelling behavior of various hydrogels. | |
3.4 Rheological behavior of PAM–Alg–PDA hydrogels
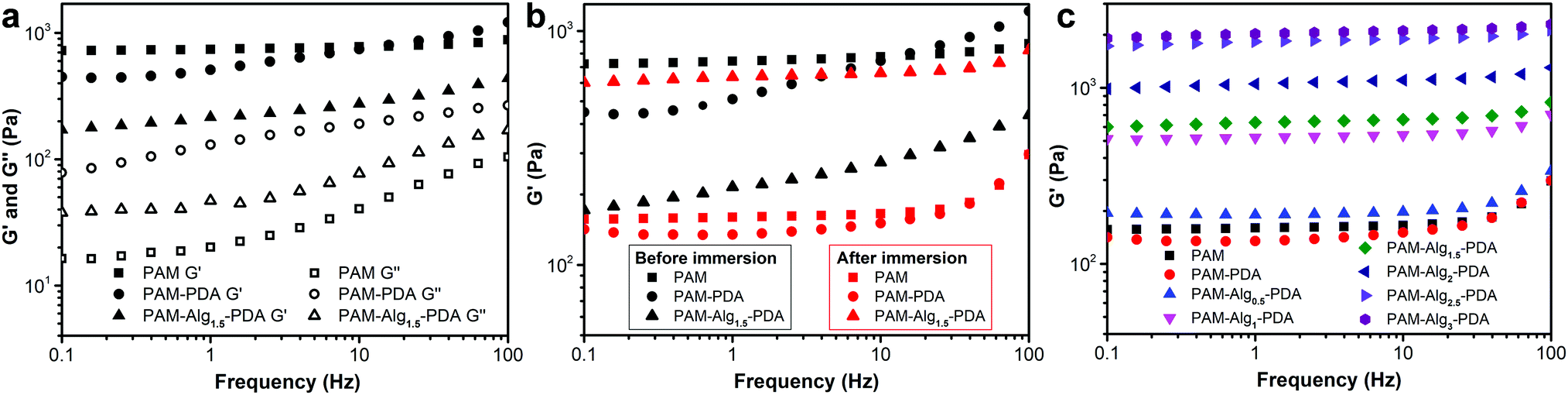
Tough mechanical properties of adhesive materials is the premise of practical applications. However, it is still challenging for previously reported underwater adhesives, particularly in seawater.22,48Fig. 5 showed the dynamic rheological behavior of the hydrogels. The storage modulus (G′) and the loss modulus (G′′) of all hydrogels increased gradually with increasing shear frequency, and G′ was much larger than G′′ in the frequency range of 0.1–100 Hz, indicating that the hydrogel systems displayed a predominantly solid-like behavior (Fig. 5a). After immersion in seawater for 3 days, the G′ of PAM and PAM–PDA hydrogels decreased significantly, suggesting that the stiffness of the hydrogel was significantly lost. However, the mechanical properties of the PAM–Alg–PDA hydrogel changed from viscoelasticity to rigidity after seawater immersion, which can be indicated by the increase of G′ (Fig. 5b). Besides, the PAM–Alg–PDA hydrogel exhibited enhanced G′ with higher Alg-Na content after seawater immersion, revealing that the construction of a double network structure contributes to a higher stiffness of the PAM–Alg–PDA hydrogel (Fig. 5c).
