
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel benzothiazole half-squaraines: model chromophores to study dye–TiO2 interactions in dye-sensitized solar cells†
Peter J.
Holliman
 *a,
Christopher P.
Kershaw
a,
Eurig W.
Jones
a,
Diana
Meza-Rojas
a,
Anthony
Lewis
a,
James
McGettrick
a,
Dawn
Geatches
b,
Kakali
Sen
b,
Sebastian
Metz
bc,
Graham J.
Tizzard
d and
Simon J.
Coles
d
*a,
Christopher P.
Kershaw
a,
Eurig W.
Jones
a,
Diana
Meza-Rojas
a,
Anthony
Lewis
a,
James
McGettrick
a,
Dawn
Geatches
b,
Kakali
Sen
b,
Sebastian
Metz
bc,
Graham J.
Tizzard
d and
Simon J.
Coles
d
aCEMEG, College of Engineering, Swansea University, Bay Campus, Swansea SA1 8EN, UK. E-mail: p.j.holliman@swansea.ac.uk; Tel: +44(0) 1792 513758
bScientific Computing Department, STFC Daresbury Laboratory, Warrington, UK
cFraunhofer Institute for Solar Energy System ISE, Heidenhofstrasse 2, D-79110, Freiburg, Germany
dUK National Crystallography Service, Chemistry, University of Southampton, University Road, Southampton SO17 1BJ, UK
First published on 20th October 2020
Abstract
We report the synthesis of 9 new half squaraine (HfSQ) dyes; 5 containing a benzothiazole moiety and 4 containing an indolenine moiety. X-ray single crystal structural and characterisation data have been correlated with device data to understand the widely reported but poorly understood influence of S heteroatoms on DSC device performance. The S heteroatom in these new dyes has also been used as an atomic probe of the dye–TiO2 interface to dye binding and orientation. Thus, for the first time, using the S heteroatom probe, angle-resolved X-ray photoelectron (AR-XPS) data have shown these dyes sit horizontally at the dye–TiO2 interface confirmed by DFT computer modelling of novel and analogous HfSQ dyes with a benzoindole backbone.
To provide affordable and secure energy in a sustainable fashion, renewable energy sources like solar cells are expected to play a vital role. Given that the cost effectiveness is a key requirement for success, dye-sensitised solar cells (DSCs) are considered to be in a strong position to contribute to this development. The first major breakthrough in dye-sensitized solar cell (DSC) technology was reported in 1991 by using a high surface area, mesoporous TiO2 photo-anode to vastly increase dye loading and photo-current.1 Recently, there have been several reports of DSC devices with efficiency (η) > 12%2–6 leading to the highest DSC efficiency report of 14.7%.7 All these preceding reports assume that optimised devices utilise a monolayer of dye molecules on the TiO2 surface, leading most to add co-sorbents (e.g. chenodeoxycholic acid – CDCA) to minimise dye aggregation.8 In addition, many recent DSC reports have used co-sensitisation of the TiO2 photo-anodes using two9 or even three10 dyes to maximise light harvesting. And yet, despite the crucial importance of the dye:TiO2 interface, the binding mechanism and dye configuration remain poorly understood.
The most studied area of DSC technology is the development of new dyes – often containing expensive metal ions. Ruthenium bipyridyl-based complexes (e.g. N3,1 N719,11 C106 (ref. 12)) were reported first, and remained the focus of research until ca. 2009, leading to efficiencies over 11%. To achieve better spectral efficiencies and cost effectiveness, interest also turned towards metal free organic dyes with higher extinction coefficients (ε) such as perylene,2,3 porphyrin4 and carbazole/coumarin dyes5 generally based around a donor–spacer–acceptor motif to improve electron injection into TiO2.6,13 All these dyes possess a group that enables effective adsorption to the TiO2 surface. Generally, these “chemical linkers” are carboxylate based allowing the dye to chemisorb to TiO2 through ester-like linkages.14,15 In this context, the TiO2 adsorption of Ru-bipy dyes has been studied with IR data suggesting bidentate binding16 of two of the four available carboxylate groups.17 By comparison, atomistic modelling correlated with vibrational spectroscopy suggests dye binding through three of the four available carboxylate groups.18 In addition, dye HOMO–LUMO levels are often calculated using techniques such as density functional theory (DFT) where the energy level data are used to understand electronic spectra, and the HOMO–LUMO locations indicate the potential for electron injection into TiO2.19.
DFT calculations are computationally time intensive. Generally, single dye molecules are modelled in the gas phase,20–22 or attached to a few TiO2 units,22–24 or as cluster models18,25–30 to represent interactions of dyes with TiO2 surfaces. As such, fewer DFT studies have been reported where the energy levels of the entire dye:TiO2 system are considered.31 By comparison, in terms of experimental data and from the perspective of the mesoporous TiO2 photo-anode, reports have included studies to optimise particle crystallinity,32 surface area and electrode thickness,33 and processing.34
Overall, the surface interaction of dyes with TiO2 surfaces remains poorly understood, including dye orientation, surface organisation and inter-molecular interactions (e.g. between dyes and co-sorbents). It is well established that having different molecular species on the photo-anode can have a profound effect on device performance; negatively for aggregated dyes,35 or positively on adding the CDCA.8,9,36 Given the strong effect they have on the device performance, these topics are astonishingly rarely studied.19
In this paper we address the dye–TiO2 orientation issue through a systematic study using a half-squaraine (HfSQ) dye chromophore where we have incorporated a sulphur atom into the indole moiety as an atomic tag. We have studied this class of dyes previously and found them to be synthetically versatile, reasonably efficient in DSC devices for single (η = 5.0%)37 or double linker group (η = 5.5%).38 We have also found these dyes to be positively influenced by CDCA co-sorbent and capable of effective co-sensitisation with squaraine dye (η = 6.1%).37 Here, we have synthesised two novel but analogous families of HfSQ dyes using benzyl indoles linked to squaric acid moieties. One family of dyes has a sulphur heteroatom opposite the indole nitrogen (dyes (4), (6–8), (16)) whilst the second family has a dimethyl-derivatised carbon in this position (dyes (11), (12), (14), (15), (17)). These data have been correlated with DFT calculations of selected dyes on anatase TiO2 (1 0 1) surfaces. The overall aim of the paper is to report the first, combined synthetic-theoretical–experimental-device study of the orientation and surface organisation of dye molecules on TiO2 surfaces.
Results and discussion
Dye synthesis
The synthetic routes used to produce the Hf-SQ dyes in this work are show in Scheme 1. The overall aim of the synthetic work was to produce dyes based on the same half-squaraine chromophore, either with a C(CH3)2 group or a sulphur atom opposite the indole nitrogen. In part, this was to compare the effect of a sulphur atom within the dye structure on device performance because previous work suggests a sulphur moiety in the main dye structure interacts with triiodide electrolytes thereby increasing recombination and reducing cell efficiency.39 Hence, here we have synthesized dyes that are structurally analogous apart from the variation of a S atom or a C(CH3)2 moiety in the indole ring. However, we have also used this S atom as an atomic tag to study dye orientation on a TiO2 surface using a combination of experimental data (angle resolved XPS) and theoretical DFT modelling.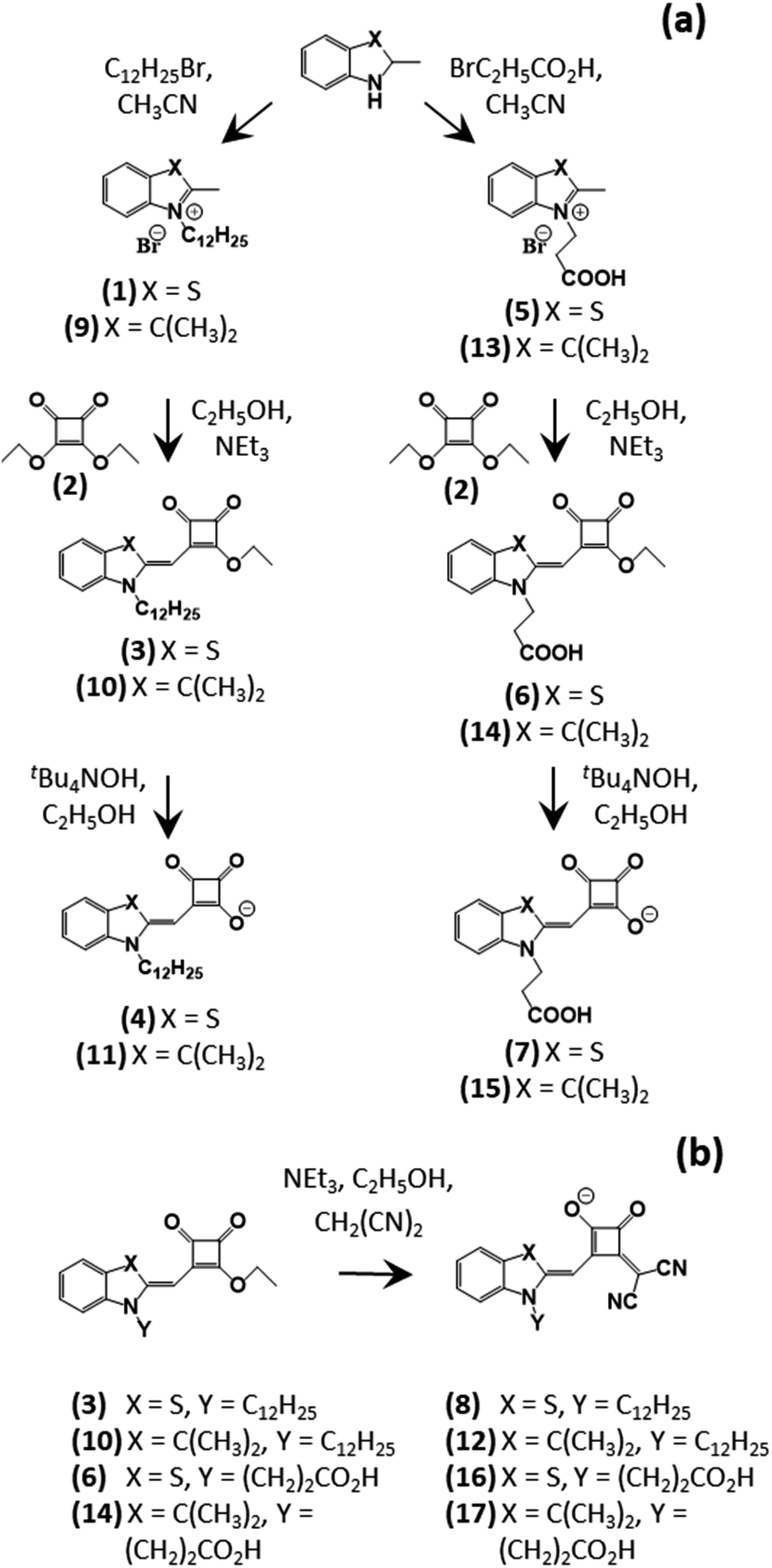 | ||
| Scheme 1 Synthetic pathways to (a) half-squaraine (HfSQ) dyes and (b) vinyl dicyano HfSQ dye derivatives. | ||
Scheme 1 shows three main synthetic routes to the various dyes. Materials (1), (5), (9), (10), (11), (13) and (15) have all been synthesised previously37,38 and our characterisation data match the published data. Dyes (4) and (11) have an acyloin linker on the squaric acid group and these were produced by first adding an alkyl chain to the N atom of a benzyl indole (containing a sulphur atom or methylene unit, respectively), followed by reaction with squaric acid ester and finally de-esterification with base to produce the carboxylic acid dyes. The second route followed a similar strategy but produced dyes (7) and (15) with 2 linkers; a carboxylic acid and an acyloin group. This was achieved by reacting 3-bromopropionic acid with the initial indole and then reacting this with squaric acid ester and de-esterifying in the same way. The final synthetic strategy was to convert the squaraine group of the 4 esterified dyes (i.e. the sulphur/methylene indoles along with N-indole alkyl/propionic acid dyes) into vinyl dicyano acyloin moieties ((8), (12), (16), (17)). The 1H and 13C NMR confirm the identity of the precursors and final dyes with the expected number of signals and coupling constants.
