
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic conductive and photocatalytic properties of cementitious materials: calcium silicate hydrate and calcium aluminoferrite
Masahiro
Nagao
 a,
Kazuyo
Kobayashi
a,
Yongcheng
Jin
b,
Ippei
Maruyama
ac and
Takashi
Hibino
*a
a,
Kazuyo
Kobayashi
a,
Yongcheng
Jin
b,
Ippei
Maruyama
ac and
Takashi
Hibino
*a
aGraduate School of Environmental Studies, Nagoya University, Nagoya 464-8601, Japan. E-mail: hibino@urban.env.nagoya-u.ac.jp
bSchool of Materials Science and Engineering, Ocean University of China, Qingdao 266100, China
cGraduate School of Engineering, The University of Tokyo, Tokyo 113-8656, Japan
First published on 27th July 2020
Abstract
Few studies have reported the ionic conductive and photoactive properties of cementitious materials, owing to the compositional and structural variations of cement paste. The present report focuses on calcium silicate hydrate (C-S-H) and tetracalcium aluminoferrite (C4AF), prepared individually from pure constituent elements. C-S-H is an ionic conductor with a conductivity ranging from 10−4 to 10−3 S cm−1 in humidified air at temperatures between 60 and 100 °C. The hydroxide ions present in the mesopores of C-S-H as basic O–H groups were determined to be the major charge carriers, and the faradaic efficiency for the water oxidation reaction was estimated to be approximately 100%. C4AF is a mixed ionic n-type electronic conductor, particularly at high oxygen partial pressures. The conductivity of this compound was several times lower than that of C-S-H under the same humidity and temperature conditions, not only because of the bulk resistance but also the grain boundary resistance. C4AF and the hydrates exhibited photovoltaic and photocatalytic responses for water splitting and methylene blue degradation, respectively. These results indicate the important contribution of C-S-H and hydrated C4AF to the electrochemical and photochemical properties of cement paste.
Introduction
Advanced cementitious materials are being actively developed to enhance the performance1 and durability of concrete.2,3 Great attention is devoted to eco-cementitious materials that can reduce environmental impact and coexist with the environment.4–6 However, data on the electrochemical and photochemical properties of cement paste are lacking in the guidelines for the design and operation of these materials. Degradation of reinforced concrete is triggered by deterioration of the cement paste and corrosion of the steel beams and columns, which are mainly due to the carbonation of and salt damage to concrete induced by the external environment.7,8 However, the degradation mechanism related to the internal conditions of concrete is not well understood. Additionally, building finishing materials play a very important role in creating a comfortable indoor environment and keeping building exteriors clean, but these functionalities are conventionally achieved by coating the concrete surface with a photocatalyst.9,10It has been demonstrated that the ionic conduction in a hardened cement paste is driven by the humidification of a thin cement-based membrane.11 When the cement paste was moistened, the electrical resistance decreased by two orders of magnitude, the extent of which became more significant as the temperature increased. However, since there are many constituent cations and anions in cement paste, it cannot be determined which ions conduct charge carriers in the solid. Calcium silicate hydrate (C-S-H), the main hydration product of Portland cement, as well as sodium and potassium hydroxides (NaOH and KOH), are believed to be ion-conductive materials. However, calcium hydroxide (Ca(OH)2) is a minor ionic conductor, because of its low solubility product. If the ionic conductivity of C-S-H is comparable to those of alkali metal hydroxides, then the pH of the solution in the pores of the cement paste would be determined not only by the amounts of alkali metal hydroxides but also the amount of C-S-H, thus affecting the corrosion behavior of steel.
Iron-based oxides such as Fe3O4, CuFe2O4, and ZnFe2O4 have been studied as photocatalysts.12,13 Unlike the commonly known titanium dioxide (TiO2), these metal oxides show visible-light-induced catalytic activity, which enables the use of white LED lamps as the light source for various photoinduced reactions. Hirabayashi et al. reported the photocatalytic activity of brownmillerite-type Ca2Fe2O5, which is similar in structure and composition to tetracalcium aluminoferrite ((CaO)4·Al2O3·Fe2O3; C4AF), one of the main components of Portland cement.14 C4AF undergoes hydration to form ettringite (katoite) in the presence (absence) of gypsum (CaSO4).15,16 It is also reported that C4AF exists as Fe-siliceous hydrogarnet during hydration in the coexistence of silicate.17,18 It is meaningful to investigate the photochemical properties of these products; however, we consider it as future work, because the crystalline structure and solid solution state of the hydration products cannot be precisely controlled in the synthesis, thus making it difficult to obtain pure and reproducible material at this moment. (Even if a certain product is obtained, it may not be accepted as a satisfactory representative of various products of C4AF.) According to the literature, the degree of hydration of C4AF remains in the 0.6–0.8 range 1 year after the start of hydration.19 Moreover, as will be discussed later, a C4AF-rich cement clinker can be used to enhance the photocatalytic activity of the cement paste, which would further increase the amount of unreacted C4AF in the concrete. For these reasons, an easily synthesizable and highly pure C4AF was used as a research target in this study.
The goal of this study was to investigate the electro- and photo-chemical properties of cementitious materials. C-S-H and C4AF were prepared from pure constituent elements. The experimental conditions were adjusted by controlling the temperature and humidity of the sample during the measurements. In the electrochemical measurements, the alternating current (AC) conductivity was compared with the direct current (DC) conductivity to separate the electrical resistance components. Various galvanic cells were constructed to inspect the electrochemical behavior of the conductive ions in the solid. In the photochemical measurements, photovoltaic water splitting and photocatalytic degradation of methylene blue (MB) were performed under visible light irradiation. The relationships between the function and structure of C-S-H and C4AF were discussed.
Experimental
Synthesis of C-S-H and C4AF
Ca(OH)2 (Kanto Chemical, 96%) and fumed silica (Evonik, Aerosil 200) were used for the synthesis of C-S-H, and Ca(CO)3 (Kanto Chemical, 99.5%), Al2O3 (Showa Denko, AL-160), and Fe2O3 (Kanto Chemical, 99%) were used for the synthesis of C4AF. These reagents were all used without further purification. Ca(OH)2 and fumed silica powders were mixed at a Ca/Si molar ratio of 1.4 in deionized water and then stirred in a sealed container at 70 °C for 7 days. The product was filtered in an argon (Ar) glove box to prevent carbonation of the product. The filtrate was immediately transferred to a vacuum dryer. The appropriate amounts of Ca(CO)3, Al2O3, and Fe2O3 powders were mixed with a mortar and pestle. The mixture was pressed into pellets and then calcined in an alumina crucible at 1350 °C for 10 h. The product was ground into a powder using a stirred ball mill. To increase the specific surface of C4AF, this sample was optionally treated using a planetary ball mill at a rotation speed of 150 rpm. The C-S-H and C4AF membranes used for, respectively, electro- and photo-chemical measurements were prepared by binding 1.00 g of the powder with 0.07 g of polytetrafluoroethylene (PTFE) and then cold-rolling it to a thickness of 0.5 mm and a diameter of ca. 17 mm.Characterization
All measurements were conducted on the synthesized C-S-H and C4AF powders. X-ray powder diffraction (XRD) patterns were collected on a MiniFlex II (Rigaku) using CuKα radiation over a 2θ range from 10° to 60°. Transmission electron microscopy (TEM) images were obtained using a JEM-2100Plus (JEOL) at an accelerating voltage of 200 kV with a beam current of 92 μA. Nitrogen (N2) and krypton (Kr) isotherms were recorded at a relative pressure of 0 to 0.98 for N2 and 0 to 0.68 for Kr at liquid N2 temperatures using a BELSORP 18PLUS-HT (Nihon Bell). The samples were degassed at 150 °C for 5 h before measurements. The pore size distribution was calculated using the grand canonical Monte Carlo (GCMC) technique. The Brunauer–Emmett–Teller (BET) method was employed to determine the specific surface areas from multi-point N2 adsorption data obtained by using an adsorption/desorption apparatus in the case of C-S-H and one-point desorption data obtained by using a conventional flow apparatus equipped with a thermal conductivity detector (TCD) in the case of C4AF. Simultaneous thermogravimetry (TG) and differential thermal analysis (DTA) were performed using a DTG-60 (Shimadzu). The samples were heated in flowing air from room temperature to 900 °C at a rate of 10 °C min−1. Infrared spectra were measured using a Fourier transform-IR (FTIR) system (FT/IR-460 JASCO). Spectral analysis was performed in the 4000–400 cm−1 range with a resolution of 1 cm−1. The temperature-programmed desorption (TPD) technique using CO2 as probe molecule was employed to determine the basicity of C-S-H and C4AF. 0.2 g of the powder were placed at the center of a quartz tube, and CO2 was adsorbed by supplying 100% CO2 for 1 h at room temperature. After purging with Ar, the furnace temperature was raised from room temperature to 900 °C at a rate of 10 °C min−1. The concentration of CO2 (m/z = 44) in the outlet gas from the quartz tube was monitored using a mass spectrometer (ThermoStar, Pfeiffer Vacuum).Electrochemical measurements
Pt/C electrodes (area: 0.5 cm2, Pt loading: 2.0 mg cm−2, ElectroChem) were attached to both surfaces of the membrane sample and then fixed in graphite holders. The cell was humidified by simultaneously supplying deionized water (0.13 mL min−1) and high-purity air (10 mL min−1, Taiyo Nippon Sanso) to the bottom holder. The cell temperature was adjusted using a plate heater with a thermocouple. The DC resistance was measured by varying the current density from 2 to 10 mA cm−2. The current was interrupted for 0.0002 s at intervals of 0.002 s using a current pulse generator (Nikko Keisoku). The voltage signal from the cell was captured by a digital oscilloscope workstation (Nicolet). The AC impedance was measured by a potentiostat/galvanostat (Solartron Analytical 1280) and an impedance/gain-phase analyzer (Solartron Analytical 1260A) with an amplitude voltage of 60 mV in the frequency range of 0.1 to 106 Hz. Impedance measurements were also performed at various partial pressures of oxygen (PO2), which were obtained by mixing oxygen (O2) or hydrogen (H2) with Ar. The PO2 of the mixture was monitored using a zirconia O2 sensor. In water vapor concentration cells, an air – deionized water mixture and unhumidified air were fed to the bottom and top holders, respectively. In the oxygen gas concentration cell, i.e., the fuel cell, a mixture of H2 and deionized water and a mixture of air and deionized water were fed to the bottom and top holders, respectively. The voltage generated from the cell was recorded using an HE-104 electrometer (Hokuto Denko). Water vapor electrolysis was performed in flowing Ar by using the Pt/C electrode on the top holder as the cathode. H2 (m/z = 2) generated from the cathode at a constant current density was detected using the mass spectrometer.Photochemical measurements
A white LED lamp composed of 16 units (ODS-LS-16, OHM) was used as a visible light source. The average intensity of the lamp was 4.8 W m−2, which was measured using a light irradiance meter (T-10A, Konica Minolta). Water splitting was conducted by fabricating an H-type glass-made reactor consisting of a C4AF membrane anode and a Pt plate cathode immersed in a 1 M NaOH electrolyte solution. The anode surface was irradiated with the lamp at a distance of ca. 15 cm from the front. Cyclic voltammetry (CV) profiles were collected between 0 and +1.6 V at a scan rate of 10 mV s−1. Then, impedance spectra were acquired in a manner similar to that used for the AC resistance measurements, except that a bias voltage of +1 V was applied to the cell. Photocatalytic degradation of MB was conducted in a 100 mL Pyrex beaker that was irradiated from the top, with the lamp positioned ca. 15 cm above the beaker. A total of 0.1 g of the C4AF powder was dispersed in 10 mL of 10 ppm MB solution (Kanto Chemical), whose pH was maintained at 7.2 by adding a phosphate buffer solution (Nacalai Tesque). Absorption spectra of the solution were recorded using a UV-visible spectrophotometer (JASCO UV 360) with a quartz cell of 10 mm path length. The absorbance of MB was acquired between 200 and 800 nm (fixed 1 nm width) at specific irradiation time intervals.Results and discussion
Characterization of C-S-H and C4AF
An XRD pattern with no detectable Ca(OH)2, CaCO3, or SiO2 peaks was obtained for the C-S-H sample, as shown in Fig. 1a. The crystalline phase was identified as calcium silicate hydrate (Ca1.5SiO3.5·xH2O; PDF 00-033-0306) on the basis of the three peaks at 2θ = 29°, 32°, and 50°.20,21 However, this assignment may not be correct, because the preparation of C-S-H at a Ca/Si molar ratio as high as 1.4 causes the formation of a Ca-rich phase (probably amorphous Ca(OH)2 or nanocrystalline portlandite), which results in a smaller Ca/Si molar ratio in the case of C-S-H, as compared to that of the starting composition.22–25 Different morphologies are observed for C-S-H products in hardened Portland cements, depending on the composition and preparation conditions of C-S-H.24,26 The TEM image in Fig. 1b indicates that C-S-H had a plate- or sheet-like morphology, 50–100 nm in thickness and 400 nm in length. In addition, fine dark spots, assignable to Ca-rich phases, were seen on the surface of the C-S-H plates. These observations are consistent with the results reported by Richardson, who prepared C-S-H at a Ca/Si molar ratio of 1.5.27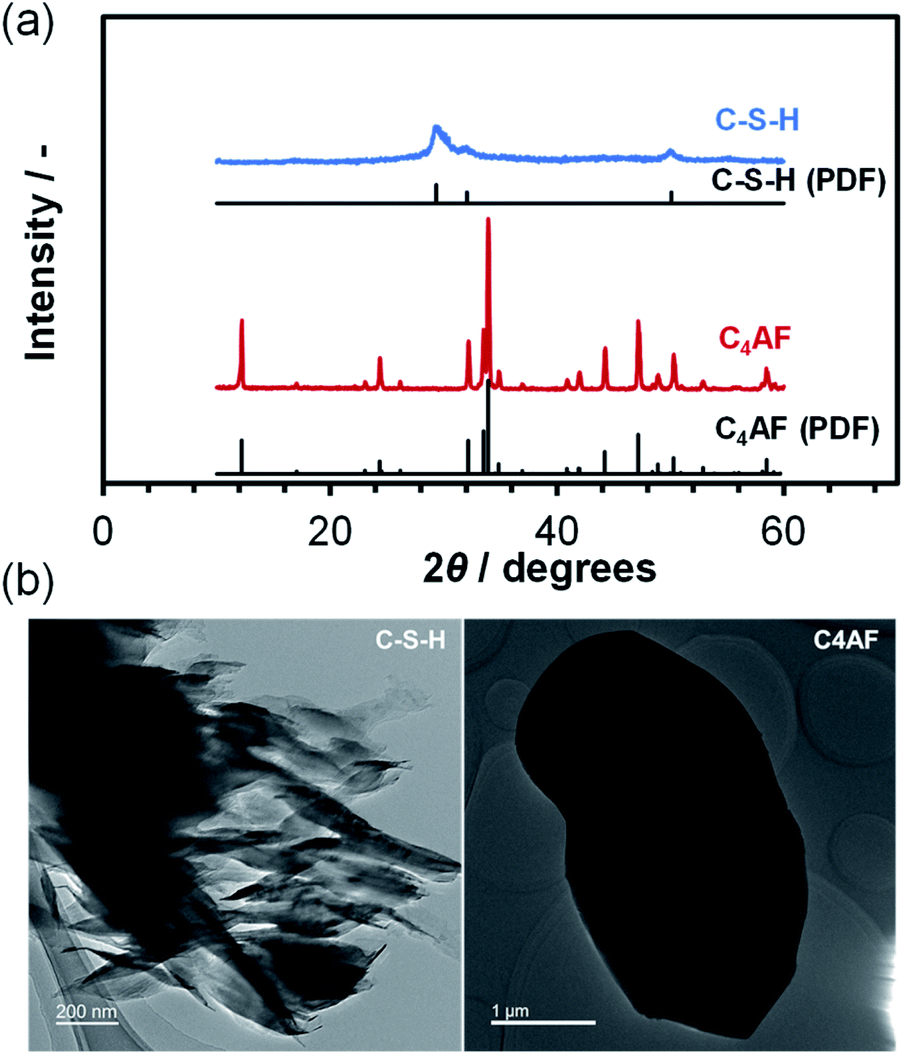 | ||
| Fig. 1 (a) XRD patterns for C-S-H and C4AF. The PDFs of Ca1.5SiO3.5·xH2O and Ca2FeAlO5 are included for reference. (b) TEM images of C-S-H and C4AF. | ||
Fig. 1a also shows the XRD pattern for the C4AF sample, which is indexed to brownmillerite (Ca2FeAlO5; PDF 01-071-0667),28 with an average crystallite size of 446 Å. This pattern is almost identical to that refined previously by Rietveld analysis,11 where the octahedral and tetrahedral layers, occupied by Fe3+ and Al3+ ions, respectively, were alternately connected in the brownmillerite structure. It appears from the TEM image in Fig. 1b that the C4AF particle is a coarse crystal with microdomains, rather than an aggregate of crystallites. The C4AF particle size of approximately 2–4 μm is comparable to the values observed for Portland cement clinker.29
N2 adsorption/desorption isotherms of the C-S-H and C4AF samples were measured (see Fig. 2a). The C-S-H sample showed a type-IV sorption behavior, with an H3-type hysteresis loop, which is characteristic of slit-like pores, giving rise to capillary condensation of the adsorbates.30,31 The BET specific surface area and average pore diameter were calculated to be 125 m2 g−1 and 19 nm, respectively. The extent of N2 adsorption on the surface of the C4AF sample was significantly lower. Similarly, the adsorption volume obtained for this sample from the Kr sorption isotherm was smaller (see Fig. 2a), confirming that the surface area of C4AF available for gas adsorption was small. These results suggest that C-S-H is composed of aggregates of plate-like particles, while C4AF is a bulky, non-porous material. This agrees with the structural data obtained by XRD and TEM observations (Fig. 1).