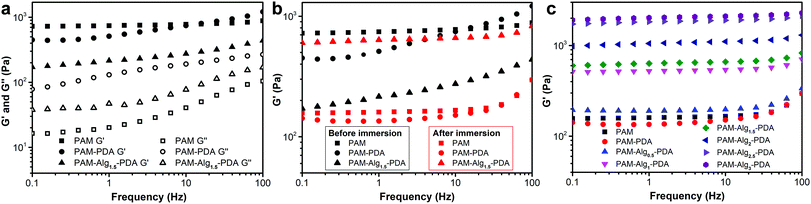 |
| | Fig. 5 The dynamic rheological behavior of various hydrogels. (a) The storage modulus (G′) and loss modulus (G′′) of hydrogels before immersion in artificial seawater; (b) comparison of G′ of hydrogels before and after immersion in seawater for 3 days; (c) the change in G′ of hydrogels with the concentration of Alg-Na immersed in artificial seawater for 3 days. | |
3.5 Mechanical property of PAM–Alg–PDA hydrogels
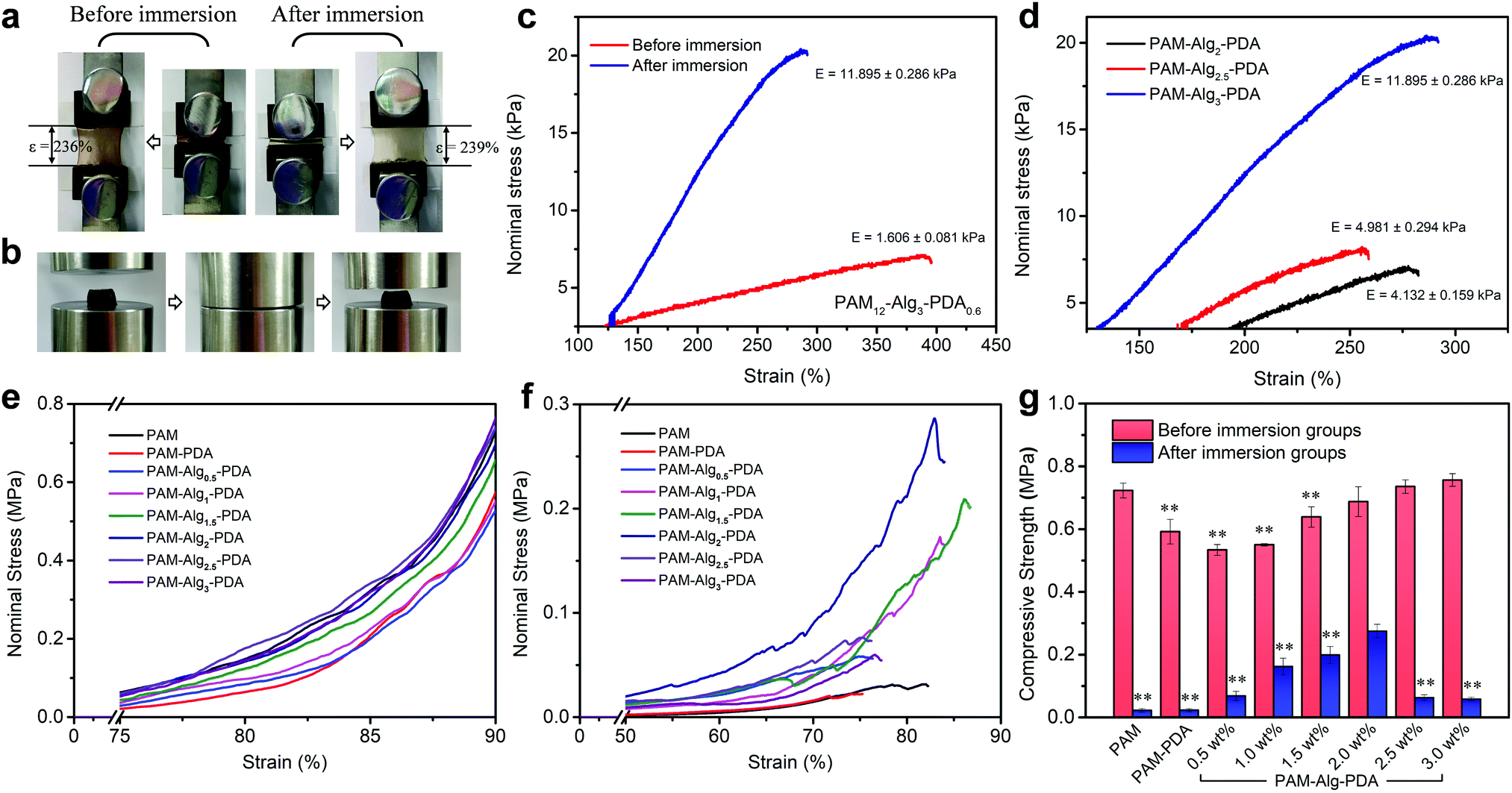
Hydrogels exhibited very low stiffness before immersion in seawater, which is greatly related to the low concentration of crosslinking agent (mMBAm:mAm = 0.8%). The tensile strain of the PAM12–Alg1.5–PDA0.6 hydrogel did not change significantly before immersion (stored at 25 °C for 3 days) and after immersion in seawater for 3 days (Fig. 6a). After immersion in seawater, the elastic modulus of the PAM–Alg–PDA hydrogels was increased through the completely constructed double network (Fig. 6c). And the elastic modulus increased with higher Alg-Na concentration, accompanied by decreased tensile strain (Fig. 6d). Moreover, compared with one before immersion, the tensile loading-unloading curve indicated that the PAM–Alg–PDA hydrogel after immersion in seawater has a weak mechanical hysteresis effect (Fig. S8, ESI†).
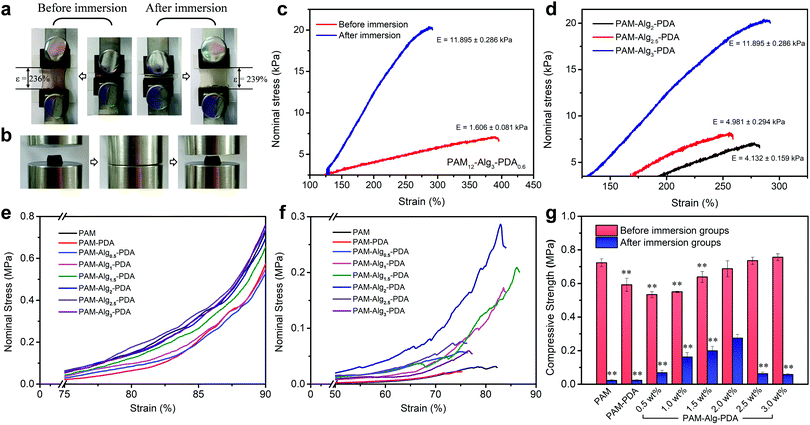 |
| | Fig. 6 The mechanical properties of various hydrogels. (a) Tensile strain of PAM12–Alg1.5–PDA0.6 hydrogel did not change significantly before immersion (stored at 25 °C for 3 days) and after immersion in seawater for 3 days; (b) PAM12–Alg1.5–PDA0.6 hydrogels retain their original shape after being compressed to 90% deformation before immersion; (c) tensile stress–strain curves of PAM12–Alg3–PDA0.6 hydrogel before and after immersion in seawater for 3 days; (d) tensile stress–strain curves of PAM–Alg–PDA hydrogels with different concentrations of Alg-Na after immersion in seawater for 3 days; compressive stress–strain curves of hydrogels with different concentrations of Alg-Na before immersion (e) and after immersion in artificial seawater for 3 days (f), respectively; (g) compressive strength of the PAM–Alg–PDA hydrogel with different concentrations of Alg-Na, PAM–PDA hydrogel and PAM hydrogel (*p < 0.05, **p < 0.01, vs. the maximum compressive strength in the same test environment). | |
In addition, the compressive mechanical properties of the hydrogels were also characterized by a mechanical testing machine. The stress–strain curve showed the non-linear mechanical behavior of the hydrogel during compression, and the Young's modulus increased with the compression ratio both before and after immersion in artificial seawater (Fig. 6e and f). The higher compressive strength of the hydrogel material has a positive effect on persistent underwater adhesion. All prepared hydrogels can withstand the compression stress of MPa grade, and they retain their original shape after being compressed to 90% deformation (Fig. 6b). However, excessive hydration softening of single network hydrogels leads to a lack of bulk cohesion, and then results in a significant decrease in compressive strength. Construction of double network hydrogels is undoubtedly an advisable choice, because their unique network structure and effective energy dissipation mechanism allow the hydrogel adhesive to bear a greater force and greater shape deformation.33 The PAM–Alg–PDA hydrogels can reduce the loss of compressive strength after swelling in seawater versus a single network hydrogel. The compressive strength could be modulated over a broad range (0.06–0.27 MPa) to match various types of adherends by varying the content of Alg-Na (Fig. 6g). Thus, the incorporation of Alg used as a second network could improve the bulk compressive capacity of PAM–PDA hydrogels under seawater, due to the chelation with divalent cations in seawater.49 Besides, the