Whilst indoles (1) and (9) and the propionic acid-derivatised indoles (5) and (13) only differ by swapping a S atom for a C(CH3)2, the S compounds has drastically lower yields (ca. 20%) compared with their C(CH3)2 counterparts (ca. 90%). Changing the solvent from anhydrous CH3CN to anhydrous CH3CH2OH doubled the S indole yields to ca. 40%, which we ascribe to improved reactant solubility. The remaining difference in yields is ascribed to the S atom shifting electron density to make the indole N less likely to undergo nucleophilic substitution. Interestingly, the opposite effect is observed when these alkylated indoles ((1), (5), (9), (13)) react with squaric acid to form half-squaraine dyes. Thus, the sulphur dyes had much better yields than their C(CH3)2 counterparts. Thus, the S dyes exhibited a rapid colour change to red with no blue impurity (full squaraine dye) while the C(CH3)2 dyes turned green due to a mixture of half- and full squaraine (see ESI Fig. 1†). Hence, the full squaraine by-product lowered the yield which was confirmed by thin layer chromatography (TLC). Finally, de-esterification of (3) and (10) using tBu4NOH produced the long chain dyes (4) and (11), respectively. Easy purification was possible as the starting material was soluble in petroleum ether:ethyl acetate whereas the product dissolved in CH2Cl2:CH3OH. By comparison, the esters and acids of dyes (6)/(7) and (14)/(15) had similar solubilities making them difficult to separate. However, using flash chromatography increased the acid yield thereby improving purification of the desired product.
Dyes (11) and (15) have been previously reported and have been confirmed by 1H, 13C NMR and mass spectrometry. To the best of our knowledge dyes (4), (6–8), (12), (14), (16) and (17) have not been reported in the literature and were identified using several analytical techniques. The resonances in the 1H NMR spectra suggest that (8), (12), (16) and (17) have all been isolated as the triethylammonium salt (e.g. quintet and triplet at ca. 3.2 and 1.3 ppm, which integrate to 6 and 9 protons respectively, and at 46 and 9 ppm in the 13C NMR data). These are caused by the ethyl group of this cation. The other proton signals and coupling constants are similar to those reported for the previous half squaraine dyes.38 High resolution mass spectrometry identifies the M− ions that correspond to the target molecules in non-salted form and infrared spectroscopy confirms the presence of nitrile and carbonyl groups at 2852, 2198, 1738, 1644 (m) and 1484 (m) cm−1 (please see data in Experimental section).
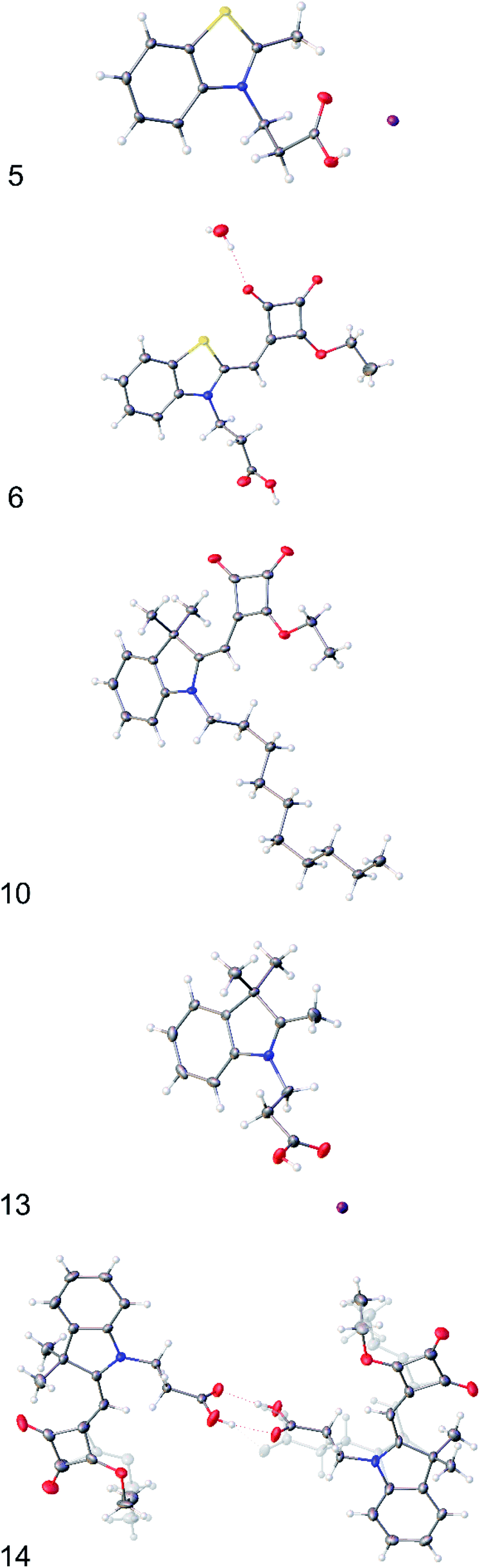 | ||
| Fig. 1 X-ray crystal structures of (5), (6), (10), (13) and (14). Displacement ellipsoids are shown at 50% probability and minor disorder components are shown ‘ghosted’. | ||
Spectroscopic analysis
Fig. 2 shows the UV-Vis spectra of HfSQ dyes in solution and after sorption onto TiO2 electrodes. The solution spectra (Fig. 2a) show that the absorption maxima of the sulphur-containing dyes (dotted + circle lines) are bathochromically shifted relative to the C(CH3)2 dyes (solid lines). Modification of the squaric acid moiety by replacing a carbonyl group with a vinyl dicyano group causes a further bathochromic shift, which is particularly clear for the sulphur-containing vinyl dicyano dyes (8) and (16) compared to (4) and (7), respectively. This reflects a smaller HOMO–LUMO gap for these dyes when in solution. Higher ε are observed for the esterified squaraines; e.g. (3), (10) and (14), where ε = 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–71000 M−1 cm−1, a similar behaviour was observed previously for other half-squaraine dyes.37,38 For the de-esterified (carboxylate) dyes, the S-containing versions ((4), (6) and (7)) all have lower ε compared to their C(CH3)2 counterparts ((11), (14) and (15)), respectively. The exceptions here are the vinyl dicyano modified dyes where the sulphur-containing versions ((8) and (16)) have higher ε than the analogous C(CH3)2 dyes ((12) and (17)), respectively. This is very interesting as it shows that sulphur plays an important role in the photochemistry and is involved in the HOMO and/or LUMO of these dyes.
000–71000 M−1 cm−1, a similar behaviour was observed previously for other half-squaraine dyes.37,38 For the de-esterified (carboxylate) dyes, the S-containing versions ((4), (6) and (7)) all have lower ε compared to their C(CH3)2 counterparts ((11), (14) and (15)), respectively. The exceptions here are the vinyl dicyano modified dyes where the sulphur-containing versions ((8) and (16)) have higher ε than the analogous C(CH3)2 dyes ((12) and (17)), respectively. This is very interesting as it shows that sulphur plays an important role in the photochemistry and is involved in the HOMO and/or LUMO of these dyes.
 | ||
| Fig. 2 UV-Vis spectra of (a) HfSQ dyes in solution and (b) dyes adsorbed onto a transparent TiO2 surface. For clarity, structurally related dyes have same colour and C(CH3)2 dyes have solid lines, whilst S dyes have dashed lines with circle markers. | ||
Fig. 3 shows the electron density distribution of the HOMO and LUMO for selected half-squaraine dyes adsorbed on a TiO2 surface with the HOMO–LUMO energy gaps listed in Table 1. The data have been generated using Kohn–Sham orbitals so the data show relative rather than absolute energies. The data show several important features. Firstly, the data in Fig. 3 clearly show that the HOMO is located on the dye and that the LUMO is located on TiO2. This is essential for electron injection from the dye into the TiO2; otherwise the devices would not work. In addition, because we have modelled the entire dye:TiO2 system rather than just the gas phase dye molecules, the data also show how sterically close the HOMO is to the dye surface. For most of the dyes, the HOMO is also situated on, or close, to a linker group so that charge separation occurs from this into the TiO2 rather than from one part of the dye to another as is often presented when the modelling of gas phase dyes is presented. In terms of the HOMO–LUMO gaps (Table 1) and UV-Vis spectra, although the energies of the theoretical data (ESI Fig. 11†) differ from the experimental, they show similar trends. So, Table 1 shows that, as expected, the DFT HOMO–LUMO decreases on dye adsorption to the TiO2 surface. Thus, compared to either pristine TiO2 or isolated dye molecules, the dye:TiO2 system is more semi-conducting which benefits device performance. Dye (16) is the most red-shifted, dyes (4), (6) and (7) absorb at similar wavelengths, and dyes (14) and (15) are most blue shifted. The simulated spectra of (16) has one dominant π–π*, whereas the rest have a low intensity n–π* and a dominant π–π* band. Solvatochromic shifts in protic solvents like alcohols are different for n–π* and π–π* transitions which could explain the differences with the experimental data in ethanol. However, the DFT data do not highlight consistent differences caused by the S-atom alone.