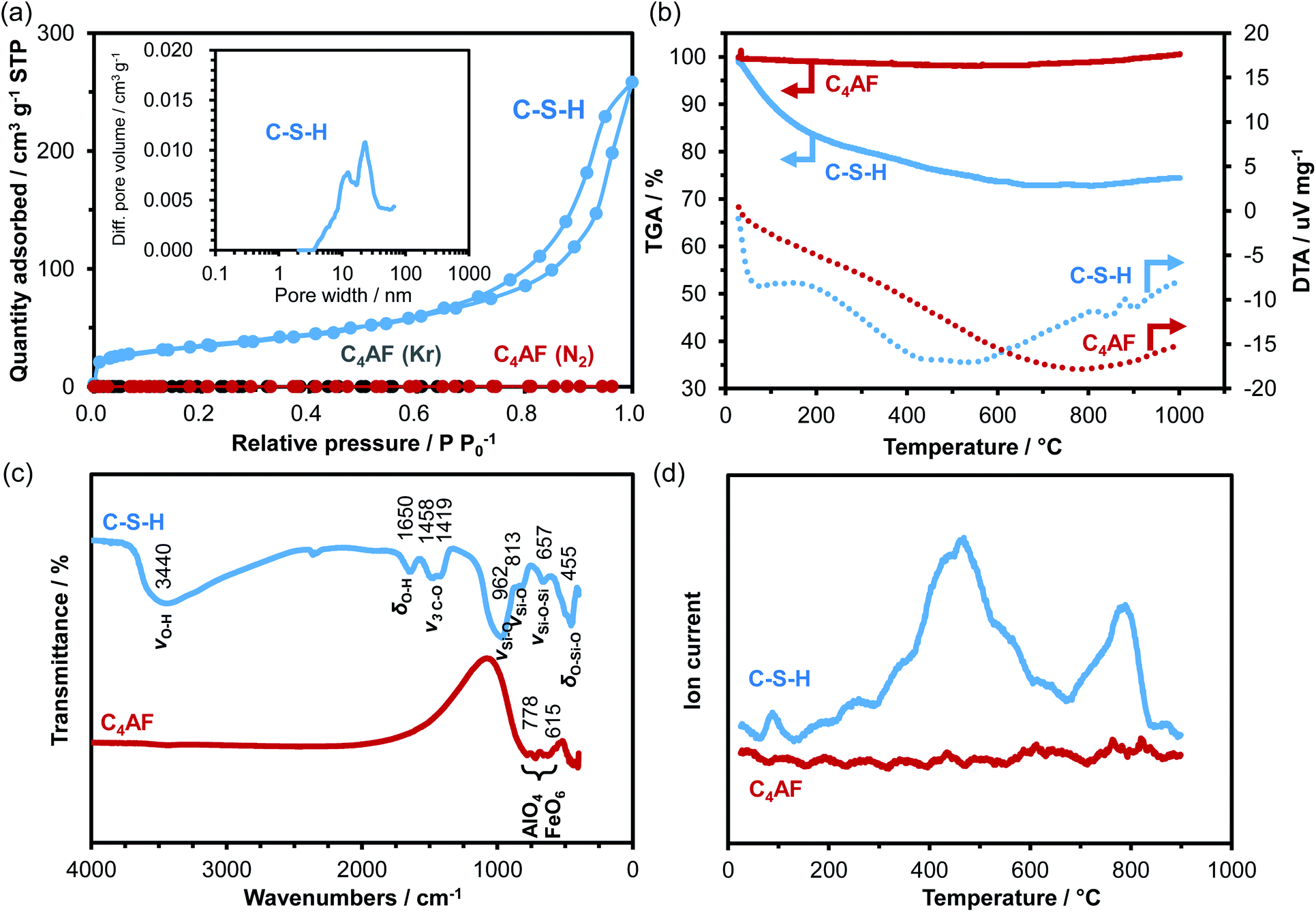 | ||
| Fig. 2 (a) Adsorption/desorption isotherms of nitrogen for C-S-H and C4AF and of krypton for C4AF. The inset is the pore size distribution of C-S-H. (b) TG and DTA curves and (c) FTIR spectra of C-S-H and C4AF. (d) Desorption profiles of CO2 for C-S-H and C4AF. | ||
The degrees of hydration of the C-S-H and C4AF samples were compared by thermal analysis (see Fig. 2b). The TGA curve for C-S-H indicated a weight loss of approximately 25% up to 600 °C, while the DTA curve exhibited two endothermic peaks at ca. 70 and 500 °C, attributable to dehydration and dehydroxylation, respectively.32,33 No weight loss was found above 600 °C, which is indicative of the absence of CaCO3 in the C-S-H sample. In contrast, the weight of the C4AF sample remained almost unchanged during heating from room temperature to 1000 °C, indicating that the as-synthesized sample was scarcely hydrated.
The results of FTIR analysis in Fig. 2c reveal the surface structures of the C-S-H and C4AF samples. The peak positions of all the bands observed for the C-S-H sample were in close agreement with the values reported by Lodeiro et al.34 The broad and strong peak at 3440 cm−1 is assigned to the stretching vibration mode of O–H groups. The peak at 1650 cm−1 is correlated to the bending vibration mode of O–H groups. These peaks are attributed to the O–H groups of both C-S-H and Ca-rich phase. The peaks of the carbonate group at 1458 and 1419 cm−1 correspond to the stretching vibrations of C–O bond groups. The appearance of these peaks is attributed to the adsorption of atmospheric CO2 on the C-S-H surface occurring during the measurements. The four peaks at 1000–450 cm−1, typical of silicates, are ascribed to the asymmetric stretching vibration of Si–O–Si bonds for the first three peaks and to their bending vibration for the last peak. Meanwhile, the C4AF sample showed only two peaks at 778 and 615 cm−1 corresponding to internal modes of AlO4 tetrahedra and FeO6 octahedra in the brownmillerite structure,35,36 which demonstrates the absence of any functional groups on the C4AF surface.