concentration of Am and DA also affected the compressive properties of hydrogels before and after immersion (Fig. S8, ESI†). ANOVA with Tukey's HSD test showed partially statistical differences between mean compressive strength of hydrogels with different Am, DA and Alg-Na contents, respectively (Tables S7–S9, ESI†). Although adhesives formulated at 1.5 wt% of Alg content demonstrated a good balance of surface adhesive and mechanical strength in an adhesion test under seawater for 24 h as described above, the increased compressive strength via strengthened chelation between the Alg network and seawater of the adhesive polymer revealed that hydrogels prepared at higher concentrations of Alg (e.g., 2 wt%) may have more advantages in long-term adhesion.
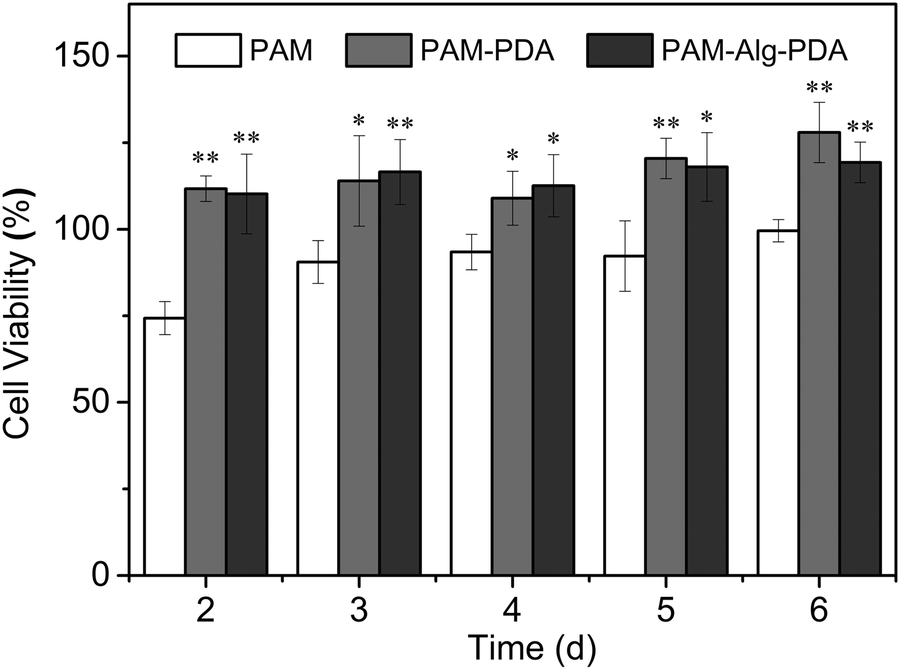
3.6 Cytocompatibility of PAM–Alg–PDA hydrogels
Marine environment is closely related to human health, so the adhesive system used in seawater must be environmentally friendly and free of cytotoxicity. Zooxanthellae is a symbiotic dinoflagellate residing inside many invertebrates (e.g., coral, actiniaria) in the ocean.50 The cell viability of zooxanthellae cultured with hydrogels was evaluated through the CCK-8 assay (Fig. 7). The PAM hydrogel exhibited slight cytotoxicity at all time points. Nevertheless, the introduction of PDA containing abundant hydroxyl and amine groups into polymer systems is often beneficial for the adhesion, spreading and growth of cells, showing enhanced cytocompatibility.51,52 It was observed that the cells in the PAM12–Alg1.5–PDA0.6 hydrogel groups moved in a similar circular motion to those in the control groups. Besides, there was no significant difference between the cell viability of PAM–Alg–PDA and PAM–PDA hydrogels (P > 0.05). Consequently, it can be considered that the PAM–Alg–PDA hydrogel system in this study has excellent cytocompatibility, and even can actively promote zooxanthellae cell proliferation.
