
A bioinspired hydrogen bond crosslink strategy toward toughening ultrastrong and multifunctional nanocomposite hydrogels†
Fengcai
Lin
a,
Zi
Wang
a,
Jingsi
Chen
b,
Beili
Lu
 a,
Lirong
Tang
a,
Xuerong
Chen
a,
Chensheng
Lin
c,
Biao
Huang
*a,
Hongbo
Zeng
*b and
Yandan
Chen
*a
a,
Lirong
Tang
a,
Xuerong
Chen
a,
Chensheng
Lin
c,
Biao
Huang
*a,
Hongbo
Zeng
*b and
Yandan
Chen
*a
aCollege of Material Engineering, Fujian Agriculture and Forestry University, Fuzhou 350108, China. E-mail: fjaucyd@163.com; bhuang@fafu.edu.cn
bDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: hongbo.zeng@ualberta.ca
cFujian Key Laboratory of Developmental and Neural Biology, College of Life Sciences, Fujian Normal University, Fuzhou 350108, China
First published on 16th March 2020
Abstract
Developing physical hydrogels with advanced mechanical performance and multi-functionalities as alterative materials for load-bearing soft tissues remains a great challenge. Biological protein-based materials generally exhibit superior strength and toughness owing to their hierarchical structures via hydrogen-bonding assembly. Inspired by natural biological protein materials, tannic acid (TA) is exploited as a molecular coupling bridge between cellulose nanocrystals (CNCs) and poly(vinyl alcohol) (PVA) chains for the fabrication of a bio-based advanced physical hydrogel via strong multiple H-bonds. When exposed to mechanical stress, the sacrificial H-bonds can dissipate energy effectively on the molecular scale via dynamic rupture and reformation, endowing these biomimetic hydrogels with remarkable toughness, ultrahigh strength, large elongation, and good self-recoverability, which are much superior to those of most hydrogen bond-based hydrogels. Moreover, the characteristics of TA endow these biomimetic hydrogels with versatile adhesiveness and good antibacterial properties. This work presents an innovative biomimetic strategy for robust biocompatible hydrogels with superior mechanical strength and functionalities, which holds great promise for applications in tissue engineering and biomedical fields.
1. Introduction
Hydrogen bonding (H-bonding), especially cooperative H-bonding, has been recognized as being crucial for the formation and stabilization of hierarchical structures of biosystems,1–3 and is also one of the most essential physical interactions for constructing and toughening biological hydrogels.4–6 Although a single H-bond is usually considered to be weak, multiple H-bonds can synergistically direct the assembly of building blocks into complex structures.7 The dynamic nature of H-bonds enables them to break and reform during the deformation of materials, which can effectively dissipate energy and endow the protein-based materials with exceptional strength, toughness, and resilience,8,9 providing inspiration for the development of soft materials that behave as natural tissues.Hydrogels hold great promise for mimicking the functions of biological soft tissues, due to their porous 3D structure, ability to retain large amounts of water or biological fluids, excellent stretchability, good flexibility, and biocompatibility. In particular, the hydrogels based on poly(vinyl alcohol) (PVA), a biocompatible, nontoxic and hydrophilic polymer with abundant hydroxyl groups, have been recognized as one of the most promising candidates for tissue engineering.10 However, the low mechanical strength and poor toughness of pure PVA hydrogels impede their further application as load-bearing biomaterials. Traditionally, the incorporation of nanomaterials such as hydroxylated boron nitride nanosheets (OH-BNNSs),11 carbon nanotubes (CNTs),12,13 and graphene oxide (GO)14,15 into a PVA matrix is considered to be an effective way to improve the mechanical properties. Nevertheless, the lack of hydrophilicity of nanofillers limited their role only as nanoreinforcements without participating in the construction of polymer networks. Moreover, these incorporated additives are either high-cost, nondegradable or have limited biological activity,14,16 which weakens the green feature of the PVA hydrogels.
Recently, the H-bond crosslink strategy has been extensively used to construct PVA hydrogels with exceptional mechanical performance. H-bonding can dramatically increase the mechanical strength of PVA hydrogels without sacrificing the extensibility and toughness because of its directionality, versatility and reversibility.17 Recent studies showed that small molecules with abundant hydroxyl groups or amino groups can serve as H-bond cross-linkers within the PVA matrix. For instance, Wang's group developed a novel series novel of H-bonded supramolecular hydrogels from PVA and glycerol, which exhibit extraordinary toughness, strength and robustness.18,19 In these H-bonded PVA–glycerol hydrogels, the small glycerol molecules served as bridge molecules to connect PVA chains into bundles via multiple H-bonding interactions. The easy breakage of the H-bonds during deformation, slippage, dislocation of the PVA chains as well as the easy reconstruction of H-bonds between PVA chains and glycerol endow the hydrogels with remarkable energy dissipation capacity. Likewise, tannic acid (TA) was also exploited as a strong H-bond cross-linker to prepare a tough PVA–TA hydrogel with shape memory properties.20 Inspired by spider silk, Song et al. demonstrated the preparation of a strong and tough PVA matrix cross-linked by small organic cross-linkers, such as four pyrimidine-derived multiamines and melamine, via the H-bond self-assembly.21–23 On the other hand, super-strong and tough hydrogels can also be constructed by using H-bonding between PVA chains and polymers, such as poly(acrylic acid) or poly(acrylamide-co-acrylic acid).6,24 Therefore, H-bond crosslinking is an attractive strategy for the development of toughened, ultra-strong and multifunctional PVA hydrogels.
Cellulose nanocrystals (CNCs), as a kind of easily available biomass, are rod-like nanoparticles with at least one dimension <100 nm, which can be extracted from renewable natural resources in larger volumes.25 Owing their sustainability, biocompatibility, high aspect ratios, as well as high strength (∼7.6 GPa) and stiffness (∼138 GPa),26 CNCs have been recognized as an ideal reinforcing element in composites27–29 and employed as a biologically safe reinforcement to combine with the PVA hydrogels toward biomimetic soft materials. Based on the hydrogen bonds formed through the cyclic freezing–thawing method, the reinforcing effects of CNCs on the mechanical properties of PVA hydrogels have been investigated.30,31 However, due to the lack of an effective energy-dissipation mechanism, the mechanical performance, especially toughness, of these hydrogels still cannot be comparable to that of biological tissues.
Tannic acid (TA) is a water-soluble natural polyphenol that can also be abundantly extracted from sustainable plant sources; hence, it possesses biological activity and fully biodegradable feature. The TA molecule consists of a central glucose unit core with all five hydroxyl moieties esterified with two gallic acid molecules (Scheme 1a), which can easily couple with other materials via multiple interactions, such as hydrogen bonds, ion coordinate bonds, and hydrophobic interactions.32,33 Thus, TA has been successfully employed to construct a variety of advanced functional materials for bioengineering applications as they are nontoxic to in vitro cultured cells. Previous studies confirmed the formation of multiple H-bonds between the functional groups (containing –N– or –O– units) of polymers and the phenolic hydroxyl groups of TA.34–39 Jin's group developed a series of supramolecular hydrogels based on TA and various polymers via H-bonds and ionic interactions, which exhibited multiple functionalities such as mechanical tenability, rapid self-healing, and pH responsiveness.38,40 However, leveraging noncovalent interactions provided by TA as a multiple dynamic associate moiety to complex nature polysaccharide and biopolymers into ultrastrong multifunctional hydrogels has not yet been reported.
 | ||
| Scheme 1 (a) Chemical structure of TA, CNC and PVA. (b–e) Illustration of the fabrication of TA–PVA/CNC hydrogels and their possible formation mechanism. (f–k) The hydrogen bonds between TA and CNC, CNC and CNC, PVA and CNC, TA and PVA, TA and TA, as well as PVA and PVA, respectively. | ||
Inspired by the multiple H-bonded self-assemblies of biological protein-based materials, in this work, TA is exploited as a molecular coupling bridge to connect CNC nanoparticles and PVA chains to design a bio-based advanced physical hydrogel via multiple H-bonds specifically, CNCs are incorporated into a physically cross-linked PVA network to construct a nanocomposite PVA/CNC hydrogel, which is combined with TA via extensive H-bonding interactions between TA and both PVA chains and CNC nanoparticles (Scheme 1). The cellulose nanocrystals with high stiffness and numerous hydroxyl groups not only act as a nano-reinforcement but also participate in the crosslinking of the H-bonded structure. When exposed to mechanical stress, the well-dispersed rigid CNC could maintain the configuration of hydrogels, resist stress concentration, as well as transfer energy from the PVA chains to the CNC to prevent crack propagation of the composite hydrogels. Meanwhile, the sacrificial H-bonds can dissipate energy effectively on the molecular scale via dynamic rupture and reformation, endowing these nanocomposite hydrogels with remarkable toughness, ultrahigh strength, large elongation, and good self-recoverability. Moreover, the synthesized TA–PVA/CNC hydrogels exhibit multiple functionalities including good self-recovering and healing abilities, repeatable and versatile adhesiveness to various substrates as well as good biocompatibility and antibacterial properties. This work demonstrates a method to transform normal nanocomposite hydrogels to advanced hydrogels with superior mechanical performance and versatile functionalities, which presents promising applications requiring load-bearing biomaterials such as artificial muscles, tendons and ligaments.
2. Results and discussion
2.1. Design strategy for TA–PVA/CNC hydrogels based on the H-bond assembly
Scheme 1a presents the chemical structures of TA, CNC, and PVA in the study. TA molecule possesses a five-polyphenol-arm structure with 25 hydroxyl groups, which are capable of forming H-bonds with abundant hydroxyl groups on the CNC and PVA chains. We found that both CNC and PVA solutions could form stable hydrogels respectively when they were mixed with TA solutions under ambient conditions (Fig. S2, ESI†). This indicates that the self-assembled structures are most likely contributed by multiple H-bonding interactions since hydroxyl groups are the only functional groups of TA, CNC and PVA. Accordingly, supertough and multifunctional TA–PVA/CNC hydrogels were synthesized by post-crosslinking PVA/CNC nanocomposite hydrogels with TA via a simple soaking strategy (Scheme 1b–d). Specifically, a series of PVA/CNC nanocomposite hydrogels with different CNC contents (CCNC) were prepared via the cyclic freezing–thawing process based on PVA and CNC mixtures. After three freezing–thawing cycles, the crystalline domains of PVA chains were associated with the H-bonds between CNC and PVA molecules to form the first physically crosslinked network of PVA/CNC hydrogels (Scheme 1c). The obtained nanocomposite hydrogels were immersed in TA (CTA) aqueous solutions of desired concentration to form a second noncovalent network based on intermolecular multiple H-bonds between TA and the PVA/CNC matrix, resulting in tough TAx–PVA/CNCy hydrogels (where x and y are the concentrations of TA and CNC, respectively). As depicted in Scheme 1e–g, TA acts as a molecular coupling bridge to connect CNC nanoparticles and PVA chains to construct a densely H-bond crosslinked network, which is similar to the multiple H-bonding interactions in biological protein-based materials. Simultaneously, CNCs with high stiffness and numerous hydroxyl groups can be used as nano-reinforcements, and participate in the H-bonding crosslinking of the structure. Therefore, the prepared hydrogels based on H-bonds are expected to be ultrastrong, supertough and display versatile functions.After treating the PVA/CNC hydrogel with TA solution, the obtained TA–PVA/CNC hydrogels exhibited different degrees of shrinking and it is significantly affected by the CCNC and CTA as shown in Fig. 1a and Fig. S3 (ESI†). Meanwhile, as summarized in Table S1 (ESI†), the water content of TA–PVA/CNC hydrogels is also affected by the CCNC and CTA. The TA macromolecule exhibited much stronger H-bonding interactions with the –OH groups of the PVA/CNC matrix than water molecules, leading to the significant dehydration and shrinkage of TA–PVA/CNC hydrogels. The volume shrinkage and the solid content increase of the nanocomposite hydrogels resulted in a much denser physically crosslinked network, which endowed the hydrogels with outstanding mechanical performance. Interestingly, the resulting TA–PVA/CNC hydrogels not only presented an attractive free-shapeable property to adapt to any customized shapes (such as a Christmas tree, a Dolphin, and a Star), but also were tough enough to withstand large deformation without any damage and quickly restored to their original shape (Fig. 1b and Movie S1, ESI†). As shown in Fig. 1c, the TA–PVA/CNC hydrogels could also be easily bended to “TA”, “PVA” or “CNC” shapes and stretched to a greatly large strain (∼1100%) (Movie S2, ESI†), indicating the excellent ductility and stretchability of these physical hydrogels. The excellent mechanical performance was also demonstrated by the large deformations of the twisted, knotted and crossover stretched hydrogels. In addition, the hydrogel with a diameter of 3 mm and a weight of 0.81 g could easily lift up a steel block (4.23 kg) with ∼5000 times its own weight (Fig. 1d). Moreover, these physical hydrogels quickly recovered to their original shape after the high compression stress was removed, suggesting that the hydrogels possessed good shape-recovery properties (Fig. 1e and Movie S3, ESI†).
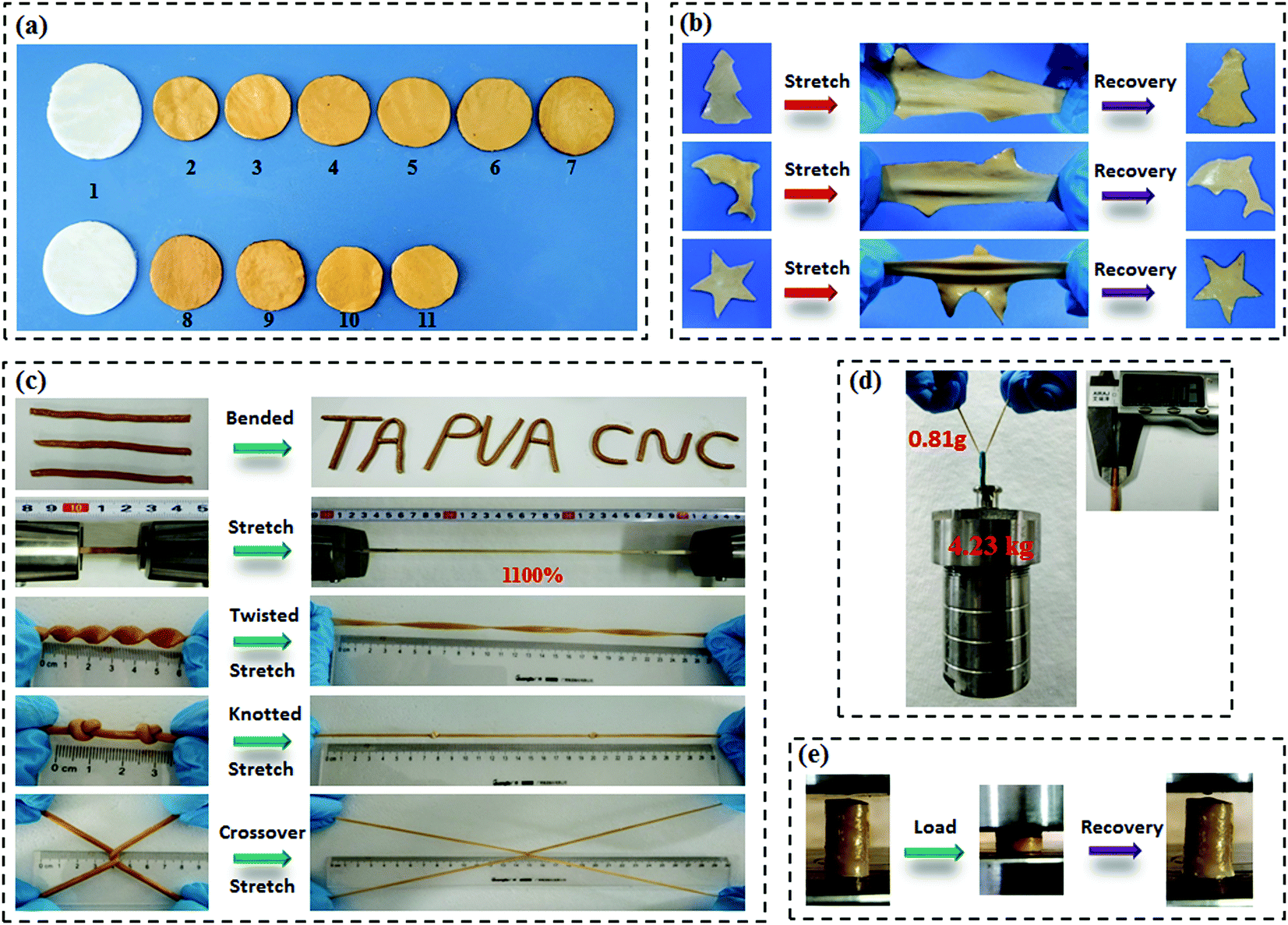 | ||
| Fig. 1 (a) Photographs of the PVA/CNC hydrogel (a1), TA10–PVA/CNCx (where x = 0, 5, 10, 15, 20, and 25 wt%) (a2–a7), and TAy–PVA/CNC20 (where y = 5, 10, 15, 20 wt%) (a8–a11). (b) Free-shapeable properties of the TA–PVA/CNC hydrogels: a Christmas tree (top), a Dolphin (middle), and a Star (bottom) could rapidly recover to its original shape after removal of the stretch force. (c) Outstanding mechanical performance of the TA15–PVA/CNC20 hydrogel: bending, stretching, twisting, knotting, and crossover stretching. (d) TA15–PVA/CNC20 cylindrical hydrogel with a diameter of 3 mm and a weight of 0.81 g lifted up a steel block of 4.23 kg in weight. (e) Compression and rapid recovery of the TA15–PVA/CNC20 hydrogel. | ||
To evaluate the H-bonding interactions between TA and the PVA/CNC matrix, FTIR measurements were performed (Fig. 2 and Fig. S4, ESI†). According to previous reports,20,41,42 the formation of H-bonding between TA (or cellulose nanofiber) and PVA chains reduces the force constants of the –OH groups of PVA, leading to the shift of their vibration frequencies to a lower wavenumber. Stronger H-bonding results in a more pronounced shift in vibrational frequency. As presented in Fig. 2a and b, the stretching vibrations of both hydroxyl groups (νO–H) and C–OH groups (νC–OH) in the TA–PVA/CNC hydrogels exhibited a more obvious redshift when CCNC and CTA increased. To be more specific, the PVA-gel showed typical νO–H at 3463 cm−1 and νC–OH at 1125 cm−1, which shifted to a lower wavenumber of 3448 cm−1 and 1112 cm−1 after being treated with TA (10 wt% TA solution), indicating the formation of H-bonds between PVA and TA (Fig. 2a1 and a2).20 As the CCNC increased from 5 to 25 wt%, a broader absorption band with a significant redshift of νO–H was observed, from 3433 cm−1 for TA10–PVA/CNC5 to 3412 cm−1 for TA10–PVA/CNC25. In addition, the νC–OH of TA–PVA/CNC exhibited a redshift analogous to νO–H. The absorption peak of C–OH shifted from 1107 cm−1 (from PVA) and 1046 cm−1 (from CNC) for TA10–PVA/CNC5 to 1088 cm−1 and 1032 cm−1 for TA10–PVA/CNC25, respectively. Besides, as shown in Fig. 2b1 and b2, the TA–PVA/CNC hydrogels (CCNC = 20 wt%) prepared with different CTA exhibited a similar trend. The broadened and pronounced redshift of the absorption bands in FTIR spectra with increased CTA demonstrated the enhanced H-bonding interactions between pyrogallol/catechol groups in TA molecules and –OH groups in the PVA/CNC matrix, which endowed the TA–PVA/CNC hydrogels with excellent mechanical performance. Moreover, the characteristic vibration peaks of TA appeared at 1721 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1536 cm−1, 1443 cm−1 (aromatic C–C), and 750 cm−1 (phenolic hydroxyl) (Fig. 2a and b) in all hydrogels, further indicating the successful synthesis of TA–PVA/CNC hydrogels.35
