
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting Hansen solubility parameters to tune porosity and function in conjugated microporous polymers†
Jie
Chen
 a,
Ting
Qiu
c,
Wei
Yan
d and
Charl F. J.
Faul
*b
a,
Ting
Qiu
c,
Wei
Yan
d and
Charl F. J.
Faul
*b
aCollege of Environment and Resources, Fuzhou University, Fuzhou 350116, P. R. China
bSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: charl.faul@bristol.ac.uk
cCollege of Chemical Engineering, Fuzhou University, Fuzhou 350116, P. R. China
dDepartment of Environmental Science and Engineering, Xi'an Jiaotong University, Xi'an 710049, P. R. China
First published on 2nd September 2020
Abstract
Here we expand our recently reported Bristol–Xi'an Jiaotong (BXJ) approach using simple salts to fine-tune the porosity of conjugated microporous materials synthesized by various reaction approaches, including Buchwald–Hartwig (BH), Sonogashira–Hagihara, oxidative coupling and Suzuki cross-coupling. The surface area and the porosity of the produced conjugated microporous polyanilines (CMPAs) acquired from the non-salt-added BH coupling are optimized by the addition of inorganic salts. BXJ-salt addition provides a facile route to radically improve the BET surface area from 28 to 901 m2 g−1 for PTAPA and from 723 m2 g−1 to 1378 m2 g−1 for PAPA in a controllable manner. In addition, the surface area shows a gradual decrease with an increase in the ionic radius of salts. We furthermore show high compatibility of this approach in the synthesis of typical CMPs, further increasing the surface area from 886 to 1148 m2 g−1, 981 to 1263 m2 g−1, and 35 to 215 m2 g−1 for CMP-1, PTCT and p-PPF, respectively. More importantly, the BXJ approach also allows the broad PSD of the CMPs to be narrowed to the microporous range only, mimicking COFs and MOFs. With the porosity optimized, CO2 uptakes are dramatically improved by >300% from 0.75 mmol g−1 to 2.59 mmol g−1 for PTAPA and from 2.41 mmol g−1 to 2.93 mmol g−1 for PAPA. Careful addressing of Hansen solubility parameters (HSPs) of solvents and resulting polymers through salt addition has the potential to become an important design tool for the preparation of fully tuneable porous materials. We are currently exploring further methods to tune both structure and function in a wide range of organic porous materials.
Introduction
Rapid consumption of fossil fuels has caused excessive and uncontrolled CO2 emissions, resulting in global climate change and wide-ranging environmental challenges.1 As such, methodologies focusing on CO2 capture are of great interest to meet these growing global environmental problems.2,3 Increasing effort has been focused on the search for materials for CO2 sequestration, and a wide variety of advanced porous materials have been developed. Such materials include crystalline polymers (covalent organic frameworks (COFs),4 metal–organic frameworks (MOFs),5 covalent triazine frameworks (CTFs)6) as well as amorphous organic polymers (porous organic cages (POCs),7 polymers of intrinsic microporosity (PIMs),8 porous coordination polymers (PCPs),9 hyper-crosslinked polymers (HCPs),10 porous aromatic frameworks (PAFs),11 and conjugated microporous polymers (CMPs)3,12–15). Among them, CMPs possess attractive intrinsic properties, including low skeleton density, high chemical stability, rich microporosity and large surface area, exhibiting considerable advantage over other organic polymers, especially over MOFs and COFs, in terms of the stability.16,17 Additionally, they exhibit unique extended π-conjugation and rich heteroatom content throughout the porous 3D networks.17 These intrinsic properties enable CMPs to be suitable for application in many areas of interest, especially in CO2 capture, in which the micropores and heteroatoms contribute to their high affinity towards CO2, thus increasing their adsorption capacity and selectivity.15A wide range of coupling chemistries, such as Sonogashira–Hagihara,12 Yamamoto,18 Suzuki,19 Friedel–Crafts,20 and oxidative coupling,21 have been devoted to construct novel CMPs. Recently, Buchwald–Hartwig (BH) coupling has been applied in CMP design and synthesis, where carbon–nitrogen bonds can be formed in a facile manner using a Pd catalyst and a base.15 A simple route was therefore developed to form N-rich redox-active CMPs, i.e., 3D analogues of the well-known redox-active polymer poly(aniline) (PANi). However, using BH coupling generally resulted in products with low porosity.14,15 Moreover, such materials possessed unexpectedly low micropore volumes and broad pore size distributions (PSDs) when compared, for example, with structurally similar poly(aryleneethynylene)s (PAEs).14,15 To address this unexpected challenge, we have shown in a recent paper22 that the surface area of a model BH-synthesized CMP, polytriphenylamine (PTPA), could be fine-tuned and optimized by the addition of inorganic salts to the reaction mixtures. This approach, termed the Bristol–Xi'an Jiaotong (BXJ) approach, yielded PTPA with a 20 times higher surface area than the standard reaction. The initially broad PSD of PTPA could additionally be tuned by the BXJ approach to a narrow micropore range distribution, closely mimicking that of COFs and MOFs. Our developed approaches therefore present an opportunity to tune surface area and porosity, potentially for a range of amorphous polymers. However, this potentially impactful methodology has only been applied in one specific instance in our first published study,22 and its general applicability to create a design tool for wider application remains to be fully explored.
Thus, we describe here a series of BXJ-assisted conjugated microporous polyanilines (CMPAs) synthesized by BH coupling chemistry.23,24 We explored different linkers with longer length or with more polymerization nodes (i.e., A3 + B3, as depicted in Scheme 1) compared to that used in PTPA to fully confirm the generality of our BXJ approach in the synthesis of amorphous CMPs using BH coupling. Additionally, application of the BXJ approach to other typical coupling reactions, including Sonogashira–Hagihara, Suzuki and oxidative coupling, were studied to further optimize the porosities of these typical model materials (i.e.,CMP-1, p-PPF and PTCT).
