
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel confined in 2D earth-abundant oxide layers for highly efficient and durable oxygen evolution catalysts†
Yayun
Pu
ab,
Matthew J.
Lawrence
 a,
Veronica
Celorrio
c,
Qi
Wang
d,
Meng
Gu
d,
Zongzhao
Sun
b,
Leonardo Agudo
Jácome
e,
Andrea E.
Russell
f,
Limin
Huang
*b and
Paramaconi
Rodriguez
*a
a,
Veronica
Celorrio
c,
Qi
Wang
d,
Meng
Gu
d,
Zongzhao
Sun
b,
Leonardo Agudo
Jácome
e,
Andrea E.
Russell
f,
Limin
Huang
*b and
Paramaconi
Rodriguez
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: P.B.Rodriguez@bham.ac.uk
bDepartment of Chemistry, Southern University of Science and Technology, No. 1088 Xueyuan Blvd, Shenzhen, Guangdong 518055, China. E-mail: huanglm@sustech.edu.cn
cDiamond Light Source Ltd, Diamond House, Harwell Campus, Didcot, OX11 0DE, UK
dDepartment of Materials Science and Engineering, Southern University of Science and Technology, No. 1088 Xueyuan Blvd, Shenzhen, Guangdong 518055, China
eBundesanstalt für Materialforschung und-prüfung (BAM), Department Materials Engineering, Unter Den Eichen 87, 12205 Berlin, Germany
fSchool of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 22nd June 2020
Abstract
Low cost, high-efficiency catalysts towards water splitting are urgently required to fulfil the increasing demand for energy. In this work, low-loading (<20 wt%) Ni-confined in layered metal oxide anode catalysts (birnessite and lepidocrocite titanate) have been synthesized by facile ion exchange methodology and subjected to systematic electrochemical studies. It was found that Ni-intercalated on K-rich birnessite (Ni-KMO) presents an onset overpotential (ηonset) as low as 100 mV and overpotential at 10 mA cm−2 (η10) of 206 mV in pH = 14 electrolyte. By combining electrochemical methods and X-ray absorption and emission spectroscopies (XAS and XES), we demonstrate Ni sites are the active sites for OER catalysis and that the Mn3+ sites facilitate Ni intercalation during the ion-exchange process, but display no observable contribution towards OER activity. The effect of the pH and the nature of the supporting electrolyte on the electrochemical performance was also evaluated.
1. Introduction
The availability of a carbon neutral sustainable energy supply has become remarkably urgent in recent decades. Electrochemical water splitting, using renewable energy sources, offers an attractive route to chemical energy storage. However, one of the current limitations for the implementation of this technology is related to the inefficiency of the oxygen evolution reaction (OER) catalyst.1–3 Metal oxide catalysts based on precious metals such as IrO2 and RuO2 have been commonly used. However, their high cost and low abundance is a significant hindrance. Therefore, in the last decade there has been an enormous effort from the scientific community towards the development of inexpensive and earth-abundant OER catalysts.4–9Amongst a number of different catalysts reported, layered metal oxide/hydroxide nanoparticles of earth abundant elements are of particular interest in electrochemical water splitting. Compared with the more typically employed RuO2 and IrO2 electrocatalysts, the M-based oxides/hydroxides (M = Ni, Co, Fe, Mn), as well as their oxyhydroxide and double metal hydroxides, are attractive as they possess extremely high surface areas, providing a large number of exposed active sites, and enhanced electronic conductivity after exfoliation.10–14 Such layered oxide/hydroxides consist of MO6 host layers stabilized by charge-balancing ions accommodated into the interlayer spacing. The M–O layers fulfill both roles of skeleton, for architecture, and active sites, leading to insufficient atomic efficiency. To enhance the performance and efficiency of these catalysts previous reports have presented the preparation of ultrathin two-dimensional (2D) nanosheets, aiming at the exposure of more active sites.5,8 Improving the atomic efficiency of active atoms, such as Ru, Ir, Ni or Co, is economically attractive for large-scale industrial applications.15,16 In the literature a variety of strategies have been implemented, including synthesis of nanoclusters and even single atoms anchored on carbon-based and metal oxides-based supports.17,18 Although, to date, very little attention has been dedicated to the ions in the channels formed between the 2D layered oxide structures. Catalytic ions in between the layers can be arranged in-plane and immobilized by the opposite charge of the 2D host layer (Fig. 1a).19 The 2D channels fundamentally act as ideal electrochemical nanoreactors providing ample interlayer spacing for free transportation of reactants and products and confinement of the under-coordinated active atoms.20,40 Previous work by Thenuwara et al., show that Ni-intercalated on birnessite is a potential catalyst for the OER at pH = 14.40
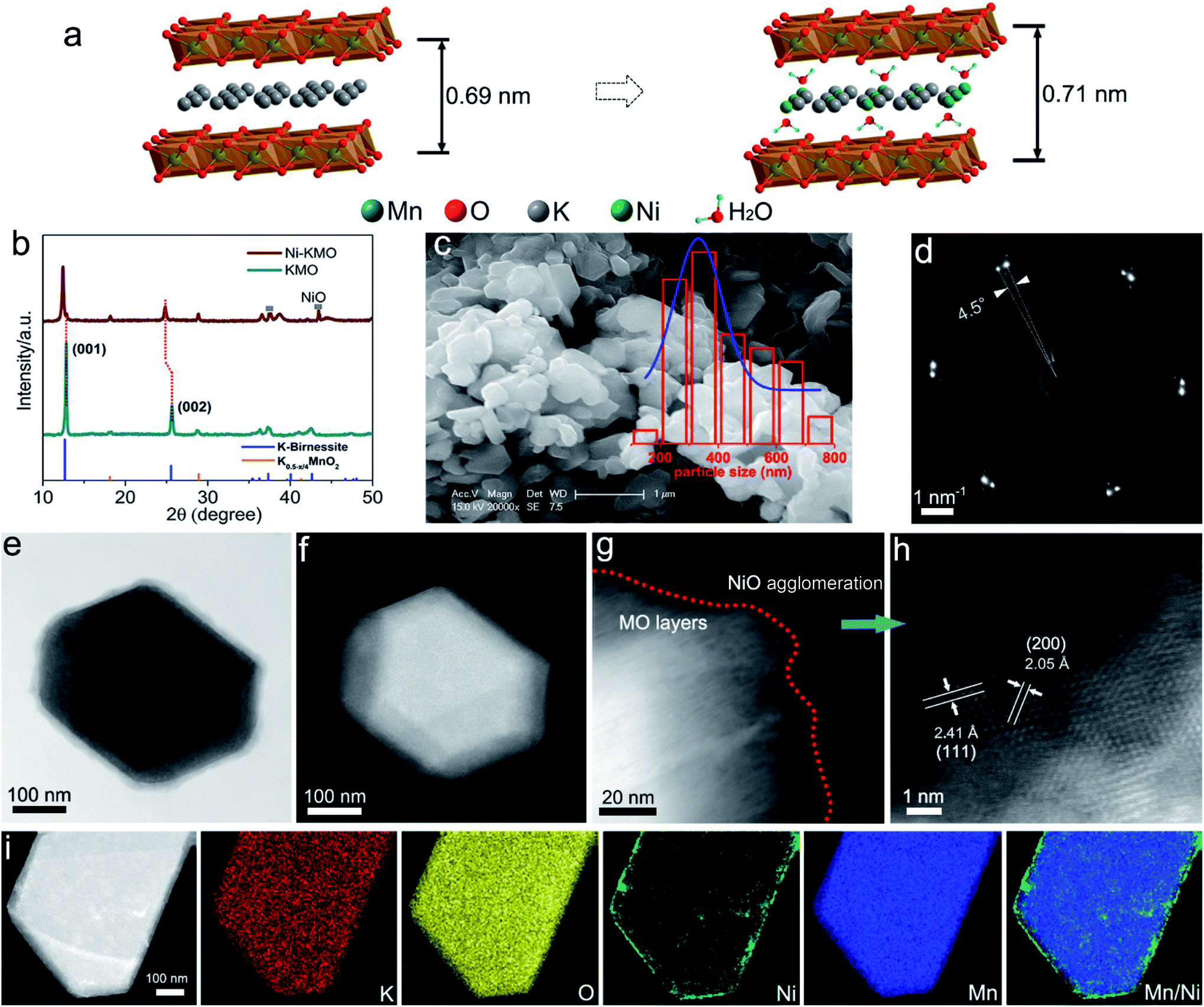 | ||
| Fig. 1 (a) Schematic illustration of the impact of Ni intercalation into layered K-birnessite manganese oxide. (b) X-ray diffraction patterns of Ni-KMO and pristine KMO. (c) SE-SEM image of Ni-KMO nanoplates. The particle size distribution histogram is inset. (f) SAD diffraction pattern (ZA = [001]Ni-KMO) of particle in (e). (e) Bright field (BF) and (f) HAADF-STEM images of Ni-KMO nanoparticle seen along [001] zone axis (ZA). HAADF-STEM images (g), and (h) enlarged Ni agglomeration at the edge of a Ni KMO nanoplate. (i) HAADF-STEM image of single Ni-KMO nanoplate with corresponding EDS elemental mapping. | ||
An important parameter affecting the electrochemical performance of catalysts during the OER is the pH. It is well known that the pH and composition of the electrolyte have a significant impact on the performance and durability of the catalyst during the OER. Establishing correlations between pH and measured voltammetric profiles facilitates the deduction of the active sites for OER and the evolution of adsorption/desorption processes and species on the surface. Takashima et al. reported that the instability of active Mn3+ at pH <9 is the key factor in the observed poor activity of δ-MnO2 toward the OER, which requires additional potential to electrochemically oxidize Mn2+ to Mn3+.21 Koper's group proposed the surface deprotonation mechanism of NiOOH during OER catalysis based on the pH sensitivity observed.22 With that in mind, the catalytic activity of confined catalysts in layered structures, where reactant diffusion is rigorously restricted, would be strongly affected by the pH dominated deprotonation kinetics.
The manuscript describes the synthesis and full characterization of Ni ions confined to the inter-layer space in 2D layered MnO nanoparticles prepared via a facile ion exchange strategy using NiCl2 as a precursor. The characterization of the materials using XAS/XES provided comprehensive understanding of the role of Mn3+ in the Ni intercalation process, the oxidation state of the species and the different Ni coordination/structures obtained for Ni-KMO and Ni-HMO. We demonstrate the catalytic activity towards the OER and the long term durability of the catalyst. In addition, we also present the reaction mechanism for the OER on the Ni-confined catalyst based on an exhaustive study of the catalyst under different conditions, e.g. pH and scan rate.
2. Results and discussion
2.1 Characterization of Ni-confined KMO
Ni-exchanged K0.45MnO2 (Ni-KMO) was prepared by mixing K-rich birnessite (K0.45MnO2) and NiCl2 solution at ambient temperature, as described in the Experimental section (ESI†). Fig. 1b presents the XRD patterns of the as-prepared KMO and after Ni exchange. From the XRD pattern it was determined that the K-birnessite possessed a layered structure with an interlayer spacing of 0.69 nm (AMCSD 0001302).23 The same layered structure was retained after the Ni exchange process, with XRF measurements suggesting that Ni cations partially replaced K ions in the interlayer spacing (Table 1). Such intercalation resulted in the enlargement of the interlayer distances (001) and (002) along the c axis, demonstrated by the decrease in 2θ values for those diffraction signals at 2θ = 12.43° and 24.84°. The SEM (Fig. 1c) and STEM images (Fig. 1d, e and g–i) of Ni-KMO show plate-like nanoparticles with facets with dimensions between 300–400 nm with near-identical morphology to that of KMO (Fig. S1†). The bright field (BF) and high angle annular darkfield (HAADF) images in Fig. 1e and f show a single nanoparticle comprising stacked layers of KMO. In Fig. 1d, the selected area diffraction (SAD) pattern shows a splitting of the reflections, which can be interpreted as an in-plane rotation of 4.5° (marked at the Ni-KMO (110) reflection) between two sets of layers in the nanoparticle. The HAADF-STEM image in Fig. 1h suggests that the Mn–O framework retained a layered structure, which is consistent with the XRD results. In addition to the Ni atoms inserted into the interlayer channels, aggregation of Ni at the edges of the nanoplate crystals, generating a few nanometer-thick region, is observed in the HAADF images and EDS elemental maps (Fig. 1i (contrast the Ni and Mn HAADF images) and Fig. S2 and S3†). The additional XRD signals observed for Ni-KMO at 37.4° and 43.4°, which can be assigned to (111) and (200) of NiO (JCPDS 73-1519), are in agreement with STEM results.| Samples | Ni (at%) | K (at%) | Mn (at%) | Ti (at%) |
|---|---|---|---|---|
| a TGA was performed to determine the water amount in Ni-intercalated layered metal oxides (Fig. S24). | ||||
| Ni0.16K0.2MnO2·0.7H2O | 11.67 ± 0.18 | 14.46 ± 0.2 | 73.87 ± 0.37 | — |
| Ni0.33K0.04MnO2·0.9H2O | 24.1 ± 0.4 | 2.69 ± 0.09 | 71.26 ± 0.5 | — |
| Ni0.033Ti0.91O2·0.6H2O | 3.60 ± 0.07 | — | — | 96.40 ± 0.07 |
2.2 XAS analysis
The adsorption energies of *OH and O* are predominantly dependent on the electronic configuration of the Ni surfaces.24 Therefore, the local environment of the Ni sites and associated species determine the corresponding electrochemical activity of these materials. X-ray absorption spectroscopy (XAS) was implemented to determine the oxidation state and coordination environment of Ni and the Mn-oxide host structure and to enable correlation of these parameters with the underlying OER performance of Ni-KMO. The characteristic XANES spectra of metallic Mn, standard Mn-oxide compounds with oxidation states spanning the range Mn+2 to Mn+4 and the synthesized X–K(H)MO nanoplates (where X= H or Ni) are presented in Fig. 2a.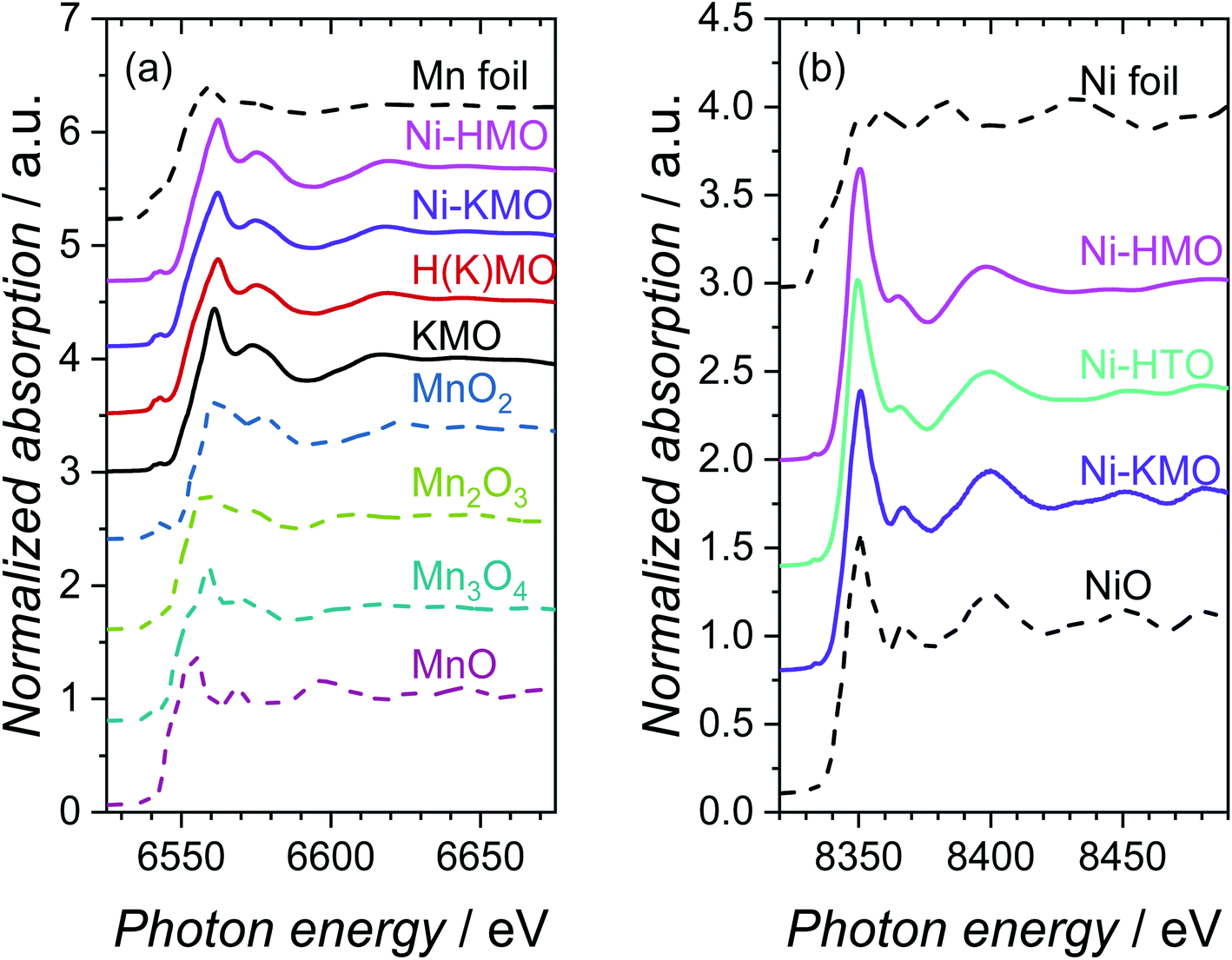 | ||
| Fig. 2 Normalized XANES spectra measured at the (a) Mn K-edge and (b) Ni K-edge for KMO, H(K)MO, Ni-KMO, Ni-HMO and Ni-HTO catalysts, in addition to relevant standard oxide compounds and metal foils. References, represented by dashed lines, are shown for comparison. | ||