O), 1536 cm−1, 1443 cm−1 (aromatic C–C), and 750 cm−1 (phenolic hydroxyl) (Fig. 2a and b) in all hydrogels, further indicating the successful synthesis of TA–PVA/CNC hydrogels.35
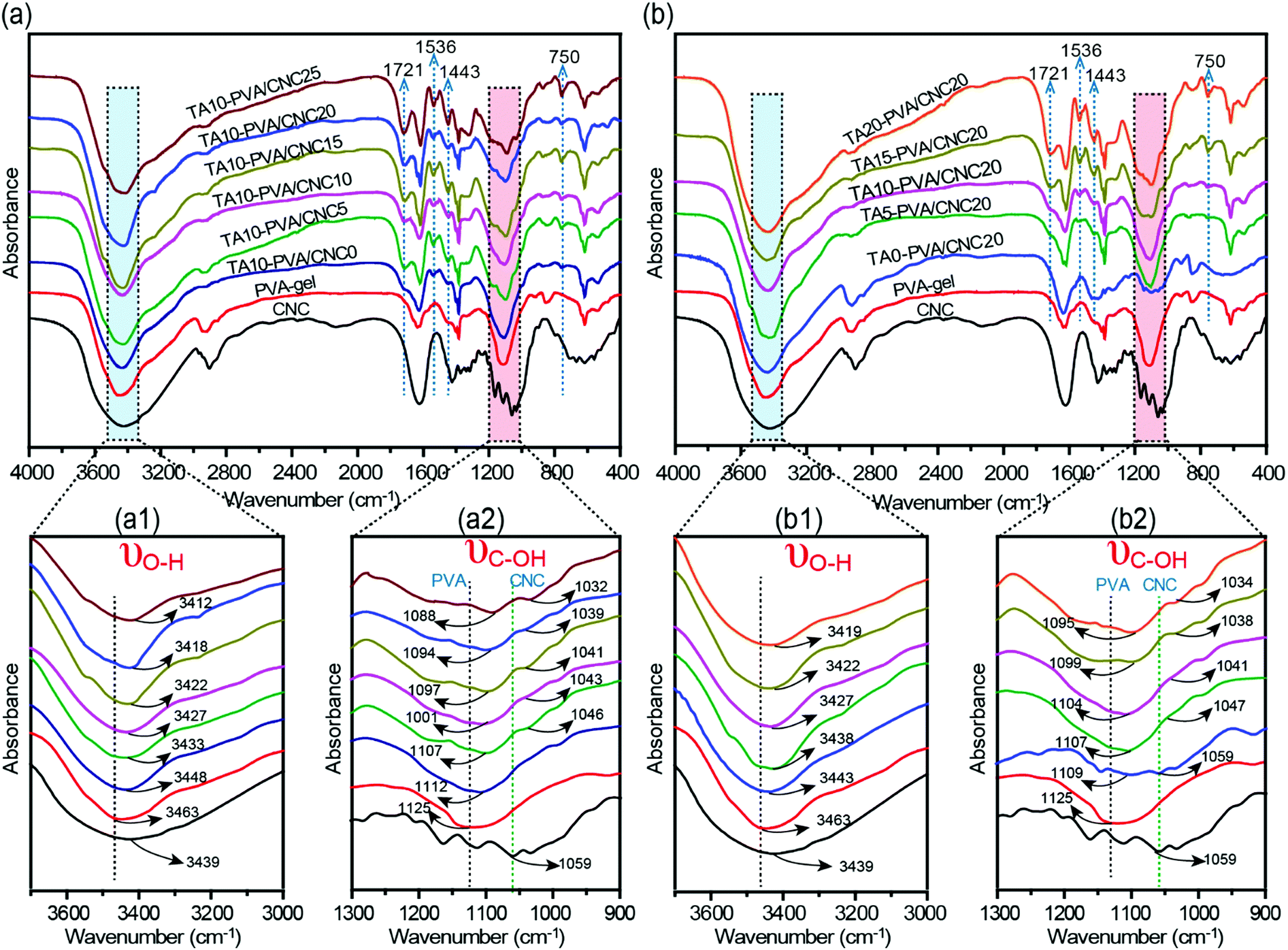 | ||
| Fig. 2 (a and b) FTIR spectra of CNC, PVA-gel, and TA–PVA/CNC hydrogels showing the changes in O–H and C–OH stretching vibration. | ||
The noncovalent interactions between TA and the PVA/CNC matrix were further investigated by the O1s and C1s XPS spectra of TA0–PVA/CNC20 and TA10–PVA/CNC20, as shown in Fig. S5 (ESI†). The O1s spectrum of TA0–PVA/CNC20 was deconvoluted into two peaks at binding energies of 532.18 eV for C–O (from CNC or PVA) species and 532.86 eV for O–C–O/CO (from CNC) species (Fig. S5a, ESI†).43 After being treated with TA, the binding energies of C–O and O–C–O/CO shifted upward to 532.31 eV and 533.24 eV (Fig. S5b, ESI†), respectively, because the formation of hydrogen bonds induced the electron transfer from the O of C–O or O–C–O/CO to the HO– of TA and changed the binding energy.44 Additionally, the C1s spectra of TA10–PVA/CNC20 (Fig. S5d, ESI†) also exhibited increased binding energies of 286.18 eV for C–O species and 288.73 eV for CO/O–C–O species, compared to those in the TA0–PVA/CNC20 spectrum (Fig. S5c, ESI†), further evidencing that the chemical and bonding environment of oxygen functional groups in the hydrogel changed after being treated with TA.45 The results confirmed the existence of H-bonding interactions between TA and the PVA/CNC matrix, which was consistent with the FTIR analysis.
2.2. Mechanical properties of the TA–PVA/CNC hydrogels
Considering the diverse physiological loads that tissue replacement materials may encounter, both the typical tensile and compression tests were conducted on the PVA-gel, TA0–PVA/CNC20, TA10–PVA/CNC0 and TA10–PVA/CNC20 hydrogels (Fig. 3). The PVA-gel exhibited a rather ductile behavior with poor tensile strength (0.48 MPa) at low fracture elongation (267.74%) and small compressive stress (4.0 MPa) at a fracture strain of 87%. With the introduction of CNCs along with TA, the tensile stress and fracture strain of the obtained TA10–PVA/CNC20 hydrogel significantly increased to 8.4 MPa and 1089% (Fig. 3a), respectively. The tensile elastic modulus and toughness were dramatically improved to 1.43 MPa and 50.03 MJ m−3 (Fig. 3b), respectively, which were 13 and 87 times higher than those of the PVA-gel. Meanwhile, the compressive strength of the TA10–PVA/CNC20 hydrogel could reach up to 51.6 MPa at a strain of 90% without fracture and the compression modulus and toughness sharply increased by 1138% and 786%, compared to those of the PVA-gel (Fig. 3c and d). However, when the PVA-gel was solely combined with CNC or TA, the mechanical strength and toughness of the resultant hydrogels were only improved to a certain extent (Fig. 3a–d and Fig. S6, ESI†), intuitively suggesting that the synergistic interactions between the CNC nano-reinforcement, TA macromolecular and PVA chains were essential for the mechanical strength and toughness improvement. This is possibly due to the abundant hydroxyl groups of CNC, TA and PVA, which could be easily assembled together via noncovalent interactions. In the TA10–PVA/CNC20 hydrogel, TA acted as a molecular coupling bridge between CNC and PVA chains, forming a much stronger dynamic network via multiple H-bonds compared to the hydrogels without TA. Meanwhile, the well dispersed rigid CNC not only acted as a nano-reinforcement but also participated in the multiple H-bonding assembly, which was well-involved in the formation of a physical cross-linked network. The newly formed strong noncovalent interactions significantly reduced the interchain distance and contributed to a much denser microstructure of the TA10–PVA/CNC20 hydrogel, resulting in the great improvement of its mechanical properties. As shown in Fig. 3e–g, both the PVA-gel and TA0–PVA/CNC20 hydrogel exhibit loose microstructures with large pore size. After being treated with TA solution (CTA = 10.0 wt%), the obtained TA10–PVA/CNC20 hydrogel displays a highly compact structure, further indicating that strong intermolecular interactions occurred between TA and the PVA/CNC matrix.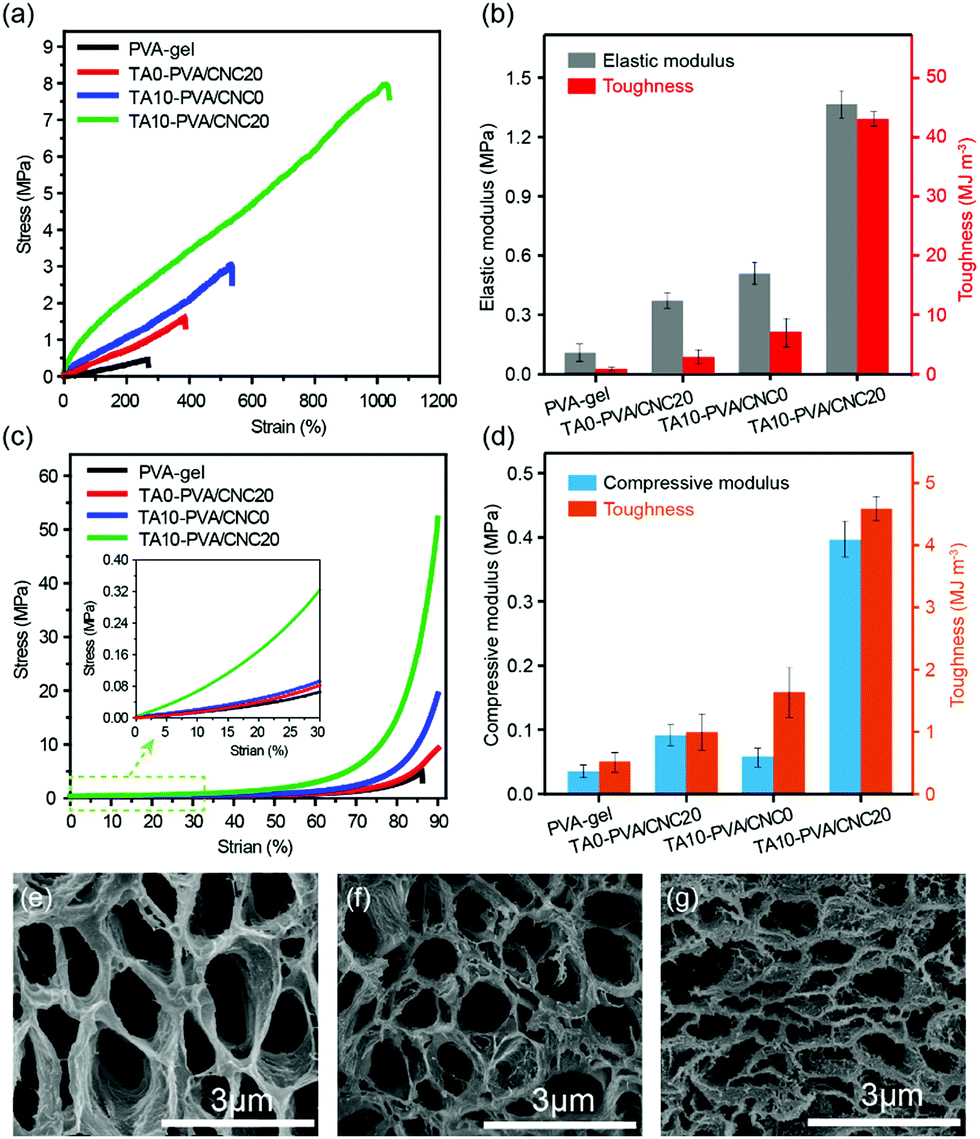 | ||
| Fig. 3 Typical tensile (a) and compressive (c) stress–strain curves and the corresponding elastic modulus and toughness (b and d) of the PVA-gel, TA0–PVA/CNC20, TA10–PVA/CNC0, and TA10–PVA/CNC20 hydrogels. SEM image of the PVA-gel (e), TA0–PVA/CNC20 (f), and TA10–PVA/CNC20 (g) hydrogels. | ||
To gain insight into the strengthening mechanism of the TA–PVA/CNC hydrogel, the PVA-gel, TA0–PVA/CNC20 hydrogel and TA10–PVA/CNC20 hydrogel were treated with 1.0 M urea aqueous solution (which is commonly considered as a H-bond breaker) for 2 h at 50 °C, and then the tensile tests were conducted. As illustrated in Fig. S7 (ESI†), the tensile strength of the urea-treated TA10–PVA/CNC20 hydrogel reduced significantly to 3.54 MPa which was only 46.7% of the original specimen, while the urea-treated PVA-gel was unchanged in strength. This phenomenon was attributed to the disruption of the extensive H-bonds between TA and the PVA/CNC matrix after urea treatment, and urea displayed little effect on the crystalline domains of the cross-linked PVA-gel. In the curves of the TA0–PVA/CNC20 hydrogel, a slight decrease of the tensile strength was due to the breakage of H-bonds between CNC and PVA chains. These results indicated that the strong and dynamic multiple H-bonding interactions between TA and the PVA/CNC matrix were the main driving force for the outstanding mechanical performance of the biomimetic TA–PVA/CNC hydrogel.
The mechanical performance of the TA–PVA/CNC nanocomposite hydrogels can be adjusted flexibly by varying the mass concentration of CNC (CCNC) and TA (CTA). With CTA fixed at 10 wt%, the effects of CCNC on the tensile properties of the TA–PVA/CNC hydrogels were investigated and are shown in Fig. 4a–c and Table S2 (ESI†). When the PVA-gels (tensile strength 0.48 MPa and toughness 0.57 MJ m−3) were treated with TA, the tensile strength and toughness of TA10–PVA/CNC0 (without CNC) increased to 2.88 MPa and 7.32 MJ m−3, implying that noncovalent interactions occurred between PVA and TA. Introducing more CNCs in the matrix created more crosslinking points to the H-bonded network, which could dramatically increase the mechanical properties of the TA–PVA/CNC hydrogel. With the CCNC increasing to 20 wt%, the tensile strength of the TA–PVA/CNC hydrogel reached up to 7.58 MPa at a fracture strain of 1034.31%. Meanwhile, the elastic modulus and toughness increased significantly from 0.67 MPa and 7.32 MJ m−3 to 1.68 MPa and 42.09 MJ m−3, respectively. When the CCNC increased to 25 wt%, the tensile stress and strain decreased slightly, which was probably due to the partial aggregation of CNC nanoparticles.46 On the other hand, with the CCNC (20 wt%) being constant, the tensile strength and toughness of the TA–PVA/CNC hydrogel increased sharply with the increase of CTA (Fig. 4d–f and Table S2, ESI†). The maximum tensile strength of 8.71 MPa with a fracture strain of 1107.55% could be achieved when the CTA increased to 20 wt%. Higher CTA led to stronger H-bonding interactions and more densely crosslinked networks, which could effectively dissipate energy and delay the network fracture, endowing the TA–PVA/CNC hydrogels with high mechanical strength and toughness. Similarly, the TA–PVA/CNC hydrogels demonstrated an enhanced compression strength as the CCNC and CTA increased (Fig. 4d, e and Table S2, ESI†), and a maximum stress of 67.90 MPa (at 90% strain) with a compression modulus of 2.34 MPa was achieved for TA20–PVA/CNC20.
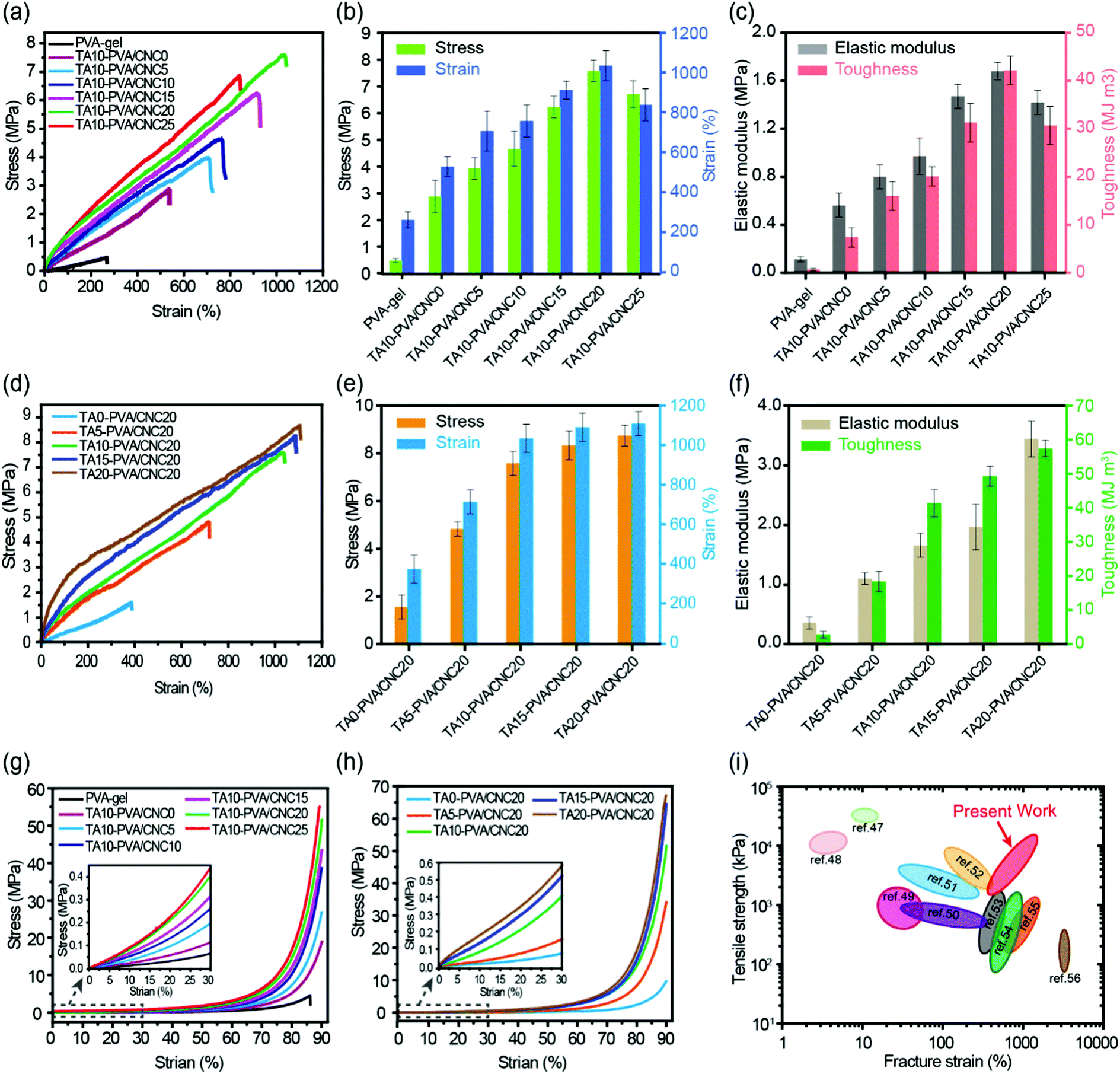 | ||
| Fig. 4 Typical tensile stress–strain curves (a), the stress and strain (b) and the corresponding elastic modulus and toughness (c) of the TA10–PVA/CNCy hydrogels with varied CNC mass concentration. Tensile stress–strain curves (d), the stress and strain (e) and the corresponding elastic modulus and toughness (f) of the TAx–PVA/CNC20 hydrogels with different TA mass concentration treatment. Compressive stress–strain curves of the TAx–PVA/CNCy hydrogels with varied CNC mass concentration (g) and different TA mass concentration treatment (h). Comparison of tensile strength and fracture elongation of TA–PVA/CNC hydrogels with other reported hydrogen bond-based physical hydrogels (i).47–56 | ||
Fig. 4i presents a chart for comparing the mechanical properties of the TA–PVA/CNC hydrogels with other hydrogen bond-based hydrogels.47–56 Most physical hydrogels developed via the hydrogen bond cross-linking strategy only showed improvement in either the mechanical strength or fracture strain. Impressively, the obtained TA–PVA/CNC hydrogels based on the bio-inspired H-bonding assembly integrated both high strength and excellent elongation. Thus, the mechanical properties of the bio-based hydrogels prepared in this study outperformed most previously reported H-bonded hydrogels, which made them feasible to be used as load-bearing biomaterials.
2.3 Toughness and self-recovery of the TA–PVA/CNC hydrogels
The densely multiple H-bond crosslinked network in TA–PVA/CNC hydrogels not only strikingly reinforces their mechanical strength but also significantly toughens the hydrogels by providing effective energy dissipation. Cyclic loading–unloading tests were performed to investigate the energy dissipation capacity and coefficient of the TA–PVA/CNC hydrogels (TA15–PVA/CNC20 was chosen for further investigation). The pronounced hysteresis in Fig. 5a indicated that the TA15–PVA/CNC20 hydrogels could dissipate energy effectively, and the hysteresis energy increased sharply with the rise of strain which is essential for tough hydrogels. As is shown in Fig. S8a (ESI†), the energy dissipation of the TA15-PVA/CNC20 hydrogel increased dramatically from 0.96 MJ m−3 at a strain of 100% to 11.89 MJ m−3 at a strain of 600% with a corresponding dissipation coefficient reaching up to 88.59%. Furthermore, the stress–strain curves and energy dissipation at 200% strain of the PVA-gel, TA0–PVA/CNC20 and TA15–PVA/CNCN20 are displayed in Fig. 5b and Fig. S8b (ESI†). An obviously large hysteresis loop was observed in TA15–PVA/CNCN20, which showed much more efficient energy dissipation than both the PVA-gel and TA0–PVA/CNC20. The obvious difference in hysteresis loops manifested that the reversible multiple H-bonds between TA and the PVA/CNC matrix played a vital role in the energy dissipation of the TA15–PVA/CNCN20 hydrogel. During the loading process, the TA–PVA/CNC hydrogel deformed and the partial slippage of PVA crystalline domains was most likely to dissipate a small portion of energy firstly, followed by the breakage of a dynamic multiple H-bonding network contributing to the significant energy dissipation. The low mechanical strength of the pure PVA-gel should be attributed to the finite extensibility of PVA chains between crystalline domains, resulting in the easy fracture of the hydrogel when an external load was applied.27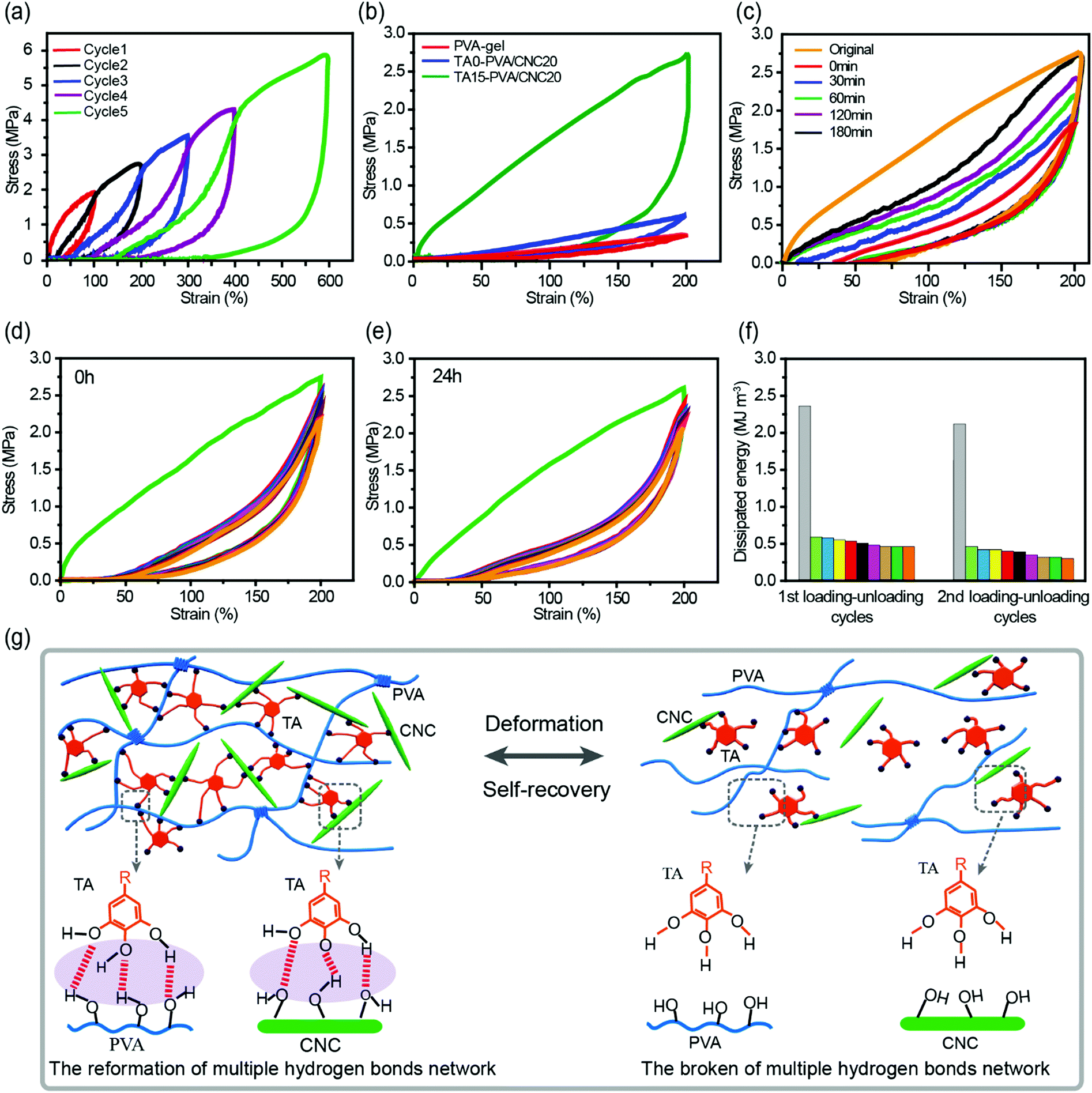 | ||
| Fig. 5 (a) Cyclic tensile loading–unloading curves of TA15–PVA/CNC20 under different strains (100%, 200%, 300%, 400%, and 600%). (b) The tensile loading–unloading curves of the PVA-gel, TA0–PVA/CNC20, and TA15–PVA/CNC20 at a strain of 200%. (c) Cyclic tensile loading–unloading curves at 200% strain of the TA15–PVA/CNC20 hydrogel at different resting times. Ten successive loading–unloading cycles of the (d) as-prepared and (e) recovered (24 h at room temperature) TA15–PVA/CNC20. (f) The corresponding dissipated energy in every cycle calculated from (d) and (e), respectively. (g) Schematic for the reversible hydrogen bonds formed between the pyrogallol/catechol groups of TA and the PVA/CNC matrix. | ||
The toughness of nanocomposite hydrogels must be closely related to the microstructure. The microscopic structures of the PVA-gel, TA0–PVA/CNC20, and TA15–PVA/CNC20 nanocomposite hydrogels were observed by SEM. As shown in Fig. S9a and d (ESI†), the pristine PVA hydrogel presented a porous network structure with a flat and smooth pore wall. This loose microarchitecture, which is generally brittle and fractures easily, results in the poor energy dissipation capacity of the PVA-gel. After introducing CNCs, TA0–PVA/CNC20 still shows a highly porous structure, but the network structure is more compact than that of the PVA-gel due to the H-bonding interactions between PVA chains and CNC nanoparticles (Fig. S9b, ESI†). Moreover, as shown in Fig. S9e (ESI†), CNCs and PVA are well mixed without obvious aggregation or separation, indicating a uniform dispersion of CNC within the PVA/CNC hydrogel matrix. This result is probably due to the fact that both CNC and PVA are hydrophilic and can form strong H-bonding interactions. After treating with TA solution, the resultant TA15–PVA/CNC20 hydrogel displays a highly compact structure (Fig. S9c, ESI†). Compared with the TA0–PVA/CNC20 hydrogel, the wall of the TA15–PVA/CNC20 matrix seemed thickened (Fig. S9f, ESI†). Some TA nanoparticles were found to appear in the pore wall, indicating that TA crosslinking interaction occurred in the internal structure of the PVA/CNC hydrogel, which could significantly drive close the interchain distance of the hydrogel matrix via strong intermolecular interactions. Due to the synergistic effect produced by H-bonded and compact network structures as well as the nano-reinforcement effects of CNCs, the resulting TA15–PVA/CNC20 hydrogel possesses a remarkable energy dissipation capacity. Specifically, after introducing the rigid CNC and then crosslinked by TA, the hydrogel was assembled based on multiple H-bonds, where the easy rupture and reformation of the H-bonded network provided an effective energy dissipation mechanism toward excellent mechanical properties and toughness (Fig. 5g). Meanwhile, the well-dispersed rigid CNC could maintain the configuration of hydrogels, resist stress concentration, as well as transfer energy from the PVA chains to the CNC nanorods to prevent crack propagation of the composite hydrogels.57
Cyclic tensile tests with different relaxation times and strains of 200% were performed to evaluate the self-recovery behavior of the TA–PVA/CNC hydrogels at room temperature (Fig. 5c and Fig. S8c, ESI†). Subsequent loading–unloading cycles without resting time produced a loop with much smaller hysteresis than the original (orange and red curves in Fig. 5c), which was attributed to the serious deformation and breakage of the PVA crystalline domains and H-bonding network. However, with the increase of relaxation time, the hysteresis loops gradually grew and approached that of the original. After resting for 180 min, the recovery ratios of elastic modulus and dissipated energy reached 61.8% (0.59 MPa) and 66.2% (1.57 MJ m−3) (Fig. S8c, ESI†). The good self-recovery ability manifested that the fractured internal networks could be repaired via rebuilding the dynamic H-bonds between TA and the PVA/CNC matrix (Fig. 5g), which is important for the applications of hydrogels exposed to successive loading.
Owing to the self-recovery of the TA15–PVA/CNC20 hydrogel, it exhibited a favorable antifatigue property against repeated deformation as illustrated by 10 successive loading–unloading tests (200% strain) of the as-prepared and recovered (resting 24 h) TA15–PVA/CNC20 samples. Fig. 5d shows that the as-prepared TA15–PVA/CNC20 hydrogel dissipated a large amount of energy during the first cycle, but showed a substantial decrease and displayed similar hysteresis energy during the following cycles, which indicated that the internal fracture of the first physical networks of PVA crystalline domains in the nanocomposite hydrogel could not be quickly reconstructed. After resting for 24 h at room temperature, the tensile strength (2.61 MPa) and hysteresis energy (2.08 MJ m−3) of the recovered TA15-PVA/CNC20 hydrogel were comparable to those of the original sample (tensile strength = 2.74 MPa and dissipated energy = 2.35 MJ m−3, Fig. 5e and f). The eminent self-recovery and fatigue resistance properties of the TA15–PVA/CNC20 hydrogel were further evaluated by 50 continuous compressive loading–unloading cycles at a strain of 50% (Fig. S8d–f, ESI†). The compression stress–strain curves for 2–50th cycles were almost superimposable (Fig. S8d, ESI†). Although the maximum stress after 50 successive compressive–relaxation cycles decreased, it remained to be more than 1.27 MPa (87.6% of first cycle at 50% strain) and the dissipated energy still remained to be more than 25 kJ m−3 (Fig. S8f, ESI†). Delightfully, it was noted that our biomimetic TA–PVA/CNC hydrogels exhibited a better compression strength (1.27–1.45 MPa at a strain of 50%) than artificial cartilage (0.24–0.85 MPa at a strain of 40–60%)58 under continuous loading–unloading cycles, showing great potential for load-bearing soft tissue replacement.
The above results suggested that the multiple H-bonds between TA molecules and the PVA/CNC matrix were strong enough to serve as special dynamic junction pints of the hydrogels. When the external load was applied, the easy breakage of hydrogen bonds acted as the sacrificial bond to dissipate energy. After the external loading was removed, the temporarily fractured hydrogen bonds could be reconstructed without any external stimulus, which endowed the TA15–PVA/CNC20 nanocomposite hydrogel with good antifatigue ability (Movie S4, ESI†).
2.4. Healing behavior of the TA–PVA/CNC hydrogels
The dynamic nature of H-bonds can also endow the TA–PVA/CNC nanocomposite hydrogels with healing ability. As shown in Fig. 6a, a strip TA15–PVA/CNC20 hydrogel was cut into two halves and then the cut surfaces were put together without applying any pressure. After adding a droplet of TA solution (15 wt%) on the contact surfaces, the sample was allowed to stand for 24 h at room temperature. The healed hydrogel could lift a weight of 100 g. The cutting and healing processes of TA–PVA/CNC hydrogels are provided in Movie S5 (ESI†). Besides TA solution, the fractured hydrogels could also be healed with the assistance of Fe3+ solution (0.3 mol L−1) and the healed hydrogels could load a 200 g weight (Fig. 6a and Movie S6, ESI†). The stress–strain curves and self-healing efficiencies of the original and healed TA15–PVA/CNC20 hydrogels are presented in Fig. 6b and c. After healing with the assistance od TA, the hydrogel reached a stress of 1.4 MPa with a fracture strain of 214.7%. The healing efficiency was 17.1% based on stress and 19.7% based on the fracture strain. These results indicated that the reversible H-bonds between the catechol (pyrogallol) groups of the TA molecule and hydroxyls of the PVA/CNC matrix could be reformed via TA assistance, leading to the spontaneous self-healing of the nanocomposite hydrogel. Moreover, the healing efficiency of the hydrogel could be further improved by coordination bonds in the presence of Fe3+ ions. With Fe3+ assistance, the fracture stress and strain of the TA–PVA/CNC hydrogel recovered to 30.5% and 33.8% of the original value, respectively. The healing efficiency of the TA15–PVA/CNC20 hydrogel was higher than that of TA induced healing behavior, which was attributed to the synergistic effects of dynamic coordination bonds and hydrogen bonds within nanocomposite hydrogels. When the two separate TA15–PVA/CNC20 hydrogels were brought into contact, the added Fe3+ ions could gradually diffuse toward the freshly cut interface and form the dynamic coordinate bonds with the catechol groups of TA molecules within hydrogels to rebuild the hydrogel network structure between the two separate pieces. At the same time, the fracture hydrogels lead to the formation of fresh catechol groups (originating from the contained TA of the TA15–PVA/CNC20 hydrogel) that located on neighboring fracture surfaces, which could form new hydrogen bonds with CNC or PVA chains when the fracture surfaces were brought into contact. Subsequently, the synergistic effects of dynamic coordination bonds and hydrogen bonds could further facilitate the reconstruction of hydrogel networks, resulting in promoting hydrogel healing behavior. Healing behaviors associated with the coordination bond interactions in TA–PVA/CNC nanocomposite hydrogels will be further explored in our future work.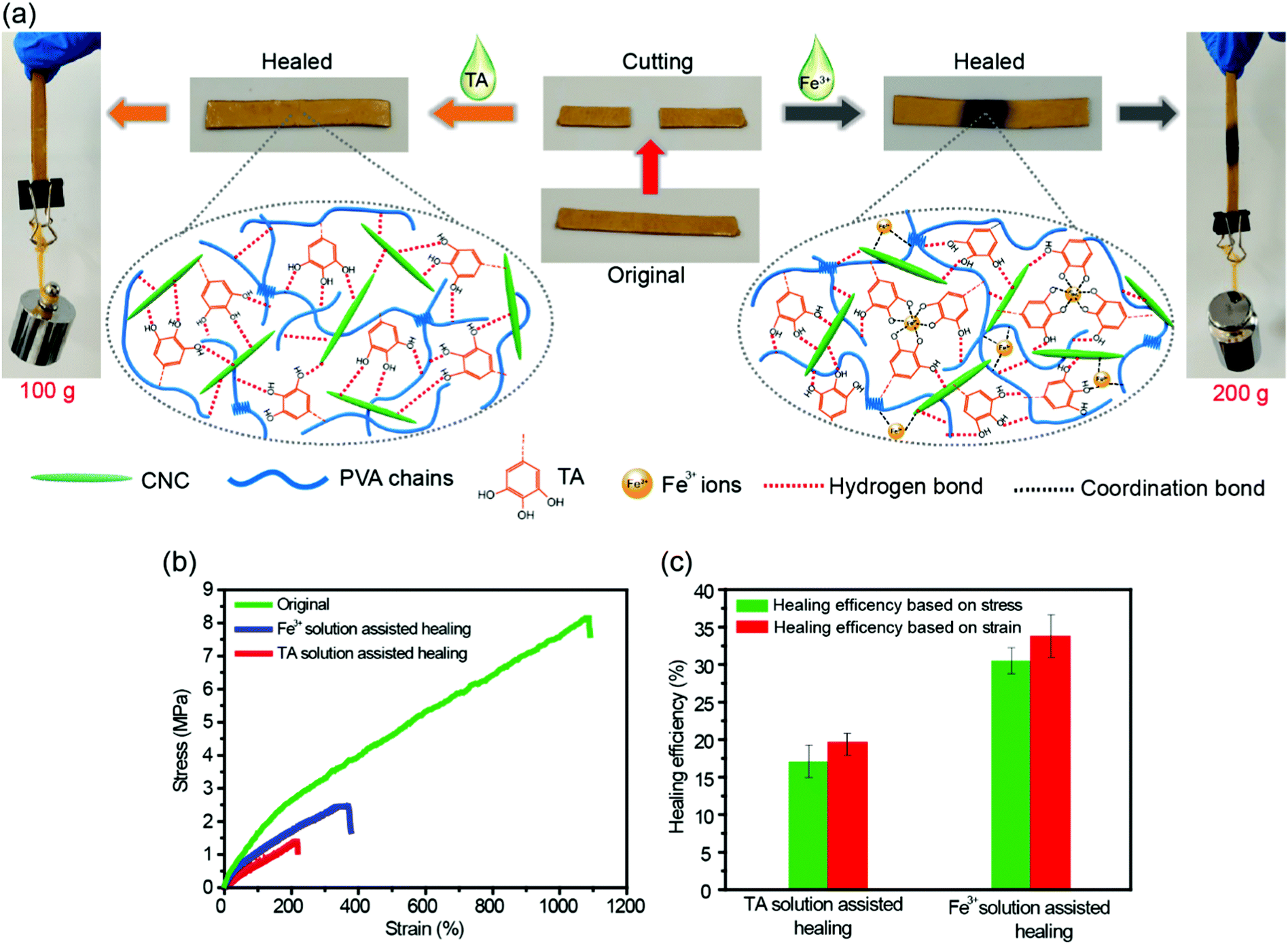 | ||
| Fig. 6 Demonstration of TA solution (15 wt%) and Fe3+ solution (0.3 mol L−1) assisted healing process of the TA15–PVA/CNC20 hydrogel and its possible mechanism (a). Stress–strain curves of the TA15–PVA/CNC20 hydrogel before and after healing assisted by TA solution (15 wt%) and Fe3+ solution (0.3 mol L−1) (b). Healing efficiency based on fracture stress and strain of the TA15–PVA/CNC20 hydrogel (c). | ||
2.5. Adhesiveness of the TA–PVA/CNC hydrogels
Combining excellent mechanical and high adhesive properties into hydrogels is still a major challenge.59,60 In the present work, the synthesized TA–PVA/CNC physical hydrogels simultaneously possess both superior mechanical performance and good adhesiveness to a variety of substrates, attributed to the numerous catechol groups of TA molecules imitating the mussel adhesion mechanism. Fig. 7a indicates that TA15–PVA/CNC20 exhibits good adhesiveness to both hydrophobic and hydrophilic surfaces, including polytetrafluoroethylene (PTFE), plastics, glass, wood, aluminum, and stainless steel. Moreover, the adhesive TA15–PVA/CNC20 hydrogel can easily adhere to biological tissues, including cartilage, tendon, weasand, heart, muscle, and bone, which is crucial for biological applications (Fig. 7b). As shown in Fig. 7c, the adhesive hydrogel can tightly adhere to the human skin and accommodate the wrist movements, which can be repeatedly peeled off from the skin without any residue and anaphylactic reaction. Tensile adhesion tests (Fig. 7e) were conducted to quantitatively determine the adhesion strength of the TA15–PVA/CNC20 hydrogel to different surfaces and the results are depicted in Fig. 7f. Among all substrates, the TA–PVA/CNC hydrogel exhibited the highest adhesion strength of 112.4 kPa to the wood surface, followed by stainless steel (101.7 kPa), aluminum (92.5 kPa), porcine skin (87.7 kPa), glass (82.4 kPa), plastic (68.2 kPa), and PTFE (59.8 kPa). | ||
| Fig. 7 Demonstration of the strong adhesiveness of the TA15–PVA/CNC20 hydrogel to various material surfaces (a) and tissues (b). (c) The TA15–PVA/CNC20 hydrogel repeatedly adhering to and moving with the wrist. Exhibition of no residual or irritation behavior to skin after 10 cycles of repeated adhering–stripping process. (d) The adhesion mechanism between the TA15–PVA/CNC20 hydrogel and different surfaces. (e) Schematic illustration of the hydrogel tensile adhesion tests. (f) Adhesive strength of the TA15–PVA/CNC20 hydrogel to different substrates tested by the tensile adhesion tests. (g) Repeatable adhesion behavior of the adhesive TA15–PVA/CNC20 hydrogel to different substrates tested by cyclic tensile adhesion tests. | ||
On the basis of the chemical structure of the hydrogel, the good adhesiveness originated from physical interactions, such as hydrogen bonding, metal coordination, π–π stacking, hydrophobic interactions, and several synergetic interactions simultaneously existing between the TA–PVA/CNC hydrogel and substrate surface (Fig. 7d).61,62 The TA15–PVA/CNC20 hydrogel on the surface of wood showed the highest adhesion strength, which resulted from the synergy of hydrogen bonding, π–π stacking, and hydrophobic interactions between hydrogels and the functional groups (hydroxy, carboxyl, and aromatic rings) on the wood surfaces.63,64 In addition, the coarser surfaces may also play a certain role in the adhesion to wood as compared with other substrates. The good adhesion of the TA15–PVA/CNC20 hydrogel to stainless steel and aluminum should be due to the metal coordination and hydrogen bonding, and the hydrogen bonding and hydrophobic interaction were probably the two major physical interactions in the adhesion of the TA15–PVA/CNC20 hydrogel to glass, plastic, and PTFE.65,66 For biological tissues, except for the hydrogen bonding interactions, the free catechol groups in the TA15–PVA/CNC20 hydrogel also could easily bind with amine or thiol groups (from peptides and proteins) on tissue surfaces via cation–π or π–π interactions.67–69 Moreover, due to the reversibility of the physical interactions, our TA15–PVA/CNC20 hydrogel exhibited a repeatable and durable adhesive behavior. As shown in Fig. 7g, after three cycles of adhesion tests, the TA15–PVA/CNC20 hydrogel still maintained the original adhesive strength on different substrates. Thus, it was envisioned that a high mechanical strength with superior and repeatable adhesion to various materials and tissues would give TA–PVA/CNC hydrogels an opportunity to be widely applied as tissue scaffolds, biosensors, wearable devices, etc.