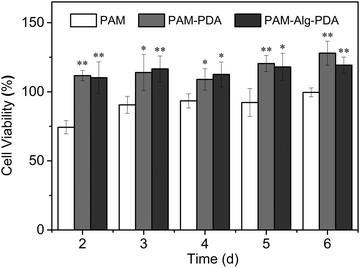 |
| | Fig. 7 The cell viability of various hydrogels for zooxanthellae (*p < 0.05, **p < 0.01, vs. the cell viability of the PAM hydrogel at the same time point). | |
4 Conclusions
In conclusion, a bio-inspired synergy strategy based on mussels and algae was proposed to design a tough double network hydrogel adhesive with high adhesive strength under wet conditions, especially in seawater. PDA nanoparticles were in situ-formed other than chemically grafted onto Alg or PAM chains, and distributed evenly throughout this double network hydrogel. The combination of the PDA nanoparticles and double network in the PAM–Alg–PDA hydrogels led to excellent underwater adhesion for various substrates in the artificial seawater environment, and the adhesion ability was related to the content of Alg-Ca or Alg-Mg. The synergistic effect of the adhesive surface and the dissipative matrix contributed to higher adhesion and mechanical strength for the hydrogel in seawater. Besides, the hydrogel adhesive has excellent cytocompatibility for zooxanthellae, due to high cellular affinity of PDA. The coordination of adhesion and strength of adhesive has been achieved in this strategy of construction of a bioinspired hybrid hydrogel in seawater. It would serve to develop flexible materials which have strong and durable adhesive performance for wet environment applications in the biomedical field.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the Special Project on Development of National Key Scientific Instruments and Equipment of China (2011YQ03013403), the National Natural Science Youth Foundation of China (81600407), Suzhou medical apparatus and new medicine Fund (ZXY201440), and the Fundamental Research Funds for the Central Universities.
References
- X. Zhu, C. Wei, F. Zhang, Q. Tang and Q. Zhao, Macromol. Rapid Commun., 2019, 40, 1800758 CrossRef PubMed.
- M. V. Rapp, G. P. Maier, H. A. Dobbs, N. J. Higdon, J. H. Waite, A. Butler and J. N. Israelachvili, J. Am. Chem. Soc., 2016, 138, 9013–9016 CrossRef CAS PubMed.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- P. Rao, T. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1801884 CrossRef PubMed.
- Y. Zhao, Y. Wu, L. Wang, M. Zhang, X. Chen, M. Liu, J. Fan, J. Liu, F. Zhou and Z. Wang, Nat. Commun., 2017, 8, 2218–2225 CrossRef PubMed.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CrossRef CAS PubMed.
- J. K. Park, J. D. Eisenhaure and S. Kim, Adv. Mater. Interfaces, 2019, 6, 1801542 CrossRef.
- Y. Xu, K. Liang, U. Wajeeh, Y. Ji and J. Ma, Carbohydr. Polym., 2018, 190, 324–330 CrossRef CAS PubMed.
- X. Li, W. Li, Z. Q. Liu, X. Wang, H. Guo, R. Wang, X. Guo, C. Li and X. Jia, J. Appl. Polym. Sci., 2018, 135, 46579–46586 CrossRef.
- X. Li, T. Zheng, X. Liu, Z. Du, X. Xie, B. Li, L. Wu and W. Li, Langmuir, 2019, 35, 4995–5003 CrossRef CAS PubMed.