 | ||
| Fig. 3 DFT-calculated HOMO (green: isovalue 0.04 e Å−3) and LUMO (blue: isovalue 0.02 e Å−3) of horizontally oriented dyes on TiO2. | ||
| Dye | V oc (V) | J sc (mA cm−2) | Fill factor | η (%) | DFT HOMO–LUMO energy (eV) | |
|---|---|---|---|---|---|---|
| M | M + S | |||||
| 14 | 0.49 | 7.72 | 0.58 | 2.2 | 2.36 | 2.09 |
| 6 | 0.40 | 5.36 | 0.56 | 1.2 | 2.27 | 1.29 |
| 11 | 0.47 | 9.34 | 0.59 | 2.6 | — | — |
| 4 | 0.50 | 8.68 | 0.51 | 2.2 | 2.35 | 1.38 |
| 12 | 0.52 | 3.84 | 0.58 | 1.2 | — | — |
| 8 | 0.47 | 6.32 | 0.63 | 1.9 | — | — |
| 15 | 0.52 | 10.13 | 0.57 | 3.0 | 2.41 | 1.33 |
| 7 | 0.42 | 7.07 | 0.52 | 1.5 | 2.42 | 2.10 |
| 17 | 0.58 | 7.43 | 0.53 | 2.3 | — | — |
| 16 | 0.35 | 2.92 | 0.49 | 0.5 | 3.07 | 1.75 |
| N719 | 0.67 | 15.02 | 0.61 | 6.1 | — | — |
Fig. 2b shows the UV-Vis data for dyes absorbed onto transparent, mesoporous TiO2 films. The data show peak broadening compared to dyes in solution. Interestingly, all the HfSQ dyes (other than (11) and (12)) show greater absorbance compared to N719. In part, this can be explained by higher ε. However, dyes (6) and (17) have similar ε to N719 yet show much higher absorbance, which appear to reflect higher dye loading on the TiO2 surface. The low absorbance of (12) might be due to it having the lowest ε of all the dyes (10, 835 M−1 cm−1). However, dye (11) has lower absorbance despite an ε = 38218 M−1 cm−1. This suggests the low absorbance of (11) and (12) is due to low dye uptake, which might be because (11) and (12) both adsorb mainly using the single linker of the acyloin moiety, while the C(CH3)2 group and the alkyl chain on indole N stabilise the dissolved dye and don't interact strongly with the TiO2 surface.
Attenuated total reflectance (ATR)-infrared spectroscopy of pure dye and TiO2 adsorbed dye powders has been used to study the interaction between the dyes and TiO2. Focussing on the key features of the spectra, the O–H stretch (>3000 cm−1) becomes less intense on dye adsorption and the sharp, intense carbonyl stretch (ca. 1750 cm−1) also becomes less intense and shifts slightly (ca. 1764 cm−1). This suggests the formation of an ester link between the TiO2 surface and the dye. A N–H peak is also observed for the TiO2-sorbed dyes. Considering there is only a single nitrogen atom this suggests that the dyes orient horizontally and the N atom is interacting with O–H groups on the TiO2 surface.
Device testing
Table 1 shows I–V data for all HfSQ devices. To study the influence of S atom on device performance, dyes (4), (6–8) and (16) were synthesized with a S atom incorporated into the indolenine group, whilst dyes (11), (12), (14), (15) and (17) were identical but with a C(CH3) group. One hypothesis was that, if the dyes (6), (7) and (16) oriented themselves vertically so that the S atom became situated at the furthest distance away from the TiO2 surface, this might produce a positive interaction with the triiodide electrolyte increasing the speed of dye regeneration and therefore improving DSC device performance. However, there is no evidence for this in the device data (η = 0.5–1.5%). By comparison, if dyes (4) and (8) oriented themselves vertically by adsorption through the squaraine moiety, then the S atom should sit much closer to the TiO2 surface. Whilst the S atom should still interact positively with the triiodide, it would also increase the concentration of iodide near the TiO2 surface, which would increase recombination by pulling injected electrons out of the TiO2. The performance data doesn't support this hypothesis as dyes (4) and (8) are more efficient (η = 2.2 and 1.9% versus 0.5–1.5%) and exhibit higher Voc (0.47–0.50 V versus 0.35–0.42 V) compared to dyes (6), (7) and (16). Furthermore, on average the device performance for S dyes were lower than the average power conversion efficiency (PCE) which for the S dyes is η = 1.3% compared to η = 2.2% for the C(CH3)2 dyes. Also, (15), which has no S, is the best dye (η = 3.0%), whereas the S-containing (16) is the worst (η = 0.50%) one. The consistently lower Voc for the S dyes suggests that, for all these dyes, the S atoms are located close to the TiO2 surface which increases recombination from iodide into the TiO2. Thus, the device testing data suggest that, when designing DSC dyes, it is important to ensure that sulphur atoms are not located close to the TiO2 surface because these do seem to increase recombination with the electrolyte.Five groups of dye with single or double linkers have been tested; single linkers include carboxylate from the indole N (14) and (6), squaraine (11) and (4), vinyl dicyano-modified squaraine (12) and (8), the double linkers include carboxylate plus squaraine linkers (15) and (7), and carboxylate plus vinyl dicyano-modified squaraine linkers (17) and (16). From these examples, the most efficient dye is (15) which has 2 linker groups and the highest photocurrent (10.12 mA cm−2). The least efficient dye was (16) which had the lowest photocurrent. This dye also had 2 linker groups but one was the vinyl dicyano-modified squaraine, which we previously found binds very poorly to TiO2.37 This was further evidenced by dyes (11) and (12) which differed in the presence of a vinyl dicyano group on the squaraine moiety for (12). Dye (12) bound so weakly to TiO2 that it was desorbed by ethanol washing (ESI Fig. 13†). These trends also apply to the other dyes; i.e. dyes binding through the squaraine moiety give rise to higher photocurrents than those binding either through the carboxylate or vinyl-dicyano-modified linker (or combinations of these 2 linkers). This is most likely because dyes bound through the acyloin moiety follow the typical structure of organic dyes (donor–π bridge–acceptor–linker–TiO2), whereas the dyes bound through the carboxylate linker follow a slightly different architecture with the linker group in the centre of the dye rather than on the end.
Fig. 4 shows the spectral response of the HfSQ dyes. Dyes (15) and (4) show the highest external quantum efficiency (EQE) (ca. 80%) between 400 and 500 nm, which correlates with higher Jsc (10.13 and 8.68 mA cm−2) for these dyes. The data also show that all the vinyl dicyano-modified dyes have their spectral response red shifted by ca. 60 nm. Of these, dye (17) responds most strongly (EQE ca. 65%) between ca. 480 and 550 nm (where solar intensity is highest) corresponding to Jsc of 7.43 mA cm−2. The lower EQE for dye (12) (ca. 20%) is in line with the lower light absorption for this dye which we ascribed to low dye uptake. We have observed this effect before despite seeing effective electron injection for vinyl dicyano-modified HfSQ dyes and linked this with dye desorption in the electrolyte solution.37
 | ||
| Fig. 4 EQE of DSC devices using HfSQ dyes. For clarity, structurally related dyes have same colour and C(CH3)2 dyes have solid lines, whilst S dyes have dashed lines with circle markers. | ||
Dye orientation studies (DFT modelling)
Using density functional theory (DFT) we have gained insight into the orientation and binding of single dye molecules on the (1 0 1) surface of anatase TiO2, at zero kelvin (0 K). We explored six of the nine dyes: four benzothiazole – (4), (6), (7), and (16); two dimethyl-derivatised – (14) and (15), and found the dye molecules preferentially adsorb to lie near-planar and horizontal to the surface.From a sub-section of orientations – including vertical and edge-on – we allowed the dye molecules to relax to their energy minima; further details of the modelling method and DFT parameters are in the Modelling procedure section and the ESI S2.† We calculated absolute adsorption energies and found the lowest energy minima (i.e. preferentially adsorbed orientations) corresponded to molecules lying horizontally to the surface; additionally, we simulated the corresponding angle resolved-XPS data to compare with experimental data.
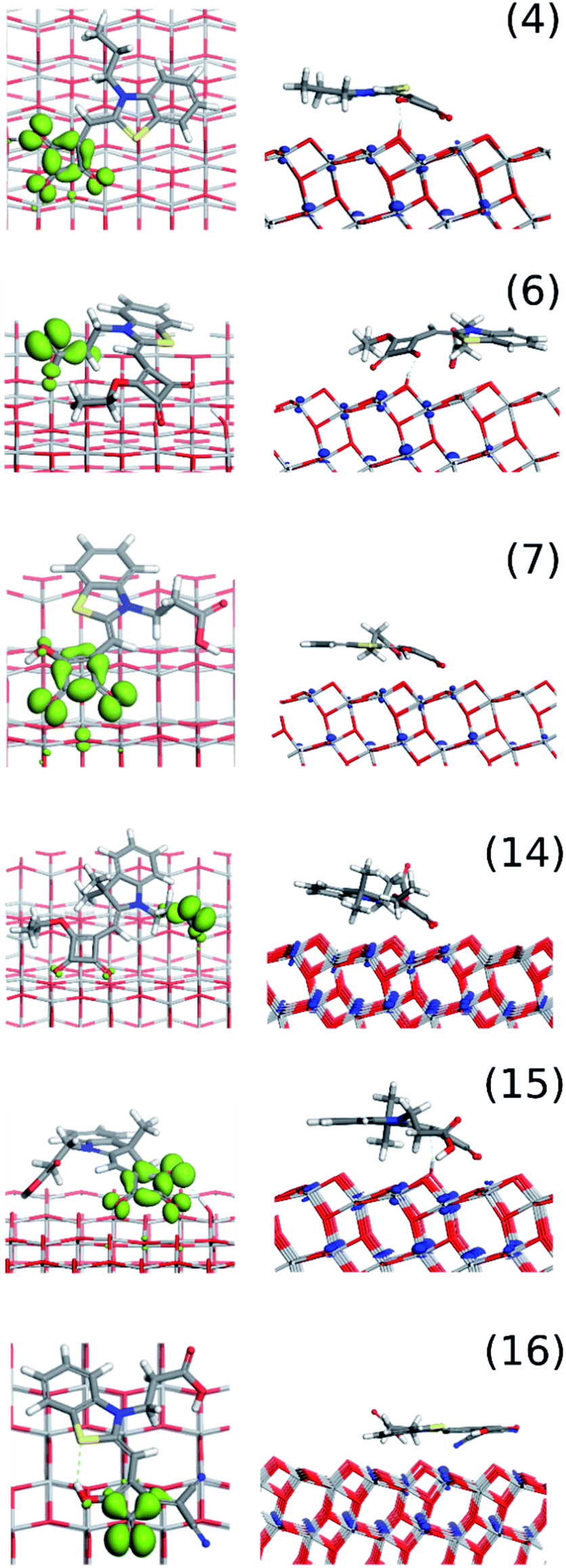
The DFT modelling of the six individual dyes as neutral gas phase molecules is in good agreement with the available crystal structure data (Fig. 1) for all the dyes (Fig. 5). The common surface-bonding motif to under-coordinated Ti sites (i.e. Ti5c) is via the linker groups: squaraine, carboxylic acid and the cyano group. Carboxylic acid and squaraine – when not directly bonded to the surface – can form hydrogen-bonds to 2-coordinated surface oxygen. Table 2 provides the details of the surface bonding motifs and the orientations are given in Fig. 5. These hydrogen bonds in a physical system are dynamic and are influenced by ambient energetics; the hydrogen will ‘hop’ between the surface and dye. The simulated hydrogen bonds range in length from 1.76 Å to 2.16 Å, implying they are relatively strong. These secondary bonding phenomena enhance stability of these dye configurations on the TiO2 surface. All six dyes exhibit all or a selection of bonding features: linkers, hydrogen bonds and electrostatic interactions between the planar rings of the dye and the TiO2 surface.
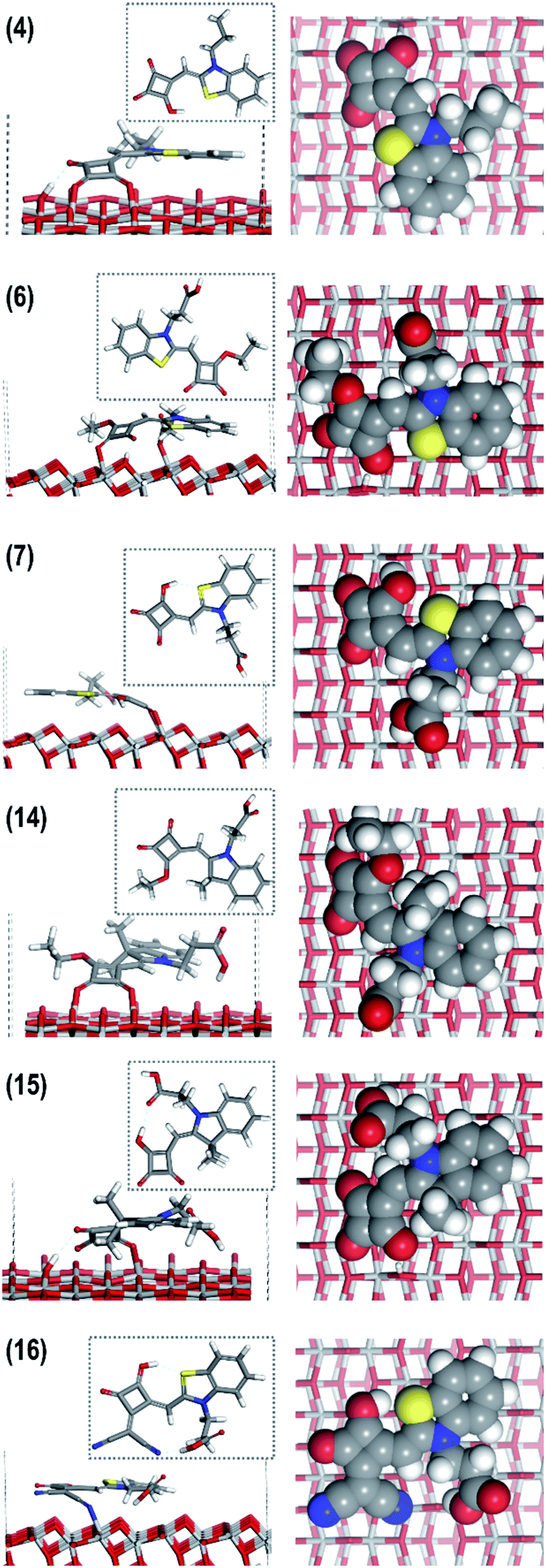 | ||
| Fig. 5 DFT modelling of the relaxed configurations of the most probable orientations of (from top to bottom) dyes (4), (6), (7), (14–16). Left: gas phase neutral dyes; middle: dye on anatase (1 0 1) TiO2 surface, where ‘most probable’ means largest (in magnitude) adsorption energy; right: plan views of dye molecules showing their van der Waals outline mode on the TiO2 surface. | ||
| Dye | Group opposite indole N | Number of (linking modes)a | Approx. angle to TiO2 surface (degrees) | Projected surface area of dye on surface (Å2) | |
|---|---|---|---|---|---|
| Squaraine moiety | Planar rings | ||||
| a Sq (O): squaraine O bonds directly to Ti5c; COO: carboxylic O bonds directly to Ti5c; Sq (O)–Surf (HO): hydrogen bond between squaraine O and hydroxylated surface; Sq (OH)–Surf (O): hydrogen bond between hydroxylated squaraine and surface O; COOH–Surf (O): hydrogen bond between carboxylate group and surface; Cyano (N): cyano-N bonds directly to Ti5c. | |||||
| (4) | S | 2 × Sq (O) | 40 | 10–15 | 72 |
| 1 × Sq (O)–Surf (HO) | |||||
| (6) | S | 1 × Sq (O) | 45 | 15–20 | 73 |
| 1 × COO | |||||
| 1 × Sq (O)–Surf (HO) | |||||
| (7) | S | 2 × Sq (O) | 30 | 0–5 | 86 |
| 1 × Sq (OH)–Surf (O) | |||||
| 1 × COOH–Surf (O) | |||||
| (14) | C(CH3)2 | 2 × Sq (O) | 35 | 15–20 | 119 |
| 1 × COOH–Surf (O) | |||||
| (15) | C(CH3)2 | 2 × Sq (O) | 30 | 0–5 | 83 |
| 1 × Sq (O)–Surf (HO) 1 × COOH–Surf (O) | |||||
| (16) | S | 1 × Cyano (N) | 5 | 5–10 | 109 |
| 1 × COOH–Surf (O) | |||||
The images on the right of Fig. 5 show the dyes in plan view, which gives an indication of the surface area they occupy and what the electrolyte or hole transport material “sees”. We estimated the surface area of TiO2 covered by a single dye, and found dyes occupy a surface area between 72–119 Å2. For dye (4) the surface area occupation in a physical system could be larger because it was truncated for modelling (see ESI S2†). If an average 10 μm thick mesoporous, anatase TiO2 photoanode film has a density of 3.78 g cm−3, this represents 3.78 mg of TiO2 per cm2. If the TiO2 has a surface area of 50 m2 g−1, there should be enough surface area for 2.56 × 1017 molecules of (6), 1.59 × 1017 molecules of (14) and in-between for (7), (15), (16). We emphasize that these are crude approximations based on 0 K, DFT-optimized geometries, allowing for complete occupation of the available surface area (Table 2).
Simulated AR-XPS
The DFT calculations enabled the probing of the orientation of the dyes on the surfaces at 0 K, but the experimental temperature was ambient and so higher (ca. 298 K) and could provide sufficient kinetic energy to the dyes to change their orientation. The adsorption energies of these orientations would be close to the 0 K-calculated adsorption energies. Although probing the size and effect of the thermodynamic energy is outside the scope of this work, instead we considered a selection of the DFT-optimised orientations with the largest (in magnitude) adsorption energies for each dye and distinctly different orientation (e.g. horizontal, vertical and/or ‘side’) and calculated AR-XPS data. As stated in the DFT modelling section, irrespective of the chemical nature of the dyes, the horizontal orientation resulted in the most favourable adsorption energy; the corresponding AR-XPS data are provided in Fig. 6. The AR-XPS data for the other orientations of each dye are provided in the ESI.† | ||
| Fig. 6 Angle resolved-XPS for the dye orientations on TiO2 with the largest (in magnitude) adsorption energy. The y-axis shows the perpendicular distance above the highest Ti in the surface in atomic units of: indole nitrogen (N), cyano nitrogen (Ncn), sulphur (S), oxygens of the squaraine moiety (Os), oxygens of the carboxyl group (Oc). (In dyes other than (16) the dicyano moiety is replaced by an oxygen i.e. Os.) The AR-XPS graphs of the additional low-energy configurations are in ESI S2.† | ||
The calculated AR-XPS show a smaller spread of data for the dyes containing the S atom probe – (4), (6), (7) and (16) – due to these dyes orienting themselves more horizontally to the surface than the dyes without the probe – (14) and (15). In the horizontally-orientated dyes the S and N atoms lie within 1–2 a.u. (z-distance) of one another, whereas in the vertically-orientated dyes they lie within 3–5 a.u. (z-distance) of one another (see ESI†). These variations in z-distances between S and N (corresponding to horizontal and vertical dye orientations) is a clear marker for experimental probing.
Experimental dye orientations (angle resolved XPS)
Data for dyes (4), (7), (11) and (16) are shown in Fig. 7. The data are presented with the vertical axis (ln(I/Io)) representing the vertical distance moving away from the surface. The different functional groups are then presented starting with the C 1s signals, then the N 1s, then the O 1s, then S if it is present and finally Ti. The labels show the element first, followed by the chemical environment. For example, C(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is a carbon signal for a carbon in a carbonyl or squaraine moiety whilst C(CxHy) is a carbon from an alkyl chain etc.
O) is a carbon signal for a carbon in a carbonyl or squaraine moiety whilst C(CxHy) is a carbon from an alkyl chain etc.
 | ||
| Fig. 7 Atomic positions in the z-axis (perpendicular to TiO2 single crystal surface) of dyes (a) (4), (b) (7), (c) (15) and (d) (16) calculated from angle resolved XPS data. | ||
Looking at dye (4) first, the data show signals for oxygen (metal oxide: MOx) and Ti (TiO2) species at the lowest point (ca. −2.5) for the titania single crystal as expected. The data then show C–O and CO signals (ca. −1.7) in line with the acyloin linker moiety being the closest part of the dye to the TiO2 surface. Interestingly, the signals corresponding to the other part of the dye molecule (including reduced N and S atoms from the indole moiety) are at comparable distances (−1.5 to −1.0) to the Cx–Hy alkyl group furthest from the TiO2 (−1.0). This suggests that the dye is orientated approximately horizontally to the TiO2 surface. This is an important result because, although the referenced prior work discussed in this paper does not explicitly state a perpendicular dye orientation, where graphics are presented, they all show a perpendicular dye orientation relative to TiO2. In fact, with the exception of some reports of lateral anchoring dyes,40–42 most DSC papers do not state any dye orientation at all (i.e. either perpendicular/flat). These experimental data for a TiO2 single crystal dyed from solution over a period of hours are also in agreement with the DFT modelling data (Fig. 5) suggest that (4) positions itself horizontally to the surface with the alkyl group furthest from the TiO2 surface. Interestingly, NR3 and NR4 signals are also observed for dye (4). These signals are assigned to tertiary butyl amine and ammonium ions from the dye counterion which are present because Bu4NOH is used as the de-esterification base during synthesis (Scheme 1). Similar signals are also observed for the other dyes.
By comparison, dye (7) has 2 linker groups but still shows a similar trend to (4) with metal oxide signals at the lowest point (−2.5), then C–O and CO signals for the 2 linker groups (−1.5 to −1.0) and then signals for other parts of the dye (including S atom) in a horizontal line at around −0.7. The structure obtained from DFT calculations (Fig. 5) also reveals that the alkyl group of the carboxylate side chain is within the same plane as the bulk of the rest of the dye molecule and corroborates the AR-XPS data.
Dye (15) follows the same trend as the other (4) and (7) with metal oxide signals followed by C–O and CO linker groups and the CxHy alkyl group showing the same pattern despite this dye containing no S atom. The DFT data support experimental observations for the horizontal orientation (Fig. 5).
Finally, dye (16) contains a vinyl dicyano unit on the squaraine unit, a S atom in the indole and 2 linker groups. As such, the AR-XPS data contains more signals complicating interpretation, although it is clear that the metal oxide signals are the lowest followed by the C–O and CO linker groups as expected. Two S signals are observed for (16); one is organic S for the indole as expected, whilst the second peak is shifted towards where sulphate would typically appear. We ascribe this to some initial dye degradation via oxidation. The data for (16) also differ from the other dyes in that the S and N signals are further away from the surface than the CxHy unit of its carboxylate side chain. This corresponds to the fact that the alkyl group that forms part of the second linker group points towards the TiO2 surface. This is again in agreement with the DFT modelling. The AR-XPS data for all the dye systems consistently indicate horizontal orientation of the dye which is important new information which should be considered for future DSC dye design. Our DFT calculations provide us the additional validation that the largest adsorption energy for these dyes are those with horizontal orientation (Fig. 5). Hence providing support that horizontal orientation of the dyes is favourable. A further support to this observation comes from the simulated AR-XPS data too. Though an exact one-to-one mapping of the simulated (Fig. 6) and experimentally observed AR-XPS data (Fig. 7) is not possible technically, however, the trends on the position of the atoms are in agreement with horizontal orientation of the dyes on the surfaces. Modelling studies of dyes on clusters also indicate orientation of dyes other than vertical ones.25,27
Dye loadings have also been measured. The data show the lowest loadings for dyes (4), (11) and (12) with loadings of 3.6, 0.2 μg cm−2 and below detection limit, respectively. Dyes (6), (8) and (17) show the highest dye loadings at 38.1, 24.5 and 32.9 μg cm−2, respectively. Dyes (7), (14), (15), and (16) show interim dye loadings of 9.4, 9.0, 14.3 and 17.0 μg cm−2, respectively. Whilst the low dye sorption of (12) was noted during the device testing, there is little correlation between dye loading and device performance. For example, dye (16) has a dye loading consistent or higher with most of the other dyes and very similar to dye (15). However, dye (16) has by far the lowest DSC device performance while dye (15) has the highest. So whilst it is clear that, for successful DSC device operation, dyes must chemisorb to the TiO2 surface, increasing device efficiency does not rely on simply increasing the dye loading.
Experimental
Instrumentation and chemicals
All chemicals were purchased from Aldrich and used as supplied unless otherwise stated. Anhydrous solvents were used as supplied. NMR spectra were recorded on a Bruker AC500 at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts (δ) are given in ppm relative to (CH3)4Si and J values (in Hz) refer to JH,H unless otherwise stated. Mass spectra were recorded at the Mass Spectrometry Service at Swansea University. Infrared spectra were recorded on a PE1600 series FTIR spectrometer using an ATR attachment. UV-visible spectroscopy was measured on a Perkin Elmer spectrometer.X-ray photoelectron spectra (XPS)
XPS data were recorded on dyed planar single crystal TiO2 samples using a Kratos Axis Supra XPS with a monochromated Al Kα source and large area slot mode detector (300 × 800 μm analysis area). Data were recorded using a charge neutralizer to limit differential charging and binding energies were calibrated to the main hydrocarbon peak (BE 284.8 eV). Survey scans were performed using a pass energy of 160 eV and high-resolution spectra were recorded using a 0.1 eV step size and a pass energy of 40 eV at angles of 0, 40, 55, 63 & 70° relative to the substrate plane. Data were fitted using CasaXPS Version 2.3.23rev1.1K software with Shirley backgrounds.X-ray crystallography
In each experiment a suitable crystal was selected and mounted on a MITIGEN holder in perfluoroether oil on a Rigaku FRE+ equipped with either HF Varimax confocal mirrors (6) or VHF Varimax confocal mirrors (5, 10, 13 and 14) and an AFC12 goniometer and HG Saturn 724+ detector (5, 6, 10 and 13) or HyPix 6000 detector (14). The crystal was kept at T = 100(2) K during data collection. Using Olex2 (ref. 43) the structure was solved with the ShelXT44 structure solution program, using the Intrinsic Phasing solution method. The model was refined with ShelXL45 using Least Squares Minimisation. All non-hydrogen atoms were refined anisotropically and all hydrogen atom positions were calculated geometrically except those bonded to heteroatoms which were located from the difference map and refined using a riding model. The solvent water molecule present in (6) was refined with rigid body constraints.576 measured reflections, 2789 independent reflections [Rint = 0.0203], 2606 reflections used, wR2 (all data) = 0.0406, R1 = 0.0168 (I > 2(I)), CCDC 1908056.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, Dcalc./g cm−3 = 1.461, μ/mm−1 = 0.231; T/K = 100(2), θmax = 27.484°, 10889 measured reflections, 3776 independent reflections [Rint = 0.0429], 2693 reflections used, wR2 (all data) = 0.1309, R1 = 0.0531 (I > 2(I)), CCDC 1908057.
, Z = 2, Dcalc./g cm−3 = 1.178, μ/mm−1 = 0.075; T/K = 100(2), θmax = 27.485°, 15143 measured reflections, 5441 independent reflections [Rint = 0.0428], 3949 reflections used, wR2 (all data) = 0.1478, R1 = 0.0486 (I > 2(I)) CCDC 1908058.
790 measured reflections, 3452 independent reflections [Rint = 0.0760], 2583 reflections used, wR2 (all data) = 0.0628, R1 = 0.0336 (I > 2(I)), CCDC 1908059.
507 measured reflections, 8244 independent reflections [Rint = 0.0257], 7642 reflections used, wR2 (all data) = 0.0887, R1 = 0.0344 (I > 2(I)), CCDC 1908060.
, Z = 2, Dcalc./g cm−3 = 1.461, μ/mm−1 = 0.231; T/K = 100(2), θmax = 27.484°, 10889 measured reflections, 3776 independent reflections [Rint = 0.0429], 2693 reflections used, wR2 (all data) = 0.1309, R1 = 0.0531 (I > 2(I)), CCDC 1908057.
, Z = 2, Dcalc./g cm−3 = 1.178, μ/mm−1 = 0.075; T/K = 100(2), θmax = 27.485°, 15143 measured reflections, 5441 independent reflections [Rint = 0.0428], 3949 reflections used, wR2 (all data) = 0.1478, R1 = 0.0486 (I > 2(I)) CCDC 1908058.
790 measured reflections, 3452 independent reflections [Rint = 0.0760], 2583 reflections used, wR2 (all data) = 0.0628, R1 = 0.0336 (I > 2(I)), CCDC 1908059.
507 measured reflections, 8244 independent reflections [Rint = 0.0257], 7642 reflections used, wR2 (all data) = 0.0887, R1 = 0.0344 (I > 2(I)), CCDC 1908060.
Preparation of 3-((3-dodecylbenzothiazol-2-ylidene)methyl)-4-ethoxycyclobut-3-ene-1,2-dione (3)
2.00 g (5.02 mmol) of 3-dodecyl-2-methylbenzothiazol-3-ium bromide (1) and (0.86 g, 5.05 mmol) of 3,4-diethoxy-3-cyclobutene-1,2-dione (2) and triethylamine (2 ml) were dissolved in ethanol (25 ml) and was vigorously refluxed for 30 min. The solvent was removed in vacuo and the crude product was purified by column chromatography (SiO2) with ethyl acetate and petroleum ether (80:20) as eluent. Yield (1.03 g, 46.61%).
1H NMR (400 MHz, DMSO): δ 7.81 (1H, d, J 7.8), 7.48 (1H, d, J 8.2), 7.40 (1H, t, J 7.7), 7.22 (1H, t, J 7.6), 5.54 (1H, s), 4.75 (2H, q, J 7.0), 4.16 (2H, t, J 7.3), 1.72–1.59 (2H, m), 1.43 (3H, t, J 7.1), 1.22 (16H, s), 0.84 (3H, t, J 6.7).
13C NMR (126 MHz, DMSO): δ 180.93, 180.47, 154.81, 136.34, 122.14, 122.05, 118.71, 117.22, 106.15, 74.26, 64.90, 41.05, 27.16, 24.85, 24.78, 24.71, 24.58, 24.52, 22.16, 22.01, 17.94, 11.17, 9.37.
MS (FTMS+) [M+] calc. = 441.13, [M+] observed = 441.13, m/z.
FT-IR (ATR) ν/cm−1 2957 (m), 2920 (s), 2851 (s), 1724 (s), 1675 (s), 1470 (m), 741 (s), 718 (s).
UV-visible λmax 441 nm (47804 M−1 cm−1 ± 2175 M−1 cm−1) in ethanol.
Preparation of 3-((3-dodecylbenzothiazol-2-ylidene)methyl)-4-hydroxycyclobut-3-ene-1,2-dione (4)
3-((3-Dodecylbenzothiazol-2-ylidene)methyl)-4-ethoxycyclobut-3-ene-1,2-dione (3) (1.00 g, 2.27 mmol) was dissolved in ethanol (10 ml) and was heated to reflux. Tetrabutylammonium hydroxide solution (2 ml, 40%) was added and the solution was refluxed for 1 h and the resulting mixture was purified by column chromatography (SiO2) with ethyl acetate and petroleum ether as eluent to remove impurities. Pure product was then be run off the column with a dichloromethane: methanol mix (90:10). Yield (0.29 g, 19.46%).
1H (400 MHz, DMSO) δ 7.44 (1H, d, J 7.6), 7.17 (1H, t, J 7.7), 7.03 (1H, d, J 8.1), 6.91 (1H, t, J 7.5), 5.39 (1H, s), 3.83 (2H, t, J 7.5), 1.23 (14H, s), 0.85 (3H, t, J 6.7).
13C (101 MHz, DMSO) δ 208.61, 194.54, 179.30, 147.80, 142.16, 126.31, 126.00, 121.17, 120.79, 109.26, 81.67, 57.52, 43.94, 31.29, 28.99, 28.95, 28.83, 28.69, 26.23, 25.79, 23.06, 22.09, 19.21, 13.95, 13.49.
MS (FTMS−) [M − TBA]− calc. = 412.19, [M − TBA]− observed = 412.20, m/z.
FT-IR (ATR) ν/cm−1 3334 (br), 2958 (m), 2923 (s), 2872 (m), 2851 (m), 1748 (s), 1670 (s), 1524 (m).
UV-visible λmax 446 nm (31225 M−1 cm−1 ± 231 M−1 cm−1) in ethanol.
3-(2-((2-Ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)benzothiazol-3-yl)propanoic acid (6)
2.00 g (6.62 mmol) of 3-(2-carboxyethyl)-2-methylbenzothiazol-3-ium bromide (5), 1.13 g (6.64 mmol) of 3,4-diethoxy-3-cyclobutene-1,2-dione (2) and triethylamine (2 ml) were dissolved in ethanol (25 ml) and refluxed under N2 for 30 min. After cooling the solvent was removed was removed in vacuo and the crude product was purified by column chromatography (SiO2) with dichloromethane and methanol (90:10) as eluent. (Yield 1.46 g, 63.76%).
1H NMR (400 MHz, DMSO) δ 7.80 (1H, d, J 7.8), 7.48 (1H, d, J 8.2), 7.39 (1H, t, J 7.3), 7.21 (1H, t, J 7.3), 5.59 (1H, s), 4.75 (2H, q, J 7.1), 4.37 (2H, t, J 7.2), 2.67 (2H, t, J 7.2), 1.43 (3H, t, J 7.1).
13C NMR (101 MHz, DMSO) δ 185.22, 184.68, 172.19, 171.89, 158.97, 140.63, 127.11, 125.74, 123.53, 123.33, 112.10, 79.27, 69.29, 41.19, 31.15, 15.69.
MS (FTMS+) [M + H]+ calc. = 346.07, [M + H]+ observed = 346.07, m/z.
FT-IR (ATR) ν/cm−1 2925 (br), 1770 (s), 1710 (s), 1676 (s), 1496 (s), 1410 (s).
UV-visible λmax 424 nm (18411 M−1 cm−1 ± 510 M−1 cm−1) in ethanol.
Preparation of 3-(2-((2-hydroxy-3,4-dioxocyclobut-1-en-1-yl)methylene)benzothiazol-3-yl)propanoic acid (7)
3-(2-((2-Ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)benzothiazol-3-yl)propanoic acid (6) (1.00 g, 1.45 mmol) was dissolved in ethanol (10 ml) and heated under reflux. Tetrabutylammonium hydroxide (1 ml, 40%) was added and the mixture was refluxed for 1 h. After cooling the solvent was removed in vacuo and the solvent was removed in vacuo and the resulting mixture was purified by column chromatography (SiO2) with dichloromethane and methanol (90:10) as eluent. After purification the solution was concentrated, and the pure product precipitated with ethanol. (Yield 0.32 g, 39.51%).
1H NMR (500 MHz, DMSO) δ 7.44 (1H, d, J 7.6), 7.18 (1H, t, J 7.7), 7.09 (1H, d, J 8.1), 6.93 (1H, t, J 7.4), 5.42 (1H, s), 4.09 (2H, t, J 7.4), 2.58 (2H, t, J 7.4).
13C NMR (126 MHz, DMSO) δ 209.39, 195.06, 179.61, 172.84, 147.78, 142.22, 126.71, 126.50, 121.63, 121.44, 109.87, 82.37, 58.00, 31.36, 23.53, 19.68, 13.96.
MS (FTMS−) [M − H]− calc. = 316.03, [M − H]− observed = 316.03, m/z.
FT-IR (ATR) ν/cm−1 2958 (m), 2930 (s), 1750 (s), 1523 (m), 1424 (s).
UV-visible λmax = 446 nm (36409 M−1 cm−1 ± 455 M−1 cm−1) in ethanol.
Preparation of 3-(dicyanomethylene)-2-((3-dodecylbenzothiazol-2-ylidene)methyl)-4-oxocyclobut-1-en-1-olate (8)
A mixture of 3-((3-dodecylbenzothiazol-2-ylidene)methyl)-4-ethoxycyclobut-3-ene-1,2-dione (3) (0.5 g, 1.13 mmol) and malononitrile (85 mg, 1.25 mmol) were dissolved in ethanol (15 ml). Triethylamine (0.20 ml, 1.47 mmol) was added dropwise to the solution and it was allowed to stir for 2 h at room temperature. After the reaction had completed the solvent was removed in vacuo and the product was obtained as an orange solid after purification by column chromatography (SiO2), first with ethyl acetate and petroleum ether as eluent to remove impurities. Pure product was then be run off the column with a dichloromethane: methanol mix (80:20). Yield (0.20 g, 38.46%).
1H NMR (500 MHz, DMSO) δ 7.64 (1H, dd, J 7.8, 0.6), 7.28 (2H, ddd, J 20.9, 11.0, 4.2), 7.11–7.00 (1H, m), 5.89 (1H, s), 3.90 (2H, t, J 7.6), 1.70–1.58 (2H, m), 1.40–1.33 (3H, m), 1.33–1.28 (3H, m), 1.23 (13H, s), 0.85 (3H, t, J 6.9).
13C NMR (126 MHz, DMSO) δ 190.96, 186.34, 174.44, 168.48, 153.89, 141.82, 127.05, 126.44, 122.62, 122.29, 119.86, 118.95, 111.01, 81.88, 68.86, 46.20, 45.32, 35.51, 31.78, 29.48, 29.34, 29.18, 26.50, 22.58, 16.18, 14.42, 9.08.
MS (TOF MS ES−) [M]− calc. = 460.21, [M]− observed = 460.21, m/z.
FT-IR (ATR) ν/cm−1 3448 (br), 2920 (s), 2852 (s), 2196 (s), 2180 (s), 1744 (m), 1624 (m), 1486 (s), 1356 (s), 738 (s).
UV-visible λmax 480 nm (28148 M−1 cm−1 ± 881 M−1 cm−1) in ethanol.
Preparation of 2-(2-((1-dodecyl-3,3-dimethylindolin-2-ylidene)methyl)-3-hydroxy-4-oxocyclobut-2-en-1-ylidene)malononitrile (12)
A mixture of 3-((1-dodecyl-3,3-dimethylindolin-2-ylidene)methyl)-4-ethoxycyclobut-3-ene-1,2-dione (10) (0.50 g, 1.11 mmol) and malononitrile (95 mg, 1.44 mmol) were dissolved in ethanol (15 ml). Triethylamine (0.2 ml, 1.44 mmol) was added dropwise to the solution and it was allowed to stir for 2 h at room temperature. After cooling the solvent was removed in vacuo and the product was obtained as an orange solid after purification by column chromatography (SiO2), first with ethyl acetate as eluent to remove impurities and then with dichloromethane:methanol (80:20) to obtain the pure product. Yield (0.28 g, 53.85%).
1H NMR (500 MHz, DMSO) δ 7.32 (1H, dd, J 7.3, 0.9), 7.20 (1H, td, J 7.7, 1.2), 6.97 (1H, d, J 7.9), 6.94 (1H, td, J 7.5, 0.6), 5.92 (1H, s), 3.73 (2H, t, J 7.5), 1.62 (2H, dd, J 14.5, 7.2), 1.56 (6H, s), 1.38–1.26 (5H, m), 1.22 (14H, s), 0.85 (3H, t, J 6.9).
13C NMR (126 MHz, DMSO) δ 192.24, 185.73, 177.64, 167.90, 163.06, 143.52, 140.60, 127.96, 122.18, 121.34, 119.64, 118.64, 108.20, 84.70, 46.79, 42.49, 36.20, 31.75, 29.44, 29.31, 29.27, 29.18, 29.14, 28.41, 27.54, 26.39, 26.35, 22.55, 14.42.
MS (TOF-MS-ES−) [M]− calc. = 470.28, [M]− observed = 470.28, m/z.
FT-IR (ATR) ν/cm−1 3382 (br), 2921 (s), 2852 (s), 2198 (s), 1738 (s), 1644 (m), 1484 (m), 1316 (s).
UV-visible λmax 460 nm (10835 M−1 cm−1 ± 52 M−1 cm−1) in ethanol.
Preparation of 3-(2-((2-ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)-3,3-dimethylindolin-1-yl)propanoic acid (14)
2.00 g (6.41 mmol) of 1-(2-carboxyethyl)-2,3,3-trimethyl-indol-1-ium bromide (13), 1.09 g (6.41 mmol) of 3,4-diethoxy-3-cyclobutene-1,2-dione (2) and triethylamine (2 ml) were dissolved in 25 ml of ethanol and refluxed under nitrogen for 30 min. The solvent was removed in vacuo and the crude product was purified by column chromatography (SiO2) with dichloromethane and methanol (90:10) as eluent. (Yield 0.80 g, 35.09%).
1H NMR (400 MHz, DMSO) δ 7.42 (1H, d, J 7.3), 7.28 (1H, t, J 7.7), 7.18 (1H, d, J 7.9), 7.06 (1H, t, J 7.4), 5.39 (1H, s), 4.81 (2H, q, J 7.1), 4.15 (2H, t, J 7.0), 2.61 (2H, t, J 7.0), 1.54 (6H, s), 1.44 (3H, t, J 7.1).
13C NMR (101 MHz, DMSO) δ 192.08, 188.09, 186.61, 172.75, 167.35, 142.02, 140.26, 127.79, 122.53, 121.90, 109.25, 81.08, 69.82, 47.37, 45.66, 30.95, 26.49, 15.68.
MS (FTMS+) [M+] calc. = 355.14, [M+] observed = 355.04, m/z.
FT-IR (ATR) ν/cm−1 3425 (br), 2976 (m), 2933 (m), 2682 (w), 1770 (vs), 1722 (s), 1702 (s), 1535 (s), 1309 (s) 1175 (s), 1035 (s), 930 (s), 754 (s).
UV-visible λmax 423 nm (51672 M−1 cm−1 ± 1519 M−1 cm−1) in ethanol.
Preparation of 3-(2-((2-hydroxy-3,4-dioxocyclobut-1-en-1-yl)methylene)-3,3-dimethylindolin-1-yl)propanoic acid (15)
3-(2-((2-ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)-3,3-dimethylindolin-1-yl)propanoic acid (14) (0.5 g, 1.41 mmol) was dissolved in ethanol (10 ml) and heated to reflux. Tetrabutylammonium hydroxide solution (1 ml, 40%) was added and the solution was allowed to reflux for 1 h. After cooling the solvent was removed in vacuo and the resulting mixture was purified by column chromatography (SiO2) with dichloromethane and methanol (90:10) as eluent. The solution was then concentrated, and pure product precipitated after ethanol was added. (Yield 0.04 g, 5.00%).
1H NMR (500 MHz, DMSO) δ 7.23 (1H, d, J 7.2), 7.14 (1H, t, J 7.5), 6.84 (2H, dd, J 15.7, 7.9), 5.50 (1H, s), 3.99–3.92 (2H, m), 2.45 (2H, m), 1.55 (6H, s).
13C NMR (101 MHz, DMSO) δ 195.84, 179.17, 157.83, 144.00, 140.32, 127.77, 122.06, 120.09, 107.19, 84.78, 57.99, 49.06, 46.20, 28.00, 23.53, 19.68, 13.96.
MS (FTMS−) [M−] calc. = 326.10, [M+] observed = 326.10.04, m/z.
FT-IR (ATR) ν/cm−1 2960 (m), 2932 (w), 2874 (m), 1752 (vs), 1733 (m), 1546 (s), 1310 (vs).
UV-visible λmax 423 nm (42671 M−1 cm−1 ± 824 M−1 cm−1) in ethanol.
Preparation of 2-((3-(2-carboxyethyl)benzo[d]thiazol-2-ylidene)methyl)-3-(dicyanomethylene)-4-oxocyclobut-1-en-1-olate (16)
A mixture of 3-(2-((2-ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)benzothiazol-3-yl)propanoic acid (6) (0.50 g, 1.45 mmol) and malononitrile (106 mg, 1.6 mmol) were dissolved in ethanol (15 ml). Triethylamine (0.22 ml, 1.10 mmol) was added dropwise to the solution and it was allowed to stir for 2 h at room temperature. After cooling the solvent was removed in vacuo and the product was obtained as an orange solid after purification by column chromatography (SiO2) with dichloromethane and methanol (80:20) as eluent to obtain the pure product. (Yield 0.25 g, 47.17%).
1H NMR (500 MHz, DMSO) δ 7.63 (1H, d, J 7.7), 7.35–7.24 (2H, m), 7.07 (1H, t, J 7.2), 5.85 (1H, s), 4.15 (2H, t, J 7.1), 2.64 (2H, t, J 7.0).
13C NMR (101 MHz, DMSO) δ 190.77, 185.86, 174.21, 167.88, 153.05, 141.13, 126.49, 125.87, 122.22, 121.79, 120.16, 119.32, 118.37, 110.85, 81.53, 9.03.
MS (FTMS+) [M + H]+ calc. = 366.05, [M + H]+ observed = 366.05, m/z.
FT-IR (ATR) ν/cm−1 3056 (br, w), 2198 (s), 2178 (s), 1749 (m), 1737 (m), 1716 (s), 1663 (s), 1500 (s), 1411 (s), 1359 (s), 1308 (s), 1164 (s).
UV-visible λmax 478 nm (35374 M−1 cm−1 ± 386 M−1 cm−1) in ethanol.
Preparation of 2-((1-(2-carboxyethyl)-3,3-dimethylindolin-2-ylidene)methyl)-3-(dicyanomethylene)-4-oxocyclobut-1-en-1-olate (17)
A mixture of 3-(2-((2-ethoxy-3,4-dioxocyclobut-1-en-1-yl)methylene)-3,3-dimethylindolin-1-yl)propanoic acid (14) (0.5 g, 1.41 mmol) and malononitrile (102 mg, 1.55 mmol) were dissolved in ethanol (15 ml). Triethylamine (0.22 ml, 1.10 mmol) was added dropwise to the solution and it was allowed to stir for 2 h at room temperature. After cooling the solvent was removed in vacuo and the pure product was obtained after column chromatography (SiO2) with dichloromethane and methanol (80:20) as eluent. (Yield 0.12 g, 22.64%).
1H NMR (500 MHz, DMSO) δ 7.31 (1H, d, J 7.2), 7.19 (1H, t, J 7.6), 7.02 (1H, d, J 7.8), 6.93 (1H, t, J 7.2), 5.84 (1H, s), 3.97 (2H, t, J 6.5), 2.93 (2H, dd, J 14.0, 6.8), 1.56 (6H, s).
13C NMR (126 MHz, DMSO) δ 185.73, 177.82, 167.87, 162.84, 143.23, 140.48, 133.47, 127.93, 122.11, 121.40, 119.61, 118.57, 108.57, 84.82, 46.75, 45.74, 27.49, 9.38.
MS (FTMS+) [M]− calc. = 374.11, [M]− observed = 374.11, m/z.
FT-IR (ATR) ν/cm−1 3412 (br, w), 3056 (br, w), 2198 (m), 1742 (m), 1544 (w), 1486 (m), 1312 (vs), 742 (s).
UV-visible λmax 456 nm (22544 M−1 cm−1 ± 491 M−1 cm−1) in ethanol.
Modelling procedure
For optimisation of the unit cell of TiO2, the Brillouin zone integrations were performed on a 7 × 7 × 9 Monkhorst–Pack53 grid with 16 symmetry constraints, and for the surface-plus-molecule and gas-phase systems the single sampling point corresponded to the gamma point. We used the pairwise, semi-empirical dispersion correction (SEDC) term of Tkatchenko and Scheffler54 when modelling the TiO2 surface, molecule, and surface-plus-molecule systems to account for long-range dispersions. Following the work of Martsinovich et al.55 no Hubbard value was applied to Ti, which is further justified by our focus being the orientation of dye molecules on an anatase surface (rather than band gaps for example).
For all model systems (unless stated otherwise) the geometry was optimized using the method of Broyden–Fletcher–Goldfarb–Shanno (BFGS)56 and the self-consistent electronic minimisation method was density mixing. Further convergence details per BFGS iteration are as follows: electronic energy tolerance: 10−8 eV; energy change per ion: dE/ion 5 × 10−6 eV; maximum force: |F|max 0.01 eV Å−1; change in displacement: |dR| 5 × 10−4 Å. All calculations were non-spin polarised.
For the three-layered slab the vacuum space above the surface was tested, to determine the optimum c-length that would reduce artefactual electrostatic interactions between periodic images. A c-length of 15 Å was found to be well within the energy convergence criteria of 5 meV, although the cell was further increased to 20 Å to accommodate the dye molecules.
Finally, the optimum supercell size was determined whereby the three-layered slab of 20 Å (c-length), was increased both in the x- and y-directions. The dye molecule with the largest planar length (molecule 16) was placed on top of the surface of variously-sized slabs, and total energies were calculated without relaxation of the model system's atomic structure. Maximum forces were converged for 2X–4Y supercells producing a final, (1 0 1) anatase TiO2 slab of dimensions: 20.86 Å × 15.23 Å × 29.02 Å (see ESI Fig. S2†).
To obtain a representative structure of a dye molecule on the surface, as many configurations as possible should be checked. However, this requires a faster methodology than DFT, that can take the energy of the relative configurations into account. We used ‘AutoDock’ (release 4.2.6)66,67 as a cheap pre-screening methodology for this, acknowledging its limited application to solid-state systems. We chose five or six different dyes configurations from the pre-screening results, which were placed on clean TiO2 (1 0 1) surfaces and optimised using DFT. The surface-plus-dye configurations with the lowest energy (i.e. largest-in magnitude-adsorption energy) were identified as the most probable orientations of the dyes on the TiO2 surface. For further details see ESI S2.†
For each of the relaxed surface-plus-dye models we calculated the perpendicular, z-distance of oxygens, nitrogen and sulphur atoms above the highest surface-Ti; the results comprise simulated angle-resolved X-ray photoelectron spectroscopy (AR-XPS) for comparison to experimental results.
Finally, we calculated the absolute adsorption energies of the molecules on the dye surfaces to determine the most probable orientation of the dye in the TiO2 surface.
Device manufacture and testing
DSC devices were prepared using 1.0 cm2 (2 × 0.5 cm) TiO2 working electrodes by doctor blading one layer of transparent paste (Ti-Nanoxide T/SP, Solaronix) onto TEC8™ FTO glass (NSG). The TiO2 layer was sintered (500 °C, 30 min) before TiCl4:THF2(aq) treatment (50 mM, 70 °C, 30 min), rinsing with de-I H2O and resintering (500 °C, 30 min). To prepare counter electrodes, after washing (iPrOH) and air drying TEC8™ glass (NSG) Pt colloid (Pt-1, Dyesol) was deposited and then sintered (400 °C, 30 min). After sealing the counter and working electrodes together with Surlyn™ (DuPont) at 120 °C, the devices were fast dyed according to the method described previously68 after which acetonitrile-based electrolyte was added (50 mM I2, 50 mM LiI, 50 mM guanidinium thiocyanate, 800 mM 1-methyl-3-propylimidazolium iodide, 300 mM benzimidazole and 50 mM tBP). Conductive Ag paint (Agar) was applied to ensure good electrical contact. Device performance was measured using I–V data using a Class AAA Solar Simulator at 100 mW cm2 or 1 Sun between 0 and 1 V (Oriel Sol3A). Spectral response was measured in DC mode using a Keithley 2400 sourcemeter at a scan rate 0.1 V s−1. Lamp calibrated to 1 Sun (100 mW cm2) using a KG5 IR-filtered silicon cell (Newport Oriel 91150-KG5).To study dye loading, one layer of transparent paste (Ti-Nanoxide T/SP, Solaronix) was doctor bladed onto a series of plain glass slides (1.0 cm × 5 cm). The TiO2 layer was sintered at 500 °C for 30 min. After cooling, the TiO2 coated slides were placed in dye solutions (0.5 mmol) for 18 hours. The slides were then rinsed with ethanol and dried under N2 before the dyes were desorbed with tetrabutylammonium hydroxide solution (1.0 M in methanol). Solutions were then measured using UV-Vis spectroscopy.
Conclusions
This paper shows, we believe, the first combined theoretical (DFT) and experimental (AR-XPS) study of dye:TiO2 orientation for a family of structurally related dyes. As such, the data need to be considered in the context of the strengths and limitations of the methods used. For example, atomistic modelling provides detailed insight into the structural features and energetic trends of a dye on an ideal TiO2 surface; AR-XPS provides structural information of a non-ideal, i.e., “real” dye–TiO2 device. However, the DFT calculations are limited to single dye molecules on the surface and carried out at 0 K; the AR-XPS measurements are an average of many dye molecules on TiO2 surfaces at ambient temperature. Nevertheless, bearing these factors in mind, we believe that this paper shows, for the first time, that organic DSC dye molecules orient themselves horizontally on TiO2 surfaces rather than perpendicularly as is widely implied in the prior DSC literature.Though these findings have important implications for the future design of DSC dyes both in the context of liquid and solid-state DSC devices, they warrant further theoretical modelling of higher concentration of dyes on the surface as the orientations can alter with concentration of dye on the surface. These findings also have potential implications for dye loadings because the space occupied on the surface is directly related to the orientation of the dyes. An in-depth understanding of the orientation of multiple dyes will facilitate future design of co-sensitised dyes, devices with co-adsorbents, and the donor–spacer–linker approach to dye-design. Ultimately, consideration of dye surface area will add an extra dimension to the dye design process that already includes rapid injection, higher extinction coefficients, and more stable dyes.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge funding from the EPSRC EP/P030068/1 (PJH, DMR), EP/P03165X/1 (DG, KS, SM), EP/M015254/2 (EWJ), EU via ERDF for SPARC II (CK), EP/N020863/1 (JM) and the National Mass Spectrometry Facility at Swansea University. We also acknowledge EPSRC funding for the National Crystallography Centre at Southampton University, and Swansea University College of Engineering AIM Facility, which was funded by EPSRC (EP/M028267/1) and the European Regional Development Fund (80708). We acknowledge use of Hartree Center resources in this work. The Science and Technology Facilities Council (STFC) Hartree Center is a research collaborator in association with IBM providing High Performance Computing platforms funded by the U.K.'s investment in e-Infrastructure. The Center aims to develop and demonstrate next generation software, optimized to take advantage of the move toward exa-scale computing.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- Z. Yao, M. Zhang, R. Li, L. Yang, Y. Qiao and P. Wang, Angew. Chem., Int. Ed., 2015, 54, 5994 CrossRef CAS.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, T. Kyomen and M. Hanaya, Chem. Commun., 2015, 51, 6315 RSC.
- Y. Ren, D. Sun, Y. Cao, H. N. Tsao, Y. Yuan, S. M. Zakeeruddin, P. Wang and M. Grätzel, J. Am. Chem. Soc., 2018, 140, 2405 CrossRef CAS.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894 RSC.
- S. M. Abdalhadi, A. Connell, X. Zhang, A. A. Wiles, M. L. Davies, P. J. Holliman and G. Cooke, J. Mater. Chem. A, 2016, 4, 15655 RSC.
- P. J. Holliman, M. L. Davies, A. Connell, B. Vaca Velasco and T. M. Watson, Chem. Comm., 2010, 46, 7256 RSC.
- P. J. Holliman, M. Mohsen, A. Connell, M. L. Davies, K. Al-Salihi, M. B. Pitak, G. J. Tizzard, S. J. Coles, R. W. Harrington, W. Clegg, C. Serpa, O. H. Fontes, C. Charbonneau and M. J. Carnie, J. Mater. Chem., 2012, 22(26), 13318 RSC.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CrossRef CAS.
- Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Grätzel, Inorg. Chem., 1999, 38, 6298 CrossRef CAS.
- P. J. Holliman, B. Vaca Velasco, I. Butler, M. Wijdekop and D. Worsley, Int. J. Photoenergy, 2008, 1–8 CrossRef.
- K. S. Finnie, J. R. Bartlett and J. L. Woolfrey, Langmuir, 1998, 14, 2744 CrossRef CAS.
- C. Pérez Lyon, L. Kador, B. Peng and M. Thelakkat, J. Phys. Chem. B, 2006, 110, 8723 CrossRef.
- F. De Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2010, 114, 6054 CrossRef CAS.
- P. J. Holliman, C. Kershaw, A. Connell, E. W. Jones, R. Hobbs, R. Anthony, L. Furnell, J. McGettrick, D. Geatches and S. Metz, Sci. Technol. Adv. Mater., 2018, 19(1), 599 CrossRef CAS.
- J.-Y. Li, C.-Y. Chen, C.-P. Lee, S.-C. Chen, T.-H. Lin, H.-H. Tsai, K.-C. Ho and C.-G. Wu, Org. Lett., 2010, 12, 5454 CrossRef CAS.
- D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550 CrossRef CAS.
- J. Ho Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nüesch, F. De Angelis, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320 CrossRef.
- B. Peng, S. Yang, L. Li, F. Cheng and J. Chen, J. Chem. Phys., 2010, 132, 034305 CrossRef.
- J. Zhang, H. B. Li, S. L. Sun, Y. Geng, Y. Wu and Z. M. Su, J. Mater. Chem., 2012, 22, 568 RSC.
- C. I. Oprea and M. A. Gîrţu, Nanomaterials, 2019, 9, 357 CrossRef CAS.
- M. Pastore and F. De Angelis, J. Phys. Chem. Lett., 2013, 4, 956 CrossRef CAS.
- C. Anselmi, E. Mosconi, M. Pastore, E. Ronca and F. De Angelis, Phys. Chem. Chem. Phys., 2012, 14, 15963 RSC.
- F. De Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2007, 129, 14156 CrossRef CAS.
- P. Persson and M. J. Lundqvist, J. Phys. Chem. B, 2005, 109, 11918 CrossRef CAS.
- F. De Angelis, S. Fantacci, E. Mosconi, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 8825 CrossRef CAS.
- A. L. Puyad, Ch R. Kumar and K. Bhanuprakash, J. Chem. Sci., 2012, 124, 301 CrossRef CAS.
- C. J. Barbé, F. Árendse, P. Comte, M. Jirousek, F. Lenzmann, V. Schlover and M. Grätzel, J. Am. Ceram. Soc., 1997, 80, 3157 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS.
- P. J. Holliman, D. K. Muslem, E. W. Jones, A. Connell, M. L. Davies, C. Charbonneau, M. J. Carnie and D. A. Worsley, J. Mater. Chem. A, 2014, 2(29), 11134 RSC.
- L. Zhang and J. Cole, J. Mater. Chem. A, 2017, 5, 19541 RSC.
- H.-P. Lu, C. Y. Tsai, W.-N. Yen, C.-P. Hsieh, C.-W. Lee, C.-Y. Yeh and E. W.-G. Diau, J. Phys. Chem. C, 2009, 113, 20990 CrossRef CAS.
- A. Connell, P. J. Holliman, M. L. Davies, C. D. Gwenin, S. Weiss, M. B. Pitak, P. N. Horton, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2014, 2(11), 4055 RSC.
- A. Connell, P. J. Holliman, E. W. Jones, L. Furnell, C. Kershaw, M. L. Davies, C. D. Gwenin, M. B. Pitak, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2015, 3, 2883 RSC.
- B. O'Regan, K. Walley, M. Juozapavicius, A. Anderson, F. Matar, T. Ghaddar, S. M. Zakeeruddin, C. Klein and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 3541 CrossRef.
- Y. Hao, X. Yang, J. Cong, H. Tian, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 4031–4033 RSC.
- Y. Ooyama, Y. Shimada, S. Inoue, T. Nagano, Y. Fujikawa, K. Komaguchi, I. Imae and Y. Harima, New J. Chem., 2011, 35, 111 RSC.
- Y. Hao, X. Yang, J. Cong, X. Jiang, A. Hagfeldt and L. Sun, RSC Adv., 2012, 2, 6011–6017 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 27, 3–8 Search PubMed.
- Dassault Systèmes, Dassault Systèmes BIOVIA, Materials Studio v2016, Dassault Systèmes, San Diego, 2016 Search PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, 864 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulous, Rev. Mod. Phys., 1992, 64, 1045 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- D. R. Hamann, M. Schlüter and C. Chiang, Phys. Rev. Lett., 1979, 43(20), 1494 CrossRef CAS.
- H. Monkhorst and J. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 0730051 CrossRef.
- N. Martsinovich, D. R. Jones and A. Troisi, J. Phys. Chem. C, 2010, 114, 22659 CrossRef CAS.
- B. G. Pfrommer, M. Côté, S. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58(8), 1200 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98(45), 11623 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97(4), 2571 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100(8), 5829 CrossRef.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356(1), 98 CrossRef CAS.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135(14), 144105 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- G. M. Morris, R. H. William Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30(16), 2785 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19(14), 1639 CrossRef CAS.
- P. J. Holliman, M. L. Davies, A. Connell, B. Vaca Velasco and T. M. Watson, Chem. Commun., 2010, 46, 7256 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1908056–1908060. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ta06016j |
| This journal is © The Royal Society of Chemistry 2020 |