The edge positions, determined by the maximum of the first derivative, for Mn foil, MnO, Mn3O4, Mn2O3 and MnO2 were observed at 6539.0, 6544.2, 6546.7, 6547.7 and 6552.5 eV, respectively, in agreement with existing literature values.25–27 The measured edge positions for KMO, H(K)MO and Ni-KMO and Ni-HMO were 6549.3, 6550.3, 6549.6 and 6552.0 eV, respectively. The normalised absorption pre-edge features of the manganese oxide derivatives and relevant Mn K-edge reference oxide compounds are presented in Fig. S4.† An increase in pre-edge peak energy is observed with an increase in Mn oxidation state (Fig. S4a†). The nature of the pre-edge features of the spectra at the Mn K-edge suggest that there is a contribution from Mn2+, Mn3+ and Mn4+ to the overall structure and the intermediate mean Mn oxidation state is between +3 and +4. Thus, X-ray emission spectroscopy (XES) was employed to obtain more accurate Mn oxidation state information. The measured XES spectra and corresponding fits, a function of the sum contributions of the standard Mnn+ oxide compounds, are presented in Fig. S5.† The mean Mn oxidation state of pristine KMO was determined as +3.9 (Table S1†). A decrease in mean oxidation state was observed upon cation exchange with both H+ and Ni2+, with the greatest decrease observed for H(K)MO. The fraction of Mn4+ is effectively the same for Ni-KMO and KMO, while the Mn3+ fraction decreases upon Ni-exchange. This suggests that Ni2+ intercalation is more favorable at those Mn3+ sites, which are commonly accompanied by defects that contribute to the overall negative charge of Mn–O layers.8 The defects with higher Gibbs energy favor the ion exchange reaction of K with Ni. The concentration of Mn3+ increases significantly in H(K)MO relative to KMO, thus more Ni cations were intercalated into Ni-HMO, as confirmed by the XRF analysis (Table 1).
The characteristic XANES spectra measured of metallic Ni, Ni-oxide reference compound, Ni-KMO, Ni-HTO and Ni-HMO are presented in Fig. 2b. The edge position of the Ni foil and NiO reference were determined as 8333.0 eV and 8345.2 eV, respectively. The absorption edges of Ni-KMO, Ni-HTO and Ni-HMO were determined as 8345.3, 8345.9 and 8346.0 eV respectively, suggesting a Ni oxidation state of +2. For Ni-KMO the XANES spectrum appears identical to that of the NiO reference, indicative of the presence of NiO structure in Ni-KMO. While slightly different spectrum of Ni-HMO compared to Ni-KMO suggesting a different Ni2+ structure, as confirmed by the fitted EXAFS results below. There are no significant differences in pre-edge features observed at the Ni K-edge (Fig. S4b†).
The Mn K-edge EXAFS Fourier transforms (FTs) of the synthesised KMO, H(K)MO, Ni-KMO and Ni-HMO nanosheets and reference samples are presented in Fig. 3a. The first peak at ∼1.5 Å (not phase shift corrected) is due to the Mn–O scattering and the second peak at ∼2.5 Å (not phase shift corrected) is due to Mn–Mn/Mn–Ni scattering (Tables S2, S3 and Fig. S6†). The two Mn scattering distances are generated by the two distinct Mn birnessite lattice sites: edges and face-centers. The Ni K-edge EXAFS FT of Ni-KMO is near-identical to the NiO reference compound. The first shell coordination at ∼1.7 Å (not phase shift corrected) corresponds to Ni–O whilst the second shell coordination at ∼2.6 Å (not phase shift corrected) corresponds to Ni–Ni scattering (Table S4†). The relative amplitude of the second shell peak for Ni-KMO compared to NiO suggests that the former comprises NiO nanoparticles. This is not the case for Ni-HTO and Ni-HMO, where the relative amplitude of the second shell peak differs from that of NiO, suggesting the formation of a different structure (Tables S5 and S7†).
 | ||
| Fig. 3 EXAFS FTs of k3χ(k) functions for KMO, H(K)MO, Ni-KMO, Ni-HMO and Ni-HMO nanosheets and standard oxide reference compounds at (a) Mn K-edge and (b) Ni K-edge. References are shown for comparison. | ||
In summary, the ex situ XAS/XES results clarify the importance of Mn3+ on the Ni-exchange intercalation. The Ni-content in each of the samples is proportional to the concentrations Mn3+ sites. For all the prepared samples, Ni is present in the oxidation state (+2), but different Ni coordination/structures were obtained for Ni-KMO and Ni-HMO.
