
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanisms of carbon dioxide reduction on strontium titanate perovskites
Umberto
Terranova
 *abc,
Francesc
Viñes
c,
Nora H.
de Leeuw
ad and
Francesc
Illas
c
*abc,
Francesc
Viñes
c,
Nora H.
de Leeuw
ad and
Francesc
Illas
c
aSchool of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: umberto.terranova@buckingham.ac.uk
bSchool of Postgraduate Medicine and Allied Health, Crewe Campus, University of Buckingham, Crewe, CW1 5DU, UK
cDepartament de Ciència de Materials i Química Física, Institut de Quìmica Teòrica i Computacional (IQTCUB), Universitat de Barcelona, c/Martí i Franquès 1-11, 08028, Barcelona, Spain
dDepartment of Earth Sciences, Utrecht University, 3584 CD Utrecht, The Netherlands
First published on 22nd April 2020
Abstract
Strontium titanate (SrTiO3) is a promising material for the light-driven conversion of carbon dioxide (CO2) into renewable fuels. However, the mechanisms of the relevant reactions are not yet well understood. In this work, we have used density functional theory calculations to explore CO2 reduction on the (001) surface of the SrTiO3 photocatalyst. Our results indicate that, in contrast to COOH, the formation of a HCOO or CO2− intermediate is thermodynamically hindered, which is consistent with the fact that formic acid (HCOOH) is not a major product in the experiments reported in the literature. We show that a pathway to carbon monoxide (CO) is instead possible, and that the formation of COOH is the rate-limiting step. Finally, we suggest that substitutional doping of Sr ions represents a promising approach to lower the energy barrier of the COOH formation.
1 Introduction
To reduce the still increasing CO2 emissions responsible for global warming and the concomitant climate change remains one of the biggest challenges of our era. The catalytic technologies currently being explored to utilise the CO2 molecule by converting it into useful hydrocarbons include heterogeneous1–4 and electrochemical5–7 catalysis. Approaches such as photocatalysis,8,9 mimicking the natural process of photosynthesis, are particularly attractive, as they are achieved at room temperature and require only the harnessing of solar light as a source of energy. However, the achievement of photocatalytic conversions suitable to industrial applications remains a difficult challenge,10 which motivates the continuous search for new and improved photocatalytic materials.Since their first use in heterogeneous catalysis in 1952,11 perovskite oxides have attracted significant interest in a diverse range of reactions,12 especially because they are cheaper and easier to prepare than metal-based catalysts such as platinum or palladium.13 Compared to other widely investigated oxide photocatalysts, e.g. ZrO2 and Ga2O3, perovskite oxides have bandgaps with a better overlap with the solar spectrum. In addition, owing to the unique flexibility of their structure, perovskite oxides have bands edges which can be tuned to facilitate both the reduction of CO2 and the oxidation of water during the photocatalytic process.9
Because of their striking photochemical stabilities, titanate perovskites are particularly promising for CO2 photocatalysis.14 Taking inspiration from leaves, Zhou et al. have developed an artificial photosynthetic system based on titanate perovskites which generates carbon monoxide and methane from CO2 and water.15 These materials have also been investigated in combination with other semiconductors, co-catalysts and dopants, in the effort to improve their conversion efficiencies. For example, Li et al. obtained CO and H2 by loading SrTiO3 with noble metals,16 while CaTiO3 samples loaded with Ag exhibited stable photocatalytic activities for CO2 reduction to CO using water as an electron donor.17 The photoreduction of CO2 on SrTiO3-based nanocomposite systems has also been demonstrated, with CH4 as the major product.18 The generation of oxygen vacancies enhances the adsorption of CO2 on SrTiO3, leading to higher yields of CH4,19 while NaTaO3 doped with Ba, Ca, and Sr has shown high activity towards CO2 reduction to CO in water.20
Detailed atomistic knowledge of the light-triggered catalytic reaction mechanism is crucial to help design better photocatalysts.21 For example, it allows us to ascertain the limiting reaction steps, featuring the higher activation energy barriers in a catalytic pathway, which elucidates ways of fine-tuning the perovskite structure to lower these activation barriers. However, in contrast to the most investigated anatase TiO2 photocatalysts,22 the elementary reaction steps occurring after the adsorption of CO2 on titanate perovskites are yet to be clarified.
Here, we provide this missing piece of information on the textbook SrTiO3 perovskite, one of the most commonly investigated members of the titanate perovskite family, which, with a tunable bandgap of 3.2 eV (ref. 23) (identical to that of anatase) and remarkable charge transport properties,14 holds great potential as a photocatalyst.19,24–27 Using simulations within the density functional theory (DFT), we first aim to achieve the description of a realistic reaction pathway for the experimentally observed reduction of CO2 to CO. Afterwards, we take advantage of the fresh insights into the system to put forward a doping approach aimed at improving the efficiency of the photocatalytic reaction.