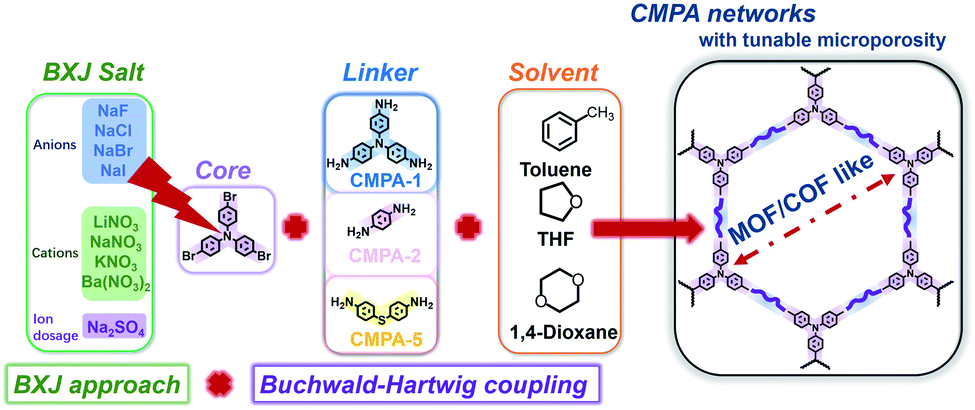 | ||
| Scheme 1 Synthetic route for the formation of salt-tuneable CMPA networks. | ||
Results and discussion
To explore the wider applicability of our BXJ approach in the synthesis of BH-prepared CMPs, salts with varying anion (i.e., NaF, NaCl, NaBr, NaI) or cation (i.e., LiNO3, NaNO3, KNO3, Ba(NO3)2) sizes were applied in the synthesis of CMPA networks. As an initial comparison we used tris(4-bromophenyl)amine as the core, and compared 1,4-phenyldiamine (used in our previous studies) with 4,4′-diaminodiphenyl sulfide (i.e., longer diphenyl linker) as linkers, leading to the formation of PTPA, and PTAPA, respectively, as shown in Scheme 1. The polymers acquired were chemically stable (i.e., were insoluble in common organic solvents and 3 M HCl or NaOH solutions), suggesting the hyper-crosslinked nature of the polymers.1 This heteroatom-containing BXJ CMP also exhibited similar multiple oxidation states as found for PTPA described in our previous work. The materials were initially a light brown colour under a N2 atmosphere during the polymerization, but gradually turned dark with increasing exposure to air, confirming the successful construction of C–N bonds and a PANi-type structure by BH coupling.22 Additionally, their molecular structures were confirmed by Fourier transform infrared (FTIR) and solid-state 13C cross-polarization magic angle spinning nuclear magnetic resonance (SS 13C CP/MAS NMR). As shown in Fig. 1 and S1,† apart from the characteristic peaks ascribed to the functional groups of the core and linkers, bands assigned to C–Br bonds (at 710, 1004, and 1070 cm−1) and those ascribed to NH2 stretching (peaks at 3400 and 3300 cm−1) are absent from the spectra of the products.15 The SS 13C CP/MAS NMR spectrum of PTAPA depicted in Fig. 2 showed the expected two main resonances at 127 and 141 ppm (assigned to the unsubstituted phenyl carbons and substituted phenyl carbons, respectively), as well as an additional peak (at 120 ppm) owing to the presence of the sulfur heteroatom.14 These results fitted well with the simulated results shown in the ESI, Fig. S2,† further confirming the successful BH coupling reactions.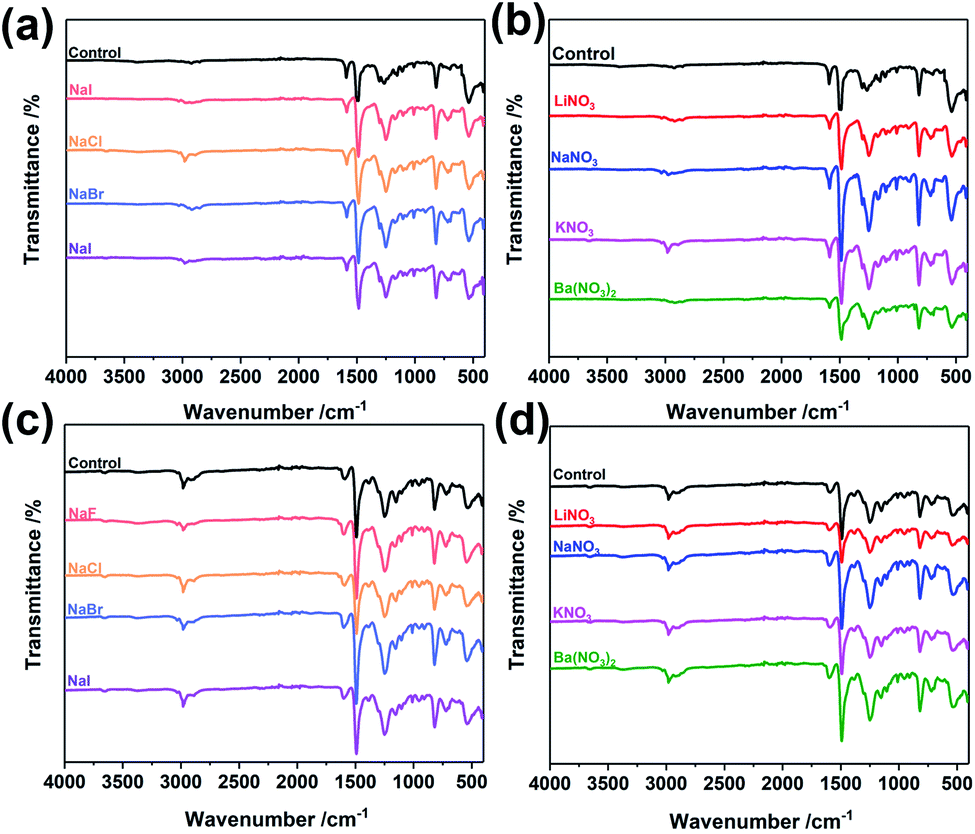 | ||
| Fig. 1 FTIR spectra of PTAPA: (a and b), before and after tuning by BXJ-Na salts; and PAPA: (c and d), before and after tuning by BXJ-NO3 salts. | ||
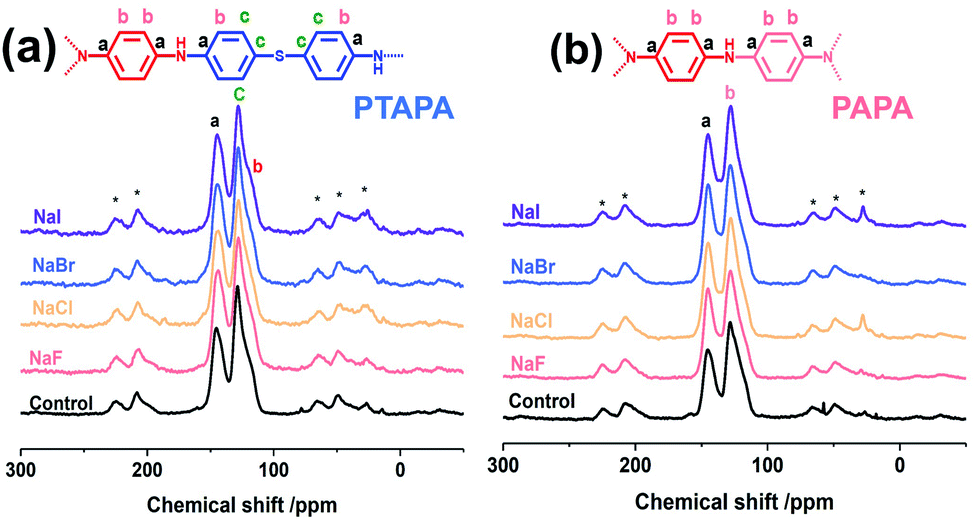 | ||
| Fig. 2 SS 13C NMR spectra of PTAPA (a) and PAPA (b) before and after tuning by BXJ-salts. | ||
Interestingly, the addition of salts during polymerization did not change the molecular structure (as shown in FTIR and SS 13C CP/MAS NMR), but led to macroscopic gelation of PTAPA during polymerization (rather than the usual precipitation when no salt was added), as was also observed in PTPA synthesis.22 These changes were also reflected in the SEM micrographs of these materials (shown in Fig. S4†), where the normally particulate-like morphology of CMPAs became almost foam-like after the addition of salts. These changes were early indicators of the positive effect the BXJ salts had on the physical properties of CMPAs produced by BH coupling.
We further investigated this influence of salts on the PTPA structures and, potentially, the porosity through detailed BET surface area analysis. PTAPA, with a longer linker than PTPA, showed a very low BET surface area of 28 m2 g−1 under standard BH cross-coupling conditions (as shown Table 1). A remarkable improvement of surface area of more than 30 times to 901 m2 g−1 was obtained with the aid of NaF. The surface area was then systematically decreased to 256 m2 g−1 by increasing the anion size of the employed sodium salts. This tuneable trend was also reflected in the change of total pore volume, with an almost 10 times increase from the initial value of 0.078 cm3 g−1 to 0.81 cm3 g−1 with the use of NaF, and gradual reduction to 0.21 cm3 g−1 (when NaI was applied). This delicate and fine control over the physical properties shows the suitability of the BXJ approach for overall structural design and optimization. General applicability was also confirmed with our BXJ nitrate salts on fine-tuning the surface area of PTAPA as shown in Table 1. A surface area of 652 m2 g−1 was effectively reached by LiNO3, and then tuned to 556 m2 g−1 by adding cations of increasing size. Note that these results were highly reproducible, with selected syntheses repeated three times (see Experimental section). However, unlike the surface area, there was no obvious trend in tuning the pore volume of PTAPA when applying salts with increasing cation radius. This outcome demonstrates the formation and control of pores with different volumes are more complex, especially with nitrate salts, as they could not be as finely controlled by the salt as the surface area. It is noteworthy that similar trends were also observed for the control of PTPA. See Table S1† for full details.
| Ion radius (Å) | Surface areaa (m2 g−1) | Total pore volumeb (cm3 g−1) | Micropore volumec (cm3 g−1) | Ultramicropore volumed (cm3 g−1) | CO2 uptake at 273 K, 1 atm (mmol g−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| a Surface area calculated from the N2 adsorption isotherm using the Brunauer–Emmett–Teller method. b The total pore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method. c The micropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for micropore (r < 2 nm) volume. d The ultramicropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for ultramicropore (r < 0.7 nm) volume. | |||||||
| Control | — | 28 | 0.078 | 0.025 | 0 | 0.75 | This work |
| NaF | 1.33 | 901 | 0.81 | 0.44 | 0.21 | 1.89 | This work |
| NaCl | 1.84 | 873 | 0.83 | 0.68 | 0.20 | 2.57 | This work |
| NaBr | 1.95 | 803 | 0.63 | 0.54 | 0.24 | 2.59 | This work |
| NaI | 2.20 | 256 | 0.21 | 0.20 | 0.085 | 0.95 | This work |
| LiNO3 | 0.76 | 652 | 0.45 | 0.43 | 0.18 | 2.10 | This work |
| NaNO3 | 1.02 | 640 | 0.33 | 0.33 | 0.19 | 1.91 | This work |
| KNO3 | 1.33 | 601 | 0.42 | 0.41 | 0.23 | 2.59 | This work |
| Ba(NO3)2 | 1.42 | 556 | 0.42 | 0.39 | 0.14 | 1.72 | This work |
| 0.33 mmol Na2SO4 | 2.35 | 130 | 0.059 | 0.059 | 0.053 | 2.38 | This work |
| 0.50 mmol Na2SO4 | 2.35 | 180 | 0.038 | 0.038 | 0.0095 | 2.61 | This work |
| 0.75 mmol Na2SO4 | 2.35 | 249 | 0.15 | 0.14 | 0.088 | 2.75 | This work |
| 1.00 mmol Na2SO4 | 2.35 | 371 | 0.23 | 0.23 | 0.14 | 2.10 | This work |
| 1.50 mmol Na2SO4 | 2.35 | 150 | 0.18 | 0.18 | 0.11 | 1.87 | This work |
| 2.00 mmol Na2SO4 | 2.35 | 131 | 0.11 | 0.11 | 0.065 | 0.81 | This work |
| Toluene/0.75 mmol Na2SO4 | 2.35 | 162 | 0.13 | 0.10 | 0.075 | 0.74 | This work |
| THF/0.75 mmol Na2SO4 | 2.35 | 249 | 0.23 | 0.23 | 0.14 | 2.75 | This work |
| Dioxane/0.75 mmol Na2SO4 | 2.35 | 418 | 0.22 | 0.22 | 0.16 | 2.65 | This work |
| CMP-1 | — | 834 | 0.53 | 0.33 | N.A. | N.A. | 25 |
| COF-1 | — | 711 | 0.32 | N.A. | N.A. | N.A. | 26 |
| CTF-1 | — | 791 | 0.40 | N.A. | N.A. | N.A. | 27 |
| MOF-1 | — | 516 | 0.29 | N.A. | N.A. | 0.86 | 28 |
In an effort to continue to further validate the applicability of our BXJ approach, we investigated the influence of the BXJ method on the BET surface area of another CMPA: PAPA, with one additional polymerization node in the linker than PTPA (see Scheme 1). Again, the addition of salts during polymerization did not change the molecular structure (as shown in FTIR and SS 13C CP/MAS NMR, Fig. 1 and 2, respectively), but led to macroscopic gelation (as found for the other systems).
Interestingly, an unexpected high surface area of 723 m2 g−1, as shown in Table 2, was acquired before salt tuning, indicating that the chosen reaction conditions were already a very good starting point for our investigations. In addition, earlier studies showed that higher connectivity in monomers often lead to higher surface area.29,30 In order to explore whether these already high values could be tuned, and further control exerted over other physical characteristics, we varied the salts using our BXJ approach. Here a very well-defined trend in the surface area was observed when sodium salts with varying anion radius were applied: the surface area was almost doubled to 1378 m2 g−1 by NaF, and then decreased with increasing anionic radius of the salts. This obtained surface is, to the best of our knowledge, one of the highest surface areas reached for BH-synthesized polymers. Nitrate salts (where the cation radius was varied) also showed regular influence on the surface area of PAPA as shown in Table 2. The initial surface area was increased slightly to 818 m2 g−1 with the decrease of cation radius. However, this influence was much less pronounced for PAPA (compared with that of PTPA and PTAPA).
| Ion radius (Å) | Surface areaa (m2 g−1) | Total pore volumeb (cm3 g−1) | Micropore volumec (cm3 g−1) | Ultramicropore volumed (cm3 g−1) | CO2 uptake at 273 K, 1 atm (mmol g−1) | Ref. | |
|---|---|---|---|---|---|---|---|
| a Surface area calculated from the N2 adsorption isotherm using the Brunauer–Emmett–Teller method. b The total pore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method. c The micropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for micropore (r < 2 nm) volume. d The ultramicropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for ultramicropore (r < 0.7 nm) volume. | |||||||
| Control | — | 723 | 0.58 | 0.47 | 0.23 | 2.41 | This work |
| NaF | 1.33 | 1378 | 0.88 | 0.47 | 0.47 | 2.51 | This work |
| NaCl | 1.84 | 777 | 0.45 | 0.21 | 0.21 | 2.28 | This work |
| NaBr | 1.95 | 742 | 0.45 | 0.27 | 0.27 | 2.29 | This work |
| NaI | 2.20 | 741 | 0.47 | 0.22 | 0.22 | 1.82 | This work |
| LiNO3 | 0.76 | 818 | 0.49 | 0.29 | 0.29 | 2.88 | This work |
| NaNO3 | 1.02 | 783 | 0.43 | 0.25 | 0.25 | 2.45 | This work |
| KNO3 | 1.33 | 765 | 0.40 | 0.20 | 0.20 | 1.63 | This work |
| Ba(NO3)2 | 1.42 | 724 | 0.36 | 0.20 | 0.20 | 2.07 | This work |
| 0.33 mmol Na2SO4 | 2.35 | 817 | 0.57 | 0.50 | 0.24 | 2.77 | This work |
| 0.50 mmol Na2SO4 | 2.35 | 830 | 0.47 | 0.46 | 0.26 | 2.58 | This work |
| 0.75 mmol Na2SO4 | 2.35 | 780 | 0.61 | 0.53 | 0.26 | 2.83 | This work |
| 1.00 mmol Na2SO4 | 2.35 | 690 | 0.39 | 0.39 | 0.22 | 1.70 | This work |
| 1.50 mmol Na2SO4 | 2.35 | 595 | 0.35 | 0.35 | 0.26 | 2.93 | This work |
| THF/0.5 mmol Na2SO4 | 2.35 | 830 | 0.47 | 0.46 | 0.26 | 2.58 | This work |
| Toluene/0.5 mmol Na2SO4 | 2.35 | 108 | 0.24 | 0.042 | 0.036 | 1.16 | This work |
| Dioxane without salts | — | 838 | 0.53 | 0.50 | 0.27 | 2.90 | This work |
| Dioxane/0.5 mmol Na2SO4 | 2.35 | 614 | 0.46 | 0.39 | 0.18 | 1.56 | This work |
| CMP-1 | — | 834 | 0.53 | 0.33 | N.A. | N.A. | 25 |
| COF-1 | — | 711 | 0.32 | N.A. | N.A. | N.A. | 26 |
| CTF-1 | — | 791 | 0.40 | N.A. | N.A. | N.A. | 27 |
| MOF-1 | — | 516 | 0.29 | N.A. | N.A. | 0.86 | 28 |
To gain insight into the influence of BXJ salts on the microporosity of these CMPAs, optimized dosages, as determined from the study of salt dosage influence on the porosity as shown in Tables 1, 2 and Fig. S6,† of various sodium salts (NaF, NaCl, NaBr, NaI) and nitrate salts (LiNO3, NaNO3, KNO3, Ba(NO3)2) were now employed in the BXJ synthesis of PTAPA and PAPA. Specifically, salt concentrations of 0.75 mmol (for PTAPA) or 0.5 mmol (for PAPA) were used and the porosities of polymers acquired were investigated by N2 adsorption and desorption isotherms, as shown in Fig. 3. For PTAPA a typical Type II isotherm was observed, as shown in Fig. 3(a) and (c), while a very broad PSD was obtained when using the standard reaction without salt as control (as shown in Fig. 3(b) and (d)). Such a broad PSD most likely results from interparticular voids or interparticulate porosity rather than from the intrinsic porosity.13,15 Type I isotherms were acquired for BXJ-PTAPA after adding both types of salt (Fig. 3(a) and (c)), with the resulting PSD becoming very narrow, showing only micropores. This result indicates that large numbers of micropores of defined size were produced by adding salts to the reaction mixture.12,31 As for PAPA, even though its isotherm showed intrinsic Type-I shape before salt tuning, the PSD was significantly narrowed after salt addition. As such, tuning the pores of amorphous CMPAs to resemble COF- or MOF-like distributions is therefore within reach with our BXJ approach (see the relevant XRD diffractograms in Fig. S7,† and transmission electron microscope (TEM) images in Fig. S8 and S9†).
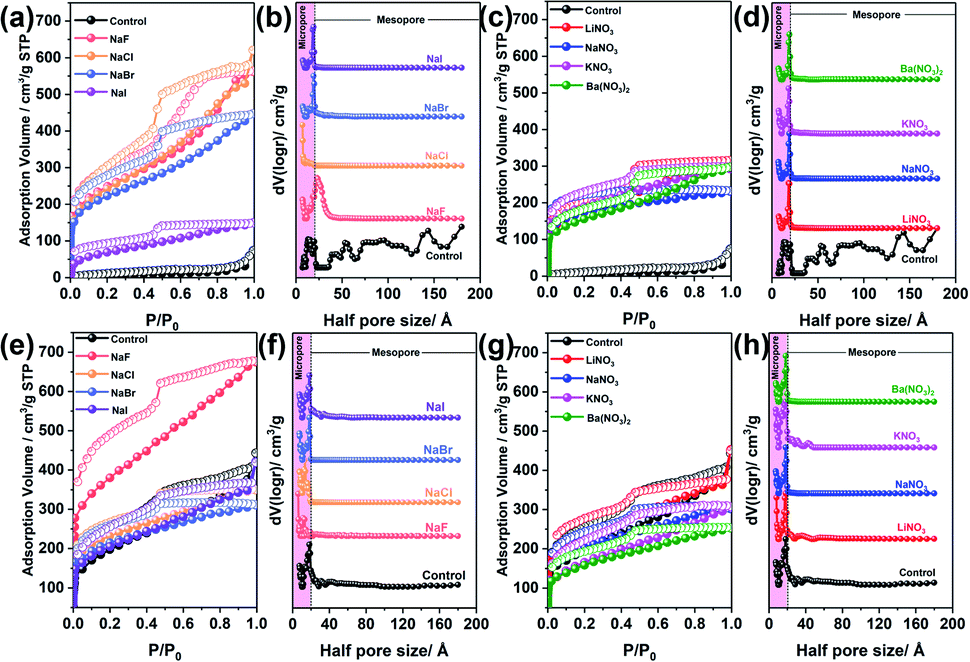 | ||
| Fig. 3 N2 adsorption and desorption isotherm and nonlocal density functional theory-pore size distribution of PTAPA ((a and b), tuned by BXJ sodium salts; (c and d), tuned by BXJ-nitrate salts) and PAPA ((e and f), tuned by BXJ sodium salts; (g and h), tuned by BXJ nitrate salts). The pink rectangular strips highlight the microporous region. | ||
To obtain a fundamental understanding of the influences of our BXJ approach on these CMPAs, we further explored the theory of Hansen solubility parameters (HSPs)32 in the context of our synthetic approach. According to the HSP theory (see detailed guide in Section S2 in ESI†), solvents are designated as “good solvents” for a polymer when the difference in their total solubility parameter |δT| < 1 (where |δT| = |δT, solvent − δT, polymer|). Solvents with a δT difference of 1 < |δT| < 3 would be classified as “intermediate solvent”, and would still be able to perform well during a polymerization. When the |δT| > 3, solvents are indicated as “poor solvents” for the resulting polymer and is not suitable for polymer synthesis.33,34 As a general guide, the early phase separation of our polymer networks (and the resulting large average diameter pores and low BET surface areas) could result from weak matching of δT, solvent with that of the growing polymer. Solvents with matching HSPs and good compatibilities (difference in |δT| < 1) with the (growing) polymer networks would, in contrast, contribute to a much later-stage phase separation, yielding polymers with uniform PSD and rich micropores (see our recent publication and the ESI† for detailed discussion).22,33,34 With this background in mind, we calculated35 the HSPs of PAPA and PTAPA, with a detailed step-by-step methodology and results presented in the ESI† (Section S3, results shown in Fig. S10, Tables S2 and S3†). The estimated total solubility parameter (δT) of PTAPA and PAPA, listed alongside that of PTPA and literature values of solvents, are summarized in Table 3.
Results in Table 3 clearly shows the reason for the BXJ tuning performance on different CMPAs. For PTAPA, the |δT| between THF and PTAPA was 3.9, indicating THF is a poor solvent for PTAPA synthesis. This explains why a low surface area of 28 m2 g−1 was acquired in THF without any salt addition. However, salts contribute to the permanent dipole interactions (δP) and the hydrogen-bonding interactions (δH) of the solvent, resulting in a decrease of |δT| and increase of compatibility of the solvent for polymer during the polymerization. A high surface area was therefore acquired for PTAPA after exploiting the BXJ approach. Additionally, the electronegativity of the ions decreases with the increase of salt ionic radius. As such, ions with larger radii should have less influence on the δP and δH components of the solvent (by adjusting the hydrogen bonding and polarity of the solvents), thus resulting in a less effective adjustment (and therefore matching) of the HSPs. The surface area of PTAPA could be therefore fine-tuned by the radii of the salts, as shown in Table 1. However, for PAPA, an intrinsic low δT was estimated, and the |δT| between THF and PAPA was only 1.4, suggesting that THF was an intermediate solvent suitable for PAPA to polymerize. This observation is consistent with the surface area data shown in Table 2, in that no or much less salts were needed to tune the solvent to be compatible for PAPA polymerization; a high surface area of 723 m2 g−1 could be reached by simply using THF, but then increased dramatically to almost double (1378 m2 g−1) by salt tuning.
For the case of PTAPA, a much larger δT of 23.4 was obtained, compared with the values of 22.6 and 20.9 for PTPA and PAPA, respectively. This obtained value validated the observations that significantly higher salt concentrations (1.00 mmol) were needed to acquire matching solvent quality for PTAPA, as observed in dosage-dependent results shown in Tables 1 and 2. These observations also explain the fact that it was easier to overtune the solvent for PAPA (with the lowest δT) compared with PTPA and PTAPA. These results further support the applicability of our BXJ approach and HSP mechanism for fine-tuning the surface area of CMPAs in a quantitative manner.
With this principle fully established, we expanded our investigations and switched solvents to dioxane to prepare PAPA (δT, dioxane = 20.5, and thus a very close match of (|δT| = 0.4) to the calculated value for PAPA). As shown in Fig. S11(d–f),†Tables 1 and 2, an increased surface area of 838 m2 g−1 was achieved by dioxane even when using the standard BH reaction without salts (compared with a value of 723 m2 g−1 acquired with THF as solvent under such conditions). Exploring properties with salt additions of 0.5 mmol Na2SO4 led to overtuning the HSPs of dioxane, and consequently to a decrease in the surface area of PAPA from 838 m2 g−1 to 614 m2 g−1.
We further expanded our BXJ approach to other typical CMP-forming reactions, including Sonogashira–Hagihara,12 oxidative,21 and Suzuki36 couplings, to attempt tuning and improving the surface area of the prototypical examples from such reactions. As a starting point we added 0.5 mmol of NaF (based on our experience for CMPA optimization as outlined above) to these reaction mixtures. Results, and comparisons to published data are shown in Fig. 4 and Table 4, clearly demonstrating the successful application of the BXJ approach in these reactions too. The surface area of poly(aryleneethynylene)s (CMP-1) acquired from Sonogashira–Hagihara reaction was increased from 886 to 1148 m2 g−1. In the case of PTCT, produced by oxidative coupling, the surface area increased from 981 to 1263 m2 g−1, while for p-PPF, synthesized by Suzuki coupling, a significant increase in surface area from 35 m2 g−1 to 215 m2 g−1 was observed, despite the fact that inorganic salts, such as CuI, FeCl3 and K2CO3 were already utilized as catalysts or bases in the original reactions. More interestingly, the PSDs of these products were generally narrowed, as shown in Fig. 4(b), (e) and (h). Improved values for micropore volumes were also observed: 0.58 to 0.80 cm3 g−1 (CMP-1), from 0.60 to 0.86 cm3 g−1 (PTCPT), and from 0.081 to 0.14 cm3 g−1 (p-PPF), respectively.
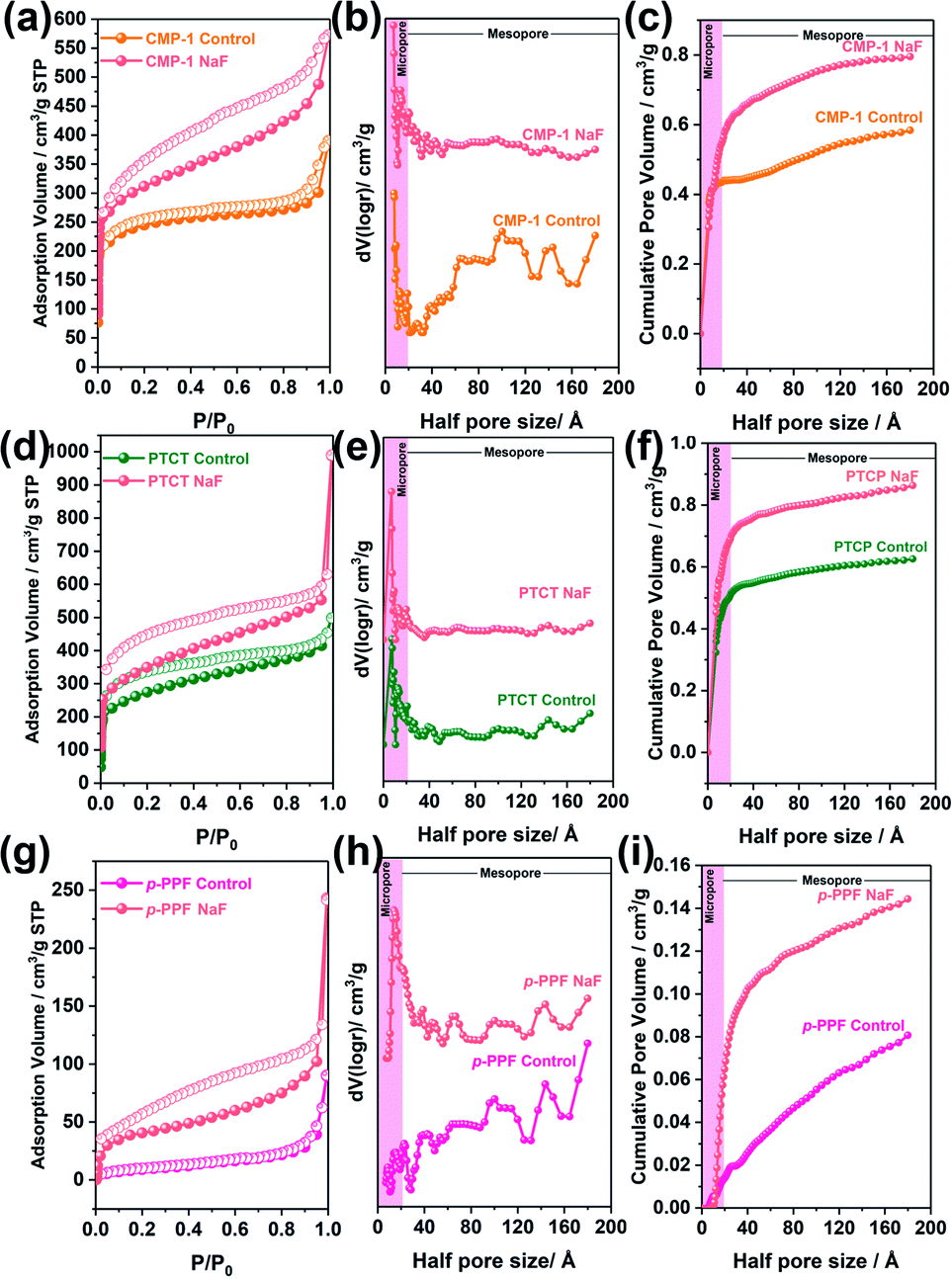 | ||
| Fig. 4 N2 adsorption and desorption isotherm, NL-DFT pore size distribution and cumulative pore volume of CMP-1 (a–c), PTCT (d–f) and p-PPF (g–i) tuned by 0.5 mmol BXJ NaF. The pink shaded areas highlight the microporous region. | ||
| Surface areaa (m2 g−1) | Total pore volumeb (cm3 g−1) | Micropore volumec (cm3 g−1) | Ultramicropore volumed (cm3 g−1) | CO2 uptake at 273 K, 1 atm (mmol g−1) | Ref. | |
|---|---|---|---|---|---|---|
| a Porosity data calculated from N2 adsorption and desorption were unavailable from the original paper published by authors.35 The authors acquired surface area from CO2 sorption isotherm due to the low porosity, and the data was not suitable for comparison. b The total pore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method. c The micropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for micropore (r < 2 nm) volume. d The ultramicropore volume calculated from the desorption branch of the N2 isotherm using the NL-DFT method for ultramicropore (r < 0.7 nm) volume. b c d | ||||||
| CMP-1 (published) | 834 | 0.47 | 0.33 | N. A. | 2.05 | 12 |
| CMA-1 (replication, without BXJ-NaF) | 886 | 0.58 | 0.44 | 0.31 | 1.10 | This work |
| CMA-1 (with BXJ-NaF) | 1148 | 0.80 | 0.56 | 0.38 | 1.62 | This work |
| PTCT (published) | 895 | 1.17 | 1.04 | N. A. | N. A. | 21 |
| PTCT (replication, without BXJ-NaF) | 981 | 0.60 | 0.52 | 0.32 | 1.94 | This work |
| PTCT (with BXJ-NaF) | 1263 | 0.86 | 0.73 | 0.46 | 2.53 | This work |
| p-PPF (published) | N. A. | N. A. | N. A. | N. A. | N. A. | 36 |
| p-PPF (replication, without BXJ-NaF) | 35 | 0.081 | 0.014 | 0 | 0.55 | This work |
| p-PPF (with BXJ-NaF) | 215 | 0.14 | 0.065 | 0 | 0.79 | This work |
These results further reinforced the adaptability of our BXJ approach to facilitate the creation of well-defined micropores and high surface area for polymers acquired by non-BH reaction routes. It is noteworthy that the porosity was enhanced by BXJ salts in a similar way as for CMPAs by influencing the phase separation and the polymerization degrees, as evidenced in the very different SEM morphologies obtained for these model networks (Fig. S12†) as well as the enhanced thermal stability (Fig. S13†) before and after salt addition. As shown for CMPAs, the molecular structures of these samples were not changed by our BXJ method either, and confirmed by FTIR study (Fig. 1, S14 and S15†).39
As the materials synthesized through the BXJ approach showed very promising microporosity, the influence on the functionality (i.e., these materials' ability to adsorb CO2 at 273 K) was investigated. Data are shown in Fig. 5, with the capacity at 1 atm summarized in Tables 1 and 2. Significant increases in CO2 sequestration capacities were acquired with our BXJ approach. CO2 uptake was dramatically improved by >300% for PTAPA from 0.75 mmol g−1 to 2.59 mmol g−1; a small enhancement from 2.41 mmol g−1 to 2.93 mmol g−1 was observed for PAPA. The influence was also clearly observed for the polymers produced by non-BH reactions, as shown in Table 4, where a maximum improvement of 47% in CO2 capacity was achieved for CMP-1.
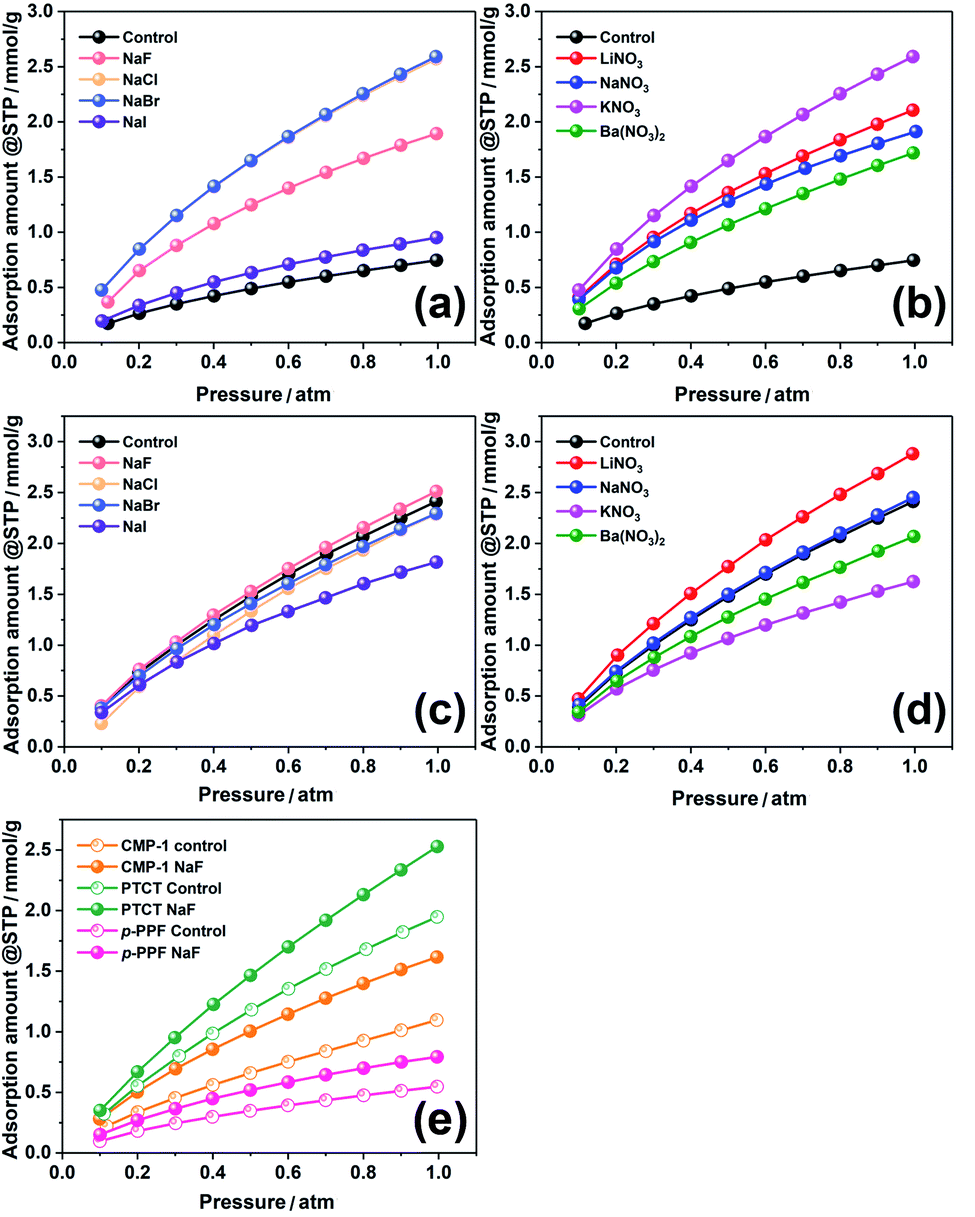 | ||
| Fig. 5 CO2 adsorption isotherm at 273 K of PTAPA (a and b), PAPA (c and d), and CMP-1, PTCT & p-PPF (e) before and after tuning by BXJ salts. | ||
However, unlike the surface area, there was no obvious trend for the CO2 uptake ability when increasing the ionic radius of anions or cations in the applied salts. The CO2 uptake shown above for PTAPA and PAPA samples seems to be unrelated to other porosity parameters such as total pore volume, micropore volume and ultramicropore volume. These observations further support the conclusion that the CO2 adsorption of such porous systems is related to a number of factors, not only surface area.37,38 By comparing the CO2 uptake ability with other polymers in Table S4,† our CMPAs, powered by the BXJ approach, exhibit very attractive CO2 sequestration capabilities, owing to the combination of their rich micropores, high surface area and significant N content.
Conclusions
Full synthetic control over the surface area and porosity of BH-produced CMPAs through our BXJ approach is reported in this paper. Using HSP theory, we showed tuneable surface area and porosity for two new CMPAs by adjusting the ionic radius of the used salts, following on our initial work with PTPA. Surface areas were dramatically increased from 28 m2 g−1 to 901 m2 g−1 for PTAPA and from 723 m2 g−1 to 1378 m2 g−1 for PAPA. With a clear understanding of the factors influencing the materials properties established, the general applicability of our approach was extended to other non-BH coupling reactions for the first time in a detailed fashion. The BXJ approach shows high adaptability for typical metal-catalyzed reactions used for the preparation of porous materials, increasing the surface area from 886 to 1148 m2 g−1, from 981 to 1263 m2 g−1, and from 35 to 215 m2 g−1 for CMP-1, PTCT, p-PPF, respectively. More importantly, the BXJ approach also allows the broad PSD of the polymers to be tuned to approach those found for COFs and MOFs. With the porosity optimized, CO2 sequestration capacities were dramatically improved by more than 300% in some cases. Salt tuning is therefore an important first step to increase our understanding and ability to tune the HSPs of solvents to adjust polymer properties and function. Our BXJ approach not only opens a facile and simple avenue for optimizing CMP properties and functionality, but also provides a rational and generalized route towards the design of novel porous materials. We are currently exploring further effective methodologies to establish design rules to optimize and tune HSPs with the aim to obtain porous materials with fully tuneable functionality for wide application.Experimental section
Chemicals
Tris(4-aminophenyl)amine, 4,4′-diaminodiphenyl sulfide, 1,3,5-triphenyl tribromide, benzene-1,4-diboronic acid, 1,4-diiodobenzene, tris(4-bromophenyl)amine, 1,3,5-triethynylbenzene, 4,6-tri(9H-carbazol-9-yl)-1,3,5-triazine (TCT), bis(dibenzylideneacetone)palladium(0) (Pd(dba)2), tetrakis(triphenylphosphine)-palladium(0), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos, 97%), sodium tert-butoxide (NaOtBu, 97%), copper iodide (CuI), potassium carbonate (K2CO3), Et3N, FeCl3, (BXJ-salt) NaF, NaCl, NaBr, NaI, LiNO3, NaNO3, KNO3, Ba(NO3)2 and Na2SO4 were of AR grades, which were purchased from Sigma-Aldrich, UK and were used as received.Synthesis of conjugated microporous polyanilines without salts (control)
A Schlenk tube was charged with tris(4-bromophenyl)amine (0.5 mmol) as core, and the following linkers: 4,4′-diaminodiphenyl sulfide (0.5 mmol to achieve a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 core to linker ratio, yielding PTAPA), or, tris(4-aminophenyl)amine (0.5 mmol to achieve a 1:1 core to linker ratio, yielding PAPA), XPhos (0.045 mmol, 4.5 mol%), Pd(dba)2 (0.03 mmol, 3 mol%) and NaOtBu (3.5 mmol, 7 equiv.). Note that a 1:1 core to linker ratio led to higher surfaces area for our CMPAs. However, when we used a 1:1.5 stoichiometry of core to linker, low surface area CMPAs were acquired, even though salts were added. This phenomenon was also observed in the synthesis of CMP-1&2, which are structurally related to our CMPAs by using 1,3,5-triethynylbenzene as the core and 1,4-diiodobenzene/4,4′-diiododiphenyl as the linker. We inferred that excessive bromo functionality would promote linkage of Br–NH2 through the BH coupling, leading to networks with higher surface area of CMPAs. Anhydrous THF (30 mL) was thereafter added under a nitrogen atmosphere. The reaction mixture was then heated under stirring and kept at 65 °C. After 48 hours, the mixture was cooled to room temperature and the product collected by centrifugation. The products were purified with CHCl3, ethanol, methanol, and boiling water (200 mL each), respectively, to remove the catalyst, impurities, and oligomers. The products were further purified by a Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively, before drying under vacuum at 60 °C for 24 h. The average yields of PTAPA and PAPA were 56.87% and 95.24%, respectively. We confirmed that the results optimized by BXJ method were highly reproducible, with similar results for BET surface area and porosity when same synthetic procedures were repeated. For example, preparation of PTAPA (tuned by Ba(NO3)2) was performed 3 times, and yielded the following values for surface area and porosity shown in Fig. S16 and Table S5.† The very similar surface areas, pore volumes and pore size distributions clearly confirmed the high reproducibility of our BXJ–BH method for CMPA synthesis.
:1 core to linker ratio, yielding PTAPA), or, tris(4-aminophenyl)amine (0.5 mmol to achieve a 1:1 core to linker ratio, yielding PAPA), XPhos (0.045 mmol, 4.5 mol%), Pd(dba)2 (0.03 mmol, 3 mol%) and NaOtBu (3.5 mmol, 7 equiv.). Note that a 1:1 core to linker ratio led to higher surfaces area for our CMPAs. However, when we used a 1:1.5 stoichiometry of core to linker, low surface area CMPAs were acquired, even though salts were added. This phenomenon was also observed in the synthesis of CMP-1&2, which are structurally related to our CMPAs by using 1,3,5-triethynylbenzene as the core and 1,4-diiodobenzene/4,4′-diiododiphenyl as the linker. We inferred that excessive bromo functionality would promote linkage of Br–NH2 through the BH coupling, leading to networks with higher surface area of CMPAs. Anhydrous THF (30 mL) was thereafter added under a nitrogen atmosphere. The reaction mixture was then heated under stirring and kept at 65 °C. After 48 hours, the mixture was cooled to room temperature and the product collected by centrifugation. The products were purified with CHCl3, ethanol, methanol, and boiling water (200 mL each), respectively, to remove the catalyst, impurities, and oligomers. The products were further purified by a Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively, before drying under vacuum at 60 °C for 24 h. The average yields of PTAPA and PAPA were 56.87% and 95.24%, respectively. We confirmed that the results optimized by BXJ method were highly reproducible, with similar results for BET surface area and porosity when same synthetic procedures were repeated. For example, preparation of PTAPA (tuned by Ba(NO3)2) was performed 3 times, and yielded the following values for surface area and porosity shown in Fig. S16 and Table S5.† The very similar surface areas, pore volumes and pore size distributions clearly confirmed the high reproducibility of our BXJ–BH method for CMPA synthesis.
Synthesis of conjugated microporous polyanilines with BXJ approach
A Schlenk tube was charged with tris(4-bromophenyl)amine (0.5 mmol) as core, and the following linkers: 4,4′-diaminodiphenyl sulfide (0.5 mmol to achieve a 1:1 core to linker ratio, yielding PTAPA), or, tris(4-aminophenyl)amine (0.5 mmol to achieve a 1:1 core to linker ratio, yielding PAPA), XPhos (0.045 mmol, 4.5 mol%), Pd(dba)2 (0.03 mmol, 3 mol%), NaOtBu (3.5 mmol, 7 equiv.) and salts (0.75 mmol for PTAPA and 0.5 mmol for PAPA of NaF, NaCl, NaBr, NaI, LiNO3, NaNO3, KNO3 or Ba(NO3)2; for salt dosage investigation, 0.33 mmol–2.00 mmol of Na2SO4 were applied). Anhydrous THF (or toluene/dioxane for solvent influence study) (30 mL) was thereafter added under the nitrogen atmosphere. The reaction mixture was then heated with a hotplate under stirring at 65 °C. After 48 hours, the mixture was cooled to room temperature. The products were then purified with CHCl3, ethanol, methanol, and boiling water (200 mL each), to remove the catalyst, impurities, and oligomers. The products were further purified by a Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively, before drying under vacuum at 60 °C for 24 h. The average yields of PTAPA and PAPA after salt tuning reached over 99.0%.
Synthesis of model CMPs by non-BH reactions
These CMPs were synthesized according to the published procedures (including Sonogashira–Hagihara,12 oxidative coupling21 and Suzuki36), and the BXJ-assisted polymers were obtained by an additional dosing of NaF (0.5 mmol).22 Specifically, for CMP-1, 1,3,5-triethynylbenzene (2.0 mmol), 1,4-diiodobenzene (2.0 mmol), tetrakis-(triphenylphosphine)palladium (100 mg), copper iodide (30 mg) with/without NaF (0.5 mmol) were placed in the mixture of toluene (2.5 mL) and Et3N (2.5 mL) under a N2 atmosphere in a Schlenk tube. The mixture was then heated to 80 °C for 72 h, after which it was cooled to RT and the resulting polymer were filtered and washed with chloroform, methanol and water (200 mL each), followed by a 72 h Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively, before drying under vacuum at 60 °C for 24 h. For PTCT, the carbazole-substituted monomer TCT (1 mmol, 1 equiv.) with/without BXJ-NaF (0.5 mmol) were placed in CHCl3 (50 mL). Anhydrous FeCl3 (3 equiv. per carbazole unit) dissolved in CH3NO2 (20 mL) was then added into the flash charged with monomer solution for a homogenous reaction. The mixture was vigorously stirred overnight, and the resulted polymer was filtered and washed with chloroform, methanol and water (200 mL each), followed by a 72 h Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively, and, being dried under vacuum at 60 °C for 24 h. For p-PPF, 1,3,5-triphenyl tribromide (0.46 mmol), 1,4-benzene diboronic acid (0.92 mmol) with/without BXJ-NaF (0.5 mmol) were dissolved in DMF (20 mL) after been degassed by four freeze–pump–thaw cycles. After which, K2CO3 (2 mmol) in water (4 mL) and tetrakis(triphenylphosphine)-palladium(0) (70 mg, 60.6 mmol) were added to this mixture, which was followed by another degassing step and purged with N2 and stirred at 150 °C in a Schlenk tube for 36 h. The mixture was cooled to RT and the resulted polymer was filtered and washed with chloroform, methanol, and water (200 mL each), followed by a 72 h Soxhlet extraction with methanol (24 h), THF (24 h) and chloroform (24 h), respectively.Characterization
A PerkinElmer Spectrum 100 spectrometer was used to acquire the FTIR spectra in the region between 4000 and 400 cm−1. The SS 13C CP/MAS NMR spectra were collected on a Varian VNMRS-600 spectrometer using a spin rate of 6800 Hz. A Quantachrome Quadrasorb instrument was applied for N2 adsorption/desorption isotherms (NAD) and CO2 adsorption study after degassing the samples under high vacuum at 150 °C for 5 h. The specific surface areas were calculation from the Brunauer–Emmett–Teller (BET) model using the adsorption branches of the N2 isotherms in the low pressure range from 0.05 to 0.20 at 77 K. The pore size distribution was calculated from the desorption branch of the N2 isotherms using the nonlocal density functional theory (NL-DFT). Scanning electron microscope (SEM) images were acquired on a JEOL 5600LV SEM microscope. Powder X-ray diffraction (XRD) patterns were obtained with a Bruker D8 Advance diffractometer using Cu Kα radiation (2θ = 5°–80°, 40 kV, 30 Ma). Transmission electron microscopy (TEM) images were recorded on a JEM model 2100 electron microscope.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. C. acknowledges the supports of Science and Technology Project of Fujian Educational Committee (Grant No. JAT190051), Fuzhou University Testing Fund of precious apparatus (Grant No. 2020T008), Research Initiation Funding of Fuzhou University (Grant No. GXRC-19051) and the Chinese Scholarship Council (CSC); T. Q. acknowledges National Natural Science Foundation of China (Grant No. 21878054); W. Y. acknowledges National Natural Science Foundation of China (Grant No. 51978569).Notes and references
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 RSC.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- W.-Y. Gao, H. Wu, K. Leng, Y. Sun and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 5472–5476 CrossRef CAS.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS.
- S. Hong, M. R. Rohman, J. Jia, Y. Kim, D. Moon, Y. Kim, Y. H. Ko, E. Lee and K. Kim, Angew. Chem., Int. Ed., 2015, 54, 13241–13244 CrossRef CAS.
- J. Weber, Q. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS.
- T. Fukushima, S. Horike, Y. Inubushi, K. Nakagawa, Y. Kubota, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 4820–4824 CrossRef CAS.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS.
- Y. Liao, S. Cai, S. Huang, X. Wang and C. F. J. Faul, Macromol. Rapid Commun., 2014, 35, 1833–1839 CrossRef CAS.
- Y. Liao, J. Weber, B. M. Mills, Z. Ren and C. F. J. Faul, Macromolecules, 2016, 49, 6322–6333 CrossRef CAS.
- Y. Liao, J. Weber and C. F. J. Faul, Chem. Commun., 2014, 50, 8002–8005 RSC.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034–2048 CrossRef CAS.
- H. Wang, Z. Cheng, Y. Liao, J. Li, J. Weber, A. Thomas and C. F. J. Faul, Chem. Mater., 2017, 29, 4885–4893 CrossRef CAS.
- J. Chen, W. Yan, E. J. Townsend, J. Feng, L. Pan, V. Del Angel Hernandez and C. F. J. Faul, Angew. Chem., Int. Ed., 2019, 58, 11715–11719 CrossRef CAS.
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem., Int. Ed., 1995, 34, 1348–1350 CrossRef CAS.
- J. Chen, Y. Wang, C. Ye, W. Lyu, J. Zhu, W. Yan and T. Qiu, ACS Appl. Mater. Interfaces, 2020, 12, 28681–28691 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., 2007, 119, 8728–8732 CrossRef.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- Z. Guo, H. Xu, S. Su, J. Cai, S. Dang, S. Xiang, G. Qian, H. Zhang, M. O'Keeffe and B. Chen, Chem. Commun., 2011, 47, 5551–5553 RSC.
- J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531–8538 CrossRef CAS.
- L. Pan, Z. Liu, M. Tian, B. C. Schroeder, A. E. Aliev and C. F. J. Faul, ACS Appl. Mater. Interfaces, 2019, 11, 48352–48362 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS.
- G. K. C. D. J. Hansen, C. Panayiotou, L. Willliams, T. Poulsen, H. Priebe and P. Redelius, Hansen Solubility Parameters, CRC Press, Boca Raton, 2007 Search PubMed.
- F. S. Macintyre and D. C. Sherrington, Macromolecules, 2004, 37, 7628–7636 CrossRef CAS.
- W. Kangwansupamonkon, S. Damronglerd and S. Kiatkamjornwong, J. Appl. Polym. Sci., 2002, 85, 654–669 CrossRef CAS.
- S. Ata, T. Mizuno, A. Nishizawa, C. Subramaniam, D. N. Futaba and K. Hata, Sci. Rep., 2014, 4, 7232 CrossRef.
- K. V. Rao, S. Mohapatra, C. Kulkarni, T. K. Maji and S. J. George, J. Mater. Chem., 2011, 21, 12958–12963 RSC.
- H. Li, X. Ding, Y.-C. Zhao and B.-H. Han, Poly, 2016, 89, 112–118 CrossRef CAS.
- E.-J. Wang, Z.-Y. Sui, Y.-N. Sun, Z. Ma and B.-H. Han, Langmuir, 2018, 34, 6358–6366 CrossRef CAS.
- Slight difference was caused by the moisture and salts trapped in the micropores.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05563h |
| This journal is © The Royal Society of Chemistry 2020 |