
Confined growth of pyridinic N–Mo2C sites on MXenes for hydrogen evolution†
Hao
Wang‡
 a,
Yanping
Lin‡
b,
Shuyuan
Liu‡
b,
Jianmin
Li
c,
Liangmin
Bu
b,
Jianmei
Chen
d,
Xu
Xiao
*e,
Jin-Ho
Choi
b,
Lijun
Gao
*b and
Jong-Min
Lee
*a
a,
Yanping
Lin‡
b,
Shuyuan
Liu‡
b,
Jianmin
Li
c,
Liangmin
Bu
b,
Jianmei
Chen
d,
Xu
Xiao
*e,
Jin-Ho
Choi
b,
Lijun
Gao
*b and
Jong-Min
Lee
*a
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, Singapore 637459, Singapore. E-mail: jmlee@ntu.edu.sg
bSoochow Institute for Energy and Materials Innovations, College of Energy, Soochow University, Suzhou 215006, China. E-mail: gaolijun@suda.edu.cn
cDepartment of Materials Science and Engineering, National University of Singapore, Singapore 117576, Singapore
dInstitute of Functional Nano and Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou 215123, China
eA.J. Drexel Nanomaterials Institute and Department of Materials Science and Engineering, Drexel University, Philadelphia, 19104, USA. E-mail: xx58@drexel.edu
First published on 2nd April 2020
Abstract
Developing low-cost and high-performance hydrogen evolution reaction (HER) electrocatalysts is a key research area for scalable hydrogen production from water electrolysis. Here, a hybrid of nitrogen-doped carbon encapsulated Mo2C nanodots on Ti3C2Tx MXene (Mo2C/Ti3C2Tx@NC) is developed through in situ polymerization of dopamine and a Mo precursor on the Ti3C2Tx MXene surface. During the annealing treatment, the polydopamine plays multiple roles in forming N-doped carbon, confining MoO42− ions into ultrasmall Mo2C nanodots, and stabilizing the MXene flakes against spontaneous oxidation. The as-synthesized hybrid exhibits excellent HER activity in acidic electrolyte with an overpotential of 53 mV at 10 mA cm−2 and excellent stability over 30 hours. The combination of experiments and simulations demonstrates that pyridinic N-doped carbon coated Mo2C nanodots serve as the active sites and Ti3C2Tx MXene facilitates the charge transfer, synergistically contributing to the superior HER performance.
Owing to the high energy density and zero carbon emission, hydrogen is considered as an optimal solution to address the energy crisis and alleviate the environmental pollution.1,2 Although industrial hydrogen production still relies on fossil fuels, water electrolysis shows promise as a clean and sustainable route for hydrogen generation.3,4 Thus, developing efficient hydrogen evolution reaction (HER) electrocatalysts is of great importance to realize scalable electrocatalytic hydrogen production. Until now, platinum (Pt) and its derivatives remain the best HER electrocatalysts with low overpotentials and fast reaction kinetics, but their low abundance and high cost severely hinder their large-scale use.5 Recently, intensive research efforts have been made to explore low-cost and earth-abundant alternatives with comparable HER performance.6–15 Among them, molybdenum carbide (Mo2C) holds great promise due to its Pt-like d-band electronic structure.16 In general, the HER activity of Mo2C mainly relies on the accessibility of active sites and conductivity. Mo2C nanoparticles, even ultrasmall nanodots, are preferable but tend to agglomerate when processed into electrodes. In this context, introducing a highly conductive substrate that could simultaneously optimize the dispersion of ultrasmall Mo2C would be of importance.17,18
Recently, an emerging family of two-dimensional (2D) transition metal carbides and nitrides, called MXenes, have attracted much attention due to their unique characteristics of metallic conductivity (up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 S cm−1), hydrophilicity and diversity stemming from various surface terminations.19,20 Typically, the most studied Ti3C2Tx MXene (Tx denotes the surface –OH, –O and –F groups) shows great promise in various applications including supercapacitors,21 sensors,22 water purification,23 electromagnetic interference shielding24 and catalysis.25–29 It would be natural to assume that coupling Mo2C and MXenes may deliver satisfactory HER performance, while it remains challenging due to the following factors. First, the synthesis of Mo2C always requires high annealing temperature. Proverbially, MXenes with high surface energy are prone to oxidation at evaluated temperatures, although protected by an inert atmosphere, leading to inferior conductivity.30 In addition, the high synthesis temperature would accelerate the agglomeration of Mo2C nanoparticles and thus result in poor accessibility of active sites.31 Accordingly, protecting MXenes from surface oxidization and Mo2C nanoparticles from aggregation during the calcination process is of great importance to realize MXene–Mo2C hybrid HER electrocatalysts.
000 S cm−1), hydrophilicity and diversity stemming from various surface terminations.19,20 Typically, the most studied Ti3C2Tx MXene (Tx denotes the surface –OH, –O and –F groups) shows great promise in various applications including supercapacitors,21 sensors,22 water purification,23 electromagnetic interference shielding24 and catalysis.25–29 It would be natural to assume that coupling Mo2C and MXenes may deliver satisfactory HER performance, while it remains challenging due to the following factors. First, the synthesis of Mo2C always requires high annealing temperature. Proverbially, MXenes with high surface energy are prone to oxidation at evaluated temperatures, although protected by an inert atmosphere, leading to inferior conductivity.30 In addition, the high synthesis temperature would accelerate the agglomeration of Mo2C nanoparticles and thus result in poor accessibility of active sites.31 Accordingly, protecting MXenes from surface oxidization and Mo2C nanoparticles from aggregation during the calcination process is of great importance to realize MXene–Mo2C hybrid HER electrocatalysts.
Here, we report a 2D coupled nanohybrid of Mo2C nanodots and Ti3C2Tx MXene encapsulated by nitrogen-doped carbon layers (denoted as Mo2C/Ti3C2Tx@NC) synthesized using molybdate ions chelated in polydopamine (Mo-PDA) as the precursor (Fig. 1). Notably, Ti3C2Tx flakes are fully capped with Mo-PDA by in situ polymerization, which could inhibit the surface oxidation of Ti3C2Tx during annealing treatment and thus retain the excellent conductivity.32 Moreover, due to the confinement effect of the Mo-PDA structure, the ultrasmall Mo2C nanodots are uniformly dispersed in the carbon matrix without agglomeration. Demonstrated by theoretical and experimental analyses, the highly accessible pyridinic N–Mo2C sites coupled with the metallic Ti3C2Tx support of Mo2C/Ti3C2Tx@NC can deliver remarkable HER activities with an overpotential of 53 mV at 10 mA cm−2, a Tafel slope of 40 mV dec−1 and excellent stability over 30 hours in acidic electrolyte, which are superior to those of reported noble metal-free electrocatalysts.