2 Models and methods
The most-stable SrTiO3 (001) surface can have two different competitive terminations, namely SrO and TiO2.28 In this study, we have focussed on the SrO termination only, since its equivalent has been shown generally to be the favoured plane in a wide diversity of perovskites.29,30 Our (001) surface model, built from an optimised primitive cell, consists of a 2 × 2 × 4 supercell, and contains 80 atoms located within 8 layers. Surface slabs were separated by a vacuum region of 15 Å along the normal direction to avoid spurious interactions between periodic images. All structures were drawn with the visualization for electronic and structural analysis (VESTA) program suite.31The adsorption energy of CO2 was evaluated according to the formula
| Eads = Eslab/mol − Eslab − Emol, | (1) |
| G = E + Fvib + pV, | (2) |
μ(T,p) = μref(T0,p0) + ΔH(T,p0) − TΔS(T,p0) + kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(p/p0), log(p/p0), | (3) |
log(p/p0) includes the effect of pressure on the chemical potential with respect to the standard one. If T0 = 0 K, μref can be approximated by the DFT energy of the molecule in the gas-phase, including the zero point energy contribution. Thus, eqn (3) allows us to calculate the chemical potential of CO2, CO, H2O and H2 at given temperature and pressure by using their respective tabulated enthalpy and entropy values at the standard ambient pressure p0.34
The substitutional energy for the dopant X (X = Ba or Ca) was calculated according to
| Esubs(X) = E(doped) + E(Sr) − E(undoped) − E(X), | (4) |
All DFT calculations were performed with the VASP 5.3 package35,36 using the Perdew–Burke–Ernzerhof (PBE) functional.37 We have employed the projector augmented wave method to model the core–electron interaction,38 treating explicitly the following electrons: Sr 4s 4p 5s; Ti 3d 4s; O 2s 2p; Ba 5s 5p 6s; Ca 3s 3p 4s. All calculations were performed with a plane wave cutoff of 500 eV, which guarantees that the absolute energies of bulk SrTiO3 are converged within 2 meV per atom. Calculations were stopped when the forces acting on the ions were less than 10−2 eV Å−1. After finding that a 6 × 6 × 6 Monkhorst–Pack grid39 ensures that the absolute energies of bulk SrTiO3 are converged to better than 1 meV per atom, we scaled the grids of the surface calculations inversely with the dimension of the unit cells to 3 × 3 × 1. We have also undertaken test calculations to make sure that the atomic relaxations in the surface slab are converged with respect to its thickness. Transition states were located with the climbing image nudged elastic band algorithm.40 The number of images used varied from one to four, depending on the elementary reaction step. We have calculated vibrational frequencies under the harmonic approximation and with Hessian matrix elements estimated from finite differences of analytical gradients with displacements of 0.015 Å in length. Only the degrees of freedom of the adsorbate species and the atoms in the topmost atomic layer of the surface have been included in the vibrational analysis.
3 Results and discussion
3.1 Adsorption of CO2
A sufficiently large number of different adsorption configurations for CO2 on the SrO-terminated SrTiO3 (001) surface have been probed to ensure that realistic minimum energy structures would be obtained. Following optimisation, all configurations relaxed towards the structure depicted in Fig. 1, where the arrangement of the atoms at the adsorption site resembles that of a CO32− carbonate ion. In particular, each carbonyl oxygen forms a bridging bond with two surface Sr ions, whereas the remaining oxygen, Os, is significantly displaced from its surface position. The C–O and C–Os bond lengths are 1.27 and 1.36 Å, respectively, while the O1–C–O2 angle of 122.4° confirms the strong interaction with the surface, as shown by the large adsorption energy of −1.82 eV. All of these values are in line with a CO2 capture and activation process.41 Both the geometry and the energetics, which are summarised in Table 1, are comparable to those found in a previous DFT investigation by Sopiha et al.,42 who also showed that the adsorption geometry of CO2 slightly changes, while its interaction becomes stronger, when the size of the surface unit cell is increased from a 2 × 2 to a 3 × 3 supercell. As the structure in Fig. 1 is the only stable on the 2 × 2 unit cell which we adopted, we assume that any reaction pathway must include it as a reactant configuration. However, we stress that other starting configurations, potentially leading to different reduction mechanisms, may also be possible at lower CO2 coverages.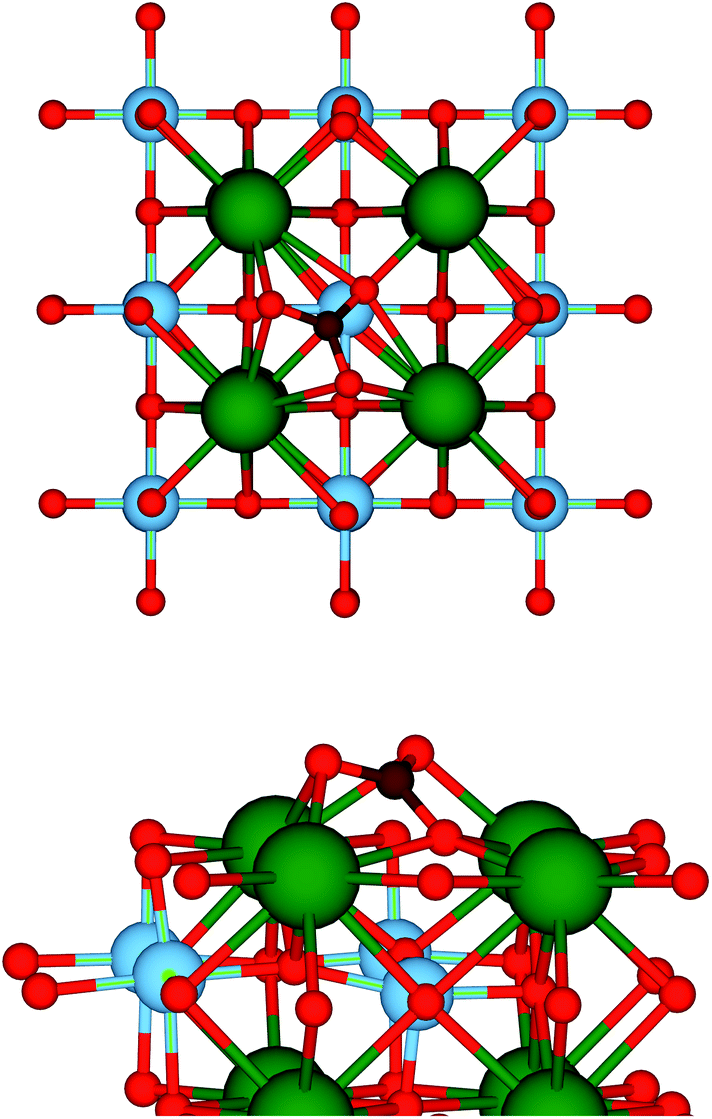 | ||
| Fig. 1 Top (upper panel) and side (lower panel) views of the adsorption structure of CO2 on the SrO-terminated (001) surface of SrTiO3. Colour code: Sr – green, Ti – blue, O – red, C – brown. | ||
| E ads (eV) | C–O1 (Å) | C–O2 (Å) | C–Os (Å) | O1–C–O2 (°) |
|---|---|---|---|---|
| −1.82 | 1.27 | 1.27 | 1.36 | 122.4 |
It is instructive, at this point, to compare the adsorption energy of this structure with that of a water molecule. Regardless of the initial configuration and in line with the literature,43 we found that water spontaneously dissociates on the SrO-terminated (001) surface of SrTiO3. The dissociated water molecule is adsorbed with an energy of −1.26 eV, which is considerably less negative than the −1.82 eV value found for CO2. As a result, we expect SrTiO3 to be preferentially covered by CO2 rather than by water, which in turn suggests that the CO2 reduction process should be more favourable than the competing hydrogen evolution reaction.10 Future detailed studies can now build on these findings to confirm the selectivity of SrTiO3 towards the reduction of CO2, rather than water.
3.2 Thermodynamics of intermediates
The photocatalytic conversion of CO2 takes place in the presence of a photoexcited electron which reduces the SrTiO3 surface. When the carbonate-like structure described above is adsorbed on the reduced surface, which we have modelled by adsorbing a H atom on SrTiO3 as in previous works,22,44 a hydrogen bond between a carbonyl oxygen and the proton on the surface is formed, which is illustrated in Fig. 2a. However, the geometry changes with respect to the neutral surface, e.g. the elongation of the two C–O bonds, are only marginal, as can be seen from the parameters reported in Table 2.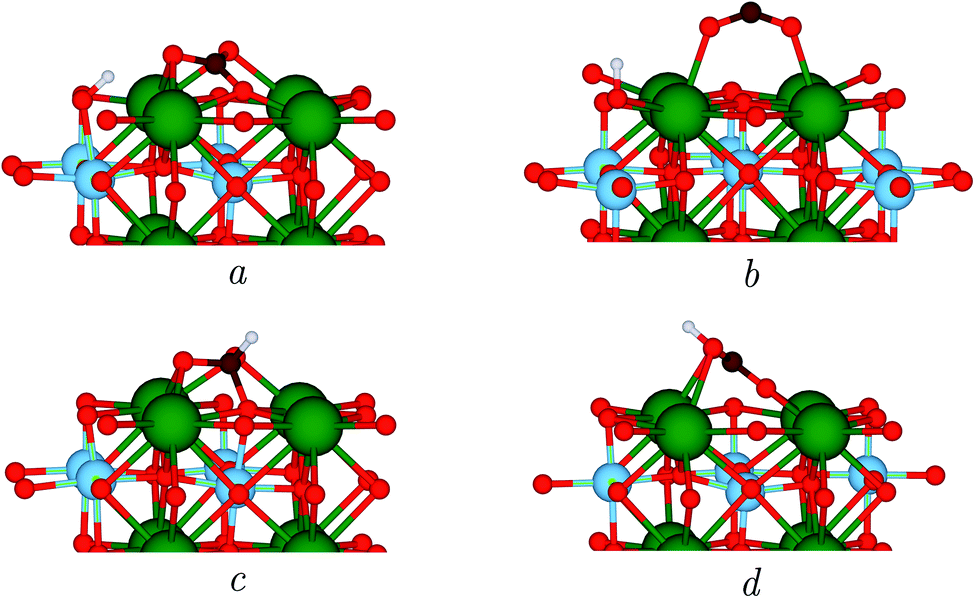 | ||
| Fig. 2 Side views of the structures discussed in the text. CO2 adsorption on a reduced surface (a); activated CO2 in a bridging bidentate configuration similar to that observed in anatase TiO2 (b); HCOO structure with hydrogenation at the C atom (c); COOH structure with formation of a O–H bond (d); Colour code: Sr – green, Ti – blue, O – red, C – brown, H – white. | ||
| Label | E (eV) | C–O1 (Å) | C–O2 (Å) | C–Os (Å) | O1–C–O2 (°) |
|---|---|---|---|---|---|
| (a) | 0.00 | 1.29 | 1.30 | 1.32 | 121.6 |
| (b) | 1.96 | 1.23 | 1.23 | — | 140.4 |
| (c) | 2.55 | 1.38 | 1.38 | 1.45 | 110.6 |
| (d) | 1.15 | 1.41 | 1.25 | 1.27 | 117.3 |
TiO2 is arguably the prototype photocatalyst,45 and many efforts have been devoted to the elucidation of the molecular mechanisms yielding fuels from CO2 using TiO2 polymorphs. He et al. concluded that the formation of an activated CO2− radical adsorbed in a bridging configuration is the first step towards hydrogenation and the formation of formic acid on anatase.22 In search of a possible similar mechanism, we have relaxed such a structure. While not stable on a neutral surface, interestingly, in the presence of a reducing electron, this bent geometry converged to one with a O1–C–O2 angle of 140.4°, see Fig. 2b and Table 2, which highlights the importance of photogenerated electrons on the activation of the molecule. However, its energy resulted to be 1.96 eV higher than that of the carbonate-like structure in Fig. 2a, which makes it an unlikely candidate as a possible intermediate, while indicating that catalytic mechanisms different than those of TiO2 are at play in SrTiO3.
Next, we assessed whether hydrogenation of CO2 could occur via a different intermediate. Thus, we detached the adsorbed H species from the surface site of Fig. 2a, creating a new H–C bond, see Fig. 2c. However, we found the energy of the resulting configuration to be 2.55 eV higher than that of the original structure, which clearly excludes its participation in any reaction mechanism.
Finally, we relaxed the structure in Fig. 2d, where we simulated the H transfer from the surface site of structure of Fig. 2a to the closest CO2 oxygen. The formation of a COOH group changes the adsorption geometry to that of a bridging bidentate, having an energy 1.15 eV higher than the initial structure, taken as reference, as shown in Table 2.
On the basis of thermodynamic (energetic) considerations, we can conclude that only the COOH intermediate can participate in the reduction of CO2, while pathways involving either CO2− and/or HCOO should be discarded. However, we cannot rule out the existence of other, in principle less favoured, pathways where two or more electrons and/or proton are transferred.22 The high energy penalty for hydrogenation through the carbon atom explains why HCOOH is not detected in CO2 photochemical reactions on SrTiO3. Instead, the COOH species that can be reached energetically points to a pathway towards CO,10 in agreement with experimental findings on titanate perovskites.15,16
3.3 Reaction pathway to CO
With the information gained in the previous section, we have derived a realistic pathway for the reduction of CO2 to CO, which we present in the top panel of Fig. 3, whereas reaction geometries are shown in Fig. 4. The formation of the COOH intermediate proceeds via TS1 and has an activation energy barrier for the hydrogen transfer of 1.22 eV. Only 0.75 eV is required to reach TS2 (where the C–O distance is 1.56 Å), break the C–O bond and form the CO and OH species at the surface. Note, however, that the barrier for CO and OH recombination is lower, which may indicate that, from a kinetic point of view, a substantial part of reactions will go backwards, with forward direction favoured by fast CO and OH diffusions. The subsequent release of CO is endothermic by merely 0.12 eV, and the process leaves a hydrogen-bonded H+ and OH− couple. In the last step of the reaction mechanism, as a result of a proton transfer in TS3, a H2O molecule forms. This water molecule interacts with the surface by means of two hydrogen bonds, and its desorption raises the energy by 0.86 eV. The entire reaction cycle, CO2 + 2H+ + 2e− → CO + H2O, is endothermic by 0.73 eV. Obviously, the energy profile provides a preliminary picture of the overall mechanism, allowing to discard the HCOO intermediate. However, to unravel the amount of CO formed and the effect of H2O requires further analysis that is beyond the scope of the present work. Note also, that the models used represent the ideal (001) surface, whereas the real material may be more complex. In any case, the present results provide some clear and unbiased hints that explain experimental observations.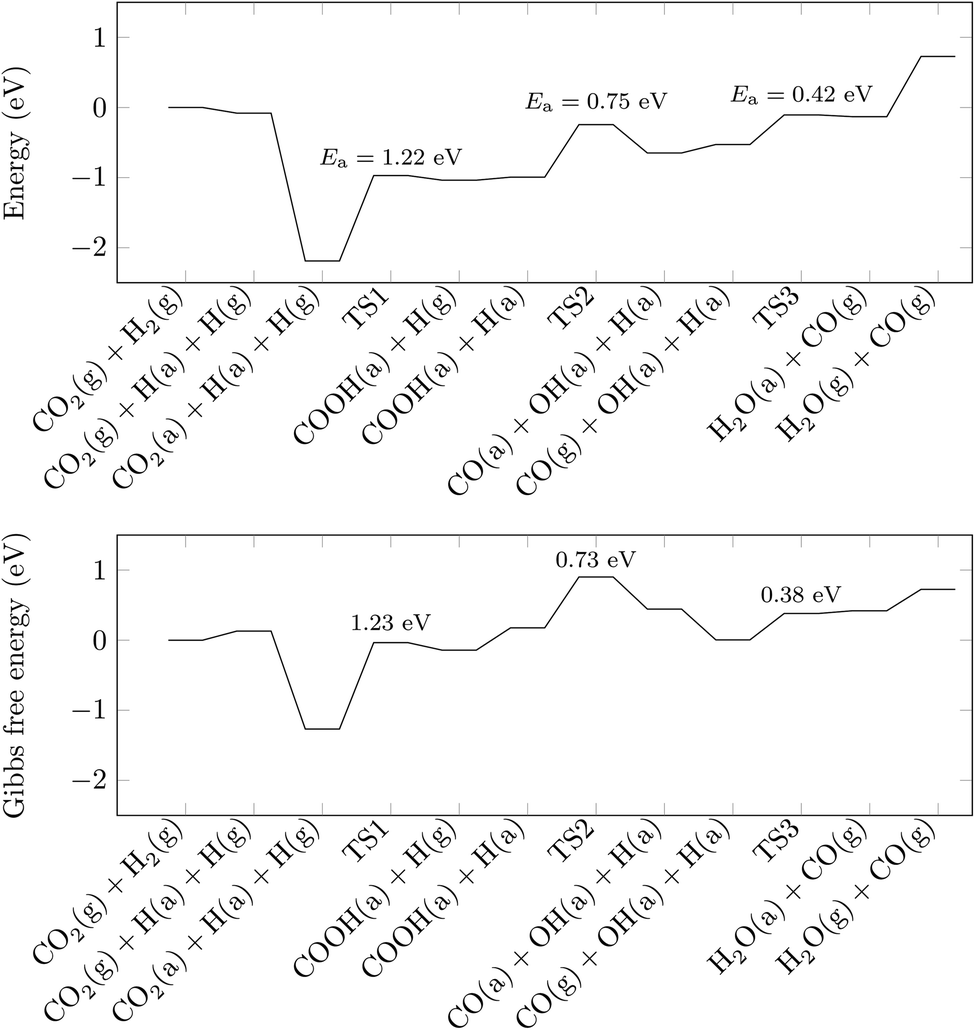 | ||
| Fig. 3 Reaction pathways for the reduction of CO2 to CO on SrTiO3. Structures are labelled according to the state of the species. (a) indicates that the species is adsorbed on the surface, while (g) indicates that the species is modelled in the gas-phase and does not interact with the surface. Transition states are indicated as TS1, TS2 and TS3. The zero of the energies are taken as those of structure CO2(g) + H2(g). Activation energies (Ea) are shown. | ||
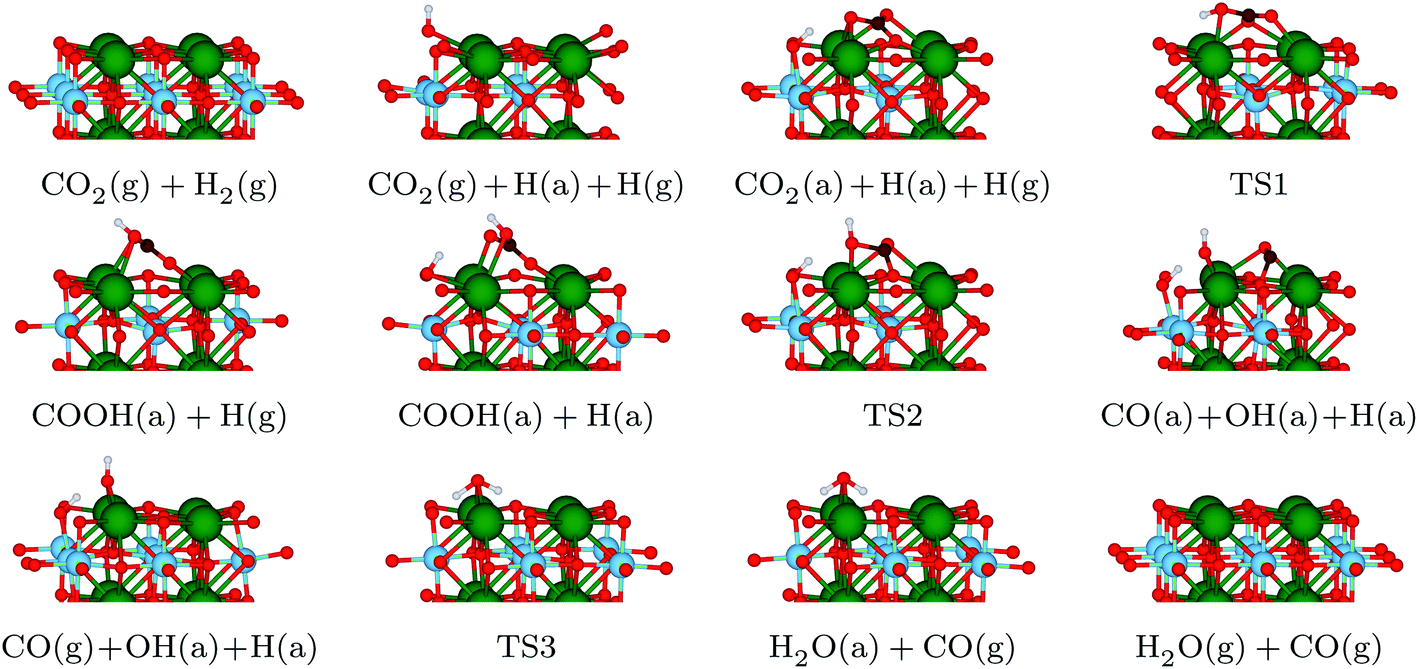 | ||
| Fig. 4 Side views of the structures participating in the reaction pathway illustrated in Fig. 3. Colour code: Sr – green, Ti – blue, O – red, C – brown, H – white. | ||
In the bottom panel of Fig. 3, we plot the Gibbs free energy of the pathway above, taking into account temperature (300 K) and pressure (1 bar) effects. The profile flattens compared to that in the top panel of Fig. 3, although the endothermicity of the reaction is not much affected. The flattening can be explained by the larger entropy contributions carried by the gaseous species compared to their adsorbed counterparts. For example, the gain due to the adsorption of CO2 increases drastically, from −2.11 to −1.40 eV, when the Gibbs free energy, rather than the DFT energy, is considered. However, the rate limiting step remains the formation of the COOH group, whose activation energy barrier remains virtually unchanged at 1.23 eV, which is in line with the low photoreduction rates typical of titanate perovskites.46 Note, however, that once pressure and temperature effects are taken into account, the thermodynamics will tend to drive the reaction backwards since now some transition states are located above the desorption limit. This is an issue deserving further attention through macroscopic simulations including all elementary steps and searching for the optimum operating conditions as well.
3.4 Substitutional doping
Having established that the formation of the COOH intermediate (more favoured than its desorption from the surface by around 0.1 eV, as shown in Fig. 3), is the rate-limiting step in the production of CO on the pure SrTiO3 (001) surface, we have carried out further calculations to explore how the activation energy of this elementary reaction step is affected by surface doping. Similar to the study by Carlotto et al.,47 we have only focussed on the top surface layer. In particular, we have considered the substitution of one of the four Sr ions by a Ba or Ca ion. We have chosen these dopants because their ionic radii in the 2+ charge state, at 149 and 114 pm, respectively, are close to the 132 pm ionic radius of Sr,48 which implies that they should be accommodated easily within the SrTiO3 material, both geometrically and in terms of charge. Moreover, BaTiO3 and CaTiO3 are the most common titanate perovskites after SrTiO3, which should ensure the successful replacement of Sr by Ba and Ca ions during the synthesis of the photocatalyst. The stability of the doped materials is suggested by the negative substitutional energies of both dopants, i.e. −0.26 and −0.09 eV, respectively, for Ba and Ca.Fig. 5 shows the activation energy barriers of the elementary steps and the difference in Gibbs free energy between the two intermediates. While Ba dopants change the thermodynamics of the reaction step without altering its kinetics, the substitution of Sr by Ca ions has the opposite effect, with the activation barrier being lowered from 1.23 to 1.15 eV. Although the effect is small, it clearly indicates that cation doping is a conceivable way to accelerate the reaction rate for CO production over the SrTiO3 photocatalyst, as shown here by Ca.
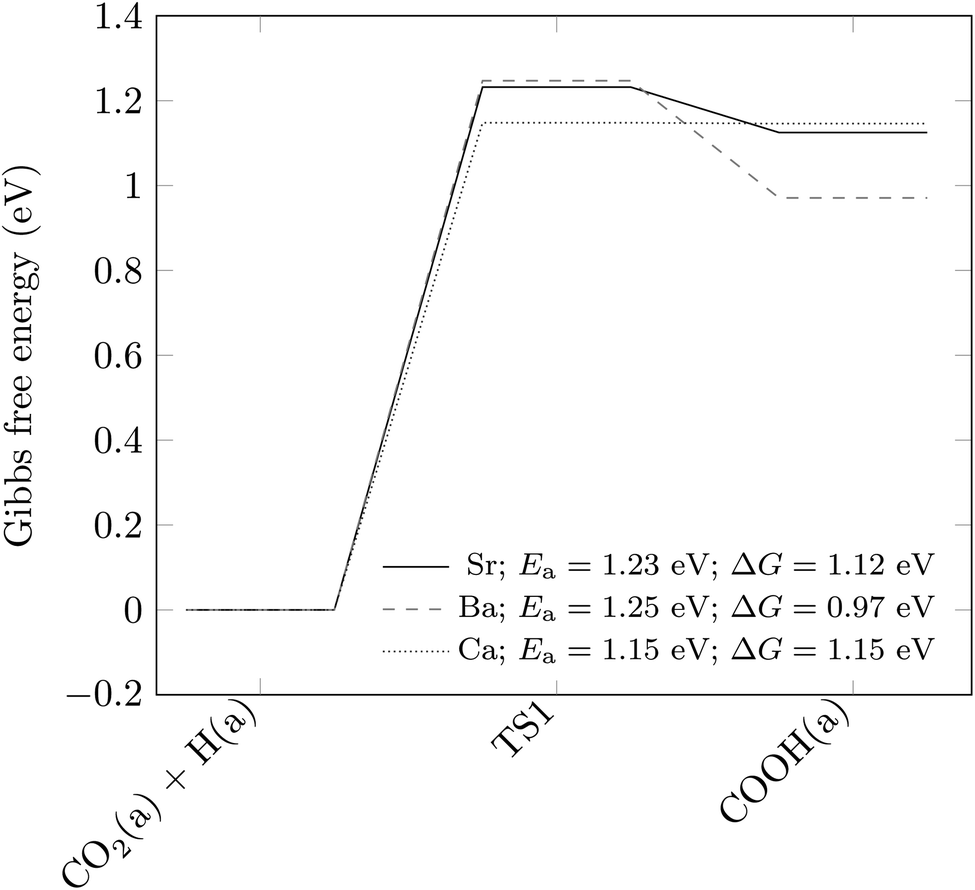 | ||
| Fig. 5 Energetics of the rate-limiting COOH formation step of Fig. 3, with and without substitutional doping of Sr ions. Structures are labelled according to the state of the species. (a) indicates that the species is adsorbed on the surface. The transition state is indicated as TS1. The zero of the Gibbs free energy is taken as that of structure CO2(a) + H(a). Activation energies (Ea) and differences in Gibbs free energies (ΔG) are also included. | ||
4 Conclusions
We have presented a theoretical investigation of the adsorption and reaction of CO2 on the SrO-terminated (001) surface of SrTiO3, a perovskite compound of significant technological interest. We have shown that the possible intermediates towards the formation of HCOOH are very high in energy, which explains why the formation of HCOOH is not normally reported in photocatalytic experiments. Instead, the reduction of CO2 to CO appears to be feasible, in agreement with experimental observations, thus pointing towards to the use of SrTiO3 as a photocatalyst for the reduction of CO2. The reaction occurs via the formation of a COOH species (the associative mechanism in the reverse water gas shift reaction3), with an activation energy of about 1.2 eV, corresponding to the rate-limiting step of the overall reaction.However, a more firm statement will require a systematic analysis of adsorption/desorption rates, the rates for diffusion of the various species and their comparison to reaction rates for all the steps in the mechanism. While such simulations are left for subsequent work, the present results provide compelling evidence that the barrier for the likely rate determining step is sensitive to the nature of the surface cations, and therefore can be lowered by means of a doping strategy, for example where smaller Ca ions substitute for Sr ions. Future experimental studies along with macroscopic simulations will be important to validate these predictions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been performed under the HPC-EUROPA3 (INFRAIA-2016-1-730897) project, with the support of the EC Research Innovation Action under the H2020 Programme. UT gratefully acknowledges the support of the computer resources and the technical support provided by the Barcelona Supercomputing Center-Centro Nacional de Supercomputación. This work was supported by the Engineering and Physical Sciences Research Council (grant EP/K009567/2), and used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk) via our membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202). FV and FI acknowledge financial support from the Ministerio de Ciencia, Innovación y Universidades MICIUN RTI2018-095460-B-I00, and María de Maeztu MDM-2017-0767 grants and, in part, Generalitat de Catalunya 2017SGR13 and XRQTC grants. FV is thankful to the Ministerio de Economía y Competitividad (MEC) for his Ramón y Cajal (RYC-2012-10129) research contract, and FI acknowledges additional support from the 2015 ICREA Academia Award for Excellence in University Research. All data created during this research are openly available from the Cardiff University Research Portal at http://doi.org/10.17035/d.2019.0086887209.References
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, A short review of catalysis for CO2 conversion, Catal. Today, 2009, 148, 221–231 CrossRef CAS
.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, Hydrogenation of carbon dioxide over Co–Fe bimetallic catalysts, ACS Catal., 2016, 6, 913–927 CrossRef CAS
- X. Liu, C. Kunkel, P. Ramírez de la Piscina, N. Homs, F. Viñes and F. Illas, Effective and Highly Selective CO Generation from CO2 Using a Polycrystalline-Mo2C Catalyst, ACS Catal., 2017, 7, 4323–4335 CrossRef CAS
- C. E. Mitchell, U. Terranova, I. Alshibane, D. J. Morgan, T. E. Davies, Q. He, J. S. Hargreaves, M. Sankar and N. H. de Leeuw, Liquid phase hydrogenation of CO2 to formate using palladium and ruthenium nanoparticles supported on molybdenum carbide, New J. Chem., 2019, 43, 13985–13997 RSC
- D. T. Whipple and P. J. Kenis, Prospects of CO2 utilization via direct heterogeneous electrochemical reduction, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS
- B. Kumar, J. P. Brian, V. Atla, S. Kumari, K. A. Bertram, R. T. White and J. M. Spurgeon, New trends in the development of heterogeneous catalysts for electrochemical CO2 reduction, Catal. Today, 2016, 270, 19–30 CrossRef CAS
- A. Roldán, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar and K. B. Holt,
et al., Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions, Chem. Commun., 2015, 51, 7501–7504 RSC
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Photocatalysis. A multifaceted concept for green chemistry, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC
- S. Zeng, P. Kar, U. K. Thakur and K. Shankar, A review on photocatalytic CO2 reduction using perovskite oxide nanomaterials, Nanotechnology, 2018, 29, 052001 CrossRef PubMed
- X. Chang, T. Wang and J. Gong, CO2 photo-reduction: insights into CO2 activation and reaction on surfaces of photocatalysts, Energy Environ. Sci., 2016, 9, 2177–2196 RSC
- G. Parravano, Ferroelectric transitions and heterogenous catalysis, J. Chem. Phys., 1952, 20, 342–343 CrossRef CAS
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, Perovskite oxides: preparation, characterizations, and applications in heterogeneous catalysis, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed
- R. Shi, G. I. Waterhouse and T. Zhang, Recent progress in photocatalytic CO2 reduction over perovskite oxides, Sol. RRL, 2017, 1, 1700126 CrossRef
- H. Zhou, J. Guo, P. Li, T. Fan, D. Zhang and J. Ye, Leaf-architectured 3D hierarchical artificial photosynthetic system of perovskite titanates towards CO2 photoreduction into hydrocarbon fuels, Sci. Rep., 2013, 3, 1667 CrossRef PubMed
- D. