
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigner DNA–silica/carbon nanotube nanocomposites for traceable and targeted drug delivery†
Yong
Hu
 and
Christof M.
Niemeyer
*
and
Christof M.
Niemeyer
*
Karlsruhe Institute of Technology (KIT), Institute for Biological Interfaces (IBG 1), Hermann-von-Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany. E-mail: niemeyer@kit.edu
First published on 26th February 2020
Abstract
Due to their unique properties like porosity, high water content, softness and biocompatibility, hydrogels are of great interest for biomedical applications such as tissue engineering and drug delivery. We describe a programmable drug delivery system that is based on highly biocompatible SiNP/CNT–DNA nanocomposites, which can be synthesized in a highly modular fashion from DNA-functionalized carbon nanotubes and silica nanoparticles via enzymatic rolling circle amplification. Specific molecular recognition properties were implemented into the materials by DNA sequence design, as demonstrated by incorporation of GC/CG-rich stem loop and aptamer motifs that enable selective binding of intercalating drugs and cell surface receptors, respectively. In a proof-of-concept study we demonstrate the utility of this approach by targeting nanocomposites loaded with the anthracycline drug doxorubicin to HeLa cancer cells. Our observation that these designer materials work more efficiently than the pure drug alone suggests that further developments of the concept might be useful to selectively trigger more complex cellular pathways.
Introduction
DNA hydrogels are 3D polymeric networks that offer excellent biocompatibility and biodegradability, tunable multifunctionality, stimulus reactivity, as well as unique molecular programmability and ability for precise molecular recognition. These properties make DNA hydrogels a powerful material for numerous applications in materials science and biomedical life sciences.1–4 For example, DNA hydrogels are well suited for drug release applications because specific cell binding motifs can be conveniently integrated into the polymer backbone by designed aptamer sequences.5 While the high water content of synthetic hydrogels is disadvantageous for drug release applications, as this often leads to a relatively fast release kinetics,6 this problem can be overcome with DNA hydrogels, since DNA intercalating drugs, such as the anthracycline doxorubicin (DOX) can be bound by intercalation into CG- or GC-rich duplex motifs of specially designed regions of the DNA backbone.5 However, in order to construct new generations of smart hydrogel systems for drug delivery, synthetic polymers are nowadays equipped with nanoparticles to provide the resulting nanocomposite hydrogels with adaptable physiochemical properties, such as mechanical, magnetic, electrical or optical features, that cannot be achieved by the polymer backbone alone.7Some examples of DNA hydrogel composites have already been described. For instance, work on DNA–carbon nanotubes (CNT) composites shows that they have good biocompatibility8,9 and DNA–quantum dot composites have been used as traceable and biocompatible nanomaterials for drug release.10 Furthermore, DNA hydrogel composites containing silica nanoparticles (SiNP) have recently been used for the bottom-up assembly of hollow spherical microstructures11 and SiNP/CNT–DNA composites were used as programmable and cell-instructive biocoatings.12 While in the latter work the outstanding attractiveness of SiNP–DNA materials for cells was used for the adjustable adhesion of eukaryotic cells, previous work has already shown that DNA-functionalized SiNP are not only highly biocompatible but are also very efficiently ingested by cells.13 This work also showed that dye-labelled SiNP are excellently suited for traceable studies of the underlying biological mechanisms. In view of the promising results on drug delivery by pure DNA hydrogels,5 we therefore wanted to investigate whether SiNP–DNA nanocomposites can also be used for traceable drug delivery and whether a further addition of CNT could influence the efficiency of drug delivery.
To produce the desired SiNP/CNT–DNA nanocomposite materials, SiNP of 80 nm diameter and CNT (0.83 nm diameter, 1 μm length) were functionalized with oligonucleotide primers (P) and the resulting particles were used for cyclization of the 98-mer oligonucleotide T that could then serve as template for enzymatic extension through rolling circle amplification (RCA, Fig. 1). The subsequent addition of dNTPs and phi29 DNA polymerase led to the formation of DNA nanocomposite networks, whereby either only SiNP or both SiNP and CNT were interwoven by DNA polymers to produce binary S or ternary SC nanocomposite materials, respectively. The same procedure was applied for the preparation of fluorescent nanocomposites by using Cy5-labeled core/shell SiNP,13 to yield the corresponding Cy5@S and Cy5@SC materials, respectively. The nanocomposite materials were obtained as viscous hydrogels, which could be dosed by pipetting and used for further investigations with living cells.
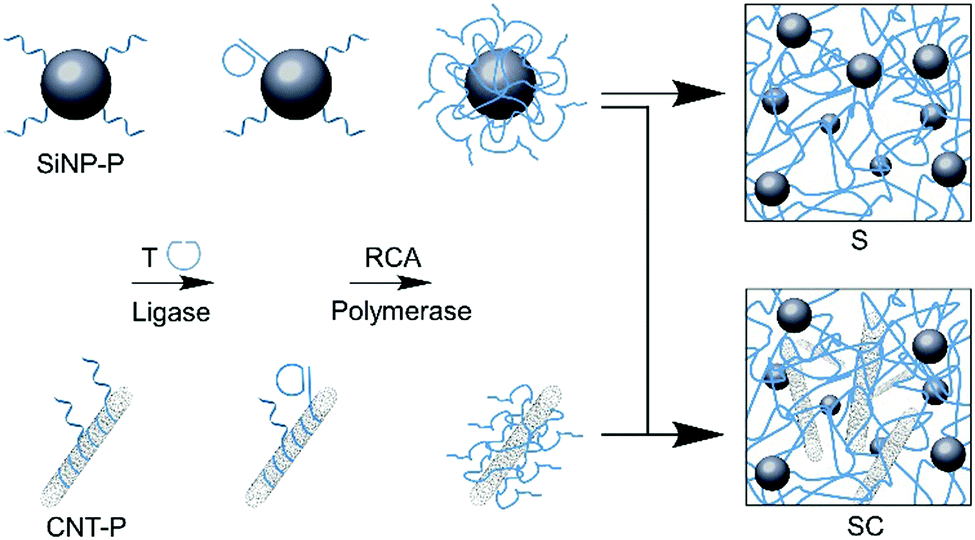 | ||
| Fig. 1 Synthesis of binary and ternary SiNP/CNT–DNA nanocomposite materials. Schematic illustration of RCA-based polymerization of nanoparticles. DNA primer (P) modified nanoparticles were used for enzymatic cyclisation of RCA template (T) and subsequent polymerization to yield the corresponding binary (S) or ternary (SC) DNA nanocomposite materials. | ||
Results and discussion
Characterization of the structural features of the S and SC materials was achieved after drying by scanning electron microscopy (SEM). The images revealed that that S and SC materials have a hierarchical porous structure (Fig. 2) where the DNA polymers wrap around and interconnect the nanoparticles embedded in the nanocomposite materials. Ultrastructural features like DNA polymers and individual CNT could clearly be observed in high resolution images of the S and SC nanocomposite materials. | ||
| Fig. 2 Scanning electron microscopy analysis of S and SC nanocomposite materials. Representative SEM images of S (a) and SC (b) nanocomposite materials at different magnifications. The right two panels of the SEM images shown in (a) and (b) are magnifications of the red circled regions in the corresponding left images. The arrows point at DNA polymers in (a) and CNT in (b), respectively. | ||
We initially investigated whether the SiNP–P, CNT–P, S and SC materials are cytotoxic to HeLa cancer cells. The analysis by a CCK-8 assay clearly showed a negligible inhibition of the proliferation of the cancer cells and thus proved the very good biocompatibility of the materials (Fig. S1, ESI†). The loading capability of the nanocomposites with the anthracycline drug doxorubicin (DOX) was then demonstrated by titration analyses of the quenching of DOX fluorescence (Fig. 3). The fluorescence of DOX is quenched when the drug intercalates into the DNA backbone.10 As shown in Fig. 3c and d, the progressive decrease in fluorescence intensity with increased amount of the DNA nanocomposite materials was observed. The obtained quenching spectra of SC are similar to those of S. The amount of loaded DOX was identical for both nanocomposite materials and was estimated in S or SC to be about 9 μg DOX per mg material. This suggests that the added CNT do not significantly affect drug loading of the materials, which is most likely due to the rather low amount of CNT (80 μg mL−1 in RCA reaction mixture) and the fact that the CNT are tightly coated with DNA polymers, shielding the CNT surface from the direct binding of DOX. To allow for saturation of S or SC with the drug for the cell studies, DOX and S/SC were mixed at pH = 7.4 and kept at room temperature for 48 hours (Fig. S2, ESI†).
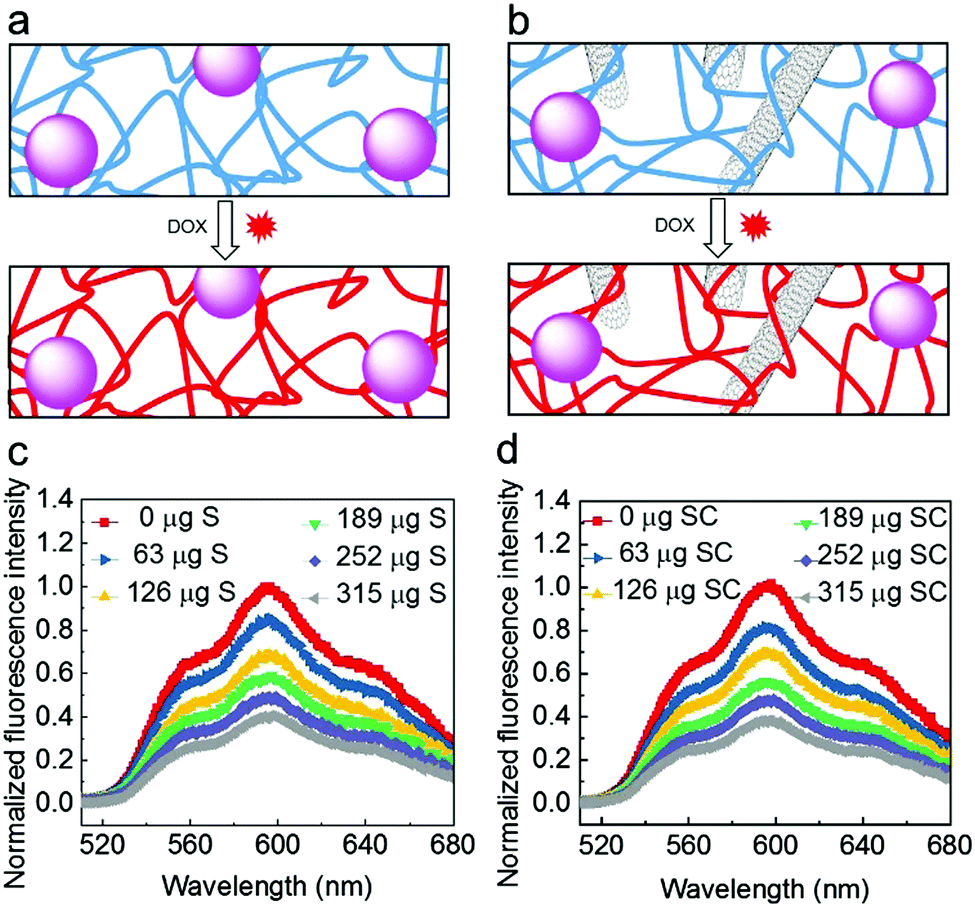 | ||
| Fig. 3 Intercalation of DOX into the DNA nanocomposites. (a and b) Schematic illustrations of DOX loading into DNA backbones of (a) S and (b) SC, respectively. (c and d) Fluorescence quenching of DOX by different amounts of (c) S and (d) SC at pH 7.4, respectively. Based on the titrational analysis, the amount of drug in the DOX-saturated nanocomposites was estimated as about 9 μg DOX per mg nanocomposite materials. | ||
To achieve efficient and selective drug delivery in tumor cells, the RCA template was equipped not only with a drug loading sequence, but also with an aptamer sequence. Hence, numerous repetitions of these sequence stretches were incorporated into the DNA backbone of the nanocomposite materials (Fig. 4). To experimentally demonstrate the selective cancer cell recognition and targeted drug delivery, we used a fluorescent material DOX/Cy5@S that contained a structured DNA backbone bearing the Sgc8 aptamer (Fig. 4a) which specifically binds to the protein tyrosine kinase-7 (PTK7) receptor.5,14 Initial studies with PTK7-positive HeLa cells showed that uptake of the fluorescent Cy5@S material was dependent on the amount of aptamer units incorporated in the material over time during the RCA synthesis (Fig. S3, ESI†).
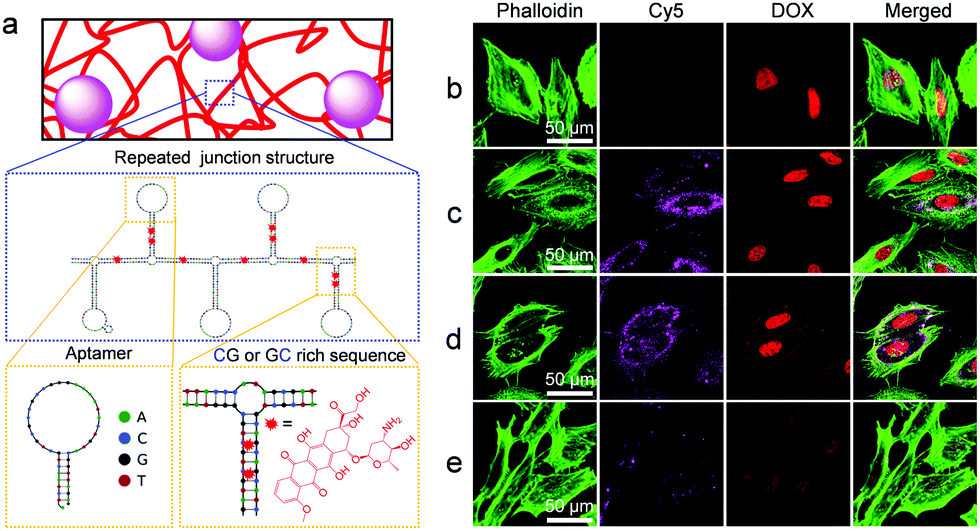 | ||
| Fig. 4 Traceable drug delivery to PTK7-positive HeLa cells. (a) Predicted secondary structure15 of the nanocomposite's DNA backbone produced by RCA. Note the presence of Sgc8 aptamer and GG/GC-rich stem loops, designed to serve as binding sites for cells and the intercalating drug DOX, respectively. (b–e) Representative confocal microscopy images of HeLa cells treated with (b) free DOX, (c) DOX/Cy5@S and (d) DOX/Cy5@SC, respectively. Note that although the images in (c) and (d) show approximately equally strong Cy5 signals, the uptake of DOX/Cy5@SC may be slightly higher than that of DOX/Cy5@S because the CNT cause a weak quenching of the Cy5 fluorescence signal. The cells in (e) were simultaneously treated with DOX/S and free Sgc8 aptamer, the latter of which inhibited uptake of the DOX/Cy5@S material. In all cases, cells were incubated with materials (38 μg mL−1 DOX equivalent) for 2 h, washed with PBS to remove unbound materials, fixed with PFA, and subjected to staining of the filamentous actin with Alexa Fluor 488 Phalloidin (green) for membrane visualization. Note that high-resolution images indicate the localization of the Cy5-labeled DNA composites in the cellular cytoplasm and the inner side of the cell membrane (Fig. S4, ESI†), which is in agreement with earlier work on pure DNA hydrogels.5 | ||
By virtue of double-stranded CG- or GC-rich drug association sites, the Cy5@S or Cy5@SC materials could be used for enrichment of intercalating drugs, such as anthracycline DOX, thus enabling the traceable drug delivery to cells. The nuclei of HeLa cells treated with DOX/Cy5@S (Fig. 4c) and DOX/Cy5@SC (Fig. 4d) for 2 h displayed comparable DOX fluorescence signal intensities as observed for the nuclei of cells treated with free DOX (Fig. 4b). These results suggested that DOX molecules were successfully released intracellularly. It is important to note that HeLa cells treated with DOX/Cy5@S or DOX/Cy5@SC (Fig. 4c and d) revealed significantly stronger fluorescence signals of Cy5@SiNP and DOX than cells treated with the composite materials and free aptamer (Fig. 4e). Following earlier work,16 the latter experiment was used as a negative control. Typical PTK7-negative cells, such as Ramos suspension cells,5 could not be used here because the separation of excess composite material that was not ingested is hardly possible. The obtained results clearly indicated that the free aptamer blocks the process of receptor-mediated endocytosis and it is consistent with the results of cellular uptake of the fluorescent S material (Fig. S3, ESI†). Hence, the results conclusively show that the cellular uptake of DOX/Cy5@S was indeed accomplished by the aptamer motifs in the DNA backbone. After cultivation for another 24 h, the cells exhibited a round shape and significant loss of filamentous actin fibers, thus indicating the induction of cell apoptosis by the ingested DOX (Fig. S5, ESI†).
Additionally, the kinetics of DOX release from the DOX/S and DOX/SC materials were investigated in different media (Fig. 5a–c). We found that DOX was not released from either DOX/S or DOX/SC in PBS (pH 7.4), acetate buffer (pH 5.0) and cell culture medium for up to 48 h at 37 °C. In contrast, DOX was quickly released in the presence of the exonuclease DNase I. Furthermore, the cytotoxicity of the DOX-loaded nanocomposites against HeLa cells was quantified after 2 h, 24 h, and 48 h incubation, respectively (Fig. 5d). We found that nearly all cells were viable after 2 h of incubation, whereas cells were killed by DOX/S or DOX/SC about 10% or 20% more effective, respectively, than by free DOX after 24 h of treatment. After 48 h of treatment, cells were killed by DOX/S or DOX/SC about 5 fold more effectively than by free DOX. These results reflect the efficient cellular uptake and potency of the DOX-loaded composite materials. Interestingly, the killing efficiency of DOX/SC was about 2 fold greater than that of DOX/S, thus suggesting that the CNT particles contribute to the cytotoxic efficacy of the DNA nanocomposites. This effect might stem from CNT's high propensity to cross cell membranes,17–19 facilitating an improved cellular uptake of the CNT-containing nanocomposite drug delivery system (see also Fig. 4) and subsequently inducing increased cytotoxicity by the internalized DOX. Altogether, the results emphasize that the novel nanocomposite materials are well suited as vehicles for the cell-specific delivery of DNA-binding drugs into cancer cells.
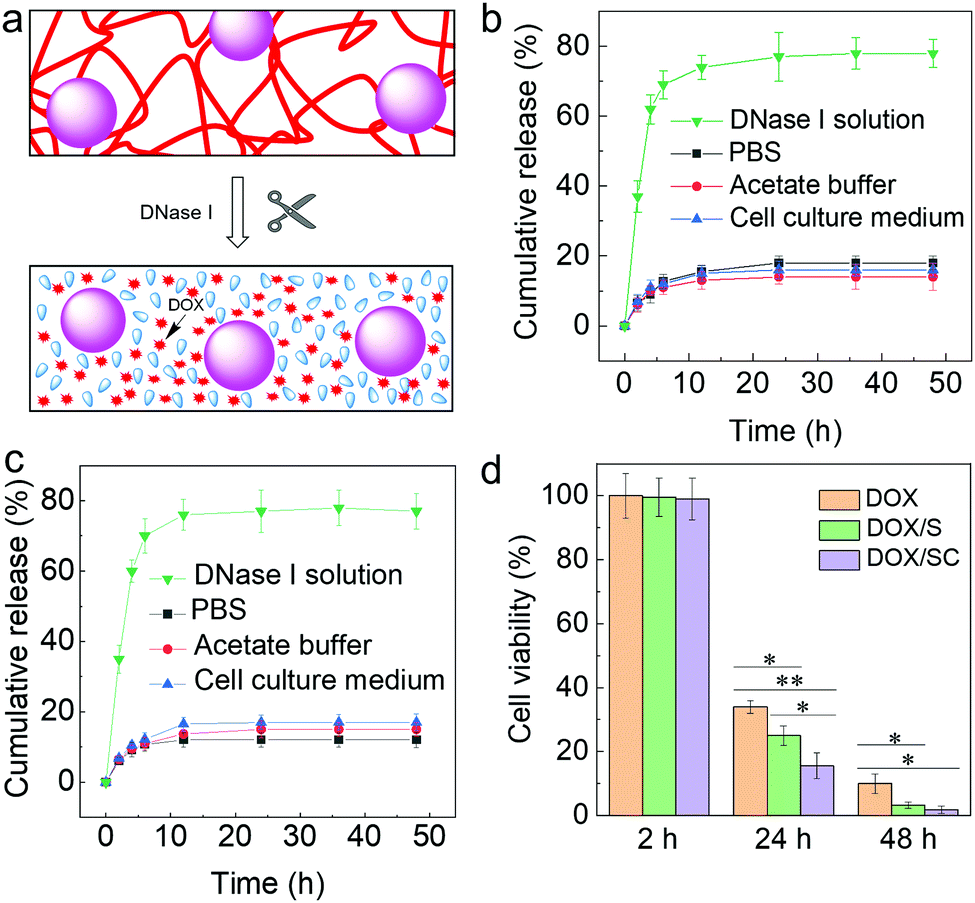 | ||
| Fig. 5 Time-dependent release of DOX. (a) Schematic drawing of degradation and release of DOX from the composite materials. (b and c) Release profiles of DOX from (b) DOX/S and (c) DOX/SC complexes in PBS (pH 7.4), acetate buffer (pH 5.0), cell culture medium, and DNase I solution at 37 °C for different times. (d) Time-dependent cytotoxicity of free DOX, DOX/S, or DOX/SC against HeLa cells, determined after 2 h, 24 h, or 48 h incubation, respectively. | ||
Conclusions
In summary, we describe a programmable drug delivery system based on highly biocompatible SiNP/CNT–DNA nanocomposites. These materials are available from DNA-functionalized CNT and SiNP via enzymatic rolling circle amplification in a highly modular synthetic procedure. Specific molecular recognition properties can be easily implemented into the materials by DNA sequence design, as demonstrated here by incorporation of GC/CG-rich stem loop and aptamer motifs that facilitate selective binding of intercalating drugs and cell surface receptors, respectively. The utility of this approach is demonstrated in a proof-of-concept study by targeting SiNP/CNT–DNA nanocomposites loaded with the anthracycline drug DOX to HeLa cancer cells. Given that the nanocomposites showed traceable and targeted drug delivery along with high biocompatibility in vitro, we believe that such systems might as well be suited for theranostic applications in vivo. Such developments would benefit from the use of near-infrared dyes (instead of the here used Cy5) and they would require the systematic assessment of e.g., the tumor uptake and excretion kinetics in higher organisms. Furthermore, the observation that these designer composite materials work more efficiently than the pure active ingredients suggests that the implementation of more complex structures, such as transmembrane receptor-activating DNA nanostructures,20 can be harnessed to trigger more complex cellular signaling pathways and cell fate decisions.Experimental section
Preparation of SiNP/CNT–DNA nanocomposite materials
Two different types of DNA nanocomposites, binary S and ternary SC materials were prepared by using SiNP (diameter 80 nm)21 and CNT (diameter 0.83 nm, length 1 μm, Sigma-Aldrich) according to previously reported procedures.12 Briefly, the linear ssDNA (T) phosphorylated at the 5′ end was circularized through hybridization with P1 attached on the surface of SiNP–P using T4 DNA ligase. To this end, linear ssDNA (T, 10 μM, 30 μL) and 10× T4 DNA ligation buffer (500 mM Tris–HCl, 100 mM MgCl2, 10 mM ATP, 100 mM dithiothreitol (DTT), 7.5 μL) were added to 60 μL SiNP–P suspension (10 mg mL−1), and the mixture was incubated at 25 °C for 3 h. After addition of 2.5 μL T4 DNA ligase (400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1, New England Biolabs), the mixture was further incubated for more than 3 h at 25 °C to ligate the nicked ends of the template, leading to the formation of particle–primer–template (SiNP–P–T) complexes. Similarly, for preparation of the CNT–P–T, 30 μL of linear ssDNA (T, 10 μM) and 60 μL of P2 wrapped CNT–P (200 μg mL−1) were used.
000 U mL−1, New England Biolabs), the mixture was further incubated for more than 3 h at 25 °C to ligate the nicked ends of the template, leading to the formation of particle–primer–template (SiNP–P–T) complexes. Similarly, for preparation of the CNT–P–T, 30 μL of linear ssDNA (T, 10 μM) and 60 μL of P2 wrapped CNT–P (200 μg mL−1) were used.
The RCA reaction mixture contained dNTPs (10 mM, 10 μL), 10× BSA (10 mg mL−1, 5 μL), 10× phi29 DNA polymerase buffer (500 mM Tris–HCl, 100 Mm MgCl2, 100 mM (NH4)2SO4, 40 mM DTT, pH 7.5, 5 μL) and phi29 DNA polymerase (10000 U mL−1, 5 μL, New England Biolabs). The polymerisation was initiated via the addition of 50 μL of the SiNP–P–T or the mixture of SiNP–P–T/CNT–P–T. After incubation at 30 °C for 48 h, the SiNP–DNA nanocomposite hydrogels or SiNP/CNT–DNA nanocomposite hydrogels were purified by carefully replacing the reaction mixture with Dulbecco's PBS for 5–7 times and the nanocomposite hydrogels were stored at 4 °C before use. With a final SiNP–P concentration of 4 mg mL−1, the formed SiNP–DNA nanocomposite hydrogel was denoted as S nanocomposite material. In the case of mixtures containing SiNP–P (4 mg mL−1) and CNT–P (80 μg mL−1), the formed SiNP/CNT–DNA nanocomposite hydrogel was denoted as SC nanocomposite material. Fluorescent S and SC nanocomposite materials were prepared using Cy5@SiNP, synthesized as previously described.13 Unless stated otherwise, fluorescent S and SC were used for fluorescence imaging.
DOX loading into SiNP/CNT–DNA nanocomposite materials
Doxorubicin hydrochloride (DOX, Sigma Aldrich) solution (29 μg mL−1) was incubated with the SiNP/CNT–DNA hydrogels (63, 126, 189, 252 and 315 μg S and SC, respectively) dispersed in 200 μL Dulbecco's PBS (Sigma Aldrich) at room temperature for 48 h with 250 rpm of shaking. The fluorescence quenching spectra of DOX were reported by measuring the remaining DOX in the supernatant using a BioTek Synergy H1 microplate reader (Ex: 475 nm). The DOX loading amount into S or SC materials was calculated as shown in eqn (1):| Loading amount = Total DOX amount − DOX amount in supernatant | (1) |
In vitro release of DOX from DOX-loaded SiNP/CNT–DNA nanocomposite materials
In a typical drug release experiment, 315 μg of DOX/S or DOX/SC were added to 1 mL of Dulbecco's PBS (pH 7.4), acetate buffer (pH 5.0), cell culture medium (Gibco Laboratories) or DNase I solution (50 U, New England Biolabs), respectively, and incubated with 250 rpm orbital shaking for variable times. The supernatant was then collected and centrifugated to remove the particles and other aggregates. The volume of the medium sample was maintained constant by replenishing the corresponding solution. The concentration of released DOX was measured by the fluorescence spectroscopy.Cell culture
Human cervical cancer (HeLa) cells were obtained from ATCC. The cells were cultured in 25 cm2 tissue culture flask (Corning Inc.) with cell culture medium, comprised of EMEM, with addition of 1% penicillin/streptomycin (Thermo Fisher Scientific) and 10% FBS (Biochrom) at 37 °C in a 5% CO2 environment. The cells were washed twice with Dulbecco's PBS (−/−) (without calcium and magnesium) and trypsinated by addition of 500 μL 0.25% Trypsin solution (Biochrom) in PBS–EDTA (PBS with 0.02% EDTA) for 3 min. The trypsin activity was blocked by addition of 4.5 mL of fresh cell culture medium. The cell concentration of the resulting suspension was determined by hemocytometer analysis.Cytotoxicity assay
CCK-8 cell viability assay was used according to manufacturer's instructions to evaluate the cytotoxicity of the various composite materials. In brief, HeLa cells in 200 μL medium at a density of 8 × 103 cells per well were seeded into wells of a 96-well plate. After incubation at 37 °C and 5% CO2 for 12 h, the adherent cells were incubated with 200 μL fresh medium containing 80 μg SiNP–P, 20 μg CNT–P, 315 μg S or SC nanocomposite materials, respectively. Cells incubated under the same conditions in the absence of materials were used as control. After another 24 h incubation, 20 μL CCK-8 (Dojindo) were added to each well, and the HeLa cells were then cultivated for an additional 4 h at 37 °C and 5% CO2. Subsequently, the absorbance at 450 nm was recorded with a BioTek Synergy microplate reader. Cell viability was calculated according to eqn (2):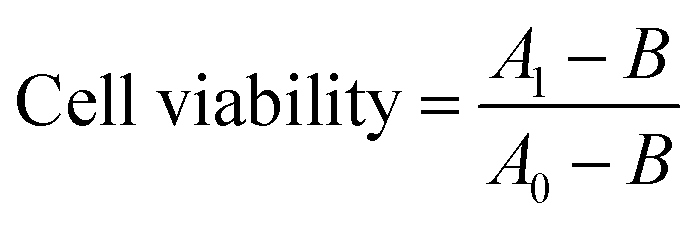 | (2) |
Imaging of cells after uptake of SiNP–DNA nanocomposite materials
To investigate the influence of aptamer formation during RCA synthesis of the nanocomposite materials, HeLa cells were seeded into a glass bottom 96-well plate (MoBiTec GmbH) at a density of 8 × 103 cells in 200 μL fresh medium and cultured at 37 °C and 5% CO2 overnight to allow the cells to adhere onto the cover slips. The medium was then replaced with 200 μL fresh medium containing the nanocomposite materials prepared via 0, 2, 4 and 6 h RCA reaction, respectively. After incubation at 37 °C and 5% CO2 for 2 h, the cells were rinsed with Dulbecco's PBS for 3 times, fixed with paraformaldehyde (PFA, 4%, Polysciences Inc.) in Dulbecco's PBS for 15 min. The filamentous actin was stained with Alexa Fluor 488® phalloidin (Thermo Fisher Scientific) and the cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific). Imaging was performed using confocal laser scanning microscopy (CLSM) on a Zeiss LSM880.Drug delivery with DOX-loaded SiNP/CNT–DNA nanocomposite materials
The targeted drug delivery and cytotoxicity of free DOX or DOX/nanocomposite complexes was evaluated by using fluorescence microscopy and CCK-8 cell viability assay, respectively. To this end, HeLa cells were seeded into a glass bottom 96-well plate as described above. The adhered cells were then treated with either free DOX, DOX/S or DOX/SC (38 μg mL−1 DOX equivalent) in cell culture medium. After incubation for 2 h, the supernatant medium containing the materials was replaced by fresh medium for microscopy inspection or cell viability evaluation. For fluorescence microscopy imaging, the cells were rinsed, fixed, stained, and imaged as described above. The viability of HeLa cells was determined either immediately or after cultivation for 24 h and 48 h, respectively, using CCK-8 assay as described above.Conflicts of interest
The authors declare that a patent application for SiNP–DNA composite materials has been filed.Acknowledgements
We acknowledge funding from the Helmholtz programme “BioInterfaces in Technology and Medicine”. Y. H. is grateful to the China Scholarship Council (CSC) for his PhD fellowship.References
- J. Li, L. Mo, C.-H. Lu, T. Fu, H.-H. Yang and W. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- D. Wang, Y. Hu, P. Liu and D. Luo, Acc. Chem. Res., 2017, 50, 733–739 CrossRef CAS PubMed.
- Y. Zhang, J. Tu, D. Wang, H. Zhu, S. K. Maity, X. Qu, B. Bogaert, H. Pei and H. Zhang, Adv. Mater., 2018, 30, 1703658 CrossRef.
- Y. Hu and C. M. Niemeyer, Adv. Mater., 2019, 31, e1806294 CrossRef PubMed.
- G. Zhu, R. Hu, Z. Zhao, Z. Chen, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 16438–16445 CrossRef CAS PubMed.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- S. Merino, C. Martin, K. Kostarelos, M. Prato and E. Vazquez, ACS Nano, 2015, 9, 4686–4697 CrossRef CAS.
- E. Cheng, Y. Li, Z. Yang, Z. Deng and D. Liu, Chem. Commun., 2011, 47, 5545–5547 RSC.
- H. Sun, J. Ren and X. Qu, Acc. Chem. Res., 2016, 49, 461–470 CrossRef CAS PubMed.
- L. Zhang, S. R. Jean, S. Ahmed, P. M. Aldridge, X. Li, F. Fan, E. H. Sargent and S. O. Kelley, Nat. Commun., 2017, 8, 381 CrossRef PubMed.
- Y. Hu, M. Grosche, S. Sheshachala, C. Oelschlaeger, N. Willenbacher, K. S. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2019, 58, 17269–17272 CrossRef CAS PubMed.
- Y. Hu, C. M. Domínguez, J. Bauer, S. Weigel, A. Schipperges, C. Oelschlaeger, N. Willenbacher, S. Keppler, M. Bastmeyer, S. Heißler, C. Wöll, T. Scharnweber, K. S. Rabe and C. M. Niemeyer, Nat. Commun., 2019, 10, 5522 CrossRef CAS PubMed.
- A. Leidner, S. Weigel, J. Bauer, J. Reiber, A. Angelin, M. Grösche, T. Scharnweber and C. M. Niemeyer, Adv. Funct. Mater., 2018, 28, 1707572 CrossRef.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS PubMed.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS.
- M. B. O’Donoghue, X. Shi, X. Fang and W. Tan, Anal. Bioanal. Chem., 2012, 402, 3205–3209 CrossRef PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- C. J. Serpell, K. Kostarelos and B. G. Davis, ACS Cent. Sci., 2016, 2, 190–200 CrossRef CAS PubMed.
- S. Kumar, R. Rani, N. Dilbaghi, K. Tankeshwar and K.-H. Kim, Chem. Soc. Rev., 2017, 46, 158–196 RSC.
- A. Angelin, S. Weigel, R. Garrecht, R. Meyer, J. Bauer, R. K. Kumar, M. Hirtz and C. M. Niemeyer, Angew. Chem., Int. Ed., 2015, 54, 15813–15817 CrossRef CAS PubMed.
- X. D. Wang, K. S. Rabe, I. Ahmed and C. M. Niemeyer, Adv. Mater., 2015, 27, 7945–7950 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tb02861g |
| This journal is © The Royal Society of Chemistry 2020 |