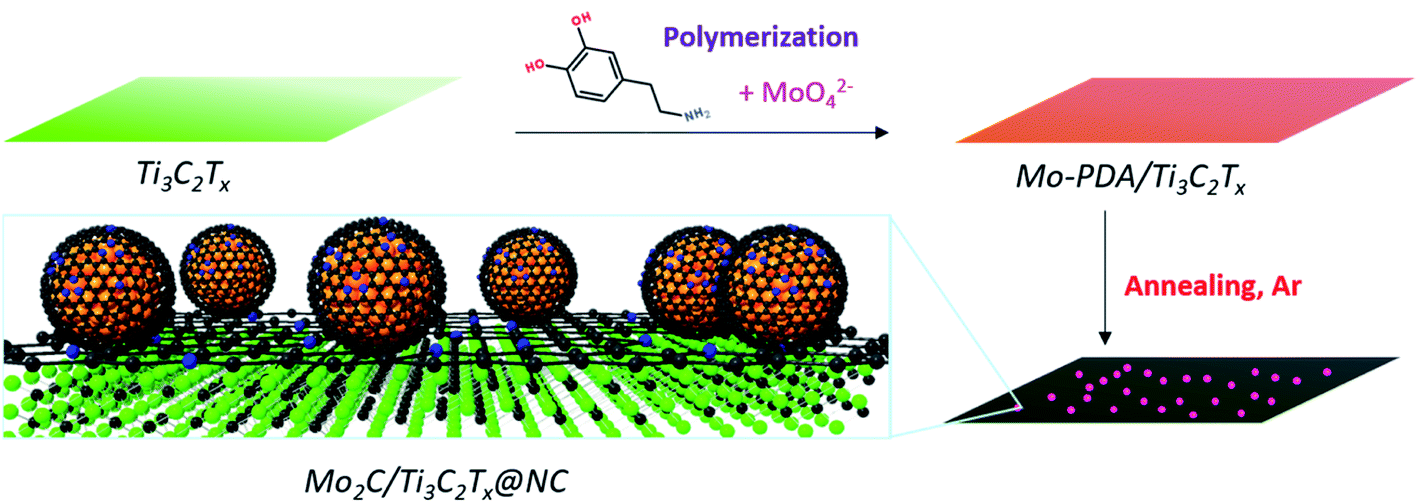 | ||
| Fig. 1 Synthetic illustration of the preparation of Mo2C/Ti3C2Tx@NC. | ||
As demonstrated previously, PDA may have a positive confinement effect on the dispersion of Mo2C nanodots, in which a hierarchical microflower carbon structure was formed (Fig. S1†). Even though Mo2C nanodots may be well dispersed, such a microflower structure would show inferior HER performance in terms of low conductivity. We assume that 2D Ti3C2Tx MXene with various terminations may be a good candidate as a metallically conductive substrate to form a flat PDA layer, realizing both well dispersed Mo2C nanodots and fast electron transport. To verify this idea, we first check the morphology of polydopamine on the MXene by in situ polymerization. The ultrathin Ti3C2Tx with a lateral size of several microns exhibits single crystallinity (Fig. S2†). By in situ polymerization of dopamine on the surface of Ti3C2Tx MXene, the polydopamine/Ti3C2Tx (denoted as PDA/Ti3C2Tx) shows a similar morphology to the pristine MXene (Fig. 2a). After annealing at 750 °C, the pristine MXene is severely oxidized with many TiO2 nanoparticles/nanorods generated on the surface (Fig. S3a†), while the N-doped carbon coated Ti3C2Tx MXene (denoted as Ti3C2Tx@NC) derived from PDA/Ti3C2Tx retains the flat surface without the presence of TiO2 particles (Fig. 2b), which is confirmed from the enlarged TEM image (Fig. S3b†). The high-resolution TEM (HRTEM) image (Fig. 2c) clearly shows that the Ti3C2Tx MXene layers are fully covered by carbon.
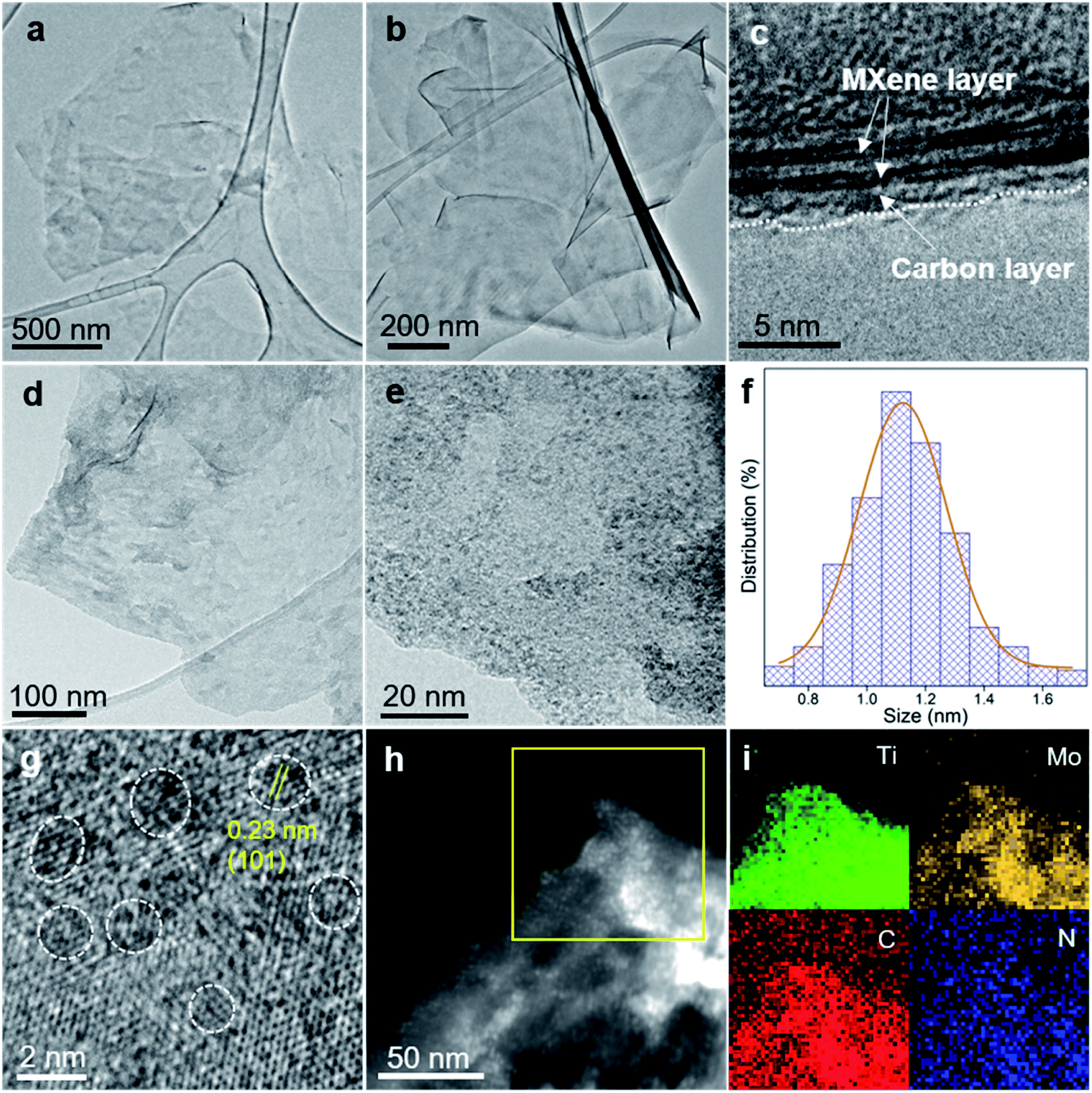 | ||
| Fig. 2 TEM images of (a) PDA/Ti3C2Tx, (b) Ti3C2Tx@NC and (d) Mo2C/Ti3C2Tx@NC. (c) HRTEM image of a Ti3C2Tx@NC edge showing the carbon layer covered Ti3C2Tx MXene structure. (e) Enlarged TEM image of Mo2C/Ti3C2Tx@NC showing the uniform dispersion of Mo2C nanodots on the surface of Ti3C2Tx MXene. (f) Size distribution of Mo2C nanodots in Mo2C/Ti3C2Tx@NC counted in (e). HRTEM image (g) and HAADF-STEM image (h) of Mo2C/Ti3C2Tx@NC. (i) EDX elemental mapping from the marked region in (h). | ||
The structures of these samples were further investigated by X-ray diffraction (XRD) and Raman spectroscopy. The pristine MXene exhibits the typical peak at ∼7° corresponding to the (002) plane of Ti3C2Tx MXene (Fig. S4a†).23 The (002) peak of PDA/Ti3C2Tx shifts to ∼6° due to the expanded interlayer spacing of Ti3C2Tx layers. The annealed pristine MXene does not show characteristic Ti3C2Tx MXene peaks but shows typical peaks of rutile TiO2 (JCPDS no. 21-1276).34 Notably, no TiO2 peaks are observed in the Ti3C2Tx@NC XRD pattern. In addition, the (002) peak of Ti3C2Tx MXene disappeared, which should be attributed to the suppressed restacking of MXene flakes by carbon layers on the surface.35 The Raman spectra (Fig. S4b†) show that all the pristine Ti3C2Tx, PDA/Ti3C2Tx and Ti3C2Tx@NC samples exhibit typical