To gain further insight into the surface structure, TPD experiments were performed using CO2 as the probe molecule: the samples were loaded with CO2 at room temperature, followed by purging with Ar until no more CO2 desorption was detected. Afterwards, the temperature was increased linearly up to 900 °C. In Fig. 2d, two large distinctive peaks are observed for the C-S-H sample, while no peak is found for the C4AF sample. This difference is due not only to the different specific surface areas of the two samples, but also to their relative basicities toward CO2. (The pH of a suspension of 0.2 g of C-S-H in 200 mL deionized water was found to be 11.9, which was higher than that of a suspension of C4AF in deionized water measured under the same conditions.) The FTIR analysis results indicate that CO2 is easily adsorbed on the C-S-H surface, but this adsorption likely does not occur on the inert C4AF surface. It is also apparent from the TPD results that C-S-H has at least two types of O–H groups that vary in terms of basicity. This strongly suggests that the Ca-rich phase present in C-S-H has O–H groups, which function as a second adsorption site for CO2.
Electrical properties of C-S-H and C4AF
A 0.5 mm-thick membrane composed of C-S-H or C4AF particles and PTFE binder was characterized through DC and AC conductivity measurements for each sample. An electrochemical cell containing the membrane electrolyte with Pt/C electrodes was placed in flowing air saturated with water vapor at temperatures between 60 and 100 °C. In the current interruption method, a current of 2 to 10 mA cm−1 was galvanostatically applied to the cell. When the current was interrupted, the cell voltage initially dropped rapidly and then decreased slowly with time (see inset in Fig. 3a). The initial voltage drop, corresponding to the product of the current (I) and ohmic resistance (R), was measured with various current densities at temperatures between 60 and 100 °C, and the results obtained for C-S-H are presented in Fig. 3a as an example. At all the tested temperatures, the IR drop and current density had an almost linear relationship, the slope of which was defined as the ohmic resistance. The C4AF membrane electrolyte also exhibited a linear relationship between the IR drop and current density (data not shown). The DC conductivity can be calculated via| σ = L/(R × S) | (1) |
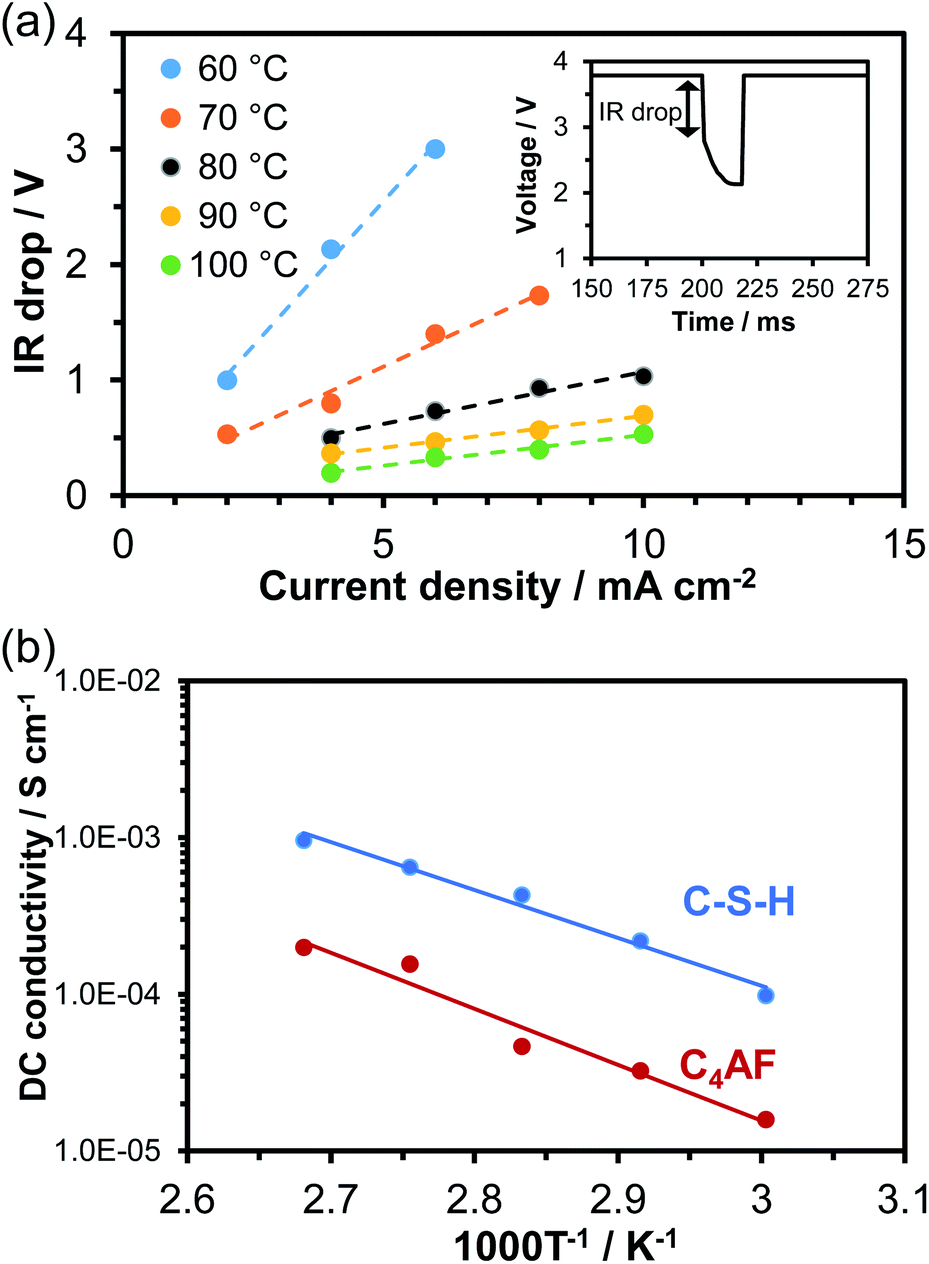 | ||
| Fig. 3 (a) IR drop as a function of the current density recorded for C-S-H at various temperatures. The inset is the transient change in voltage within a current interruption period. (b) Temperature dependence of DC conductivity for C-S-H and C4AF. | ||
Electrochemical impedance spectroscopy (EIS) is another powerful technique for measuring the ohmic resistance component of an electrolyte. The impedance spectra for cells using C-S-H and C4AF membrane electrolytes obtained at 60 °C are presented in Fig. 4a. At high frequencies, only the end section of the semicircle was observed for C-S-H, while an almost complete semicircle was obtained for C4AF. The high-frequency semicircle is often related to the ohmic resistance and the interfacial capacitance, rather than to the charge transfer resistance and the double layer capacitance, which will be discussed later. The ohmic resistance of the electrolyte was determined through equivalent circuit models, consisting of a resistor in parallel with a constant phase element (CPE) for C-S-H and a series resistor and a resistor-CPE parallel combination for C4AF, both of which were accompanied by a Warburg impedance element. The ohmic resistance of C-S-H was computed to be 602 Ω cm−2, which is in agreement with the ohmic resistance of this membrane obtained by the current interruption method. Comparatively, the ohmic resistance of C4AF was resolved into bulk and interfacial contributions: a bulk resistance of 845 Ω cm−2 and an interfacial resistance of 1900 Ω cm−2. The sum of these values, 2745 Ω cm−2, is close to the DC ohmic resistance of this membrane. Thus, the semicircle observed at high frequencies is attributable to the interfacial resistance, presumably owing to the grain boundary contribution. It is also possible that this resistance is much smaller for C-S-H than for C4AF, because C-S-H possesses a far higher specific surface area and a far larger amount of O–H groups than C4AF. Here, it should be noted that the Ca-rich phase was scattered on the C-S-H surface, as seen from the TEM image (see Fig. 1b). Thus, the Ca-rich phase does not contribute significantly to the reduction of the grain boundary resistance, due to a large gap between the phases. The ohmic resistances of these membranes above 60 °C were measured and estimated by similar procedures, and the AC conductivities were calculated viaeqn (1). The results are presented in Fig. 4b, all of which are in good agreement with the DC conductivities obtained under the same conditions. Accordingly, the higher sum of the bulk and grain boundary conductivity of C-S-H as compared to C4AF can be explained in terms of its lower grain boundary resistance. The ionic or electronic current generally passes through the grains and across the grain boundaries in polycrystalline materials (bulk conduction mechanism).37 However, recent studies suggest that ion carriers, especially protons, can migrate along the grain boundaries in nanomaterials, whose resistance is affected sensitively by the amount of adsorbed water, grain size, and porosity (interfacial conduction mechanism).38–40 Thus, these two paths for ion transport need to be considered on the basis of the conduction mechanism. In the impedance spectrum of C4AF, the grain boundary resistance was higher than the bulk resistance, and these components appeared individually. On the other hand, the impedance spectrum of C-S-H showed a single semicircle with a smaller diameter. These results, along with the characterization of the two materials, suggest that carrier transport occurs by the bulk conduction mechanism in C4AF and the interfacial conduction mechanism in C-S-H.