- M. J. Brennan, B. F. Kilbride, J. J. Wilker and J. C. Liu, Biomaterials, 2017, 124, 116–125 CrossRef CAS PubMed.
- T. Zhang, H. Yuk, S. Lin, G. A. Parada and X. Zhao, Acta Mech. Sin., 2017, 33, 543–554 CrossRef CAS.
- M. Cencer, A. Winter, Y. Liu, M. Murley, H. Meng and B. P. Lee, Biomacromolecules, 2014, 15, 2861–2869 CrossRef CAS PubMed.
- Y. Wang, X. Yang, Z. Gong, Z. Liu, T. Wang, Y. Chen, J. C. Weaver, D. K. Wainwright, C. P. Kenaley, H. Liu, J. Guan, R. J. Wood and L. Wen, Sci. Rob., 2017, 2, eaan8072–eaan8080 CrossRef.
- S. Baik, J. Kim, H. J. Lee, T. H. Lee and C. Pang, Adv. Sci., 2018, 5, 1800100 CrossRef PubMed.
- M. Cui, S. Ren, S. Wei, C. Sun and C. Zhong, APL Mater., 2017, 5, 116102 CrossRef.
- P. K. Forooshani and B. P. Lee, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 9–33 CrossRef PubMed.
- M. Krogsgaard, V. Nue and H. Birkedal, Chem. – Eur. J., 2016, 22, 844–857 CrossRef CAS PubMed.
- C. Zhang, L. Xiang, J. Zhang, L. Gong, L. Han, Z. Xu and H. Zeng, Langmuir, 2019, 35, 5257–5263 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS.
- N. F. Della Vecchia, R. Avolio, M. Alf, M. E. Errico, A. Napolitano and M. d’Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. Weng and H. Zhang, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- Z. Wang, K. Wang, Y. Zhang, Y. Jiang, X. Lu, L. Fang, D. Gan, C. Lv, H. Zhang and S. Qu, Part. Part. Syst. Charact., 2016, 33, 89–100 CrossRef CAS.
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- L. C. Bradley, N. D. Bade, D. Lee, K. J. Stebe, L. M. Mariani and K. T. Turner, ACS Appl. Mater. Interfaces, 2017, 9, 27409–27413 CrossRef CAS PubMed.
- P. J. M. Bouten, M. Zonjee, J. Bender, S. T. K. Yauw, H. van Goor, J. C. M. van Hest and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1375–1405 CrossRef CAS.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
- J. Li, J. Yang, Q. Yang, W. Whyte, B. R. Seo, J. J. Vlassak, Z. Suo, D. J. Mooney, A. D. Celiz, I. Wamala and N. V. Vasilyev, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- C. Yang, M. Wang, H. Haider, J. Yang, J. Sun, Y. Chen, J. Zhou and Z. Suo, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422 CrossRef CAS PubMed.
- J. Deng, C. Cheng, Y. Teng, C. Nie and C. Zhao, Polym. Chem., 2017, 8, 2266–2275 RSC.
- A. Cholewinski, F. Yang and B. Zhao, Mater. Horiz., 2019, 6, 285–293 RSC.
- H. Kaygusuz, G. A. Evingür, Ö. Pekcan, R. von Klitzing and F. B. Erim, Int. J. Biol. Macromol., 2016, 92, 220–224 CrossRef CAS PubMed.
- Z. Bai, W. Dan, G. Yu, Y. Chen, Y. Huang, C. Yang and N. Dan, RSC Adv., 2018, 8, 42123–42132 RSC.
- T. Chen, Y. Chen, H. U. Rehman, Z. Chen, Z. Yang, M. Wang, H. Li and H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 33523–33531 CrossRef CAS PubMed.
- Y. Liu, H. Meng, Z. Qian, N. Fan, W. Y. Choi, F. Zhao and B. P. Lee, Angew. Chem., Int. Ed., 2017, 56, 4224–4228 CrossRef CAS PubMed.
- S. Y. Oh, D. I. Yoo, Y. Shin and G. Seo, Carbohydr. Res., 2005, 340, 417–428 CrossRef CAS PubMed.
- B. Zhu and S. Edmondson, Polymer, 2011, 52, 2141–2149 CrossRef CAS.
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L. Weng, J. Xu, J. Weng, Y. Liu, F. Ren and X. Lu, NPG Asia Mater., 2017, 9, e372–e383 CrossRef CAS.
- K. Zhan, C. Kim, K. Sung, H. Ejima and N. Yoshie, Biomacromolecules, 2017, 18, 2959–2966 CrossRef CAS.
- H. Yang, J. Luo, Y. Lv, P. Shen and Z. Xu, J. Membr. Sci., 2015, 483, 42–59 CrossRef CAS.
- X. Wang, J. Shi, Z. Jiang, Z. Li, W. Zhang, X. Song, Q. Ai and H. Wu, Biomacromolecules, 2013, 14, 3861–3869 CrossRef CAS PubMed.
- A. Li, Y. Jia, S. Sun, Y. Xu, B. B. Minsky, M. A. C. Stuart, H. Cölfen, R. von Klitzing and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 10471–10479 CrossRef CAS PubMed.
- B. Jeong, B. D. Park and V. Causin, J. Ind. Eng. Chem., 2019, 79, 87–96 CrossRef CAS.
- T. Harper, R. Slegeris, I. Pramudya and H. Y. Chung, ACS Appl. Mater. Interfaces, 2017, 9, 1830–1839 CrossRef CAS PubMed.
- B. Yang, C. Lim, D. S. Hwang and H. J. Cha, Chem. Mater., 2016, 28, 7982–7989 CrossRef CAS.
- D. S. Hwang, M. J. Harrington, Q. Lu, A. Masic, H. Zeng and J. H. Waite, J. Mater. Chem., 2012, 22, 15530–15533 RSC.
- Y. Xu, Q. Liu, A. Narayanan, D. Jain, A. Dhinojwala and A. Joy, Adv. Mater. Interfaces, 2017, 4, 1700506 CrossRef.
- X. Liu, Q. Zhang, L. J. Duan and G. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 6644–6651 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS.
- K. L. Barotta, A. A. Venn, S. O. Pereza, S. Tambutté and M. Tresguerres, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 607–612 CrossRef PubMed.
- L. Zhu, J. Jiang, B. Zhu and Y. Xu, Colloids Surf., B, 2011, 86, 111–118 CrossRef CAS.
- N. G. Rim, S. J. Kim, Y. M. Shin, I. Jun, D. W. Lim, J. H. Park and H. Shin, Colloids Surf., B, 2012, 91, 189–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Components of different hydrogels and artificial seawater, morphology images and particle size of PDA nanoparticles, shear adhesion strength of PAM12–Alg1.5–PDA0.6 hydrogel on glass with different polymerization times of PDA, shear adhesion strength of PAM–PDA hydrogel with different concentrations of DA and MBAm, reversible surface adhesion of PAM12–Alg1.5–PDA0.6 hydrogel by 90-degree peeling test, gelation time of the hydrogel, tensile loading–unloading curve of the hydrogels, compressive stress–strain curves of hydrogels with different concentrations of Am and DA, adhesion strength comparison between the PAM–Alg–PDA hydrogel and other adhesives, and post hoc test to determine statistical differences of shear adhesion strength and compressive strength of multiple hydrogel groups. See DOI: 10.1039/d0tb00513d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Chunpeng
He
,
Jidong
Dai
,
Pengfei
Ren
,
Yifu
Fu
,
Faming
Wang
,
Xin
Ge
,
Tianzhu
Zhang
,
Chunpeng
He
,
Jidong
Dai
,
Pengfei
Ren
,
Yifu
Fu
,
Faming
Wang
,
Xin
Ge
,
Tianzhu
Zhang