3. Electrochemical performance of Ni-confined catalyst on KMO
3.1 Evaluation of the electrochemical activity and durability
The catalytic activity of Ni-KMO was evaluated over the pH range 8–14, to provide both assessment of the activity of the electrocatalysts for the OER and enable deeper understanding of the mechanism behind this activity. Fig. 4a–c show the stable iR-corrected voltammetric profiles of the Ni-KMO catalyst as a function of the pH, with the voltammograms shown being those obtained after 30 consecutive cycles; the evolution of the curves over the 30 scans is presented in Fig. S10.† Current densities are reported as mA cm−2 normalized by the geometric area of the electrode. As can be seen, the OER onset overpotential (ηonset) increases with decreasing pH, from 130 mV at pH 14 to 220 mV at pH 12. (Note that all voltammetric responses are reported vs. RHE and, thus, differences are not attributable to changes to the potential of the reference electrode.). At the same time, the overpotential at 10 mA cm−2 (η10) was 240 mV at pH 14, which is significantly smaller than the η10 = 1090 mV observed at pH 12. The marginal difference in measured ηonset and sharp contrast at η10 confirm the lower reaction rate of the OER at lower pH due to the lower concentration and availability of OH−. Fig. 4b summarizes the observed trends.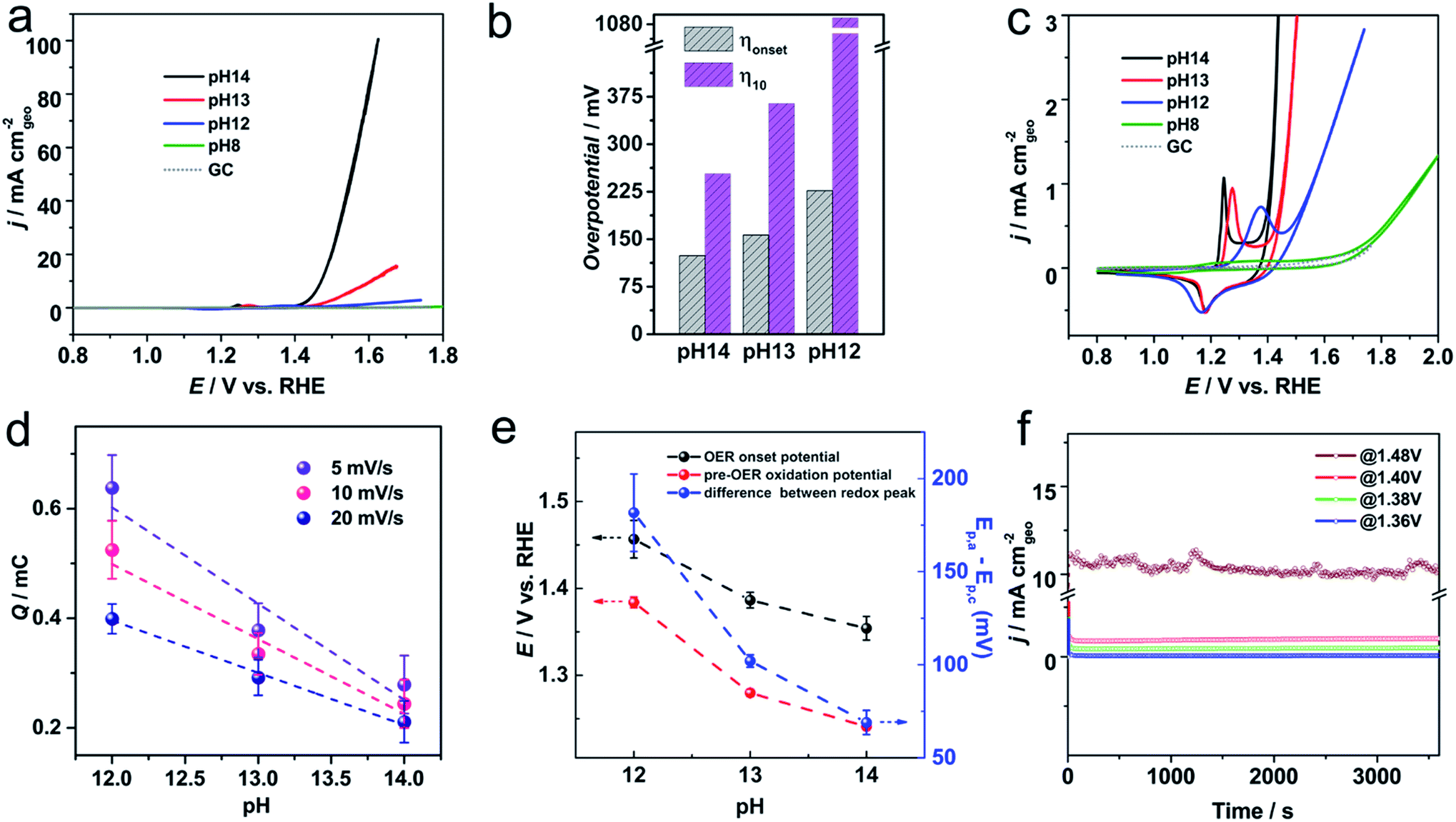 | ||
| Fig. 4 (a) Stable cyclic voltammograms of Ni-KMO at various pHs after 30 consecutive scans in NaOH electrolyte, with scan rate of 20 mV s−1. The profile of the glassy carbon (GC) support is included for comparison. All the curves are iR-corrected. (b) The onset overpotential (ηonset) and overpotential at j = 10 mA cm−2 (η10) obtained from (a). (c) Enlarged region showing the Ni2+ → Ni3+ redox peaks at various pHs. (d) Integrated charge of Ni2+/3+ oxidation peak as a function of pH at various scan rates. (e) OER onset potential, pre-OER oxidation peak potential and Ni2+/3+ ΔE variation with pH. (f) Chronoamperometry profiles of Ni-KMO at pH = 14 at various applied potentials as indicated in the figure. | ||
The redox activity of the confined Ni clusters is evident in the voltammograms as a quasi-reversible peak in the region between 1.2–1.5 V was observed at pH 14 and 12 (Fig. 4c). This peak has previously been ascribed to the Ni2+ ↔ Ni3+ couple of Ni based catalysts.15,22,28,29 Interestingly, at pH = 14 the oxidation peak of Ni2+ → Ni3+ appears at 1.24 V, which is significantly more cathodic relative to previous reports in the literature (∼1.4 V vs. RHE on carbon electrode). This unusual cathodic shift of oxidation peak to 1.24 V is beneficial for subsequent catalytic oxygen evolution. As seen in Fig. 4c, a direct relationship between the position of the Ni2+ → Ni3+ oxidation peak on the Ni-KMO and the OER onset potential was observed as a function of pH. Increasing the pH of the solution from 12 to 14 results in the shift of the Ni2+ → Ni3+ oxidation peak together with a shift of the onset potential of OER towards less positive potentials. This is a clear indicator that the Ni3+ are active sites for the OER. Moreover, the correlation between redox peaks and OER activity over multiple cycles is shown in Fig. S10.† At any one pH, the Ni2+ → Ni3+ oxidation peak shifts cathodically and the OER current density increases as a function of the number of cycles, confirming the role of Ni2+ → Ni3+ transformation on OER catalysis. This behavior is in agreement with previous work that established the relationship between the peak position of the redox couple on Ni–Fe(OOH) and the OER catalytic activity.30
Since the OER catalytic activity is directly correlated to the nature of the Ni centers, understanding the redox peak of the Ni in the cyclic voltammograms may contribute to greater understanding of the reaction mechanism as well as in the development of better catalyst. Voltammetric profiles of the Ni2+/Ni3+ redox peaks were recorded at different pHs (Fig. S11†). At pH = 14 the presence of two discrete processes, Ox1/Re1 and Ox2/Re2, were observed. Previous reports assigned the process Ox1 ↔ Re1 to the phase transition α-Ni(OH)2/γ-NiOOH, while the process labelled Ox2 ↔ Re2 has been associated with the transition of β-Ni(OH)2 to the active phase β-NiOOH.31,32 A schematic illustration of the phase transitions is presented in Fig. S12.† When the pH is decreased to 13, only one oxidation peak is observed (Ox1) and the peak labelled Re2 shifts towards more positive potentials and its current density decreases (Fig. S11†). At pH = 12, the Re2 peak is not observable and only the redox peak Ox1/Re1 is visible. The absence of the β-NiOOH transformation at pH = 12 was accompanied by a significant reduction in measured OER activity.
To explore the underlying mechanism, we thoroughly examined the relationship between the Ni2+/Ni3+ redox peaks and the scan rates. The linear relationship between peak current density and scan rate at pH = 14 was indicative that the surface reaction kinetics were dominated by charge transfer steps, whereas the linear relationship between peak current density and the square root of the scan rate at pH = 12 was indicative that the diffusion limitation was the dominating factor (Fig. S13†).
The charge associated with the redox peaks as a function of the pH is also shown in Fig. 4d and S14.† As can be seen, opposite to the literature,30 the charge associated to the redox peak of Ni2+/Ni3+ increases as the pH decreases. We further explore this phenomenon by analyzing the charges of the redox peak at pH = 12 and pH = 14 as a function of the number of cycles. Fig. S15† shows the comparison between the charge of the redox peaks of the 1st scan and 30th scan for the different pHs. The first observation is that slightly larger charges are observed at pH = 14 and pH = 13. As can be seen, the charge of the oxidation peak increases after electrochemical cycling across the pH range explored. However, the increase of the charge of the redox peak is significantly larger at pH = 12 and after 30 cycles. Charge of the redox peak at pH = 12 (399 μC) is 1.89-fold that of the same peak at pH = 14 (211 μC). According to the discussion above, and the XAS results (Section 2.2) the Ni-clusters intercalated between the KMO layers must undergo an “activation process”, which is attributed to a phase transformation of the Ni oxide species. While at pH = 14, Ni sites with OH− adsorption are fully transformed into the β-NiOOH active form, at pH = 12 most of the catalyst is only transformed into the γ-NiOOH phase. Considering that the oxidation state of the β-NiOOH is +3 and the γ-NiOOH is ∼3.7 (Fig. S15†), the transformation from Ni2+ to γ-NiOOH requires ∼1.7 fold more charges than that to the β-NiOOH. Therefore, our results suggest that at pH = 14 the active phase for the OER is the β-NiOOH and at pH = 12, the active phase for the OER is the γ-NiOOH. The unique structure of the Ni confined in 2D layer structure with flexibly adjustable interlayer spacing gives rise to such phenomenon.
In order to obtain more information on the reaction mechanism, Tafel analysis of the OER curves at different pHs was performed. Note that all voltammograms were iR corrected prior to the Tafel analysis. It has been previously proposed that the overall mechanism of OER catalysis on Ni sites in alkaline media involve four on-electron-transfer steps (eqn (1)–(4)).15,33 The Tafel plots for the OER on Ni-KMO at different pHs are presented in Fig. S16.† The Tafel slope, at pH = 14, is 60 mV dec−1 suggesting that the rate-determining step (RDS) was the deprotonation of adsorbed hydroxyl species (eqn (2)), in agreement with previous works on Ni hydroxide/oxide catalyst.34 On the other hand, a Tafel slope of 232 mV dec−1 was obtained at pH 12, suggesting that the RDS at lower pH was the adsorption of OH− species (eqn (1)). This can be rationalized by consideration of the nature of Ni sites. A higher concentration gradient is required to promote the transport of the OH− species from electrolyte through the channels of the layered oxide structure to the confined Ni nanoreactor sites. This diffusion-dependent process gives rise to the significant difference in OER activity observed for Ni-KMO with varying pHs.
| Ni* + OH− → Ni*OH + e− | (1) |
| Ni*OH + OH− → Ni*O + H2O + e− | (2) |
| Ni*O + OH− → Ni*OOH + e− | (3) |
| Ni*OOH + OH− → Ni* + O2 + H2O + e− | (4) |
The evaluation of the electrochemical performance of a catalyst should not only be based on its catalytic activity but also on the durability of the catalyst. Long-term stability of the Ni-KMO was recorded at pH = 14 via chronoamperometry at various applied potentials (Fig. 2f). As can be seen in Fig. 2f, the current density during the OER remained stable over 1 h of electrolysis at different relevant potentials. In order to gain insight into the structural and compositional stability of the catalyst, SEM and HAADF-EDS images of the Ni-KMO catalyst were collected after 10 h (Fig. S17†). Interestingly, no significant changes in the morphology or the element distribution of the catalyst were detected confirming the excellent stability of the catalyst after 10 h of electrolysis. A comparison of the long term stability, up to 23 h electrolysis in 1 M NaOH, of the Ni-KMO at 1.48 V and Ni-HTO at 1.51 V shows stable current densities for catalysis with current densities over 10 mA cm−2 (Fig. S23†).
3.2 Effect of the cations on the OER catalytic activity
Previous works have also demonstrated that in addition to the pH, the nature of the electrolyte cation also affects the observed activity and selectivity of electrochemical processes, especially in alkaline media.35,36 We investigated the effect of K+ and Na+ cations on the electrochemical activity of Ni-KMO towards the OER (Fig. 5). As can be seen, although a slightly different shape of the Ni2+ → Ni3+ redox peaks was observed, the charges for the Ni2+/Ni3+ redox peaks are almost identical, 0.261 mC in NaOH and 0.263 mC in KOH respectively. Ni-KMO presents lower ηonset and η10 for the OER in KOH (ηonset = 100 mV and η10 = 206 mV) relative to the respective values in NaOH (ηonset = 130 mV and η10 = 240 mV). It has been previously demonstrated, that the nature of the cations affects the catalytic activity of the OER and the ORR. In particular, those reports indicate that potassium enhances the catalytic activity.37–39 This effect has been associated to the non-covalent interaction between K+(H2O)x/Na+(H2O)x clusters and the active Ni-oxygenated species at the surface Ni–OH*, Ni–O*, Ni–OOH*. Fig. 5 shows that the onset of the Ni2+/Ni3+ oxidation reaction is independent of the cation present in the electrolyte (KOH or NaOH) which indicates similar adsorption reaction of OH− on Ni sites. On the other hand, at slightly more positive potentials a slight cathodic shift of OER onset in KOH suggests that the subsequent deprotonation step is favorable in KOH. Suntivich et al. suggested that charged K+(H2O)x/Na+(H2O)x clusters would modify the energy of M-oxygenated species, thus altering their stability.37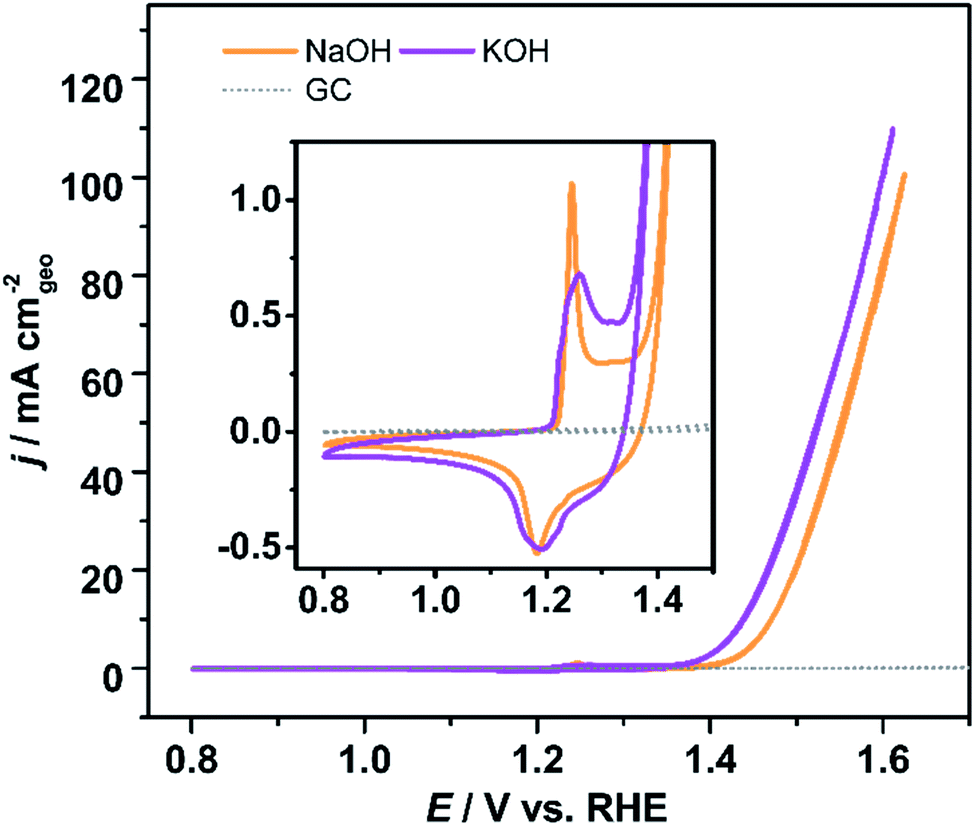 | ||
| Fig. 5 Voltammetric profiles of Ni-KMO in 1 M KOH and NaOH electrolyte, as indicated in the figure, inset is the enlarged redox features. Glassy carbon shown for comparison. Scan rate υ = 20 mV s−1. | ||
3.3 Investigation of the coordination of active Ni on the catalytic activity
In order to further investigate the specific role of Ni coordination in the performance of the catalyst, Ni-exchanged HMnO2 (Ni-HMO) and Ni-exchanged HTi0.91O2 (Ni-HTO) were prepared and evaluated towards the OER. Both materials, HMO and HTO, present the same layered structure (Fig. S18†), therefore any observable differences in the catalytic activity would suggest differences in the interaction between the Ni and species such as Mn/Ti and K/H2O around. Both Ni-HMO and Ni-HTO catalysts display notable OER activity (Fig. 6a) in comparison with the pristine HMO and HTO (Fig. S19†). As can be seen on Table S2,† Ni-KMO shows the greatest OER catalytic activity (η10 = 240 mV) of the three Ni-exchanged layered oxide catalysts in regard to geometrical activity. Despite the remarkable difference of Ni redox peaks for the 3 catalysts, the OER ηonset values are nearly identical, indicating that confined Ni is truly the active component in the observed activity of Ni-exchanged HMO and HTO. Tafel slopes of three Ni confined samples are shown in Fig. S20.† Ni-HTO with 50 mV dec−1 is smaller than Ni-KMO (60 mV dec−1) and Ni-HMO (113 mV dec−1). These data suggest that the chemical deprotonation process is the RDS for both Ni-HTO and Ni-KMO, whilst for Ni-HMO it is the adsorption of OH−. To obtain further information on the nature of the differences in the shape of the redox peaks and the catalytic activity, the Ni content in each of the catalysts was determined by XRF (Table 2). It was found that Ni-HMO had the greatest Ni loading (16 wt%), followed by Ni-KMO (8 wt%) and Ni-HTO (2 wt%). Therefore, the differences in the shape of the redox peaks could be attributed to the differences in Ni loading and coordination, but the larger catalytic activity of the Ni-KMO could not be directly correlated to the Ni loading. This implies that Ni coordination plays the predominant role in water catalysis. This assumption is in line with the previous work by Thenuwara et al., which suggested that highly ordered water molecules lower the potential barrier that facilitate the OER.40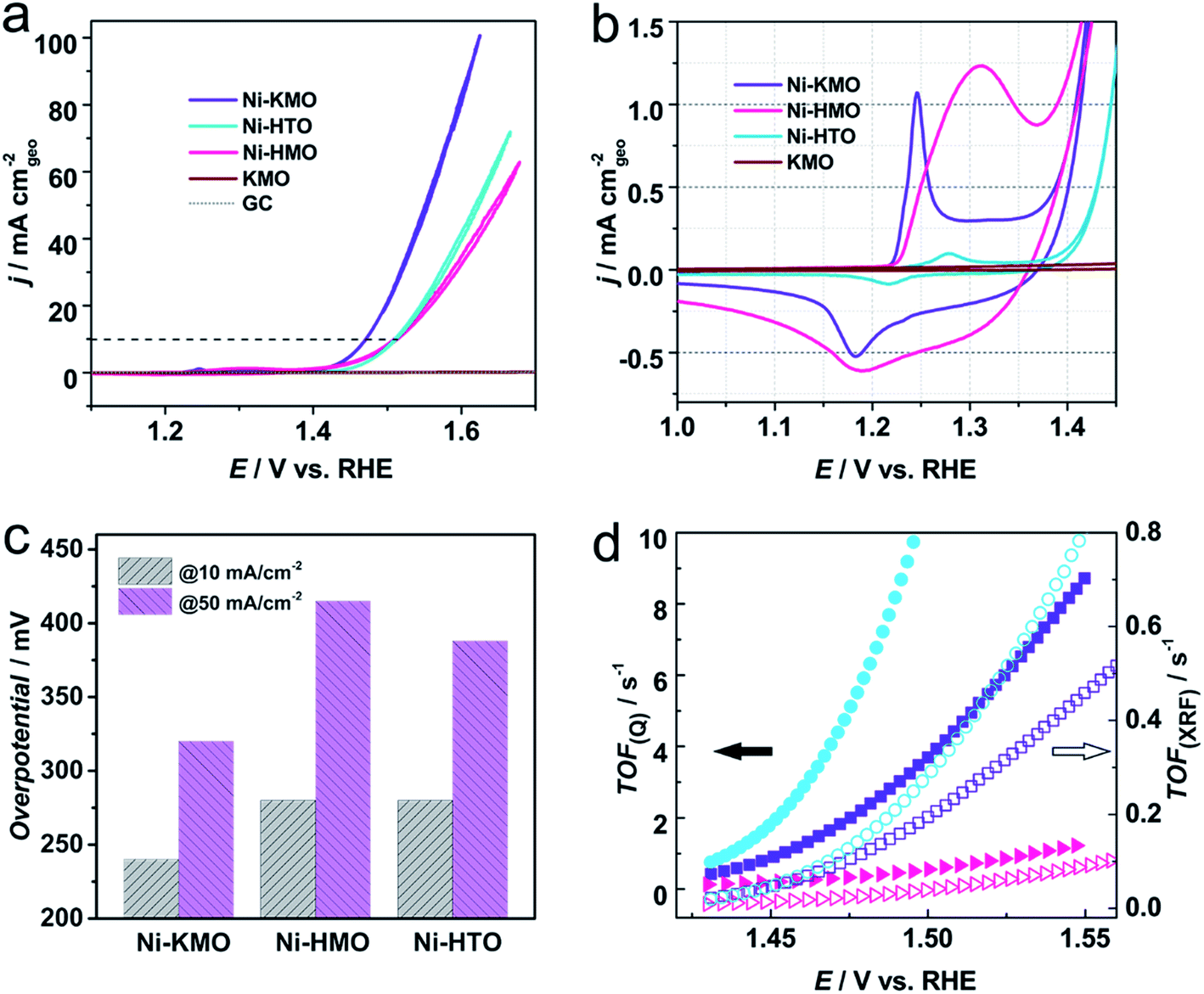 | ||
| Fig. 6 (a) iR-corrected voltammetric profiles of Ni-KMO, Ni-HMO and Ni-HTO after 30 consecutive scans at pH = 14, in 1 M NaOH at 20 mV s−1. The evolution of the voltammograms as function of the number of scans can be found in Fig. S22.† (b) Enlarged region showing the Ni2+ → Ni3+ redox peaks and OER onset. (c) η50 and η10 obtained from (a). (d) TOF as a function of potential calculated using the content of Ni content on Ni-KMO (square symbol), Ni-HMO (triangle symbol) and Ni-HTO (circle symbol) determined by the Ni+2/Ni+3 redox charge (solid symbols) and the XRF (empty symbols). | ||
| Ni0.16K0.2MnO2·0.7H2O | Ni0.33K0.04MnO2·0.9H2O | Ni0.033Ti0.91O2·0.6H2O | |
|---|---|---|---|
| η 10/mV | 240 | 280 | 280 |
| Integration charge/mC | 0.278 | 0.743 | 0.0342 |
| Corresponding Ni content/μg | 0.169 | 0.452 | 0.020 |
| Tafel slope/mV dec−1 | 60 | 113 | 50 |
| Ni content [XRF]/wt% | 8 | 16 | 2 |
| j mss at η = 240 mV/A gNi−1 | 602 | 140 | 710 |
So far we have demonstrated that the Ni sites solely give rise to the observed OER activity of the Ni-confined layered metal oxide catalysts, thus, the Ni content allows us to determine the turnover frequency (TOF) of the catalyst, a key indicator of the intrinsic activity of a catalyst. However, two possible strategies can be adopted in this case. The TOF was determined using the total Ni content obtained from XRF measurements (TOFXRF) and by use of the wt% Ni determined by the charge of Ni2+/Ni3+ redox peak (TOFQ), as previously demonstrated by Nakagawa41 and Chen42 (Fig. 6d). As shown in Fig. 6d, whilst the two methods give different absolute values of the TOF, the trend Ni-HTO > Ni-KMO > Ni-HMO is the same. Interestingly, despite the results above indicating that the Ni-KMO had the larger catalytic activity per geometrical surface area, Ni-HTO displays better TOF values (Table 2), indicating that it possesses the highest active atom efficiency. Determination of the % Ni from the integrated charges and XRF measurements both strongly support our claim of low-loading of active catalysts for OER catalysis. For example, as the XRF result is overall evaluation of Ni content in catalyst, TOF calculation based on XRF display the lower limitation. 2 wt% Ni in Ni-HTO shows a TOFXRF of 0.1090 ± 0.0067 s−1 at 1.47 V vs. RHE. While the TOFQ of Ni-HTO is 4.22 ± 0.05 s−1 based on integrated charge, which is an order of magnitude higher than TOFXRF (Fig. S21†).
The mass activities of our ion-exchanged catalysts in Fig. 7 are compared with other Ni-based catalysts previously reported in the literature, including a variety of Ni structures (Ni foam, self-supported NiOOH nanocatalysts) and shown in Table S5.† Ni-KMO and Ni-HMO were found to have the largest mass activities by a factor of 4 or more. The mass activities of these 2D Ti–O and Mn–O Ni catalysts surpasses the state-of-the-art Ni-based catalysts, such as NiV-LDH (144 gNi−1 @300 mV)43 or single Au atoms anchored on NiFe-LDH (117 gNi−1 @280 mV).44
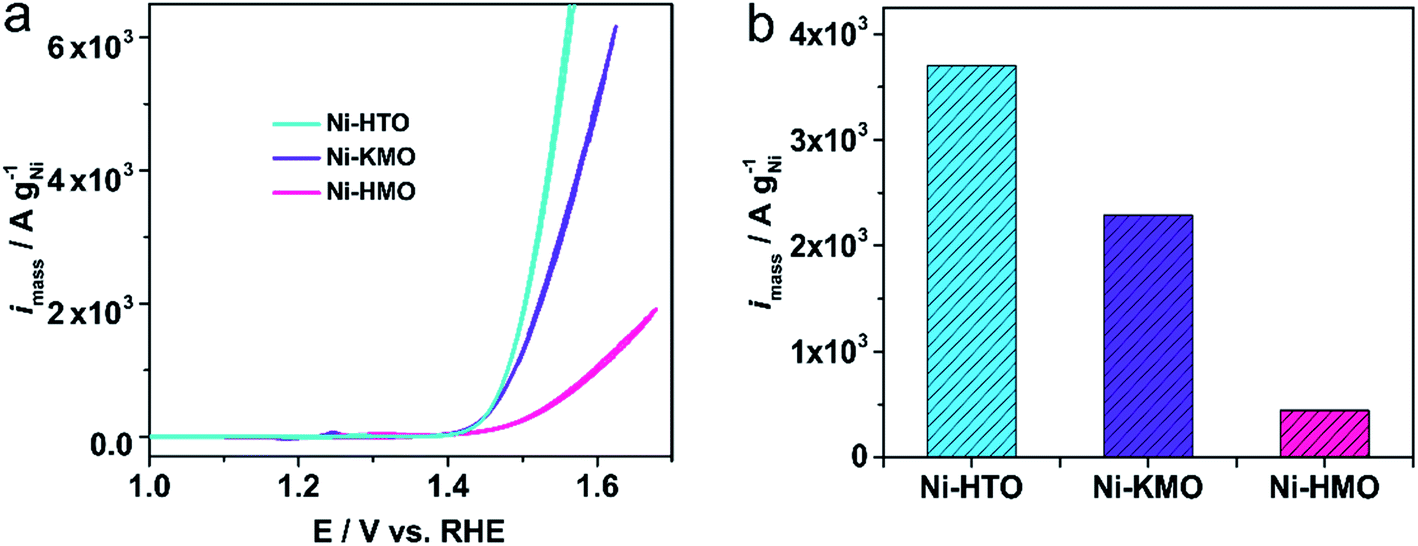 | ||
| Fig. 7 (a) Voltammetric mass activity of Ni-KMO, Ni-HMO, and Ni-HTO in 1 M NaOH. Scan rate υ = 20 mV s−1, (b) corresponding activity at 1.53 V vs. RHE. | ||
4. Conclusion
This work described the preparation and characterization of highly active OER catalysts based on the facile ion-exchange of Ni atoms into the 2D birnessite and lepidocrocite titanate structures. A direct correlation between the Ni redox peaks and the OER activity was demonstrated. After the evaluation of different catalysts as a function of the Ni content it was found that Ni-KMO and Ni-HTO show excellent mass activity of 602 A gNi−1 and 710 A gNi−1 at overpotential of 240 mV, respectively. Ni-HTO shows a 6-fold increase in catalytic activity per mass of Ni in comparison with the state-of-the-art Ni-based nanocatalysts (Au@Ni7/8Fe1/8 LDH).44 By evaluating the electrochemical performance at varying pHs in NaOH, it was found that the catalytic activity was enhanced at higher pH and in the presence of K cations, ηonset = 100 mV and η10 = 206 mV were obtained in comparison with those in the presence of Na cations (ηonset = 130 mV, η10 = 240 mV). The intercalation mechanism and oxidation state of Ni and Mn have been explored by the XAS technique. Our results demonstrate that Ni intercalation occurs at the Mn3+ sites by reducing Mn3+ to Mn2+. As mentioned above, the ex situ XAS/XES results show that the Ni-content in each of the samples is proportional to the concentrations Mn3+ sites. Even though Ni is present in the +2 oxidation state for all the samples, the Ni coordination/structures obtained for Ni-KMO and Ni-HMO were different. We associate the differences in OER activity to the differences in coordination/structures between Ni-KMO and Ni-HMO.It was also demonstrated that while the intercalated Ni is the active site for oxygen evolution reaction, Mn sites do not contribute to the OER. Thus, the evaluation of intrinsic activity, such as TOFs and mass activity, by using the Ni content offers reliable results.
In brief, highly efficient catalysts with Ni confined in 2D layered metal oxides towards OER have been synthesized by ion exchange reactions. Our synthesis protocol provides evidence for the broader applicability of our employed methodology to other layered metal oxide structures and to compositions other than manganese oxides.
Author contributions
Y. P., P. R. and L. H. conceived the experiments. All the electrochemical experiments, XRD, XRF, SEM, TGA characterization and analyses were performed by Y. P. XANES and EXAFS characterization and analyses were performed by M. J. L. and V. C. XES measurements were collected and analyzed by V. C. and A. E. R. Low magnification HAADF- and BF-STEM, and SAD characterization were performed by L. A. J. High resolution HAADF-STEM and EDX analyses were performed by Q. W., Z. S. and M. G. All authors contributed to the analysis of the results, discussion, writing and revision of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Southern University of Science and Technology (SUSTech) start fund through Shenzhen Peacock Talent program, Guangdong Innovative and Entrepreneurial Research Team Program (No. 2016ZT06N532), Guangdong Provincial Key Laboratory of Materials for Electric Power (2018B030322001) and Shenzhen Clean Energy Research Institute (CERI-KY-2019-003). P. R. acknowledges the University of Birmingham for financial support through the Birmingham fellowship program. This work was also supported by the Pico Center at SUSTech that receives support from Presidential fund and Development and Reform Commission of Shenzhen Municipality. The authors wish to acknowledge the Diamond Light Source for provision of beamtime (SP21659 and SP19850).References
- J. Kibsgaard and I. Chorkendorff, Considerations for the scaling-up of water splitting catalysts, Nat. Energy, 2019, 4(6), 430–433 CrossRef.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells, Adv. Mater., 2019, 1806296 CrossRef PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, Transition metal oxides as electrocatalysts for the oxygen evolution reaction in alkaline solutions: an application-inspired renaissance, J. Am. Chem. Soc., 2018, 140(25), 7748–7759 CrossRef CAS PubMed.
- M. P. Browne, Z. Sofer and M. Pumera, Layered and two dimensional metal oxides for electrochemical energy conversion, Energy Environ. Sci., 2019, 12(1), 41–58 RSC.
- W. Zhang and K. Zhou, Ultrathin Two-Dimensional Nanostructured Materials for Highly Efficient Water Oxidation, Small, 2017, 13(32), 1700806 CrossRef PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Trends in activity for the water electrolyser reactions on 3d M (Ni, Co, Fe, Mn) hydr(oxy) oxide catalysts, Nat. Mater., 2012, 11(6), 550 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, Cobalt–iron (oxy) hydroxide oxygen evolution electrocatalysts: the role of structure and composition on activity, stability, and mechanism, J. Am. Chem. Soc., 2015, 137(10), 3638–3648 CrossRef CAS PubMed.
- Y. Zhao, C. Chang, F. Teng, Y. Zhao, G. Chen, R. Shi, G. I. Waterhouse, W. Huang and T. Zhang, Defect-Engineered Ultrathin δ-MnO2 Nanosheet Arrays as Bifunctional Electrodes for Efficient Overall Water Splitting, Adv. Energy Mater., 2017, 7(18), 1700005 CrossRef.
- C. Luan, G. Liu, Y. Liu, L. Yu, Y. Wang, Y. Xiao, H. Qiao, X. Dai and X. Zhang, Structure effects of 2D materials on α-nickel hydroxide for oxygen evolution reaction, ACS Nano, 2018, 12(4), 3875–3885 CrossRef CAS PubMed.
- F. Song and X. Hu, Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- F. Song and X. Hu, Ultrathin cobalt–manganese layered double hydroxide is an efficient oxygen evolution catalyst, J. Am. Chem. Soc., 2014, 136(47), 16481–16484 CrossRef CAS PubMed.
- H. Wang, J. Zhang, X. Hang, X. Zhang, J. Xie, B. Pan and Y. Xie, Half-Metallicity in Single-Layered Manganese Dioxide Nanosheets by Defect Engineering, Angew. Chem., Int. Ed., 2015, 54(4), 1195–1199 CrossRef CAS PubMed.
- W. Liu, H. Liu, L. Dang, H. Zhang, X. Wu, B. Yang, Z. Li, X. Zhang, L. Lei and S. Jin, Amorphous cobalt–iron hydroxide nanosheet electrocatalyst for efficient electrochemical and photo-electrochemical oxygen evolution, Adv. Funct. Mater., 2017, 27(14), 1603904 CrossRef.
- J. Li, X. Gao, Z. Li, J. H. Wang, L. Zhu, C. Yin, Y. Wang, X. B. Li, Z. Liu and J. Zhang, Superhydrophilic graphdiyne accelerates interfacial mass/electron transportation to boost electrocatalytic and photoelectrocatalytic water oxidation activity, Adv. Funct. Mater., 2019, 29(16), 1808079 CrossRef.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, Nickel-based electrocatalysts for energy-related applications: oxygen reduction, oxygen evolution, and hydrogen evolution reactions, ACS Catal., 2017, 7(10), 7196–7225 CrossRef CAS.
- A. Bergmann, T. E. Jones, E. M. Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Unified structural motifs of the catalytically active state of Co (oxyhydr) oxides during the electrochemical oxygen evolution reaction, Nat. Catal., 2018, 1(9), 711 CrossRef CAS.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho and M. Chen, Atomically dispersed nickel–nitrogen–sulfur species anchored on porous carbon nanosheets for efficient water oxidation, Nat. Commun., 2019, 10(1), 1392 CrossRef PubMed.
- A. Munir, T. Haq, A. Qurashi, H. Rehman, A. Ul-Hamid and I. Hussain, Ultrasmall Ni/NiO Nanoclusters on Thiol-Functionalized and-Exfoliated Graphene Oxide Nanosheets for Durable Oxygen Evolution Reaction, ACS Appl. Energy Mater., 2018, 2(1), 363–371 CrossRef.
- R. Ma and T. Sasaki, Nanosheets of oxides and hydroxides: ultimate 2D charge-bearing functional crystallites, Adv. Mater., 2010, 22(45), 5082–5104 CrossRef CAS PubMed.
- D. Deng, K. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Catalysis with two-dimensional materials and their heterostructures, Nat. Nanotechnol., 2016, 11(3), 218 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts, J. Am. Chem. Soc., 2012, 134(3), 1519–1527 CrossRef CAS PubMed.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. Koper, The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation, Chem. Sci., 2016, 7(4), 2639–2645 RSC.
- J. E. Post and D. R. Veblen, Crystal structure determinations of synthetic sodium, magnesium, and potassium birnessite using TEM and the Rietveld method, Am. Mineral., 1990, 75(5-6), 477–489 CAS.
- J. C. Conesa, Electronic structure of the (undoped and Fe-doped) NiOOH O2 evolution electrocatalyst, J. Phys. Chem. C, 2016, 120(34), 18999–19010 CrossRef CAS.
- M. Belli, A. Scafati, A. Bianconi, S. Mobilio, L. Palladino, A. Reale and E. Burattini, X-ray absorption near edge structures (XANES) in simple and complex Mn compounds, Solid State Commun., 1980, 35(4), 355–361 CrossRef CAS.
- P. Ghigna, G. Flor and G. Spinolo, An Mn–K Edge XAS Investigation on the Crystal Chemistry of Cd1–δMn2Oy, J. Solid State Chem., 2000, 149(2), 252–255 CrossRef CAS.
- J.-K. Chang, M.-T. Lee and W.-T. Tsai, In situ Mn K-edge X-ray absorption spectroscopic studies of anodically deposited manganese oxide with relevance to supercapacitor applications, J. Power Sources, 2007, 166(2), 590–594 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori, Identification of highly active Fe sites in (Ni, Fe) OOH for electrocatalytic water splitting, J. Am. Chem. Soc., 2015, 137(3), 1305–1313 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni–Fe oxide water splitting electrocatalysts, J. Am. Chem. Soc., 2016, 138(17), 5603–5614 CrossRef PubMed.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, Tracking Catalyst Redox States and Reaction Dynamics in Ni–Fe Oxyhydroxide Oxygen Evolution Reaction Electrocatalysts: The Role of Catalyst Support and Electrolyte pH, J. Am. Chem. Soc., 2017, 139(5), 2070–2082 CrossRef PubMed.
- B. S. Yeo and A. T. Bell, In situ Raman study of nickel oxide and gold-supported nickel oxide catalysts for the electrochemical evolution of oxygen, J. Phys. Chem. C, 2012, 116(15), 8394–8400 CrossRef CAS.
- S. R. Mellsop, A. Gardiner, B. Johannessen and A. T. Marshall, Structure and transformation of oxy-hydroxide films on Ni anodes below and above the oxygen evolution potential in alkaline electrolytes, Electrochim. Acta, 2015, 168, 356–364 CrossRef CAS.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, Metallic nickel nitride nanosheets realizing enhanced electrochemical water oxidation, J. Am. Chem. Soc., 2015, 137(12), 4119–4125 CrossRef CAS PubMed.
- J. Liu, Y. Zheng, Y. Jiao, Z. Wang, Z. Lu, A. Vasileff and S. Z. Qiao, NiO as a bifunctional promoter for RuO2 toward superior overall water splitting, Small, 2018, 14(16), 1704073 CrossRef PubMed.
- C. Stoffelsma, P. Rodriguez, G. Garcia, N. Garcia-Araez, D. Strmcnik, N. M. Marković and M. T. Koper, Promotion of the oxidation of carbon monoxide at stepped platinum single-crystal electrodes in alkaline media by lithium and beryllium cations, J. Am. Chem. Soc., 2010, 132(45), 16127–16133 CrossRef CAS PubMed.
- G. García, C. Stoffelsma, P. Rodriguez and M. T. Koper, Influence of beryllium cations on the electrochemical oxidation of methanol on stepped platinum surfaces in alkaline solution, Surf. Sci., 2015, 631, 267–271 CrossRef.
- J. Suntivich, E. E. Perry, H. A. Gasteiger and Y. Shao-Horn, The Influence of the Cation on the Oxygen Reduction and Evolution Activities of Oxide Surfaces in Alkaline Electrolyte, Electrocatalysis, 2013, 4(1), 49–55 CrossRef CAS.
- M. Xiao, Y. Tian, Y. Yan, K. Feng and Y. Miao, Electrodeposition of Ni (OH) 2/NiOOH in the presence of urea for the improved oxygen evolution, Electrochim. Acta, 2015, 164, 196–202 CrossRef CAS.
- W. Jin, H. Du, S. Zheng, H. Xu and Y. Zhang, Comparison of the oxygen reduction reaction between NaOH and KOH solutions on a Pt electrode: the electrolyte-dependent effect, J. Phys. Chem. B, 2010, 114(19), 6542–6548 CrossRef CAS PubMed.
- A. C. Thenuwara, E. B. Cerkez, S. L. Shumlas, N. H. Attanayake, I. G. McKendry, L. Frazer, E. Borguet, Q. Kang, R. C. Remsing, M. L. Klein, M. J. Zdilla and D. R. Strongin, Angew. Chem., Int. Ed., 2016, 55(35), 10381–10385 CrossRef CAS PubMed.
- T. Nakagawa, C. A. Beasley and R. W. Murray, Efficient electro-oxidation of water near its reversible potential by a mesoporous IrO x nanoparticle film, J. Phys. Chem. C, 2009, 113(30), 12958–12961 CrossRef CAS.
- J. Y. Chen, J. T. Miller, J. B. Gerken and S. S. Stahl, Inverse spinel NiFeAlO 4 as a highly active oxygen evolution electrocatalyst: promotion of activity by a redox-inert metal ion, Energy Environ. Sci., 2014, 7(4), 1382–1386 RSC.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nickel–vanadium monolayer double hydroxide for efficient electrochemical water oxidation, Nat. Commun., 2016, 7(1), 11981 CrossRef CAS PubMed.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, Single-Atom Au/NiFe Layered Double Hydroxide Electrocatalyst: Probing the Origin of Activity for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2018, 140(11), 3876–3879 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full description of the experimental methods, XRD patterns, calculation of charge integration, Tafel plots, long-term durability experiments and XAS fitting analysis. See DOI: 10.1039/d0ta04031b |
| This journal is © The Royal Society of Chemistry 2020 |