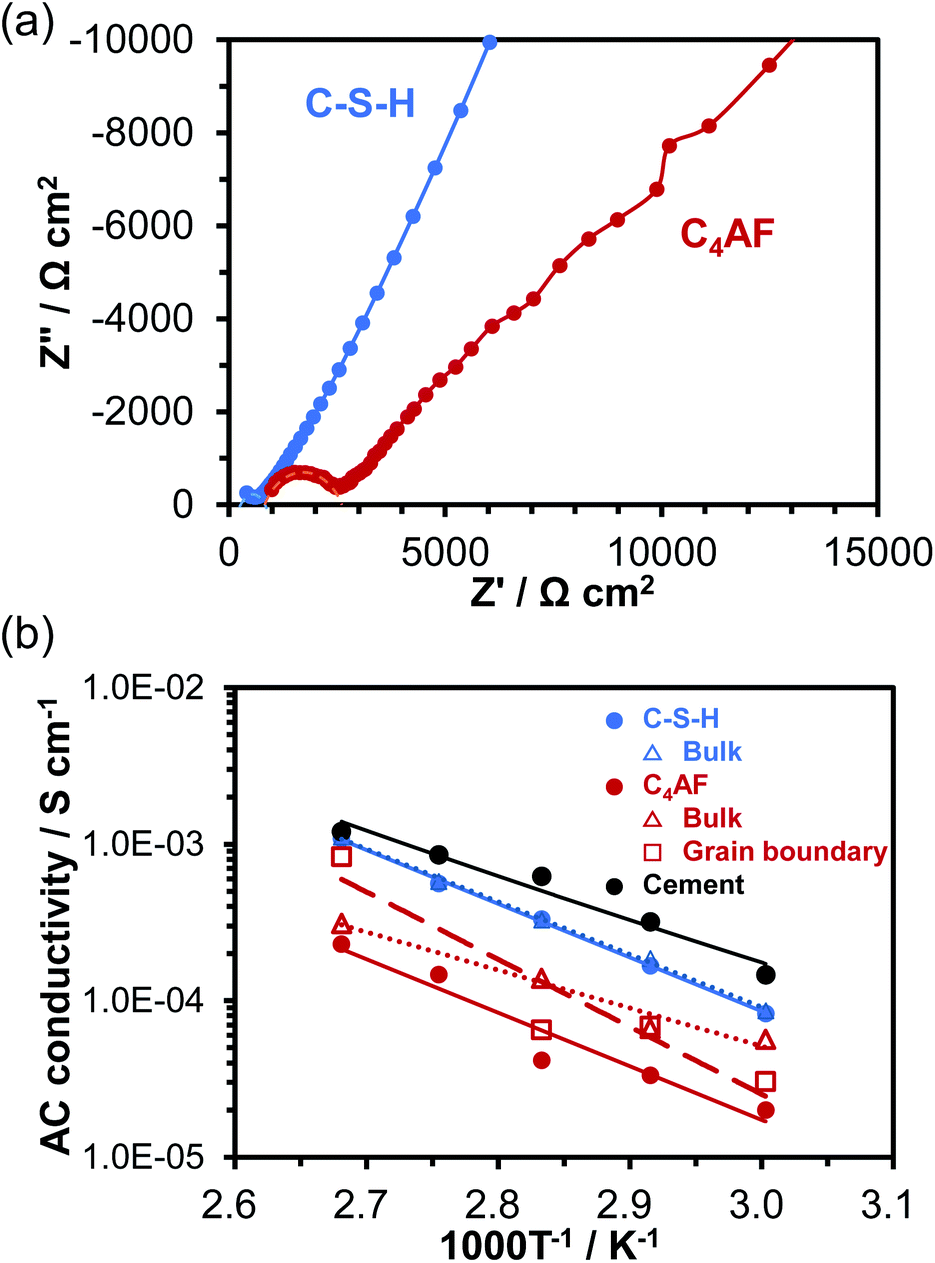 | ||
| Fig. 4 (a) Impedance spectra of C-S-H and C4AF at 60 °C. (b) Temperature dependence of AC conductivity for C-S-H and C4AF. | ||
Fig. 4b also includes the AC conductivity of the hydrated cement paste recorded in our previous study.11 A somewhat higher conductivity was obtained for the cement paste, with a slightly lower activation energy (0.57 eV) than the values for C-S-H. This difference occurs because cement paste contains alkali metal ions such as Na+ and K+, which bond to the silanol groups (Si–OH) present on the C-S-H surface.41 The effect of K+ ions on electrical conductivity will be further investigated in the next section.
Ionic conduction in C-S-H
The characterization results demonstrated that the structure and morphology of the synthesized C-S-H sample closely resembled those of the C-S-H formed in the hydrated cement paste. In addition, electrical measurements led to a new insight: that the C-S-H membrane had relatively high electrical conductivity. These findings prompted us to investigate ionic conduction in C-S-H. Fig. 5a plots the dependence of conductivity on PO2 at 100 °C. The conductivity was kept constant at 10−3 S cm−1 over the PO2 range from 1 to 10−23 atm, and exhibited no characteristics of p- or n-type electronic conductivity. Clearly, C-S-H is a pure ionic conductor under both oxidizing and reducing atmospheres. Ionic conduction in C-S-H was further investigated by fabricating two galvanic cells using the membrane as the electrolyte: wet air|electrolyte|dry air; wet H2 (PO2 = 10−23 atm)|electrolyte|wet air (PO2 = 0.21 atm). Fig. 5b plots the electromotive force (EMF) values recorded between 50 and 100 °C. The water vapor concentration cell always generated positive EMF values, which are defined as the potential of the electrode in wet air against the electrode in dry air. The pH value of the solution in equilibrium with C-S-H is reported to be in the range of ca. 10 to ca. 12, depending on the Ca/Si molar ratio of C-S-H.42,43 Assuming that hydroxide ions are the major charge carriers under such alkaline conditions, the following reaction is proposed for the wet air electrode:44| H2O + 1/2O2 + 2e− → 2OH− | (2) |
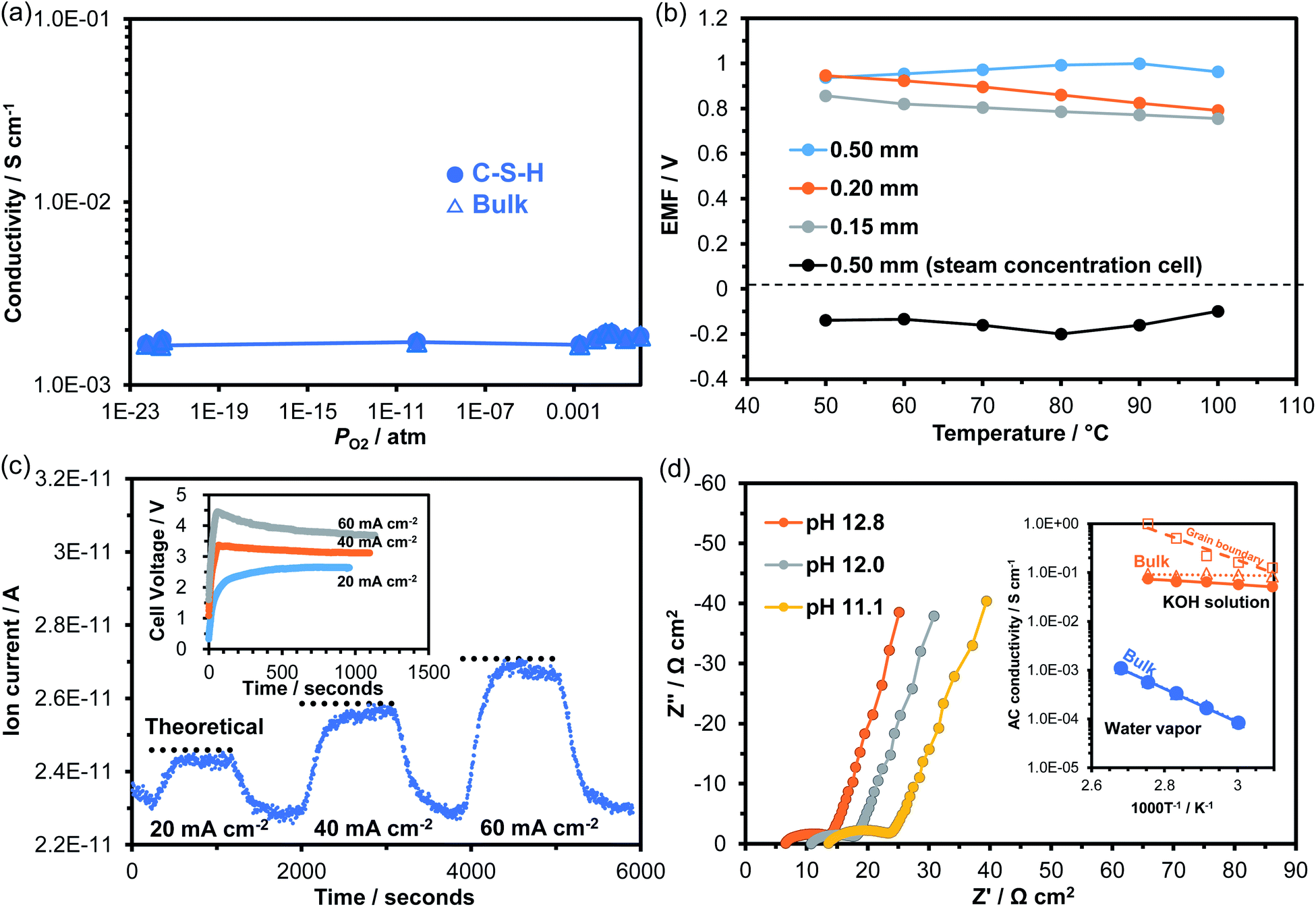 | ||
| Fig. 5 (a) Dependence of conductivity of C-S-H on partial pressure of oxygen at 100 °C. (b) EMF values obtained from the water vapor concentration cell and oxygen gas concentration cell at various temperatures. The working electrode is supplied with a flow of wet air or hydrogen. (c) Hydrogen quantity and voltage–time curves recorded during continuous electrolysis of water vapor with various current densities at 100 °C. (d) Impedance spectra of C-S-H operated in a KOH solution at varying pH at 100 °C. The inset is the temperature dependence of conductivity for C-S-H operated in the KOH solution and in water vapor. | ||
In the oxygen gas concentration cell, the H2 electrode showed opposite polarity potentials at all the tested temperatures, which can be explained through the following reaction in the hydroxide ion conductor:45,46
| H2 + 2OH− → 2H2O + 2e− | (3) |
A decrease in the EMF value with decreasing membrane thickness was observed in the oxygen gas concentration cell (Fig. 5b), which was due to gas leakage and the resultant fuel loss. Thus, the observed EMF values were lower than the theoretical values calculated by the Nernst equation, depending on the membrane state. To obtain more direct evidence of hydroxide ion conduction, water vapor electrolysis was performed using the C-S-H membrane electrolyte at 100 °C. The galvanostatic electrolysis characteristics are presented in Fig. 5c, along with the voltage–time curve at each current density. H2 was confirmed to be produced at the cathode, indicating that the H2 evolution reaction proceeded as follows:
| 2H2O + 2e− → H2 + 2OH− | (4) |
It was also confirmed that the amount of H2 formed roughly agreed with the theoretical values calculated by Faraday's law, strongly supporting hydroxide ion conduction in C-S-H. Nevertheless, degradation of H2 production was observed at a current density of 60 mA cm−2 in Fig. 5c. This was due to the application of high voltage to the cell (see inset in Fig. 5c), which may have caused decomposition of the electrolyte or corrosion of the electrode. Note that unlike the C-S-H formed in cement pastes, the C-S-H examined in this study does not contain any alkaline metal ions, thus ruling out these ions as charge carriers. However, the contribution of Ca2+ ions released from the Ca-rich phase to ionic conduction cannot be neglected.
From a practical standpoint, it is also important to determine the ionic conduction in C-S-H under alkaline conditions, because hydrated cement pastes contain alkaline metal ions.47,48 The impedance spectra of the C-S-H membrane were measured by adding KOH solutions with various pH values to the cell at 100 °C. The results are presented in Fig. 5d. All impedance spectra consisted of three components in series: ohmic resistance (intercept of the impedance line and real axis), grain boundary resistance (high-frequency part of arc), and electrode reaction resistance (low-frequency part of inclined line). Both the ohmic and grain boundary resistances decreased with increasing pH. These behaviors differ from those observed in the impedance spectrum for the alkaline-metal-free C-S-H membrane (see Fig. 4a). The difference is further emphasized by the much lower activation energy for ionic conduction (ca. 0.1 eV) estimated for the C-S-H membrane operated at a pH of 12.8, as compared to that operated in humidified air (see inset of Fig. 5d). This was due to the replacement of adsorbed water with KOH solution for the ion channels, which changes the interfacial conduction mechanism to vehicular diffusion or the Grotthuss mechanism for ionic conduction.37
These results demonstrated hydroxide ion conduction in C-S-H, which is useful for interpreting the electrochemical behavior of hydrated cement pastes. Given its relatively high ion conductivity, C-S-H can function as an electrolyte for a variety of galvanic cells formed at the cement–steel interface in reinforced concrete structures, resulting in corrosion of the steel due to self-discharge when a local cell is established. In this case, the corrosion rate is directly affected by the humidity and basicity at the cement–steel interface. Importantly, such a local cell is formed even in the absence of alkali metal and alkaline earth metal hydroxides in the cement paste. Also important is the mesoporous structure formed in C-S-H, which enhances the water adsorption capacity of this material through capillary condensation. Thus, the hydroxide ion conductivity of C-S-H is maintained high even under low relative humidity conditions, which is one of the causes of increased corrosion under such conditions.
Photoactive properties of C4AF
Although Fe-based photocatalysts have been studied extensively for their visible light response,12–14 there is no report on the photocatalytic activity of C4AF in the literature. Fig. 6a plots the dependence of C4AF membrane conductivity on PO2 at 100 °C. C4AF conductivity showed a negative correlation with PO2 at high PO2, indicating n-type electronic conductivity, but was independent of PO2 at PO2 values below 0.001 atm, suggesting that here, ion conductivity predominated. Thus, C4AF is identified as a mixed electronic and ionic conductor under oxidizing conditions. As described in the previous section, the bulk conduction was predominant in electrical conduction in C4AF. In addition, brownmillerite-type oxides possess a large number of oxygen vacancies, which potentially introduces various ionic and electronic defects not only to the surface, but also into the bulk of the oxides.49,50 The species and concentration of the ions introduced strongly depend on factors such as temperature, oxygen and water vapor partial pressures, and microstructure.40 Thus, one cannot at present determine which ions function as charge carriers; however, alkaline metal ions can be ruled out, as in the case of the C-S-H membrane.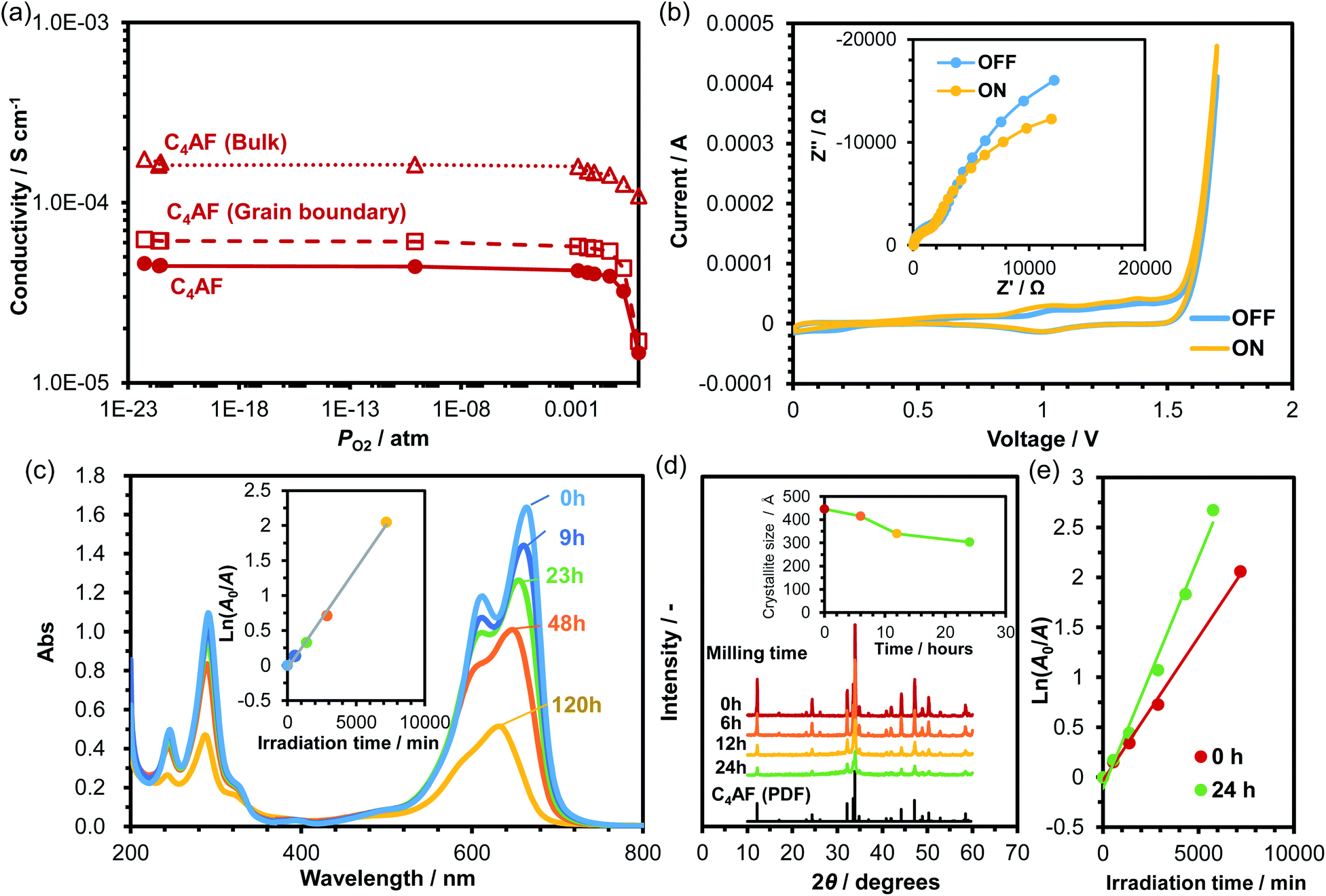 | ||
| Fig. 6 (a) Dependence of conductivity of C4AF on the partial pressure of oxygen at 100 °C. (b) CV curves for a water electrolysis cell with C4AF working electrode, Pt counter electrode, and NaOH solution electrolyte. The inset is the impedance spectra of the cell with and without irradiation. (c) Absorption spectra of MB during photocatalytic degradation under irradiation. The inset shows the natural logarithm of the relative absorbance A0/A against irradiation time. (d) XRD patterns and average crystallite size for milled C4AF. (e) Plots of the natural logarithm of the relative absorbance A0/A of MB against irradiation time for unground and ground C4AF. | ||
The electrode characteristics of C4AF as a photoanode were first validated by experimental demonstration of photovoltaic water splitting in a two-electrode cell consisting of a C4AF membrane anode, a Pt cathode, and a NaOH solution electrolyte. The CV profiles for this cell obtained under light and in the dark are presented in Fig. 6b, along with the corresponding impedance spectra recorded at a bias voltage of +1 V. Irradiation-induced enhancement effects were observed in both the cyclic voltammograms and impedance spectra, which are attributable to the decrease in the charge-transfer resistance due to irradiation. (In the impedance spectra, both the ohmic and grain boundary resistances were almost unchanged, regardless of the irradiation.) Next, the photocatalytic activity of C4AF was determined by degrading MB in deionized water under irradiation. The change in the absorbance of MB with irradiation time is shown in Fig. 6c. The peaks in the 600–700 nm range, which correspond to the conjugated π-system, and the peak at 291 nm, assignable to the aromatic ring, decreased with time. Also, the color of the MB solution changed to light purple after irradiation for 120 h. These results indicate that C4AF shows photocatalytic activity for the degradation of MB. From a plot of the natural logarithm of the relative absorbance A0/A against irradiation time (see inset of Fig. 6c), the first-order rate constant was calculated to be 3 × 10−4 min−1, which is comparable to that of α-Fe2O3 (3 × 10−4 min−1) and about half that of brownmillerite Ca2Fe2O5 (7 × 10−4 min−1).14 Note that the specific surface area of C4AF was 0.8 m2 g−1, which is lower than those of Fe2O3 (5.9 m2 g−1) and Ca2Fe2O5 (1.8 m2 g−1). We cannot further compare the photocatalytic activities of different materials, since only the rate constant and BET specific surface area have been reported in the literature. Thus, an attempt was made to increase the specific surface area of C4AF by ball milling. The particle size of C4AF decreased with grinding time, as characterized by the decrease in the XRD peak intensity and in the average crystallite size (Fig. 6d). After a 24 h grinding run, the specific surface area of C4AF increased to 2.7 m2 g−1. (No additional effect on the specific surface area was observed past 24 h.) Fig. 6e compares the rate constants for MB degradation in the case of unground and ground C4AF. The rate constant estimated for ground C4AF was 5 × 10−4 min−1, which is higher than that of Fe2O3, but lower than that of Ca2Fe2O5, despite the larger surface area of C4AF. One possible reason for the difference in photoactivity between C4AF and Ca2Fe2O5 is the occupation of the tetrahedral sites of the brownmillerite structure by Al3+ ions, which may reduce the changeable valence state of Fe3+ ions in the octahedral sites.51
The occurrence of the photocatalytic reaction on the C4AF surface is not negligible in cement clinkers, but extremely rare in cement pastes, because portions of C4AF react with gypsum (CaSO4) to form ettringite [Ca6(Al,Fe) (SO4)3·26H2O].52 However, if C4AF photocatalysis is actively applied to the removal of soil and pollutants from concrete surfaces, then it is required to increase the C4AF content of the cement clinker so as to form katoite [Ca3(Al,Fe)2(OH)12],52 which has a higher potential as a photocatalyst than ettringite, because the present XRD results showed the coexistence of the brownmillerite and hydrogarnet (katoite) phases in the photoactive sample during MB photodegradation measurements (data not shown). If realized, this may eliminate the need to coat concrete surfaces with a photocatalyst such as TiO2 for self-cleaning purposes.
Conclusions
C-S-H and C4AF were prepared, characterized, and assessed in terms of ionic conductive and photocatalytic characteristics, respectively. C-S-H was composed of aggregates of plate-like particles with a high specific surface area and a large concentration of basic O–H groups on their surface. Meanwhile, C4AF was a bulky non-porous material with a brownmillerite structure. C-S-H and C4AF showed different electrical properties at temperatures between 60 and 100 °C: bulk resistance was dominant in the total electrical resistance of C-S-H; grain boundary resistance contributed significantly to the total electrical resistance of C4AF, especially at low temperatures. Consequently, the electrical conductivity of C-S-H was much higher than that of C4AF. C-S-H was a pure ion conductor in the PO2 range of 10−23 to 1 atm, where ionic conduction occurs by the migration of hydroxide ions. The ion conductivity was determined to be 10−4 S cm−1 at 60 °C and 10−3 S cm−1 at 100 °C, which are high enough to run gas concentration cells. C4AF was a mixed ionic and n-type electronic conductor at high PO2. The Fe3+ ions in the octahedra induced photoanodic and photocatalytic activity for the water oxidation reaction and MB degradation, respectively. The ion conductivity of C-S-H and the photocatalytic activity of C4AF would contribute to the stability of concrete structures and the functionality of concrete surfaces.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by a Kakenhi Grant-in-Aid (No. 17H01895) from the Japan Society for the Promotion of Science (JSPS), and by a grant (No. JPMJCR18R2) from the CREST Program of the Japan Science and Technology Agency (JST). The authors would like to thank Prof. Hikaru Nakamura (Nagoya University) for his inspiration.References
- P. C. Aïtcin, High performance concrete, CRC Press, 1998 Search PubMed.
- M. Stefanoni, U. Angst and B. Elsener, Cem. Concr. Res., 2018, 103, 35–48 CrossRef CAS.
- P. K. Mehta, ACI Symp. Publ., 1980, vol. 65, pp. 1–20 Search PubMed.
- R. J. Flatt, N. Roussel and C. R. Cheeseman, J. Eur. Ceram. Soc., 2012, 32, 2787–2798 CrossRef CAS.
- C. Medina, I. F. Sáez del Bosque, E. Asensio, M. Frías and M. I. Sánchez de Rojas, Mater. Struct., 2016, 49, 4595–4607 CrossRef CAS.
- R. Yu, P. Spiesz and H. J. H. Brouwers, Cem. Concr. Compos., 2015, 55, 383–394 CrossRef CAS.
- M. Castellote, L. Fernandez, C. Andrade and C. Alonso, Mater. Struct., 2009, 42, 515–525 CrossRef CAS.
- D. C. MacLaren and M. A. White, J. Chem. Educ., 2003, 80, 623–635 CrossRef CAS.
- M. M. Hassan, H. Dylla, L. N. Mohammad and T. Rupnow, Constr. Build. Mater., 2010, 24, 1456–1461 CrossRef.
- A. Folli, I. Pochard, A. Nonat, U. H. Jakobsen, A. M. Shepherd and D. E. Macphee, J. Am. Ceram. Soc., 2010, 93, 3360–3369 CrossRef CAS.
- M. Nagao, K. Kobayashi, T. Hori, Y. Li and T. Hibino, Materials, 2019, 12, 3701 CrossRef CAS.
- S. Kumar, S. Tonda, B. Kumar, A. Baruah and V. Shanker, J. Phys. Chem. C, 2013, 117, 26135–26143 CrossRef CAS.
- K. Shetty, L. Renuka, H. P. Nagaswarupa, H. Nagabhushana, K. S. Anantharaju, D. Rangappa, S. C. Prashantha and K. Ashwini, Mater. Today: Proc., 2017, 4, 11806–11815 Search PubMed.
- J. Ikawa, S. Komai, D. Hirabayashi, F. Ya'ning, M. Lei and K. Suzuki, Proc. Int. Symp. EcoTopia Sci. 2007, 2007, vol. 7, pp. 959–964 Search PubMed.
- A. Cuesta, I. Santacruz, S. G. Sanfélix, F. Fauth, M. A. G. Aranda and A. G. De La Torre, Constr. Build. Mater., 2015, 101, 818–827 CrossRef.
- X. Huang, S. Hu, F. Wang, L. Yang, M. Rao and Y. Tao, ACS Sustainable Chem. Eng., 2019, 7, 6792–6801 CrossRef CAS.
- B. Z. Dilnesa, B. Lothenbach, E. Wieland, R. Dähn, A. Wichser and K. L. Scrivener, Conmod 2010, Rilem, Bagneux, 2010 Search PubMed.
- B. Z. Dilnesa, E. Wieland, B. Lothenbach, R. Dähn and K. L. Scrivener, Cem. Concr. Res., 2014, 58, 45–55 CrossRef CAS.
- I. Maruyama and G. Igarashi, J. Adv. Concr. Technol., 2014, 12, 200–213 CrossRef CAS.
- A. Hajimohammadi, T. Ngo, P. Mendis, A. Kashani and J. S. J. van Deventer, J. Cleaner Prod., 2017, 147, 330–339 CrossRef CAS.
- S. M. Park and J. G. Jang, J. Nucl. Mater., 2018, 505, 159–164 CrossRef CAS.
- I. G. Richardson, Cem. Concr. Res., 2008, 38, 137–158 CrossRef CAS.
- I. G. Richardson, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, B70, 903–923 CrossRef PubMed.
- I. G. Richardson, Cem. Concr. Res., 2004, 34, 1733–1777 CrossRef CAS.
- H. D. Megaw and C. H. Kelsey, Nature, 1956, 177, 390–391 CrossRef CAS.
- I. G. Richardson, Cem. Concr. Res., 1999, 29, 1131–1147 CrossRef CAS.
- I. G. Richardson, J. Skibsted, L. Black and R. J. Kirkpatrick, Adv. Cem. Res., 2010, 22, 233–248 CrossRef CAS.
- N. Meller, C. Hall and J. S. Phipps, Mater. Res. Bull., 2005, 40, 715–723 CrossRef CAS.
- A. Crumbie, G. Walenta and T. Füllmann, Cem. Concr. Res., 2006, 36, 1542–1547 CrossRef CAS.
- W. Gac, A. Patrykiejew and S. Sokołowski, Thin Solid Films, 1997, 298, 22–32 CrossRef CAS.
- T. Horikawa, D. D. Do and D. Nicholson, Adv. Colloid Interface Sci., 2011, 169, 40–58 CrossRef CAS.
- B. El-Jazairi and J. M. Illston, Cem. Concr. Res., 1977, 7, 247–257 CrossRef CAS.
- J. I. Bhatty, Thermochim. Acta, 1986, 106, 93–103 CrossRef CAS.
- I. García Lodeiro, A. Fernández-Jimenez, A. Palomo and D. E. Macphee, Cem. Concr. Res., 2010, 40, 27–32 CrossRef.
- I. J. Moraes, M. C. Terrile, O. R. Nascimento, M. S. Li, R. H. P. Francisco and J. R. Lechat, Mater. Res. Bull., 1992, 27, 523–529 CrossRef CAS.
- T. Nishida, S. Kubuki, M. Shibata, Y. Maeda and T. Tamaki, J. Mater. Chem., 1997, 7, 1801–1806 RSC.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- S. Kim, H. J. Avila-Paredes, S. Wang, C. T. Chen, R. A. Souza, M. Martin and Z. A. Munir, Phys. Chem. Chem. Phys., 2009, 11, 3035–3038 RSC.
- S. Miyoshi, Y. Akao, N. Kuwata, J. Kawamura, Y. Oyama, T. Yagi and S. Yamaguchi, Chem. Mater., 2014, 26, 5194–5200 CrossRef CAS.
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- S.-Y. Hong and F. P. Glasser, Cem. Concr. Res., 1999, 29, 1893–1903 CrossRef CAS.
- G. M. N. Baston, A. P. Clacher, T. G. Heath, F. M. I. Hunter, V. Smith and S. W. Swanton, Mineral. Mag., 2012, 76, 3045–3053 CrossRef.
- B. Lothenbach and A. Nonat, Cem. Concr. Res., 2015, 78, 57–70 CrossRef CAS.
- K. Tadanaga, Y. Furukawa, A. Hayashi and M. Tatsumisago, Adv. Mater., 2010, 22, 4401–4404 CrossRef CAS PubMed.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS.
- Z. F. Pan, L. An, T. S. Zhao and Z. K. Tang, Prog. Energy Combust. Sci., 2018, 66, 141–175 CrossRef.
- B. Šavija and M. Luković, Constr. Build. Mater., 2016, 117, 285–301 CrossRef.
- S. O. Ekolu, Constr. Build. Mater., 2016, 127, 306–320 CrossRef CAS.
- C. P. Jijil, S. M. Unni, K. Sreekumar and R. N. Devi, Chem. Mater., 2012, 24, 2823–2828 CrossRef CAS.
- C. P. Jijil, S. N. Bhange, S. Kurungot and R. N. Devi, ACS Appl. Mater. Interfaces, 2015, 7, 3041–3049 CrossRef CAS PubMed.
- Y. Yang, Z. Cao, Y. Jiang, L. Liu and Y. Sun, Mater. Sci. Eng., B, 2006, 132, 311–314 CrossRef CAS.
- N. Meller, C. Hall, A. C. Jupe, S. L. Colston, S. D. M. Jacques, P. Barnes and J. Phipps, J. Mater. Chem., 2004, 14, 428–435 RSC.
| This journal is © The Royal Society of Chemistry 2020 |