peaks corresponding to Ti3C2Tx MXene while annealed Ti3C2Tx shows the characteristic peaks at 237.7, 446.9 and 609.8 cm−1 of rutile TiO2.36 In addition, X-ray photoelectron spectroscopy (XPS) analyses (Fig. S5†) confirm the oxidation of Ti3C2Tx MXene into TiO2 during the annealing process. Based on the above results, it can be concluded that dopamine self-polymerized on the surface of the MXene could protect it from oxidation during the annealing treatment by in situ forming N-doped carbon layers.
Previous studies have reported that molybdate ions can react with dopamine and then polymerize into the Mo-PDA complex, which can be used to fabricate nanostructured Mo compound/carbon composites for various applications.33,37,38 Based on this inspiration, here we prepared Mo-PDA/Ti3C2Tx through a facile in situ polymerization method, as depicted in Fig. 1. The SEM and TEM images (Fig. S6†) of Mo-PDA/Ti3C2Tx show a similar ultrathin morphology to PDA/Ti3C2Tx, confirming that Ti3C2Tx flakes are tightly covered by Mo-PDA layers. Mo2C/Ti3C2Tx@NC was obtained by annealing Mo-PDA/Ti3C2Tx under Ar at 750 °C. As shown in Fig. 2d and Fig. S7,† the morphology of Mo2C/Ti3C2Tx@NC is similar to that of Ti3C2Tx@NC (Fig. 2b). The enlarged TEM image (Fig. 2e) evidences that high-density ultrasmall Mo2C nanodots with an average diameter of 1.1 nm (Fig. 2f) are uniformly anchored on the surface of Ti3C2Tx flakes. The high-resolution TEM image (Fig. 2g) exhibits the lattice fringes of ∼0.23 nm on the nanodots, corresponding to the d-spacing of the β-Mo2C (101) plane.17 The background lattices belong to Ti3C2Tx, confirming its hexagonal structure. The high-angle annular dark field scanning TEM (HAADF-STEM) image (Fig. 2h) reveals numerous bright dots decorated on the MXene sheets, further demonstrating the uniform dispersion of Mo2C nanodots. The corresponding energy dispersive X-ray spectroscopy (EDX) elemental mapping images (Fig. 2i) confirm the distribution of Ti, Mo, C and N elements on the Mo2C/Ti3C2Tx@NC surface.
Fig. 3a displays the XRD patterns of the samples. The characteristic peaks of both Mo-PDA and the MXene are observed in the XRD pattern of Mo-PDA/Ti3C2Tx. Similar to PDA/Ti3C2Tx, the (002) peaks of Ti3C2Tx negatively shift due to the expanded interlayer spacing. The absence of the (002) peak in Mo2C/Ti3C2Tx@NC results from the separation of MXene layers by the Mo2C nanodots and carbon layers. Through investigation using N2 adsorption–desorption isotherms, as shown in Fig. 3b, the Brunauer–Emmett–Teller (BET) surface areas of Mo2C/Ti3C2Tx@NC and Mo2C@NC were calculated to be 241.4 and 86.4 m2 g−1, respectively. Both samples exhibit type IV isotherms of mesoporous structures, with pores in the range from 2 to 50 nm. The larger surface area and mesoporous structure of Mo2C/Ti3C2Tx@NC could facilitate the accessibility of active sites and charge transfer during the HER process.
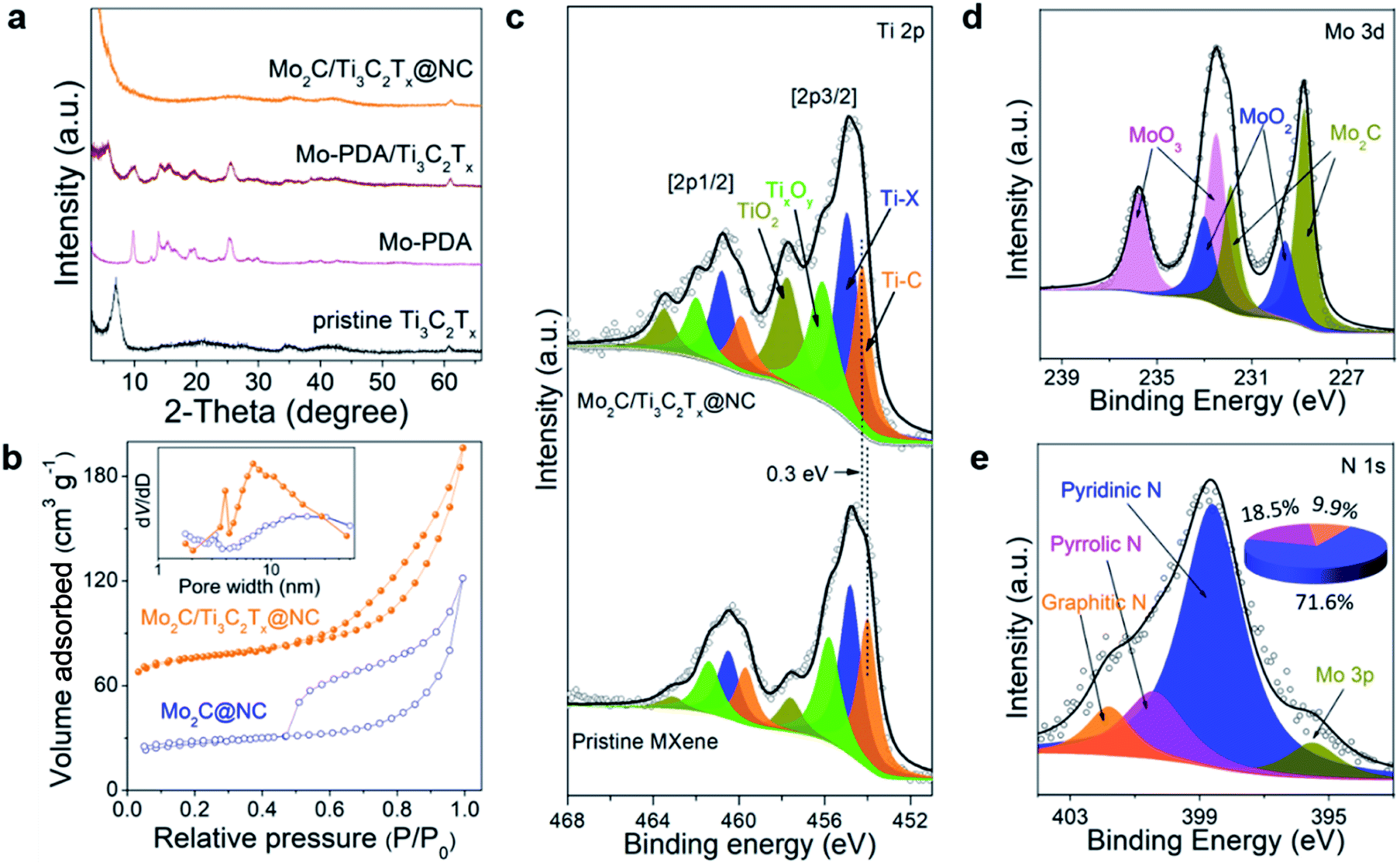 | ||
| Fig. 3 (a) XRD patterns of pristine Ti3C2Tx MXene, Mo-PDA, Mo-PDA/Ti3C2Tx and Mo2C/Ti3C2Tx@NC. (b) N2 adsorption–desorption isotherms and the corresponding pore size distributions of Mo2C@NC and Mo2C/Ti3C2Tx@NC. High-resolution XPS spectra of (c) Ti 2p, (d) Mo 3d and (e) N 1 s of Mo2C/Ti3C2Tx@NC. The Ti 2p survey of pristine Ti3C2Tx MXene is presented in (c) for comparison. The inset in (e) is the calculated contents of pyridinic N, pyrrolic N and graphitic N in Mo2C/Ti3C2Tx@NC. | ||
The valence states and compositions of Mo2C/Ti3C2Tx@NC were elucidated by XPS analyses. The XPS full scan (Fig. S8a†) evidences the existence of Ti, Mo, C, N, and O in Mo2C/Ti3C2Tx@NC. The Ti 2p core level can be fitted with four doublets for Ti 2p1/2 and Ti 2p3/2, as shown in Fig. 3c. Typically, the Ti 2p3/2 components at 454.3, 455, 456.1, and 457.8 eV are assigned to Ti–C (Ti+), Ti–X (Ti2+), TixOy (Ti3+), and TiO2 (Ti4+), respectively.22 Compared to the pristine Ti3C2Tx MXene, the binding energy of Ti–C positively shifts by about 0.3 eV, indicating the electron transfer from Ti3C2Tx to Mo2C nanodots in the hybrid.26 The deconvoluted Mo 3d XPS spectrum (Fig. 3d) reveals that the peaks at 228.8 eV and 231.9 eV are attributed to Mo2C. Meanwhile, due to inevitable surface oxidation, the peaks at 229.6, 232.5, 233, and 235.8 eV stem from Mo4+ 3d5/2, Mo6+ 3d5/2, Mo4+ 3d3/2 and Mo6+ 3d3/2, respectively.33 As shown in Fig. 3e, the deconvolution of the N1s XPS spectrum reveals the peaks at 398.6, 400.5 and 401.8 eV, which could be assigned to pyridinic, pyrrolic and graphitic N, respectively. Accordingly, the content is determined to be 71.64%, 18.43% and 9.93%, respectively. The C 1 s spectrum (Fig. S8b†) was fitted with five components at 282, 284.6, 285.5, 287.1, and 289.1 eV, stemming from Ti–C, C–C, C–N, C–O, and –COO, respectively. Upon comparison, the XPS spectra of Mo2C@NC are similar to those of Mo2C/Ti3C2Tx@NC (Fig. S9†). The above XPS analyses imply that Mo2C nanocrystals are intensely coupled on Ti3C2Tx MXene and encapsulated in the pyridinic N-rich carbon layer.
The HER performance of Mo2C/Ti3C2Tx@NC was evaluated by investigating the polarization curves in 0.5 M H2SO4 with all the potentials converted to the reversible hydrogen electrode (RHE). For comparison, Mo2C@NC, Ti3C2Tx@NC and commercial 20 wt% Pt/C catalyst were also measured. As shown in Fig. 4a, the commercial 20 wt% Pt/C exhibits the best activity with an onset overpotential of zero. Mo2C/Ti3C2Tx@NC shows a low onset overpotential of 6 mV vs. RHE (Fig. S10a†), which is very comparable to that of Pt/C but much lower than that of Mo2C@NC (53 mV) and Ti3C2Tx@NC (432 mV). Impressively, Mo2C/Ti3C2Tx@NC shows better performance than Pt/C at higher overpotentials (>106 mV). To elucidate the HER mechanism, Tafel plots of these catalysts were calculated based on the Tafel equation η = a + blog|j|, where j represents the current density and b is the Tafel slope.39 As shown in Fig. 4b, the Tafel slope of Mo2C/Ti3C2Tx@NC is determined to be 40 mV dec−1, close to that of Pt/C (31 mV dec−1), and lower than those of Mo2C@NC (67 mV dec−1) and Ti3C2Tx@NC (113 mV dec−1). The value implies that the HER operates through the Volmer–Heyrovsky mechanism on Mo2C/Ti3C2Tx@NC.40 The exchange current density (j0) for these catalysts was determined by fitting the linear portion of the Tafel plots at low current density (Fig. S10b†).41 As displayed in Fig. 4c, the j0 value of Mo2C/Ti3C2Tx@NC is 0.52 mA cm−2, smaller than that of Pt/C (0.841 mA cm−2) but much higher than those of Mo2C@NC (0.127 mA cm−2) and Ti3C2Tx@NC (2.6 × 10−5 mA cm−2), indicating the superior intrinsic activity of Mo2C/Ti3C2Tx@NC. To achieve a current density of 10 mA cm−2, the overpotentials of 27, 53, 121, and 631 mV are required for Pt/C, Mo2C/Ti3C2Tx@NC, Mo2C@NC and Ti3C2Tx@NC, respectively. As listed in Table S1,† such a low overpotential indicates that the Mo2C/Ti3C2Tx@NC exhibits one of the best HER performances among those of previously reported non-noble-metal electrocatalysts, such as MoC–Mo2C/PNCDs,9 MoS2/Ti3C2Tx,26 Co-SAC,10 and 1T′-ReSSe QDs.42 Moreover, Mo2C/Ti3C2Tx@NC also demonstrated excellent HER performance in basic (1 M KOH) and neutral (1 M PBS) electrolytes with an overpotential of 75 and 114 mV at 10 mA cm−2 and a Tafel slope of 59.2 and 80.3 mV dec−1, respectively, comparable to that of state-of-the-art non-noble-metal electrocatalysts (Fig. S11†).29,43–45
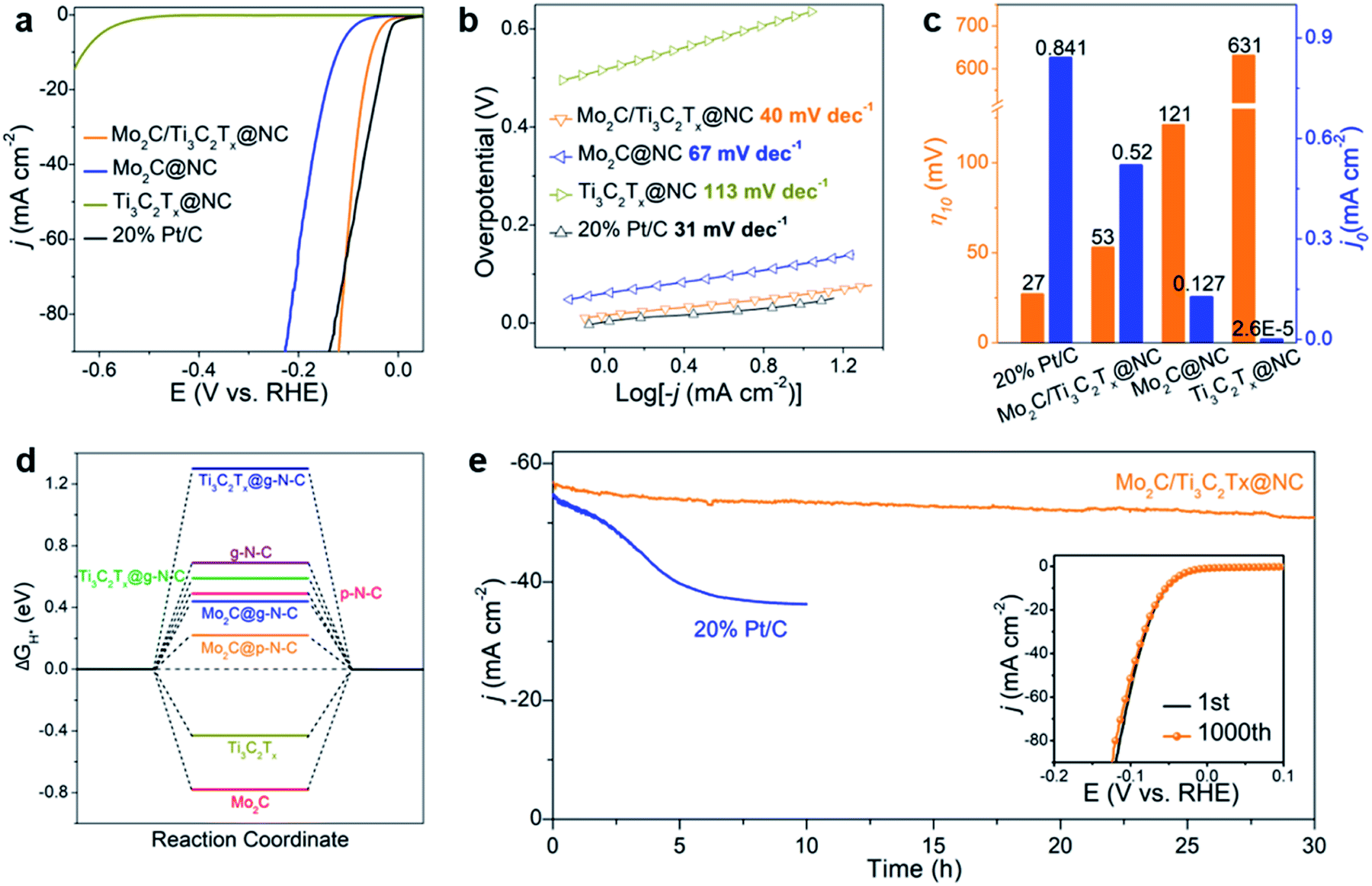 | ||
| Fig. 4 (a) Polarization curves and (b) corresponding Tafel plots of Mo2C/Ti3C2Tx@NC, Mo2C@NC, Ti3C2Tx@NC and commercial 20% Pt/C in 0.5 M H2SO4. (c) Comparison of overpotentials at 10 mA cm−2 (η10) and exchange current densities (j0) of the four catalysts. (d) Comparison of ΔGH* of p-N-C, g-N-C, Mo2C, Ti3C2Tx as well as their heterostructures. (e) Time-dependent current density curve of Mo2C/Ti3C2Tx@NC at a constant overpotential of 100 mV. The inset gives the polarization curves of Mo2C/Ti3C2Tx@NC initially and after 1000 CV cycles. | ||
The above results imply that Ti3C2Tx itself shows negligible HER activity but coupling of Mo2C with conductive Ti3C2Tx can modulate the morphological structure to optimize the HER activity. The Nyquist plots of the electrochemical impedance spectroscopy (EIS) responses (Fig. S12†) show a much smaller charge transfer resistance for Mo2C/Ti3C2Tx@NC than that for Mo2C@NC during the HER process, evidencing the faster HER kinetics due to the introduction of the conductive Ti3C2Tx support.46 To further understand the intrinsic origin, the electrochemical surface area (ECSA) and per-site turnover frequency (TOF) of Mo2C/Ti3C2Tx@NC and Mo2C@NC were typically compared. The ECSA was estimated from the electrochemical double-layer capacitance (Cdl) by performing cyclic voltammetry in the region from 0.2 to 0.3 V at rates varying from 10 to 100 mV s−1 (Fig. S13†). The ECSA of Mo2C/Ti3C2Tx@NC (603.75 cm2) is larger than that of Mo2C@NC (539 cm2), indicating that the former has a higher number of active sites. As the Ti3C2Tx@NC has very poor HER activity, the active sites should arise from Mo2C. Here, we assume that all the Mo atoms in the samples are active sites, which is determined by inductively coupled plasma optical emission spectroscopy (ICP-OES). Mo2C/Ti3C2Tx@NC has a TOF of 0.246 s−1 at an overpotential of 100 mV (Fig. S14†), much higher than that of Mo2C@NC (0.014 s−1), which is comparable to those of reported non-noble-metal HER electrocatalysts, such as 3DHP-Mo2C (0.045 s−1)47 and CoNx/C (0.39 s−1).48
The electrochemical results reveal that Mo2C/Ti3C2Tx@NC exhibits superior HER activity to that of its counterparts. To investigate the underlying mechanism, we performed density functional theory (DFT) calculations (Fig. S15–S17†). Here we considered a simple HER pathway containing three states: the initial state of H+ + e−, the intermediate state of adsorbed H (H*), and the final state of H2 molecule.39 The Gibbs free energy of the H* adsorption (ΔGH*) was calculated on the considered systems. Generally, a high-performance HER catalyst has a ΔGH* value close to zero.41Fig. 4d shows the calculated free energy diagrams for the HER. Pristine Mo2C and Ti3C2Tx MXene have a ΔGH* of −0.78 and −0.43 eV, respectively, suggesting their strong interactions with H and consequently poor HER performance. A previous study demonstrated that pyridinic N and graphitic N in carbon contribute to the HER activity of composite catalysts.49 The modeled pyridinic N doped graphene (p-N-C) and graphitic N doped graphene (g-N-C) possess a ΔGH* of 0.49 and 0.69 eV, respectively, which implies their unfavorable interactions with H and poor HER activities. Coupling N-doped carbon with Mo2C could lead to enhanced HER performance. Typically, the ΔGH* values of Mo2C@p-N-C and Mo2C@g-N-C were 0.22 and 0.44 eV, respectively, which were lower than those without N-doped carbon. Meanwhile, N-doped carbon coupled with Ti3C2Tx MXene shows high ΔGH* with unfavorable HER activity. The DFT results are in good agreement with the above experimental results, revealing that Mo2C@p-N-C serves as the active sites while the MXene support facilitates the charge transfer. Notably, Chen et al.50 reported an efficient HER catalyst of Mo single atoms anchored on N-doped carbon synthesized by an appropriate annealing method, which could be formed in the as-prepared Mo2C/Ti3C2Tx@NC. Accordingly, the Mo–N–C active site, determined as Mo1N1C2, is also considered. As shown in Fig. S18 and S19,† both Mo1N1C2 and Mo1N1C2/Ti3C2Tx exhibit extremely low ΔGH* values, suggesting that they are highly active toward the HER. The density of state near the Femi level for Mo1N1C2/Ti3C2Tx is higher than that of Mo1N1C2, indicating that the Ti3C2Tx MXenes facilitate the charge transfer during the HER process.51
The stability of HER electrocatalysts is also a critical indicator for practical applications. As shown in Fig. 4e, the time-dependent current density curve of Mo2C/Ti3C2Tx@NC at a constant overpotential of 100 mV shows a slight decrease over 30 h, which is much better than that of commercial 20 wt% Pt/C, demonstrating excellent durability. In addition, the stability of Mo2C/Ti3C2Tx@NC was studied using CV cycles in the range of 0.1 to −0.2 V vs. RHE. The polarization curve of Mo2C/Ti3C2Tx@NC after 1000 cycles (inset of Fig. 4e) shows a negligible decrease in current density compared to the initial curve, implying its long-term stability. Detailed characterization of Mo2C/Ti3C2Tx@NC after the stability test (Fig. S21†) further confirms the outstanding durability of its morphology and structure, which could be attributed to the protection of the covered carbon layers.
In controlled experiments, the effect of carbonization temperature (650 °C and 850 °C) on HER performance was investigated. The samples are named M-650 and M-850. TEM images (Fig. 5a, b and S22†) reveal that MoOx nanodots in M-650 (not completely carbonized) have an average size of 1 nm while the size of Mo2C nanodots in M-850 is 2.6 nm. It is in good agreement with a previous report which showed that the particle size would increase with the carbonization temperature.52 The XRD patterns of the two samples are similar to that of Mo2C/Ti3C2Tx@NC (here defined as M-750) except for the presence of TiO2 signals in M-850 (Fig. S23†). The XPS results (Fig. 5c) suggest that M-650 contains MoOx rather than Mo2C, which could arise from the insufficient temperature. Meanwhile, as shown in Fig. 5d, M-850 has a similar Mo 3d XPS spectrum to M-750, suggesting the formation of Mo2C nanodots. As shown in Fig. 5e, the overpotentials of M-650 and M-850 for achieving 10 mA cm−2 are 248 and 83 mV, respectively. And the corresponding Tafel slopes are 116.3 and 58.3 mV dec−1, respectively (Fig. 5f). Obviously, M-750 exhibits the best HER activity among the three samples, which could be attributed to the following two factors: the MoOx generated at a lower temperature (650 °C) has poorer intrinsic activity than Mo2C, and the higher temperature (850 °C) induces aggregation of Mo2C nanodots and partial oxidation of the MXene which would reduce the density of active sites and electron conductivity.
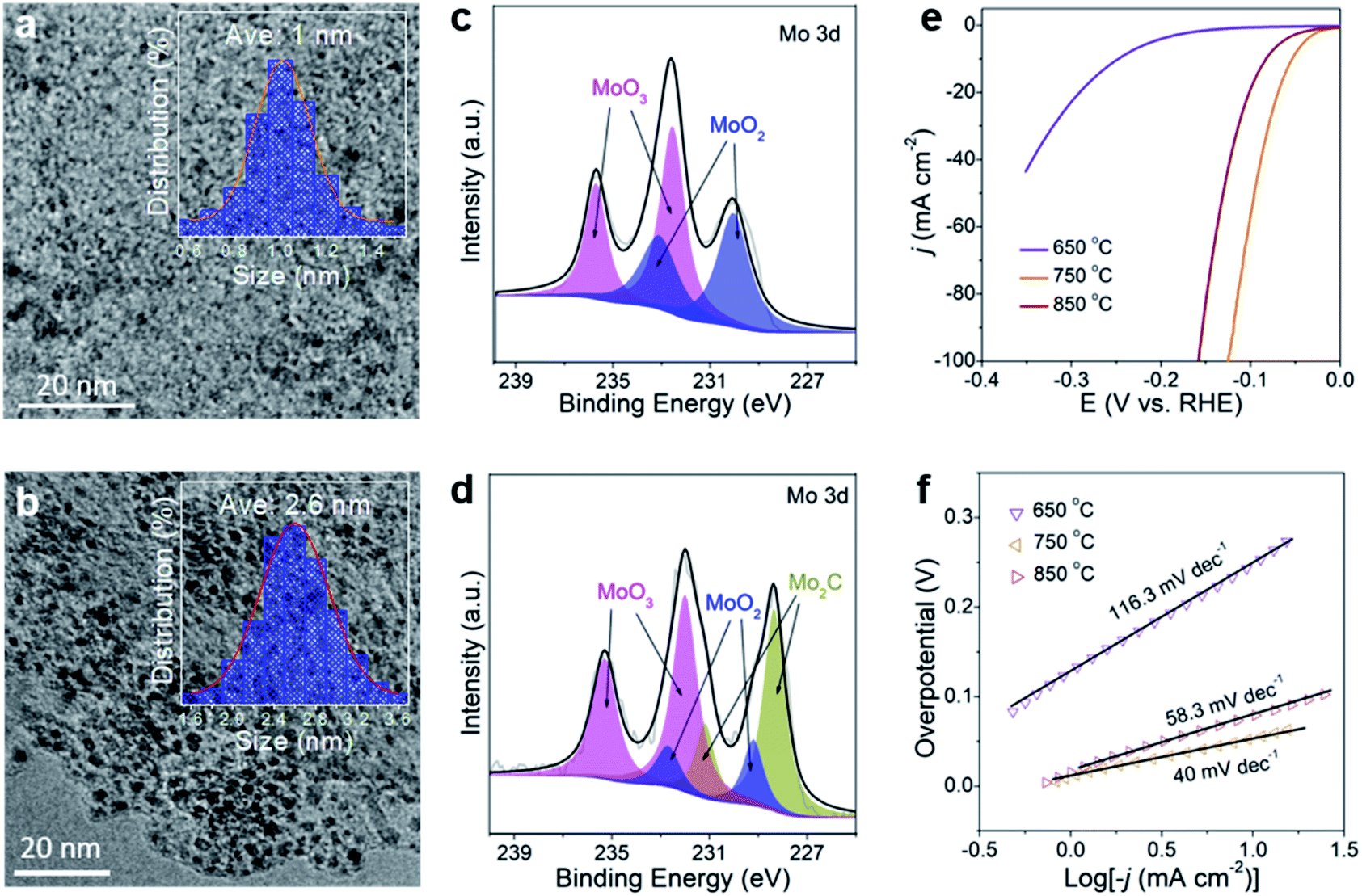 | ||
| Fig. 5 TEM images of (a) M-650 and (b) M-850. The inset gives the size distributions of nanoparticles in the samples. High-resolution XPS Mo 3d spectra of (c) M-650 and (d) M-850. Polarization curves (e) and Tafel plots (f) of M-650, M-750, and M-850. | ||
Based on the aforementioned considerations, the extraordinary HER performance of Mo2C/Ti3C2Tx@NC is postulated to arise from the following synergistic aspects: (1) in situ polymerization of dopamine retains the structural stability of Ti3C2Tx MXene at high carbonization temperature, facilitating the charge transfer; (2) polydopamine chelated MoO42− delivers highly disperse ultrasmall Mo2C nanodots on the surface of the MXene, favoring the high accessibility of active sites for the HER; (3) the pyridinic N dopants in carbon coupled with Mo2C induce activated active sites for the HER; (4) the intensely coupled Mo2C on Ti3C2Tx provides a resistance-less path favorable for fast electron transfer; (5) the geometric confinement of Mo2C and the MXene within the carbon layer guarantees the excellent stability of the HER performance during long-term operation.
Conclusions
In summary, we developed an in situ polymerization strategy to stabilize MXenes against oxidation during high-temperature treatment, which was extended to the fabrication of a hierarchical Mo2C/Ti3C2Tx@NC composite. Serving as a HER catalyst, Mo2C/Ti3C2Tx@NC exhibits excellent activity in the entire pH range and robust stability. Theoretical simulations demonstrated that the coupled Mo2C and pyridinic N doped carbon serve as the most active sites for H adsorption, while the strongly incorporated metallic Ti3C2Tx MXene provides a fast charge transfer pathway, which synergistically lead to much superior HER performance. This strategy not only provides a highly active HER catalyst, but also opens a new avenue for designing MXene-based nanocomposites that require high synthesis temperature, which could be applied in various energy-related fields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the AcRF Tier 1 grant (RG105/19) from the Ministry of Education in Singapore and the National Natural Science Foundation of China (U1401248, 11874044). The DFT calculations were supported by TianHe-1(A) at the NSCC in Tianjin. The authors also thank Prof. Yury Gogotsi from Drexel University for helpful comments on the manuscript.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16 CrossRef PubMed.
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- C. Niether, S. Faure, A. Bordet, J. Deseure, M. Chatenet, J. Carrey, B. Chaudret and A. Rouet, Nat. Energy, 2018, 3, 476 CrossRef CAS.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519 RSC.