2.6. Cell cytotoxicity and antibacterial activity
Cytotoxicity is the major concern for practical applications of biomaterials. Thereby, the cytocompatibility of the synthesized hydrogels was evaluated by comparing the attached cell number and its proliferation morphology according to previous studies.70–72 For this purpose, lentiviral-infected cells (NIH-3T3 fibroblasts) with the GFP (green fluorescent protein) were used for cytocompatibility tests. Specially, NIH-3T3 cells were seeded onto hydrogels at a density of 5 × 104 cells per dish (35 mm) and then the hydrogels with the attached live cells were further cultured for 3 days and 6 days. Subsequently, the hydrogels co-cultured with the labelled cells were carefully washed with 37% sterile PBS to remove dead cells, and then fixed with 4% paraformaldehyde solution before observation. As shown in Fig. 8a–c, only a few NIH-3T3 cells were found on the TA0–PVA/CNC20 hydrogel scaffold and the cells showed globular morphologies, indicating the poor cell attachment and low cell proliferation of TA0–PVA/CNC20. In contrast, after treating the composite hydrogels with TA, the NIH-3T3 cells could be well attached on the TA15–PVA/CNC20 scaffold (Fig. 8d and e) and the adhered cells were significantly proliferated with the incubation time increasing from 48 h (44 per mm2) to 96 h (176 per mm2) (Fig. 8f), presenting a flourishing living state on the scaffold. Meanwhile, the NIH-3T3 cells cultured on the TA15–PVA/CNC20 scaffold exhibited a more spread morphology with typical elongated filopodia (Fig. 8e and Movie S7, ESI†), demonstrating the good biocompatibility and cell affinity of the synthesized hydrogels. The significant improvement in the cell affinity of TA15–PVA/CNC20 hydrogels was attributed to the presence of TA, since it has been confirmed that the free catechol groups are favorable for cell attachment and proliferation via cation–π, or π–π interactions with the amine or thiol groups of the cell membrane.34,73–75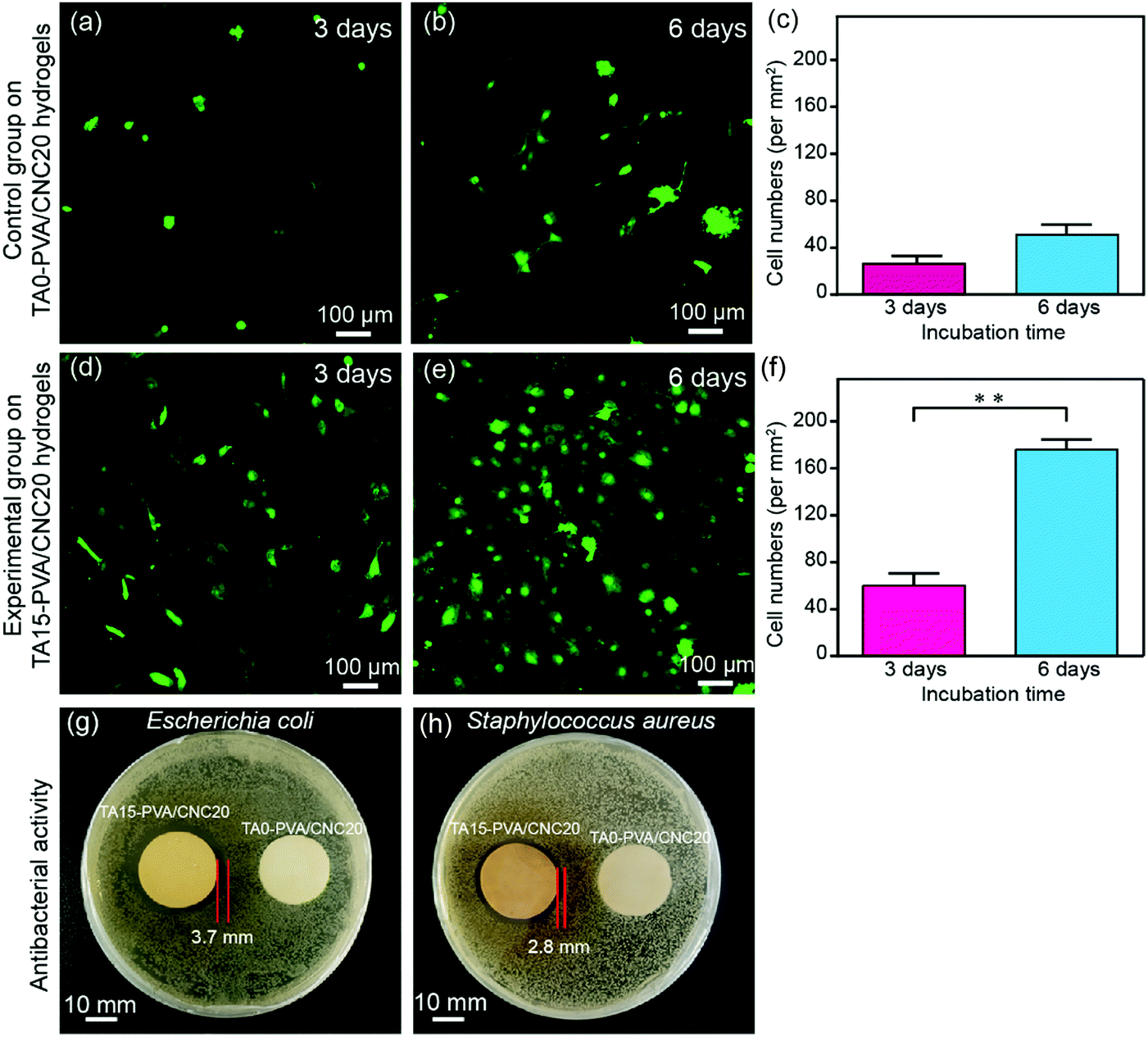 | ||
| Fig. 8 The confocal laser scanning microscopy (CLSM) images of NIH-3T3 cells cultured on the TA0–PVA/CNC20 hydrogel (a and b) and TA15–PVA/CNC20 hydrogel (d and e) for 3 days and 6 days, respectively. NIH-3T3 cell number on the TA0–PVA/CNC20 hydrogel (c) and TA15–PVA/CNC20 hydrogel (f) after culturing for 3 days and 6 days. Antibacterial activities of the TA0–PVA/CNC20 hydrogel and TA15–PVA/CNC20 hydrogel evaluated against Gram-negative viz. Escherichia coli (E. coli) (g) and Gram-positive viz. Staphylococcus aureus (S. aureus) (h). | ||
In addition, the antibacterial activity of the hydrogels is also an important property considering microbial attack during practical applications, which is rarely found in traditional biomaterials. In the TA–PVA/CNC hydrogels, TA serves as a natural antimicrobial agent and can provide resistance against microorganism attack. The antibacterial performances of the TA15–PVA/CNC20 hydrogel against Gram-negative viz. Escherichia coli (E. coli) and Gram-positive viz. Staphylococcus aureus (S. aureus) were investigated via the inhibition zone method, where the TA0–PVA/CNC20 hydrogel was used as a control sample. As shown in Fig. 8g and h, the zones of bacterial inhibition around the TA15–PVA/CNC20 hydrogel were about 3.7 mm against E. coli and 2.8 mm against S. aureus, while the TA0–PVA/CNC20 hydrogel showed virtually no zone of inhibition against either microbe, demonstrating the excellent antibacterial properties of TA15–PVA/CNC20 hydrogels. Therefore, the TA–PVA/CNC hydrogels simultaneously exhibit good biocompatibility and excellent antibacterial properties, which can be a safe candidate for tissue engineering.
3. Conclusions
In summary, TA–PVA/CNC hydrogels were successfully constructed by simply post-crosslinking the PVA/CNC composite hydrogels with TA treatment. The formation of strong multiple H-bonds between TA and the PVA/CNC matrix resulted in bio-based advanced physical hydrogels with ultrahigh tensile strength (up to 8.7 MPa), large elongation (up to 1107.6%), remarkable toughness (up to 58.2 MJ m−3), and high compressive strength (up to 67.9 MPa at 90% strain). Due to the dynamic nature of the H-bonded network, the hydrogels were endowed with excellent energy dissipation capacity, rapid self-recovering ability and good antifatigue property, which could withstand continuous deformation. More importantly, the advanced mechanical properties of these physical hydrogels can be adjusted readily in a wide range by varying the mass concentration of CNC and TA. Taking advantages of natural polyphenol TA, versatile adhesiveness and good antibacterial properties of TA–PVA/CNC hydrogels were demonstrated. Besides, the obtained TA–PVA/CNC hydrogels also exhibited a good biocompatibility as they were established based on natural plant extracts and biopolymers. This work provides new insights and approaches for the design of biocompatible hydrogels with superior mechanical performance and multi-functionalities, expanding their potential applications as biomimetic tissue scaffolds, biosensors and in other biomedical fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the generous financial support from the National Natural Science Foundation of China (Grant No. 31770611 and 31370560), the Natural Science Foundation of Fujian Province (Grant No. 2019J01388), the International Science and Technology Cooperation and Exchange Project of Fujian Agriculture and Forestry University (No. KXGH17008), the Scientific Research Foundation of Graduate School of Fujian Agriculture and Forestry University (No. 324-1122yb041), Specific research project of Guangxi for research bases and talents (AD18126005) Plan for the training of Outstanding Young Scientific Research Personnel in higher education institutions of Fujian Province (selected in 2017). F. Lin thanks the programme on Fujian Agriculture and Forestry University of Doctoral Students going aboard to be joint trained. B. Lu also thanks the support of the Outstanding Youth Scientific Research Personnel Training Plan of Colleges and Universities in Fujian Province. J. Chen and H. Zeng acknowledge the support from the Natural Sciences and Engineering Research Council (NSERC) and the Canada Research Chairs Program.References
- Y. Chen, C. Jiao, X. Peng, T. Liu, Y. Shi, M. Liang and H. Wang, J. Mater. Chem. B, 2019, 7, 3243–3249 RSC.
- J. K. Hirschberg, L. Brunsveld, A. Ramzi, J. A. Vekemans, R. P. Sijbesma and E. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed.
- J. Kříž, J. Dybal and J. Brus, J. Phys. Chem. B, 2006, 110, 18338–18346 CrossRef PubMed.
- P. A. Song, J. Dai, G. Chen, Y. Yu, Z. Fang, W. Lei, S. Fu, H. Wang and Z. Chen, ACS Nano, 2018, 12, 9266–9278 CrossRef CAS PubMed.
- P. Egan, R. Sinko, P. R. LeDuc and S. Keten, Nat. Commun., 2015, 6, 7418 CrossRef PubMed.
- T. Liu, C. Jiao, X. Peng, Y. Chen, Y. Chen, C. He, R. Liu and H. Wang, J. Mater. Chem. B, 2018, 6, 8105–8114 RSC.
- M. J. Buehler, Nano Today, 2010, 5, 379–383 CrossRef CAS.
- X. Zhang, W. Liu, D. Yang and X. Qiu, Adv. Funct. Mater., 2019, 29, 1806912 CrossRef.
- M. Cheng, Y. Wang, L. Yu, H. Su, W. Han, Z. Lin, J. Li, H. Hao, C. Tong and X. Li, Adv. Funct. Mater., 2015, 25, 6851–6857 CrossRef CAS.
- Y. Shi, D. Xiong, J. Li and N. Wang, J. Phys. Chem. C, 2016, 120, 19442–19453 CrossRef CAS.
- L. Jing, H. Li, R. Y. Tay, B. Sun, S. H. Tsang, O. Cometto, J. Lin, E. H. T. Teo and A. I. Y. Tok, ACS Nano, 2017, 11, 3742–3751 CrossRef CAS PubMed.
- J. Ren and D. Yu, Compos. Interfaces, 2018, 25, 205–219 CrossRef CAS.
- G. Zhou, J. Byun, Y. Oh, B. M. Jung, H. J. Cha, D. G. Seong, M. K. Um, S. Hyun and T. W. Chou, ACS Appl. Mater. Interfaces, 2017, 9, 4788–4797 CrossRef CAS PubMed.
- J. Chen, X. Shi, L. Ren and Y. Wang, Carbon, 2017, 111, 18–27 CrossRef CAS.
- Y. Huang, M. Zhang and W. Ruan, J. Mater. Chem. A, 2014, 2, 10508–10515 RSC.
- Q. Weng, B. Wang, X. Wang, N. Hanagata, X. Li, D. Liu, X. Wang, X. Jiang, Y. Bando and D. Golberg, ACS Nano, 2014, 8, 6123–6130 CrossRef CAS PubMed.
- P. A. Song and H. Wang, Adv. Mater., 2019, 1901244 CrossRef PubMed.
- S. Shi, X. Peng, T. Liu, Y. Chen, C. He and H. Wang, Polymer, 2017, 111, 168–176 CrossRef CAS.
- T. Liu, X. Peng, Y. N. Chen, Q. W. Bai, C. Shang, L. Zhang and H. Wang, Macromol. Rapid Commun., 2018, 39, 1800050 CrossRef PubMed.
- Y. Chen, L. Peng, T. Liu, Y. Wang, S. Shi and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 27199–27206 CrossRef CAS PubMed.
- P. A. Song, Z. Xu and Q. Guo, ACS Macro Lett., 2013, 2, 1100–1104 CrossRef CAS.
- P. A. Song, Z. Xu, Y. Lu and Q. Guo, Macromolecules, 2015, 48, 3957–3964 CrossRef CAS.
- Z. Xu, Y. Lu and Q. Guo, Compos. Sci. Technol., 2015, 118, 16–22 CrossRef.
- Z. Gong, G. Zhang, X. Zeng, J. Li, G. Li, W. Huang, R. Sun and C. Wong, ACS Appl. Mater. Interfaces, 2016, 8, 24030–24037 CrossRef CAS PubMed.
- M. Islam, L. Chen, J. Sisler and K. Tam, J. Mater. Chem. B, 2018, 6, 864–883 RSC.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- L. Wang, M. Ago, M. Borghei, A. Ishaq, A. C. Papageorgiou, M. Lundahl and O. J. Rojas, ACS Sustainable Chem. Eng., 2019, 7, 6013–6022 CrossRef CAS PubMed.
- M. Giese, L. K. Blusch, M. K. Khan and M. J. MacLachlan, Angew. Chem., Int. Ed., 2015, 54, 2888–2910 CrossRef CAS PubMed.
- M. Ago, J. E. Jakes and O. J. Rojas, ACS Appl. Mater. Interfaces, 2013, 5, 11768–11776 CrossRef CAS PubMed.
- T. Abitbol, T. Johnstone, T. M. Quinn and D. G. Gray, Soft Matter, 2011, 7, 2373–2379 RSC.
- S. Butylina, S. Geng and K. Oksman, Eur. Polym. J., 2016, 81, 386–396 CrossRef CAS.
- L. Xu, D. Pranantyo, K. G. Neoh and E. T. Kang, ACS Sustainable Chem. Eng., 2017, 5, 3055–3062 CrossRef CAS.
- R. Xu, S. Ma, P. Lin, B. Yu, F. Zhou and W. Liu, ACS Appl. Mater. Interfaces, 2017, 10, 7593–7601 CrossRef PubMed.
- V. Kozlovskaya, O. Zavgorodnya, Y. Chen, K. Ellis, H. M. Tse, W. Cui, J. A. Thompson and E. Kharlampieva, Adv. Funct. Mater., 2012, 22, 3389–3398 CrossRef CAS PubMed.
- H. Fan, J. Wang and Z. Jin, Macromolecules, 2018, 51, 1696–1705 CrossRef CAS.
- E. Costa, M. Coelho, L. M. Ilharco, A. Aguiar-Ricardo and P. T. Hammond, Macromolecules, 2011, 44, 612–621 CrossRef CAS.
- M. Shin, K. Kim, W. Shim, J. W. Yang and H. Lee, ACS Biomater. Sci. Eng., 2016, 2, 687–696 CrossRef CAS.
- H. Fan, L. Wang, X. Feng, Y. Bu, D. Wu and Z. Jin, Macromolecules, 2017, 50, 666–676 CrossRef CAS.
- A. L. Missio, B. D. Mattos, D. d. F. Ferreira, W. L. Magalhães, D. A. Bertuol, D. A. Gatto, A. Petutschnigg and G. Tondi, J. Cleaner Prod., 2018, 184, 143–151 CrossRef CAS.
- H. Fan, J. Wang, Q. Zhang and Z. Jin, ACS Omega, 2017, 2, 6668–6676 CrossRef CAS PubMed.
- Y. Chen, C. Jiao, Y. Zhao, J. Zhang and H. Wang, ACS Omega, 2018, 3, 11788–11795 CrossRef CAS PubMed.
- Z. Xu and J. Li, J. Nanosci. Nanotechnol., 2018, 18, 668–675 CrossRef CAS PubMed.
- L. Tang, F. Lin, T. Li, Z. Cai, B. Hong and B. Huang, Cellulose, 2018, 25, 4525–4536 CrossRef CAS.
- Q. Yu, Y. Zheng, Y. Wang, L. Shen, H. Wang, Y. Zheng, N. He and Q. Li, Chem. Eng. J., 2015, 260, 809–817 CrossRef CAS.
- M. Y. Lim, H. Shin, D. M. Shin, S. S. Lee and J. C. Lee, Polymer, 2016, 84, 89–98 CrossRef CAS.
- B. Lu, F. Lin, X. Jiang, J. Cheng, Q. Lu, J. Song, C. Chen and B. Huang, ACS Sustainable Chem. Eng., 2017, 5, 948–956 CrossRef CAS.
- W. Kong, C. Wang, C. Jia, Y. Kuang, G. Pastel, C. Chen, G. Chen, S. He, H. Huang and J. Zhang, Adv. Mater., 2018, 30, 1801934 CrossRef PubMed.
- F. Jiang, S. Cui, N. Song, L. Shi and P. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 16812–16821 CrossRef CAS PubMed.
- P. Wang, Y. Zhang, L. Cheng and W. Liu, Macromol. Chem. Phys., 2015, 216, 164–171 CrossRef CAS.
- B. Xu, Y. Zhang and W. Liu, Macromol. Rapid Commun., 2015, 36, 1585–1591 CrossRef CAS PubMed.
- X. N. Zhang, Y. J. Wang, S. Sun, L. Hou, P. Wu, Z. L. Wu and Q. Zheng, Macromolecules, 2018, 51, 8136–8146 CrossRef CAS.
- Y. J. Wang, C. Y. Li, Z. J. Wang, Y. Zhao, L. Chen, Z. L. Wu and Q. Zheng, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 1281–1286 CrossRef CAS.
- Z. Feng, H. Zuo, W. Gao, N. Ning, M. Tian and L. Zhang, Macromol. Rapid Commun., 2018, 39, 1800138 CrossRef PubMed.
- X. Hu, M. Vatankhah Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- R. Liu, S. Liang, X. Tang, D. Yan, X. Li and Z. Yu, J. Mater. Chem., 2012, 22, 14160–14167 RSC.
- C. Shao, H. Chang, M. Wang, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 28305–28318 CrossRef CAS PubMed.
- D. J. Huey, J. C. Hu and K. A. Athanasiou, Science, 2012, 338, 917–921 CrossRef CAS PubMed.
- H. Yuk, T. Zhang, S. Lin, G. A. Parada and X. Zhao, Nat. Mater., 2016, 15, 190–196 CrossRef CAS PubMed.
- J. Li, A. D. Celiz, J. Yang, Q. Yang, I. Wamala, W. Whyte, B. R. Seo, N. V. Vasilyev, J. J. Vlassak and Z. Suo, Science, 2017, 357, 378–381 CrossRef CAS PubMed.
- X. Liu, Q. Zhang, Z. Gao, R. Hou and G. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 17645–17652 CrossRef CAS PubMed.
- M. Krogsgaard, V. Nue and H. Birkedal, Chemistry, 2015, 22, 844–857 CrossRef PubMed.
- C. Shao, L. Meng, M. Wang, C. Cui, B. Wang, C. Han, F. Xu and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 5885–5895 CrossRef CAS PubMed.
- F. Lin, Z. Wang, Y. Shen, L. Tang, P. Zhang, Y. Wang, Y. Chen, B. Huang and B. Lu, J. Mater. Chem. A, 2019, 7, 26442–26455 RSC.
- D. Gan, W. Xing, L. Jiang, J. Fang, C. Zhao, F. Ren, L. Fang, K. Wang and X. Lu, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- X. Liu, Q. Zhang, Z. Gao, R. Hou and G. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 17645–17652 CrossRef CAS PubMed.
- L. Han, L. Yan, K. Wang, L. Fang, H. Zhang, Y. Tang, Y. Ding, L.-T. Weng, J. Xu and J. Weng, NPG Asia Mater., 2017, 9, e372 CrossRef CAS.
- L. Han, K. Liu, M. Wang, K. Wang, L. Fang, H. Chen, J. Zhou and X. Lu, Adv. Funct. Mater., 2018, 28, 1704195 CrossRef.
- M. Shan, C. Gong, B. Li and G. Wu, Polym. Chem., 2017, 8, 2997–3005 RSC.
- Z. Shi, H. Gao, J. Feng, B. Ding, X. Cao, S. Kuga, Y. Wang, L. Zhang and J. Cai, Angew. Chem., Int. Ed., 2014, 53, 5380–5384 CrossRef CAS PubMed.
- L. Han, L. Yan, M. Wang, K. Wang, L. Fang, J. Zhou, J. Fang, F. Ren and X. Lu, Chem. Mater., 2018, 30, 5561–5572 CrossRef CAS.
- F. Lin, R. Zheng, J. Chen, W. Su, B. Dong, C. Lin, B. Huang and B. Lu, Carbohydr. Polym., 2019, 205, 244–254 CrossRef CAS PubMed.
- S. Kim, H. Y. Yoo, J. Huang, Y. Lee, S. Park, Y. Park, S. Jin, Y. M. Jung, H. Zeng and D. S. Hwang, ACS Nano, 2017, 11, 6764–6772 CrossRef CAS PubMed.
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz Molina, Adv. Mater., 2013, 25, 653–701 CrossRef PubMed.
- Y. Xie, Y. Zheng, J. Fan, Y. Wang, L. Yue and N. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 22692–22702 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb00424c |
| This journal is © The Royal Society of Chemistry 2020 |