
Understanding of A-site deficiency in layered perovskites: promotion of dual reaction kinetics for water oxidation and oxygen reduction in protonic ceramic electrochemical cells†
Wei
Tang
 ab,
Hanping
Ding
*a,
Wenjuan
Bian
ab,
Wei
Wu
a,
Wenyuan
Li
c,
Xingbo
Liu
c,
Joshua Y.
Gomez
ab,
Clarita Y.
Regalado Vera
ab,
Meng
Zhou
*b and
Dong
Ding
*a
ab,
Hanping
Ding
*a,
Wenjuan
Bian
ab,
Wei
Wu
a,
Wenyuan
Li
c,
Xingbo
Liu
c,
Joshua Y.
Gomez
ab,
Clarita Y.
Regalado Vera
ab,
Meng
Zhou
*b and
Dong
Ding
*a
aIdaho National Laboratory, Idaho Falls, ID 83402, USA. E-mail: hanping.ding@inl.gov; dong.ding@inl.gov
bChemical & Materials Engineering, New Mexico State University, Las Cruces, NM 88003, USA. E-mail: mzhou@nmsu.edu
cMechanical & Aerospace Engineering Department, West Virginia University, Morgantown, WV 26506, USA
First published on 25th June 2020
Abstract
Protonic ceramic electrochemical cells (PCECs) are promising solid-state energy conversion devices which enable the conversion of energy between electricity and hydrogen at intermediate temperatures. Rapid conversion between chemical and electrical energy via PCEC technology will assist in overcoming grand challenges in energy storage. To achieve highly efficient reversible operation between hydrogen production and electricity generation, boosting water-oxidation and oxygen reduction activities of the oxygen electrode while maintaining the durable operation is one of the early-stage technical opportunities. In this study, an A-site deficient layered perovskite (PrBa0.8Ca0.2)0.95Co2O6−δ has been developed as an oxygen electrode for a PCEC which presents superior electrochemical performances. The electrolysis current density reached as high as −0.72 A cm−2 at 1.3 V, and a peak power density of 0.540 W cm−2 was obtained at 600 °C in electrolysis and fuel cell mode, respectively. The PCEC with the new electrode shows good durability under practical operating conditions for 160 hours in both operating modes with no observable degradation. The reversibility between the electrolysis and fuel cell mode is also successfully demonstrated.
Introduction
Although renewable energy harvesting is becoming more accessible, the intermittency and fluctuation of the time and site-specific wind and solar energies can restrict the electricity supplied to the grid.1 Storing the energy for peak shaving can be a feasible approach to convert excess electricity into chemical fuels that can be reversed to release chemical energy.2–4 Hydrogen is a dense energy carrier which has been widely used for various applications such as fuel cells and the chemical industry.5,6 To efficiently store electrical energy, solid oxide cells (SOCs) have been developed to convert energy between electricity and hydrogen fuel, which presents several advantages over other approaches such as using low-temperature fuel cells and electrolyzers.7–9 In the past decade, there has been significant progress in material development, electrochemical measurement, stability improvement, and stack-level studies. However, wide-spread adoption of this technology has not been achieved, due in part to the high operating temperatures (700–1000 °C) required by conventional oxygen-ion conducting electrolytes.10–13 For example, yttrium-stabilized-zirconia still poses a series of problems such as material incompatibility between compartments, relatively high temperature, fast degradation and expensive interconnects used in stacks.14,15 In addition, nickel particle coarsening and oxygen bubble generation are also identified as degradation mechanisms which contribute to electrode deterioration in electrolysis operation.16,17Protonic ceramic electrochemical cells (PCECs) are an emerging technology that produce hydrogen and generate electricity within a single device in a reduced temperature range (400–600 °C), in which a solid-state electrolyte has high proton conductivity due to lower activation energy for proton conduction.18–22 Firstly, comparing PCEC with conventional high-temperature SOCs, the intermediate temperature operation can greatly reduce the manufacturing cost as stainless steel may be utilized as interconnects and can also significantly improve the long-term operation stability.23,24 Secondly, dry hydrogen can be generated on the nickel-electrode side which avoids further gas separation process while nickel oxidation is also mitigated in fuel cell mode.25,26 In addition, the delamination of the oxygen-electrode/electrolyte interface due to high current density can be eliminated because the oxygen evolution reaction occurs on the entire electrode surface and not on the electrode/electrolyte interface particularly when a mixed proton and electron conducting electrode is utilized.27 In the past few years, there has been progressive development in material selection and improvements in the electrochemical performance of PCECs, which demonstrates the fast hydrogen production and high power density at reduced temperatures.28 However, some critical challenges still remain in terms of material activity and stability, one of which is the electrode activity at intermediate temperatures when current electrode materials particularly oxygen electrodes are relatively sluggish for the water oxidation reaction (WOR) and oxygen reduction reaction (ORR), contributing a large fraction of polarization to the total cell resistance.29,30 At present, oxygen electrodes used in PCECs are the cathodes from solid oxide fuel cells without complete consideration of the required material parameters. When the operating temperature is decreased to 500–600 °C, the oxygen electrode still contributes a great polarization resistance to both fuel cell and electrolysis operation. In addition, the chemical stability of the electrode and electrolyte/electrode bonding strength, particularly at high steam concentrations, can also challenge the performance durability. Therefore, the development of a highly active oxygen electrode with abundant active sites for catalytic reactions is essential for improving electrochemical performance with high stability for operation at high steam concentrations.31
For high-temperature SOCs, the common strategy for the selection of the oxygen electrode is to use mixed ion and electron conducting oxides which possess good electrical conductivity for extending the active triple-phase boundary area for the reaction.32–34 One of the promising electrodes is a layered perovskite structure with fast surface kinetics and bulk diffusion, e.g. PrBa0.5Sr0.5Co2−xFexO5+δ (PBSCF) which shows potential for being applied for fuel cell or electrolysis operation at reduced temperatures.35 For layered perovskite type oxides with a composition formula of LnBaCo2O5+δ (Ln = Gd, Pr, Y, La, etc.),36–38 the structure can be written as A′A′′B2O6 by doubling the unit cell of ABO3 perovskites, and consists of repetitive layers of [A′Oδ]–[BO2]–[A′′O]–[BO2], where A′, A′′ and B sites are Pr, Ba and Co respectively.39 Such ordered A cations confine and localize oxygen vacancies into A′Oδ or rare-earth layers, which helps to enhance the high concentration of disordered oxygen vacancies, fast oxide ion mobility in the layers and the high electronic conductivity.40,41 Recently, Liu et al. reported a layered perovskite PrBa0.8Ca0.2Co2O6−δ (PBCC) with excellent oxygen reduction activity and high tolerance against high CO2 concentration for solid oxide fuel cells with oxide-ion conducting doped ceria electrolyte,42 which has demonstrated the potential of this material for being utilized in PCECs. To further enhance the catalytic activity towards the WOR and ORR, increasing the intrinsic oxygen vacancy concentration by introducing cation deficiencies is a method to obtain higher oxygen diffusivity and surface defects for the reaction.43 There have been several studies to prove that the introduction of A-site deficiencies in perovskites can significantly enhance the diffusion of lattice oxygen and hence improve catalytic activity.44,45 For example, an A-site deficient (La0.6Sr0.3)CrO3 anode material shows 4 times higher peak power density than normal (La0.7Sr0.3)CrO3 at 800 °C due to the promoted ion diffusion.46 The electrochemical ORR activity has also been promoted for a cation deficient Sr0.95Co0.8Nb0.1Ta0.1O3−δ cathode which displayed about 1.5 times higher peak power density than stoichiometric SrCo0.8Nb0.1Ta0.1O3−δ because of its higher oxygen vacancy concentration.47 Another study shows that the introduction of both Pr3+ and Ba2+ deficiency into a PrBaCo2O6−δ oxygen electrode could decrease polarization resistances and activation energy and hence the electrochemical performance is significantly improved.48 Inspired by the role of A-site deficiency in changing oxygen non-stoichiometry for improving activity, in this work, a novel layered perovskite structure with A-site deficiency in PBCC, namely (PrBa0.8Ca0.2)0.95Co2O6−δ (PBCC95), was synthesized and studied as an oxygen electrode material for PCECs. When this material is integrated into PCECs, the cell demonstrates superior electrochemical performance in both fuel cell and electrolysis modes. The effects of A-site deficiency on the oxygen vacancy concentration, electrode polarization resistance and cell performance were evaluated to investigate the role of the created A-site deficiency in improving the performance and long-term stability in PCEC operation. In addition, the excellent reversible operation was also successfully demonstrated, which represents a good dynamic mode transition enabled by this new electrode with fast adaption to both the WOR and ORR at reduced temperatures.
Experimental
Materials
Praseodymium(III) nitrate hexahydrate (Pr(NO3)3·6H2O, 99.9% purity), barium nitrate (Ba(NO3)2, ≥99% purity), and calcium nitrate tetrahydrate (Ca(NO3)2·4H2O, >99% purity) were purchased from Sigma-Aldrich. Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99% purity), citric acid (C6H8O7, 99.5% purity), and glycine (NH2CH2CO2H, 99% purity) were purchased from Alfa Aesar.Synthesis of PBCC and PBCC95 electrode powder
Double-layered perovskite PBCC and PBCC95 powders were synthesized by the wet-chemistry self-combustion method. Stoichiometric amounts of metal nitrates were mixed with deionized water to form a clear precursor solution with a concentration of ∼0.05 mol L−1. Glycine and citric acid functioning as the complexing agent and fuel for the following combustion were added in the precursor nitrate solution in a mole ratio of glycine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid:cations = 2:1:1. The precursor solution was then heated on a hot plate with magnetic stirring to evaporate water. The water was totally evaporated to form a gel that was further heated to about 350 °C. Finally, the gel underwent an auto-ignition process, which produced a voluminous powder ash. The formed ash was then fired at 1100 °C for 4 h to obtain a crystallized double-layer perovskite phase.
:citric acid:cations = 2:1:1. The precursor solution was then heated on a hot plate with magnetic stirring to evaporate water. The water was totally evaporated to form a gel that was further heated to about 350 °C. Finally, the gel underwent an auto-ignition process, which produced a voluminous powder ash. The formed ash was then fired at 1100 °C for 4 h to obtain a crystallized double-layer perovskite phase.
Characterization
The phase structure of PBCC and A-site deficient PBCC95 was determined by X-ray powder diffraction (XRD, 2008 Bruker D8). The microstructure and morphology of the PBCC95 powder were examined using a scanning electron microscope (SEM, JEOL 6700F) and transmission electron microscope (TEM, JEOL 4000 EX). Energy-dispersive X-ray spectroscopy (EDX) mapping was used to investigate element distribution. Selected area electron diffraction (SEAD) patterns were applied to demonstrate the crystal nature. Thermogravimetric analysis (TGA, Q500 TA instruments) was carried out to study the weight loss for determining the behaviour of oxygen deficiency. 50 mg of PBCC95 powder was placed on an alumina holder and then heated in air from room temperature to 650 °C with a ramping rate of 10 °C min−1. For the oxygen temperature-programmed desorption (O2-TPD) experiment, 0.1 g powder was pre-treated at 500 °C in oxygen for 2 h. After cooling down, Ar gas was purged for 1 h to remove the oxygen residues on the surface. Then the desorbed oxygen species were analysed in situ with mass spectroscopy as the reactor was heated to 850 °C with a ramping rate of 4 °C min−1.Fabrication of symmetrical cells and PCECs
To prepare the symmetric cells, the BaCe0.4Zr0.4Y0.1Yb0.1O3−δ (BCZYYb4411) electrolyte pellets were first fabricated by uniaxially pressing the powders to form green pellets for high-temperature sintering at 1450 °C for 4 h to densify them. The PBCC and PBCC95 electrode slurries were prepared by dispersing the respective powders in a binder (butyl carbitol acetate) and ethanol with ball-milling for 10 min. The slurry was then mixed and deformed into a thick sticky slurry by using a planetary centrifugal mixer (Thinky mixer ARE-310). The electrolyte pellet was 0.5 inches in diameter. The 0.25 inch working electrode and counter electrode were symmetrically printed on two sides of a BCZYYb4411 electrolyte pellet in the centre. Silver paste was used as the reference electrode on the side of the working electrode with no connection to the working electrode. Silver wires were used to connect to an electrochemical workstation (Solartron 1400). The configuration of the 3-electrode symmetrical cell was shown in Fig. S1.† The electrode slurry was respectively screen painted onto both sides of the electrolyte pellet for firing at 1000 °C for 4 h to form the symmetric cell with the porous PBCC electrode with an active area of 0.178 cm2. For the preparation of full cells, the NiO/BCZYYb4411 hydrogen electrode and electrolyte green tapes were first fabricated by the tape casting method. The combined layers of the electrode support and the electrolyte membrane were laminated at 70 °C overnight to form the half-cell green tapes. The cells were cut out of the tapes with a diameter of 0.5 inches and pre-sintered at 920 °C to remove the organic solvents. Finally, the cells were sintered at 1450 °C for 6 h to densify the electrolyte. Then the PBCC (or PBCC95) electrode was sintered onto the electrolyte by firing it at 1000 °C for 4 h to obtain the final full cells with an active area of 0.178 cm2.Electrochemical performance measurement and post-test examination
The area specific resistances (ASRs) of PBCC and PBCC95 electrodes at 500–600 °C in symmetrical cells with a three-electrode configuration were measured under open circuit voltage (OCV) conditions at a bias current of −0.1 A cm−2 to study the activity in both fuel cell and electrolysis modes. The effect of current bias on the electrode resistance was studied by measuring impedance at 600 °C when the bias on the sample in oxygen was increased from 0 to −0.2 A cm−2. To investigate the ASR dependency on the oxygen partial pressure, the impedance was also measured at 600 °C when the gas condition was changed from 0.05 to 1.0 atm for oxygen partial pressure (PO2). The electrode stability was examined at a 20% steam concentration for 200 h while the symmetric cell was applied with a current density of −0.05 A cm−2 at 600 °C. For the full cell testing, the as-fabricated button cell was sealed with a ceramic sealant (Ceramabond 552) onto the testing fixture and the electrochemical performances in both electrolysis and fuel cell modes were collected by using an electrochemical work station (Solartron 1400). Electrochemical impedance spectra (EIS) was measured at OCV and 1.3 V with a frequency from 105 to 0.1 Hz and AC amplitude of 20 mV. Reversible operation between the electrolysis and fuel cell mode at 550 °C was investigated by switching the voltage to enable the dynamic operation. The long-time stability was examined for 160 h when the temperature was fixed at 500 °C with an electrolysis voltage of 1.3 V and fuel cell voltage of 0.7 V. After the electrochemical testing, the microstructure of the button cells was observed using a SEM in back scattering electron mode.Results and discussion
Characterization of PBCC95
Ordered double perovskite AA′B2O6−δ is featured with an A-site ordered in alternating octahedra along the c-axis. As shown in Fig. 1a, the Pr and Ba/Ca ions in PBCC alternately occupy the interstitial sites surrounded by CoO2 octahedra. When the A-site is deficient upon adjusting the A/A′ stoichiometry to 0.95 during synthesis to obtain PBCC95, more oxygen vacancies can be generated due to charge neutrality besides thermally activated vacancies. These extra oxygen vacancies are able to transport in the PrO layer with fast diffusion kinetics due to the ordered A-site structure, which has been extensively studied in other layered perovskites.49 To confirm the phase structure of PBCC and PBCC95, the XRD patterns were obtained (Fig. 1b) and show a pure layered perovskite belonging to space group P4/mmm which is consistent with the literature.32,50 There were no peaks shifted or corresponding to any impurities, indicating that a 5% A-site deficiency does not result in the change of the primary lattice structure. The oxygen non-stoichiometry (δ) of PBCC and PBCC95 at room temperature is determined by iodometric titration to be 0.05 and 0.3, respectively, indicating the large increase of oxygen vacancies (Fig. 1c). The oxygen non-stoichiometry at operating temperatures is calculated by obtaining the oxygen loss from the TGA result measured in air when the temperature is increased up to 650 °C (Fig. S2†). The results show that the δ value of PBCC and PBCC95 increases from 0.098 and 0.342 at 500 °C to 0.142 and 0.386 at 600 °C, respectively. Both PBCC and PBCC95 have an increased oxygen vacancy concentration at elevated temperatures and the A-site deficient PBCC95 electrode possesses higher non-stoichiometry, which facilitates the enhancement of electro-activity towards the WOR and ORR.51,52 Furthermore, the resulting catalytic activity due to the enhanced vacancy concentration has been examined by O2-TPD to analyze the activity of respective oxygen species (chemisorbed/lattice oxygen atoms) during the dissociation process in the electrode surface and bulk. Fig. 1d shows the spectrum of oxygen desorption in a temperature range of 100–850 °C. The desorption signal at temperatures below 400 °C (denoted as α-O) is related to the chemisorption of oxygen (O2− or O−) due to weak bonding with the surface.53 The second signal at a temperature higher than 400 °C is ascribed to the loss of lattice oxygen (denoted as β-O).54 The α-O peak of PBCC95 is observed at 268 °C, which is 29 °C lower than that of PBCC (297 °C), and PBCC95 also shows a lower β-O desorption peak at 764 °C compared to PBCC (803 °C). The lower oxygen desorption temperature indicates the higher oxygen diffusivity and surface activity in the PBCC95 electrode which can be obtained to improve the catalytic activity.51,55,56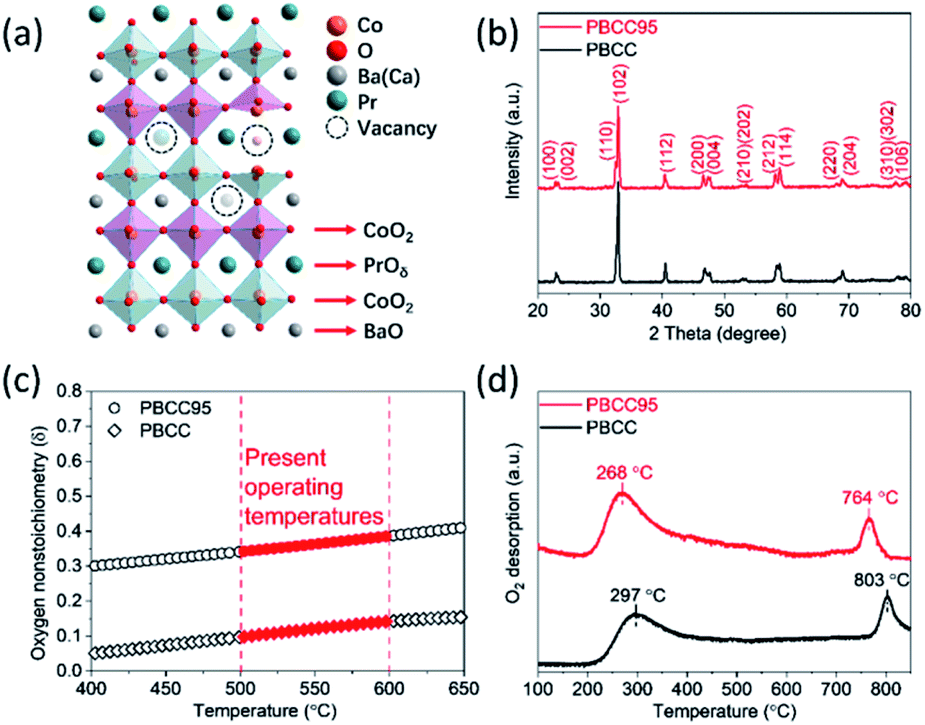 | ||
| Fig. 1 Material characterization of PBCC and PBCC95 electrodes. (a) Schematic of the crystal structure for the PBCC double perovskite with A-site deficiency; (b) XRD patterns of PBCC and PBCC95; (c) the oxygen non-stoichiometry of PBCC and PBCC95 at temperatures between 400 and 650 °C; and (d) O2-TPD profiles of PBCC and PBCC95. | ||
The morphology, lattice structure and element distribution of the synthesized PBCC95 electrode are shown in Fig. 2. After calcination at 1100 °C, the fine powders have a diameter less than 1 μm. The high resolution TEM (HR-TEM) image shows the good crystallization nature with an interplanar lattice distance of 0.383 nm (Fig. 2b), corresponding to the (002) crystal plane, which is about a half of the distance between PrO or BaO planes (0.762 nm).42 The inset shown in Fig. 2b is the SAED pattern of a single PBCC95 particle, which shows a single crystal diffraction character. The distance of the spots is measured and calculated. It is found that the diffraction pattern corresponds well to PBCC95 and this result is in good agreement with the above XRD.
 | ||
| Fig. 2 Morphology and lattice structure of the PBCC95 electrode. (a) SEM image of PBCC95 powder; (b) HR-TEM image of a PBCC95 particle; (inset) SEAD pattern of PBCC95; and (c) EDX-mapping of a PBCC95 particle. | ||
This result indicates that introducing a minor A-site deficiency has no observable effect on the double perovskite structure. The SAED pattern exhibits a single crystal structure and the interplanar lattice distance is consistent with the XRD and HR-TEM results. The uniform distribution of the cations and oxygen is confirmed by EDX mapping of a single particle (Fig. 2c).
Electrochemical performance of the PBCC95 electrode
To evaluate the improvement of the electrochemical activity for the PBCC electrode after adopting cation deficiency, the symmetric cells are prepared to compare the electrode polarization resistance (Rp) by impedance measurement with the three-electrode configuration. The 3-electrode testing can effectively mitigate the effects from the counter electrode, ensuring that a specific reaction is studied. The impedance spectra in Nyquist plots and fitting results for PBCC and PBCC95 measured at open circuit in wet oxygen (3% H2O) at 600 °C are depicted in Fig. 3a. The spectra can be separated into two arcs located in the high-frequency zone (HF, 10–500 Hz) and the low-frequency zone (LF, 0.1–10 Hz). These spectra can be fitted well to the equivalent circuit Rohm–(RHF–CPEHF)–(RLF–CPELF) as shown in the inset figure. As identified in other studies, the LF part is related to the process of oxygen diffusion while HF part originates from oxygen dissociation in the electrode.57,58 The RHF and RLF of PBCC are 0.058 Ω cm2 and 0.162 Ω cm2, respectively, while PBCC95 shows a 41% smaller RHF (0.034 Ω cm2) and 35% smaller RLF (0.106 Ω cm2) compared to PBCC. This decreasing trend of RHF and RLF indicates the enhanced kinetics and activity of the PBCC95 electrode, which results from the increased oxygen vacancies introduced by the A-site deficiency. Fig. 3b shows the Arrhenius plot of overall Rp obtained under open-circuit conditions from the fitted EIS as a function of temperature in pure oxygen. The activation energies of PBCC and PBCC95 are 1.21 eV and 1.12 eV, respectively. The activation energies are calculated using the following equation: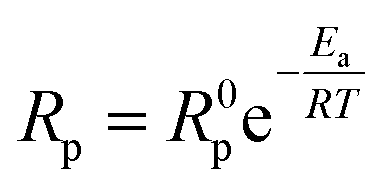 | (1) |
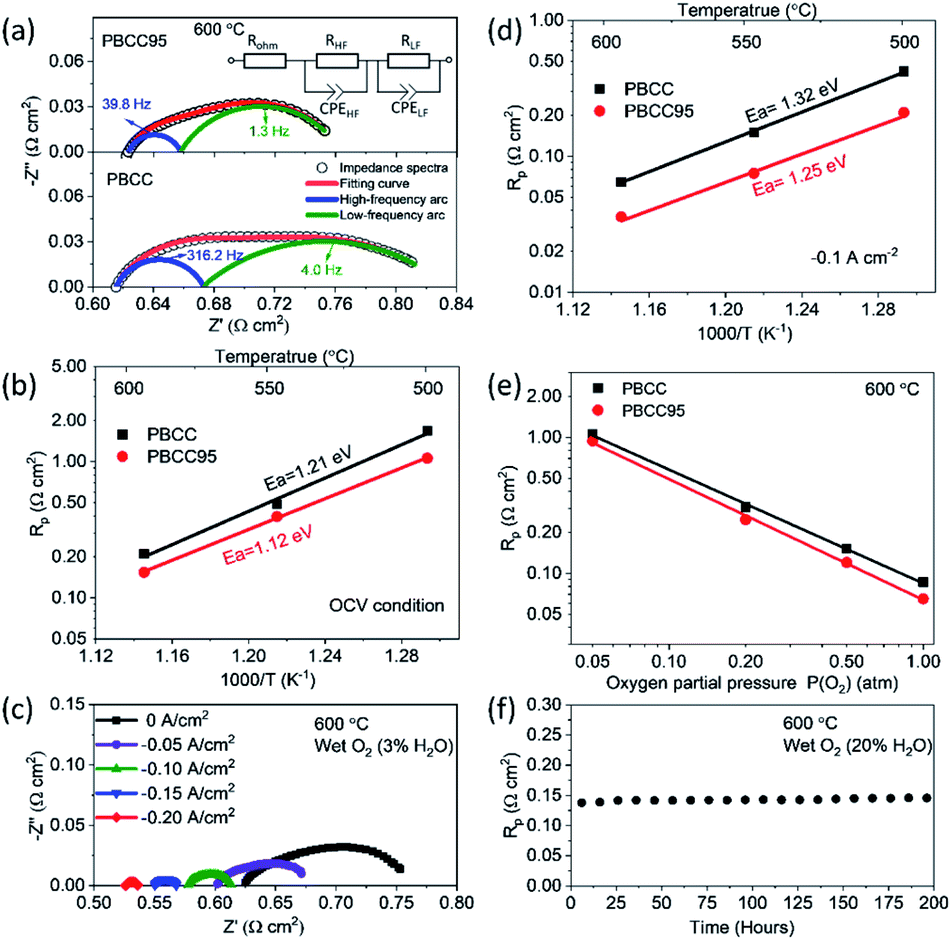 | ||
| Fig. 3 Performance of PBCC and PBCC95 in symmetric cells with a 3-electrode configuration. (a) EIS and fitted spectra of PBCC and PBCC95 tested at 600 °C; (inset) equivalent circuit used fitting; (b) Arrhenius plot of Rp for PBCC and PBCC95 between 500 °C and 600 °C under OCV condition; (c) the effect of applied electrolysis current density on the electrode polarization measured by AC impedance spectra at 600 °C for the PBCC95 electrode. (d) Arrhenius plot of Rp for PBCC and PBCC95 between 500 °C and 600 °C at an applied electrolysis current density of −0.1 A cm−2; (e) dependence of Rp as a function of PO2 tested at 600 °C and −0.1 A cm−2; (f) stability of PBCC95 electrode resistance in humidified oxygen (∼20% H2O) at 600 °C and −0.05 A cm−2. | ||
The smaller activation energy of PBCC95 indicates the higher electrochemical activity toward oxygen reduction in an equilibrium state at lower temperatures. This result has shown that the A-site deficiency can increase the catalytic activity by generating more oxygen defects in both the surface and bulk.43 Furthermore, the effect of applied electrolysis current density on EIS has been studied and shown in Fig. 3c. It can be clearly seen that the PBCC95 shows an obvious trend of decreasing Rp when the applied current density increases from 0 to −0.2 A cm−2. Fig. S3† depicts that the decrease of PBCC95 Rp results from both HF and LF arcs, indicating the enhanced electrode reaction kinetics on the surface at the applied electrolysis current density.
As the applied current largely affects the Rp, PBCC and PBCC95 oxygen electrode behaviours in electrolysis mode are also evaluated using 3-electrode symmetric cells. Fig. 3d shows the dependence of electrode polarization resistance on the operating temperature when the sample is exposed to humid oxygen (∼3% H2O) and applied with a bias current density of −0.1 A cm−2. When the temperature is increased, the Rp for PBCC increases from 0.084 Ω cm2 at 600 °C to 0.191 Ω cm2 at 550 °C and 0.298 Ω cm2 at 500 °C, respectively. For PBCC95, the comparison clearly demonstrates a decreased Rp, e.g., 0.065 Ω cm2 at 600 °C, which has decreased by 23% from 0.084 Ω cm2 for PBCC. The calculated activation energy with a bias electrolysis current density of −0.1 A cm−2 for PBCC95 is 1.25 eV, which is decreased from 1.32 eV for PBCC. The smaller activation energy of PBCC95 at a −0.1 A cm−2 applied current indicates the higher anodic electrochemical activity in the steady-state, and A-site deficient PBCC95 is kinetically more favorable than PBCC in electrolysis mode.
To evaluate the effect of other operating conditions on the oxygen electrode activity in electrolysis mode, the dependence of Rp on various oxygen partial pressure (balanced by Ar) is shown in Fig. 3e. As PO2 increased from 0.05 to 1.0 atm, the Rp of PBCC95 decreases from 0.94 to 0.065 Ω cm2 at 600 °C and an applied electrolysis current of −0.1 A cm−2. PBCC shows a similar trend of decreasing Rp from 1.06 to 0.086 Ω cm2. The result is attributed to the improved electrical conductivity at higher PO2 and indicates that a higher oxygen content is preferred in the electrolysis operation. In addition, the stability of the electrode activity at a 20% steam concentration has been examined (Fig. 3f). A constant bias current density of −0.05 A cm−2 is applied to the cell and the Rp is measured every 10 h. As can be seen, the Rp value is stable over 200 h without observable degradation, demonstrating the robust stability of the material structure and interfacial strength of this PBCC95 electrode with the electrolyte.
Electrochemical performance of the PCEC with the PBCC and PBCC95 electrode
To further demonstrate the catalytic activity improvement by the A-site deficiency manipulation, the PCECs with PBCC95 and PBCC as oxygen electrodes and BCZYYb4411 as the electrolyte have been fabricated and measured. The performance in fuel cell mode has been examined when wet hydrogen (∼3% H2O) and oxygen are fed into the anode and cathode side, respectively, to demonstrate the activity towards the ORR. The use of pure hydrogen can facilitate the examination of cell quality and performance comparison with those of other studies. As shown in Fig. 4a, the OCVs reach 1.06 V at 600 °C, 1.09 V at 550 °C and 1.12 V at 500 °C, respectively, which are close to the theoretical Nernst potentials of 1.165 V, 1.171 V and 1.178 V, respectively.59,60 These high voltages indicate high density of the electrolyte membrane and good cell sealing. The peak power densities of A-site deficient PBCC95 are 0.540, 0.354 and 0.222 W cm−2 at 600, 550 and 500 °C, respectively. The cell with the PBCC electrode shows a peak power density of 18.7%, 20.9%, and 23.8% lower than those of PBCC95 at 600, 550 and 500 °C, respectively (Fig. 4b). The higher fuel cell performance indicates the increased ORR activity due to the abundant oxygen vacancies and fast kinetics of PBCC95.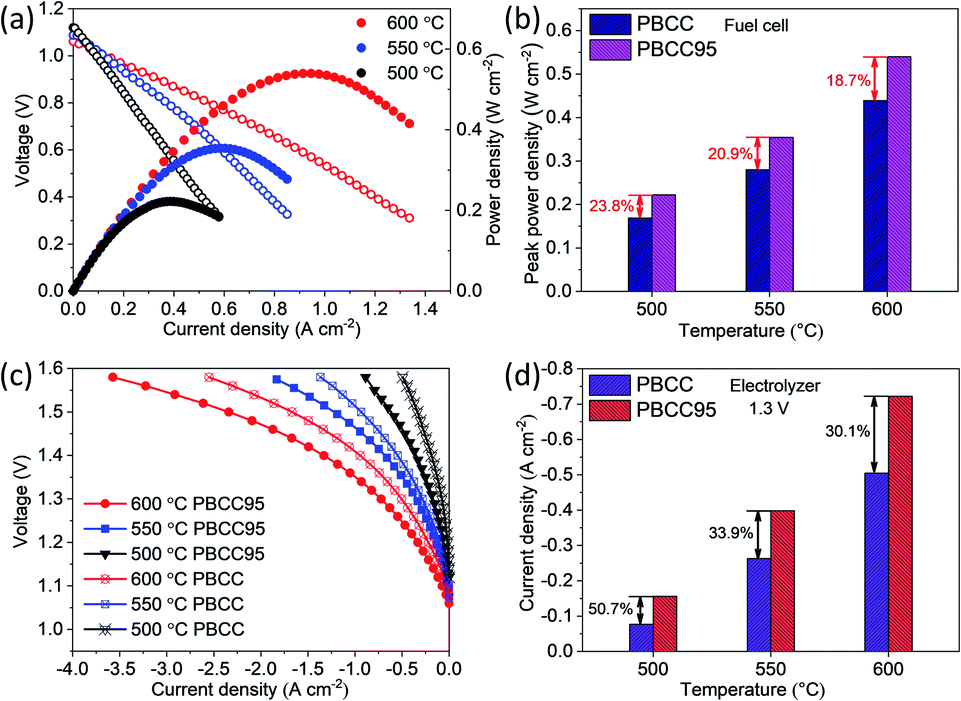 | ||
| Fig. 4 Electrochemical performances of PBCC95 and PBCC electrodes at 500, 550, and 600 °C. (a) I–V and current–power density (I–P) curves of PBCC95 in fuel cell mode; (b) peak power density comparison between PBCC95 and PBCC; (c) I–V curve of PBCC95 and PBCC electrodes in electrolysis mode; (d) electrolysis current density comparison of PBCC95 and PBCC at 1.3 V. | ||
Fig. 4c shows the current–voltage (I–V) curves of electrolysis performance in the temperature range of 500–600 °C with humidified oxygen (∼20%) for the oxygen electrode. Pure hydrogen is used in the hydrogen electrode to avoid gas separation and help maintain a high OCV. The cell with the PBCC95 electrode shows much higher performance than the one with the PBCC electrode under the same operating conditions. For example, the current densities of the PBCC95 cell at 600 °C are −0.72 A cm−2 at 1.3 V and −1.33 A cm−2 at 1.4 V, respectively, performing 30.1% higher than PBCC with −0.505 A cm−2 at 1.3 V and 22.0% higher at 1.4 V. At lower temperatures of 550 and 500 °C, the current density of the PBCC95 cell can still reach −0.39 A cm−2 and −0.16 A cm−2 at 1.3 V, which are 33.9% and 50.7% higher than PBCC performance as shown in Fig. 4d. The superior performance of this cell is among the highest performances of PCECs to date. For example, Norby et al. recently demonstrated a double perovskite as the oxygen electrode in a PCEC with a electrolysis current density less than −0.1 A cm−2 with considerable efficiency at 1.3 V and 600 °C.61 Liu et al. reported a triple-conducting Ruddlesden–Popper (R–P) structured perovskite Pr2NiO4+δ as an electrode in a PCEC with less than −0.4 A cm−2 at 1.3 V and 600 °C.62 Li et al. reported a R–P perovskite La1.2Sr0.8NiO4 in a PCEC with the current density measured to be −0.42 A cm−2 at 1.3 V and 600 °C.63 The PBCC95's performance is also comparable to that of Kim's work in which a hybrid PCEC with mixed proton and oxide-ion conducting BaCe0.7Zr0.1Y0.1Yb0.1O3−δ electrolyte and an electrolysis current density of −0.75 A cm−2 were applied at 1.3 V and 600 °C.64 Therefore, PBCC95 represents a promising oxygen electrode material for electrolysis cells. The improvement of electrolysis performance has been studied by EIS as shown in Fig. S4.† At 600 °C and 1.3 V, the Rp of the cell with the PBCC95 electrode is 0.024 Ω cm2, which is 53.8% smaller than the Rp for the PBCC electrode. Similarly, PBCC95 also performs much better than PBCC at lower temperatures. At 550 and 500 °C, the Rp is accordingly decreased by 55.8% and 52.4% respectively. Furthermore, the performance stability at different electrolysis voltages of 1.2 V, 1.3 V, 1.4 V and 1.5 V was examined at 600 °C, as shown in Fig. S5a.† The initial current densities are −0.37 A cm−2, −0.72 A cm−2, −1.33 A cm−2, and −2.36 A cm−2 respectively. At each voltage point, the cell shows a slight improvement in current density, which may be attributed to the process of electrode activation.35,65 After the durability testing, the Rp value decreases from 0.025 Ω cm2 to 0.017 Ω cm2 as shown in Fig. S5b,† indicating the improved electrode interface. The results demonstrate that PBCC95 shows better performance than PBCC in both fuel cell and electrolysis modes due to the higher catalytic activity resulting from A-site deficiencies.
To demonstrate the feasibility of the reversible operation between the electrolysis mode and fuel cell mode, it is critical to evaluate the performance of this PBCC95 electrode in a dynamic cycling transition process in a fast manner. As shown in Fig. 5a, the cell is operated at an electrolysis voltage of 1.3 V for 5 minutes to produce hydrogen, and then switched to a fuel cell voltage of 0.8 V to generate electricity for another 5 minutes at 550 °C. Then the cell is operated at higher current densities by increasing the electrolysis voltage to 1.4 V and 1.5 V and reducing the fuel cell voltage to 0.7 V and 0.6 V for examining the working flexibility. For each test, the cell is cycled 10 times. The smooth transition between each mode and the stable current densities indicate that the PBCC95 electrode can work for water splitting and oxygen reduction reactions stably and adapt to each mode quickly. When the cell is transitioned to a new mode by adjusting the voltage to three increasing electrolysis voltages and then to three decreasing fuel cell voltages, the responding current densities were still stable. This complex operation can demonstrate the high reversibility of PBCC95 in two different electrochemical reactions of water oxidation and oxygen reduction. Furthermore, the durability of the cell has been examined in both fuel cell mode and electrolysis mode at 500 °C for 160 h. As shown in Fig. 5b, in the electrolysis mode the current density at 1.3 V is about −0.16 A cm−2, while in the fuel cell mode the current density at 0.7 V is about 0.30 A cm−2, which shows good durability in both operating modes with no observable degradation.
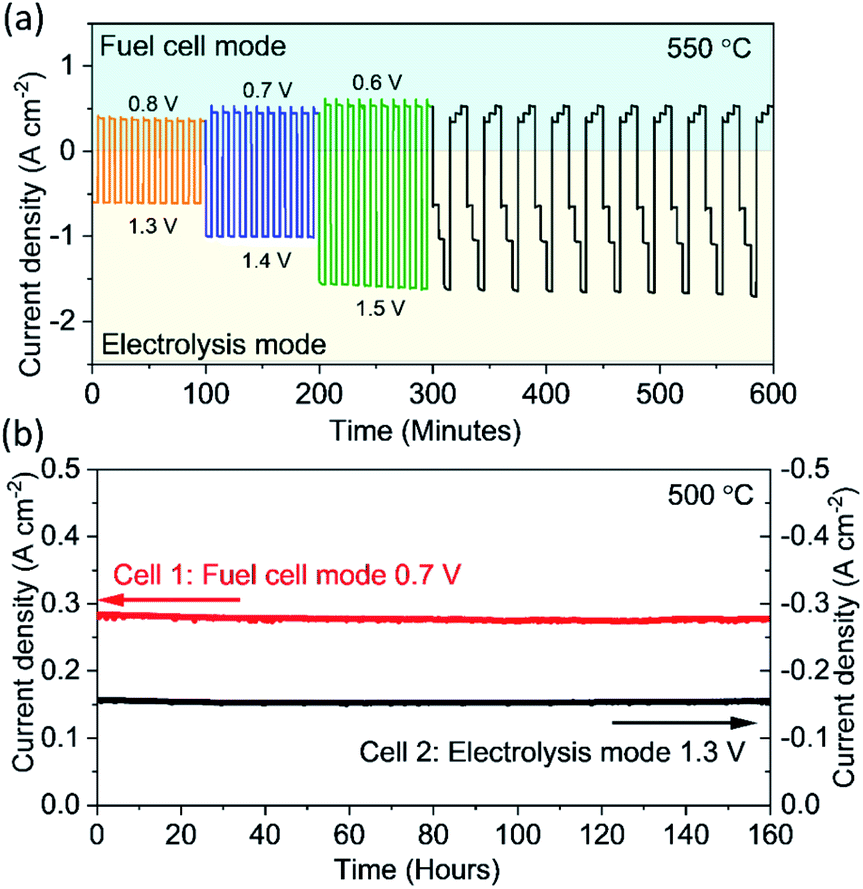 | ||
| Fig. 5 Reversible operation of the electrochemical cell and long-term stability in fuel cell and electrolysis modes. (a) Reversible operation between the electrolysis and fuel cell mode at 550 °C when the cell working voltage is transiently changed; and (b) long-term stability testing of the cell in fuel cell mode at 0.7 V and in electrolysis mode at 1.3 V and 500 °C. | ||
After all the electrochemical testing, the cell is examined by SEM and shown in Fig. 6. The BCZYYb4411 electrolyte surface (Fig. 6a) is dense without any defects (pinhole/impurity) observed, which prevents gas leakage. After the testing, the reduced hydrogen electrode is composed of porous nickel particles and dense electrolyte particles with uniform distribution between the metal and ceramic phases, as seen from the cross-sectional view (Fig. 6b). The electrolyte is fully dense with a thickness about 20 μm and adheres well to both electrodes without delamination observed at interfaces. The flower-like morphology of nickel particles in the magnified hydrogen electrode SEM image (Fig. 6c) indicates the complete reduction of NiO to Ni metal. At the oxygen-electrode/electrolyte interface (Fig. 6d), PBCC95 shows a porous structure consisting of small particles and adheres to the electrolyte membrane strongly, which ensures the fast gas diffusion and little degradation at the interface during operation.
 | ||
| Fig. 6 The morphology of the post-test cell. (a) SEM image of the electrolyte surface; (b) cross-sectional image of the interface with the PBCC95 oxygen electrode, BCZYYb4411 electrolyte, and Ni/BCZYYb4411 hydrogen electrode; (c) magnified SEM image of the hydrogen electrode with flower-like nickel particles; and (d) electrolyte/PBCC95 electrode interface. | ||
Conclusions
A-site cation ordered and deficient PBCC95 was successfully synthesized and studied as the oxygen electrode for PCECs for fuel cells and hydrogen production. This novel electrode exhibited great advantage of increased catalytic activity towards water oxidation and the ORR compared to PBCC without A-site deficiency. At intermediate temperatures, the PCEC with the BCZYYb4411 electrolyte and PBCC95 electrode showed high and stable electrochemical performance and reversible operation. A high electrolysis current density of −0.72 A cm−2 at 1.3 V and a peak power density of 0.540 W cm−2 were achieved at 600 °C. The fast reversible operation at transiently varied voltages between the electrolysis and fuel cell mode demonstrated the feasibility of this PBCC95 electrode for switching roles in water oxidation and oxygen reduction. The long-term durability testing at 500 °C proved the chemical and interfacial stability during the electrolysis and fuel cell process for 160 h.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy (USDOE), Office of Energy Efficiency and Renewable Energy (EERE), Fuel Cell Technologies Office (FCTO) under DOE Idaho Operations Office under contract DE-AC07-05ID14517.References
- S. Choi, T. C. Davenport and S. M. Haile, Protonic ceramic electrochemical cells for hydrogen production and electricity generation: exceptional reversibility, stability, and demonstrated faradaic efficiency, Energy Environ. Sci., 2019, 12(1), 206–215 RSC.
- L. Bi, S. Boulfrad and E. Traversa, Steam electrolysis by solid oxide electrolysis cells (SOECs) with proton-conducting oxides, Chem. Soc. Rev., 2014, 43(24), 8255–8270 RSC.
- W. Wu, D. Ding and T. He, Development of High Performance Intermediate Temperature Proton-Conducting Solid Oxide Electrolysis Cells, ECS Trans., 2017, 80(9), 167–173 CrossRef CAS.
- M. B. Mogensen, A. Hauch, X. Sun, M. Chen, Y. Tao, S. D. Ebbesen, K. V. Hansen and P. V. Hendriksen, Relation between Ni particle shape change and Ni migration in Ni–YSZ electrodes–a hypothesis, Fuel Cells, 2017, 17(4), 434–441 CrossRef CAS.
- B. Hua, M. Li, Y. Q. Zhang, Y. F. Sun and J. L. Luo, All-In-One Perovskite Catalyst: Smart Controls of Architecture and Composition toward Enhanced Oxygen/Hydrogen Evolution Reactions, Adv. Energy Mater., 2017, 7(20), 1700666 CrossRef.
- M. C. Tucker, Progress in metal-supported solid oxide fuel cells: A review, J. Power Sources, 2010, 195(15), 4570–4582 CrossRef CAS.
- S. Li, Y. Li, Y. Gan, K. Xie and G. Meng, Electrolysis of H2O and CO2 in an oxygen-ion conducting solid oxide electrolyzer with a La0. 2Sr0. 8TiO3+δ composite cathode, J. Power Sources, 2012, 218, 244–249 CrossRef CAS.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Enhancement of Pt and Pt-alloy fuel cell catalyst activity and durability via nitrogen-modified carbon supports, Energy Environ. Sci., 2010, 3(10), 1437–1446 RSC.
- C. Xia, F. Chen and M. Liu, Reduced-temperature solid oxide fuel cells fabricated by screen printing, Electrochem. Solid-State Lett., 2001, 4(5), A52–A54 CrossRef CAS.
- Y. Li, R. Gemmen and X. Liu, Oxygen reduction and transportation mechanisms in solid oxide fuel cell cathodes, J. Power Sources, 2010, 195(11), 3345–3358 CrossRef CAS.
- M. C. Tucker, G. Y. Lau, C. P. Jacobson, L. C. DeJonghe and S. J. Visco, Performance of metal-supported SOFCs with infiltrated electrodes, J. Power Sources, 2007, 171(2), 477–482 CrossRef CAS.
- A. B. Stambouli and E. Traversa, Solid oxide fuel cells (SOFCs): a review of an environmentally clean and efficient source of energy, Renewable Sustainable Energy Rev., 2002, 6(5), 433–455 CrossRef CAS.
- B. C. Steele, Survey of materials selection for ceramic fuel cells II. Cathodes and anodes, Solid State Ionics, 1996, 86, 1223–1234 CrossRef.
- H. Ding, D. Zhou, S. Liu, W. Wu, Y. Yang, Y. Yang and Z. Tao, Electricity generation in dry methane by a durable ceramic fuel cell with high-performing and coking-resistant layered perovskite anode, Appl. Energy, 2019, 233, 37–43 CrossRef.
- P. Patro, T. Delahaye, E. Bouyer and P. Sinha, Microstructural development of Ni-1Ce10ScSZ cermet electrode for Solid Oxide Electrolysis Cell (SOEC) application, Int. J. Hydrogen Energy, 2012, 37(4), 3865–3873 CrossRef CAS.
- S. P. Jiang and S. H. Chan, A review of anode materials development in solid oxide fuel cells, J. Mater. Sci., 2004, 39(14), 4405–4439 CrossRef CAS.
- C. Sun and U. Stimming, Recent anode advances in solid oxide fuel cells, J. Power Sources, 2007, 171(2), 247–260 CrossRef CAS.
- H. Ding, W. Wu and D. Ding, Advancement of Proton-Conducting Solid Oxide Fuel Cells and Solid Oxide Electrolysis Cells at Idaho National Laboratory (INL), ECS Trans., 2019, 91(1), 1029 CrossRef.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O'Hayre, Highly efficient reversible protonic ceramic electrochemical cells for power generation and fuel production, Nat. Energy, 2019, 4(3), 230–240 CrossRef CAS.
- D. Medvedev, J. Lyagaeva, E. Gorbova, A. Demin and P. Tsiakaras, Advanced materials for SOFC application: Strategies for the development of highly conductive and stable solid oxide proton electrolytes, Prog. Mater. Sci., 2016, 75, 38–79 CrossRef CAS.
- X. Li, N. Xu, L. Zhang and K. Huang, Combining proton conductor BaZr0.8Y0.2O3−δ with carbonate: Promoted densification and enhanced proton conductivity, Electrochem. Commun., 2011, 13(7), 694–697 CrossRef CAS.
- Z. Shao and S. M. Haile, A high-performance cathode for the next generation of solid-oxide fuel cells, in Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 255–258 Search PubMed.
- D. Han, Y. Noda, T. Onishi, N. Hatada, M. Majima and T. Uda, Transport properties of acceptor-doped barium zirconate by electromotive force measurements, Int. J. Hydrogen Energy, 2016, 41(33), 14897–14908 CrossRef CAS.
- Y. Chen, W. Zhou, D. Ding, M. Liu, F. Ciucci, M. Tade and Z. Shao, Advances in cathode materials for solid oxide fuel cells: complex oxides without alkaline earth metal elements, Adv. Energy Mater., 2015, 5(18), 1500537 CrossRef.
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Exceptional power density and stability at intermediate temperatures in protonic ceramic fuel cells, Nat. Energy, 2018, 3(3), 202 CrossRef CAS.
- J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu and G. Kim, Triple-conducting layered perovskites as cathode materials for proton-conducting solid oxide fuel cells, ChemSusChem, 2014, 7(10), 2811–2815 CrossRef CAS PubMed.
- A. V. Virkar, Mechanism of oxygen electrode delamination in solid oxide electrolyzer cells, Int. J. Hydrogen Energy, 2010, 35(18), 9527–9543 CrossRef CAS.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Enhanced sulfur and coking tolerance of a mixed ion conductor for SOFCs: BaZr0. 1Ce0.7Y0.2–xYbxO3–δ, Science, 2009, 326(5949), 126–129 CrossRef CAS PubMed.
- M. Saccoccio, C. Jiang, Y. Gao, D. Chen and F. Ciucci, Nb-substituted PrBaCo2O5+δ as a cathode for solid oxide fuel cells: a systematic study of structural, electrical, and electrochemical properties, Int. J. Hydrogen Energy, 2017, 42(30), 19204–19215 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Readily processed protonic ceramic fuel cells with high performance at low temperatures, Science, 2015, 349(6254), 1321–1326 CrossRef CAS PubMed.
- F. Li, Z. Tao, H. Dai, X. Xi and H. Ding, A high-performing proton-conducting solid oxide fuel cell with layered perovskite cathode in intermediate temperatures, Int. J. Hydrogen Energy, 2018, 43(42), 19757–19762 CrossRef CAS.
- A. Grimaud, J.-M. Bassat, M. Pollet, A. Wattiaux, M. Marrony and J.-C. Grenier, Oxygen reduction reaction of PrBaCo2−xFexO5+d compounds as H+-SOFC cathodes: correlations with physical properties, J. Mater. Chem. A, 2014, 2, 3594 RSC.
- P. I. Cowin, C. T. Petit, R. Lan, J. T. Irvine and S. Tao, Recent progress in the development of anode materials for solid oxide fuel cells, Adv. Energy Mater., 2011, 1(3), 314–332 CrossRef CAS.
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang and Y. Zhang, Self-sustainable protonic ceramic electrochemical cells using a triple conducting electrode for hydrogen and power production, Nat. Commun., 2020, 11(1), 1–11 CrossRef PubMed.
- W. Wu, H. Ding, Y. Zhang, Y. Ding, P. Katiyar, P. K. Majumdar, T. He and D. Ding, 3D Self-Architectured Steam Electrode Enabled Efficient and Durable Hydrogen Production in a Proton-Conducting Solid Oxide Electrolysis Cell at Temperatures Lower Than 600 °C, Adv. Sci., 2018, 5(11), 1800360 CrossRef PubMed.
- B. Hua, Y. Q. Zhang, N. Yan, M. Li, Y. F. Sun, J. Chen, J. Li and J. L. Luo, The excellence of both worlds: developing effective double perovskite oxide catalyst of oxygen reduction reaction for room and elevated temperature applications, Adv. Funct. Mater., 2016, 26(23), 4106–4112 CrossRef CAS.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, A perspective on low-temperature solid oxide fuel cells, Energy Environ. Sci., 2016, 9(5), 1602–1644 RSC.
- A. S. Bangwal, P. K. Jha, P. K. Dubey, M. Singh, A. Shinha, V. Sathe, P. A. Jha and P. Singh, Porous and high conducting cathode material PrBaCo2O6−δ: The bulk and surface studies for synthesis anomaly, Phys. Chem. Chem. Phys., 2019, 21, 14701 RSC.
- L. Zhang, G. Yao, Z. Song, B. Niu, W. Long, L. Zhang and T. He, Effects of Pr-deficiency on thermal expansion and electrochemical properties in Pr1−xBaCo2O5+δ cathodes for IT-SOFCs, Electrochim. Acta, 2016, 212, 522–534 CrossRef CAS.
- G. Kim, S. Wang, A. Jacobson, L. Reimus, P. Brodersen and C. Mims, Rapid oxygen ion diffusion and surface exchange kinetics in PrBaCo2O5+x with a perovskite related structure and ordered A cations, J. Mater. Chem., 2007, 17(24), 2500–2505 RSC.
- L. Zhao, J. Shen, B. He, F. Chen and C. Xia, Synthesis, characterization and evaluation of PrBaCo2−xFexO5+δ as cathodes for intermediate-temperature solid oxide fuel cells, Int. J. Hydrogen Energy, 2011, 36(5), 3658–3665 CrossRef CAS.
- Y. Chen, S. Yoo, Y. Choi, J. H. Kim, Y. Ding, K. Pei, R. Murphy, Y. Zhang, B. Zhao and W. Zhang, A highly active, CO2-tolerant electrode for the oxygen reduction reaction, Energy Environ. Sci., 2018, 11(9), 2458–2466 RSC.
- A. Donazzi, R. Pelosato, G. Cordaro, D. Stucchi, C. Cristiani, G. Dotelli and I. N. Sora, Evaluation of Ba deficient NdBaCo2O5+δ oxide as cathode material for IT-SOFC, Electrochim. Acta, 2015, 182, 573–587 CrossRef CAS.
- W. Zhou, R. Ran, Z. Shao, W. Jin and N. Xu, Evaluation of A-site cation-deficient (Ba0.5Sr0.5)1−xCo0.8Fe0.2O3−δ (x > 0) perovskite as a solid-oxide fuel cell cathode, J. Power Sources, 2008, 182(1), 24–31 CrossRef CAS.
- G. Chen, J. Sunarso, Y. Wang, C. Ge, J. Yang and F. Liang, Evaluation of A-site deficient Sr1−xSc0. 175Nb0. 025Co0.8O3−δ (x = 0, 0.02, 0.05 and 0.1) perovskite cathodes for intermediate-temperature solid oxide fuel cells, Ceram. Int., 2016, 42(11), 12894–12900 CrossRef CAS.
- Y. Sun, J. Li, Y. Zeng, B. S. Amirkhiz, M. Wang, Y. Behnamian and J. Luo, A-site deficient perovskite: the parent for in situ exsolution of highly active, regenerable nano-particles as SOFC anodes, J. Mater. Chem. A, 2015, 3(20), 11048–11056 RSC.
- X. Ding, Z. Gao, D. Ding, X. Zhao, H. Hou, S. Zhang and G. Yuan, Cation deficiency enabled fast oxygen reduction reaction for a novel SOFC cathode with promoted CO2 tolerance, Appl. Catal., B, 2019, 243, 546–555 CrossRef CAS.
- X. Jiang, Y. Shi, W. Zhou, X. Li, Z. Su, S. Pang and L. Jiang, Effects of Pr3+-deficiency on structure and properties of PrBaCo2O5+δ cathode material–A comparison with Ba2+-deficiency case, J. Power Sources, 2014, 272, 371–377 CrossRef CAS.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. Irvine and G. Kim, Layered oxygen-deficient double perovskite as an efficient and stable anode for direct hydrocarbon solid oxide fuel cells, Nat. Mater., 2015, 14(2), 205 CrossRef CAS PubMed.
- D. Chen, R. Ran, K. Zhang, J. Wang and Z. Shao, Intermediate-temperature electrochemical performance of a polycrystalline PrBaCo2O5+δ cathode on samarium-doped ceria electrolyte, J. Power Sources, 2009, 188(1), 96–105 CrossRef CAS.
- Y. Guo, H. Shi, R. Ran and Z. Shao, Performance of SrSc0. 2Co0. 8O3−δ+ Sm0.5Sr0. 5CoO3−δ mixed-conducting composite electrodes for oxygen reduction at intermediate temperatures, Int. J. Hydrogen Energy, 2009, 34(23), 9496–9504 CrossRef CAS.
- Y. Zhu, J. Sunarso, W. Zhou, S. Jiang and Z. Shao, High-performance SrNb0.1Co0.9−xFexO3−δ perovskite cathodes for low-temperature solid oxide fuel cells, J. Mater. Chem. A, 2014, 2(37), 15454–15462 RSC.
- M.-H. Castaño-Robayo, R. Molina-Gallego and S. Moreno-Guáqueta, Ethyl acetate oxidation over MnOx-CoOx. Relationship between oxygen and catalytic activity, CT&F, Cienc., Tecnol. Futuro, 2015, 6(2), 45–56 Search PubMed.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Self-Assembled Triple-Conducting Nanocomposite as a Superior Protonic Ceramic Fuel Cell Cathode, Joule, 2019, 3(11), 2842–2853 CrossRef CAS.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Synthesis, characterization and evaluation of cation-ordered LnBaCo2O5+δ as materials of oxygen permeation membranes and cathodes of SOFCs, Acta Mater., 2008, 56(17), 4876–4889 CrossRef CAS.
- J. Zhang, D. Tan, Q. Meng, X. Weng and Z. Wu, Structural modification of LaCoO3 perovskite for oxidation reactions: The synergistic effect of Ca2+ and Mg2+ co-substitution on phase formation and catalytic performance, Appl. Catal., B, 2015, 172, 18–26 CrossRef.
- A. Leonide, Y. Apel and E. Ivers-Tiffee, SOFC modeling and parameter identification by means of impedance spectroscopy, ECS Trans., 2009, 19(20), 81 CrossRef CAS.
- R. Mohammadi, M. Ghassemi, Y. M. Barzi and J. Pirkandi, The effect of mass transfer on electrochemical impedance of a solid oxide fuel cell anode, J. Solid State Electrochem., 2014, 18(10), 2815–2827 CrossRef CAS.
- Y. Li, S. Choi and S. Rajakaruna, An analysis of the control and operation of a solid oxide fuel-cell power plant in an isolated system, IEEE Transactions on Energy Conversion, 2005, 20(2), 381–387 CrossRef.
- H. Shimada, X. Li, A. Hagiwara and M. Ihara, Proton-conducting solid oxide fuel cells with yttrium-doped barium zirconate for direct methane operation, J. Electrochem. Soc., 2013, 160(6), F597–F607 CrossRef CAS.
- E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Mixed proton and electron conducting double perovskite anodes for stable and efficient tubular proton ceramic electrolysers, Nat. Mater., 2019, 18(7), 752 CrossRef PubMed.
- W. Li, B. Guan, L. Ma, S. Hu, N. Zhang and X. Liu, High performing triple-conductive Pr2NiO4+δ anode for proton-conducting steam solid oxide electrolysis cell, J. Mater. Chem. A, 2018, 6(37), 18057–18066 RSC.
- S. Yang, Y. Wen, J. Zhang, Y. Lu, X. Ye and Z. Wen, Electrochemical performance and stability of cobalt-free Ln1. 2Sr0.8NiO4 (Ln= La and Pr) air electrodes for proton-conducting reversible solid oxide cells, Electrochim. Acta, 2018, 267, 269–277 CrossRef CAS.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Hybrid-solid oxide electrolysis cell: A new strategy for efficient hydrogen production, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- Z. Pan, Q. Liu, M. Ni, R. Lyu, P. Li and S. H. Chan, Activation and failure mechanism of La0.6Sr0.4Co0.2Fe0.8O3−δ air electrode in solid oxide electrolyzer cells under high-current electrolysis, Int. J. Hydrogen Energy, 2018, 43(11), 5437–5450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta05137c |
| This journal is © The Royal Society of Chemistry 2020 |