- Y. Zang, S. Niu, Y. Wu, X. Zheng, J. Cai, J. Ye, Y. Xie, Y. Liu, J. Zhou and J. Zhu, Nat. Commun., 2019, 10, 1217 CrossRef PubMed.
- H. Jin, X. Liu, S. Chen, A. Vasileff, L. Li, Y. Jiao, L. Song, Y. Zheng and S.-Z. Qiao, ACS Energy Lett., 2019, 4, 805 CrossRef CAS.
- H. Wang, L. Ouyang, G. Zou, C. Sun, J. Hu, X. Xiao and L. Gao, ACS Catal., 2018, 8, 9529 CrossRef CAS.
- X. F. Lu, L. Yu, J. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900699 CrossRef PubMed.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. L. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. J. Sun and H. Wong, Adv. Energy Mater., 2019, 9, 1803689 CrossRef.
- Z. Fang, L. Peng, Y. Qian, X. Zhang, Y. Xie, J. J. Cha and G. Yu, J. Am. Chem. Soc., 2018, 140, 5241 CrossRef CAS PubMed.
- V. Jose, E. Edison, W. W. Manalastas Jr, S. Sreejith, J. M. Vianney Nsanzimana, M. Srinivasan and J.-M. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 39798 CrossRef CAS PubMed.
- H. Wang, Q. Yi, L. Gao, Y. Gao, T. Liu, Y.-B. Jiang, Y. Sun and G. Zou, Nanoscale, 2017, 9, 16342 RSC.
- H. Wang, X. Xiao, S. Liu, C.-L. Chiang, X. Kuai, C.-K. Peng, Y.-C. Lin, J. Zhao, J.-H. Choi, Y.-G. Lin, J.-M. Lee and L. Gao, J. Am. Chem. Soc., 2019, 141, 18578 CrossRef CAS PubMed.
- J. Yang, A. R. Mohmad, Y. Wang, R. Fullon, X. Song, F. Zhao, I. Bozkurt, M. Augustin, E. J. Santos and H. S. Shin, Nat. Mater., 2019, 18, 1309 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Catal. Today, 2005, 105, 66 CrossRef CAS.
- H. Wang, C. Sun, Y. Cao, J. Zhu, Y. Chen, J. Guo, J. Zhao, Y. Sun and G. Zou, Carbon, 2017, 114, 628 CrossRef CAS.
- Z. Zhou, Z. Yuan, S. Li, H. Li, J. Chen, Y. Wang, Q. Huang, C. Wang, H. E. Karahan and G. Henkelman, Small, 2019, 15, 1900358 CrossRef PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- K. Maleski, C. E. Ren, M.-Q. Zhao, B. Anasori and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2018, 10, 24491 CrossRef CAS PubMed.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78 CrossRef CAS PubMed.
- S. J. Kim, H.-J. Koh, C. E. Ren, O. Kwon, K. Maleski, S.-Y. Cho, B. Anasori, C.-K. Kim, Y.-K. Choi and J. Kim, ACS Nano, 2018, 12, 986 CrossRef CAS PubMed.
- W. Bao, X. Tang, X. Guo, S. Choi, C. Wang, Y. Gogotsi and G. Wang, Joule, 2018, 2, 778 CrossRef CAS.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. M. Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985 CrossRef CAS.
- J. Liu, Y. Liu, D. Xu, Y. Zhu, W. Peng, Y. Li, F. Zhang and X. Fan, Appl. Catal., B, 2019, 241, 89 CrossRef CAS.
- X.-D. Zhu, Y. Xie and Y.-T. Liu, J. Mater. Chem. A, 2018, 6, 21255 RSC.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086 CrossRef CAS PubMed.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 9, 1901333 CrossRef.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13, 3448 CrossRef CAS PubMed.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337 CrossRef CAS PubMed.
- M. Han, X. Yin, X. Li, B. Anasori, L. Zhang, L. Cheng and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2017, 9, 20038 CrossRef CAS PubMed.
- M. Yu, S. Zhou, Z. Wang, J. Zhao and J. Qiu, Nano Energy, 2018, 44, 181 CrossRef CAS.
- X. Wang, Z. Wang, M. Zhang, X. Jiang, Y. Wang, J. Lv, G. He and Z. Sun, J. Alloys Compd., 2017, 725, 1166 CrossRef CAS.
- Y. Yang, M. Luo, Y. Xing, S. Wang, W. Zhang, F. Lv, Y. Li, Y. Zhang, W. Wang and S. Guo, Adv. Mater., 2018, 30, 1706085 CrossRef PubMed.
- J. Zhang, L. Zhou, Q. Sun, H. Ming, L. Sun, C. Wang, Y. Wu, K. Guan, L. Wang and J. Ming, Chem.–Eur. J., 2019, 25, 8813 CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, 23 CrossRef.
- Z. Lai, A. Chaturvedi, Y. Wang, T. H. Tran, X. Liu, C. Tan, Z. Luo, B. Chen, Y. Huang and G.-H. Nam, J. Am. Chem. Soc., 2018, 140, 8563–8568 CrossRef CAS PubMed.
- X. Zhang, X. Yu, L. Zhang, F. Zhou, Y. Liang and R. Wang, Adv. Funct. Mater., 2018, 28, 1706523 CrossRef.
- I. K. Mishra, H. Zhou, J. Sun, F. Qin, K. Dahal, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2246 RSC.
- K. Liang, S. Pakhira, Z. Yang, A. Nijamudheen, L. Ju, M. Wang, C. I. Aguirre-Velez, G. E. Sterbinsky, Y. Du and Z. Feng, ACS Catal., 2018, 9, 651 CrossRef.
- J. Liang, C. Ding, J. Liu, T. Chen, W. Peng, Y. Li, F. Zhang and X. Fan, Nanoscale, 2019, 11, 10992 RSC.
- H. Ang, H. Wang, B. Li, Y. Zong, X. Wang and Q. Yan, Small, 2016, 12, 2859 CrossRef CAS PubMed.
- H.-W. Liang, S. Brüller, R. Dong, J. Zhang, X. Feng and K. Müllen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- X. Wang, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 31, 1803625 CrossRef PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Zhu, Y. Wang, J. Dong, S. Tian, W.-C. Cheong, S. Lu, L. Zheng, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 16086 CrossRef CAS PubMed.
- M. Yu, S. Zhou, Z. Wang, J. Zhao and J. Qiu, Nano Energy, 2018, 44, 181 CrossRef CAS.
- Y. Liu, G. Yu, G. D. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, Angew. Chem., Int. Ed., 2015, 54, 10752 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta01697g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |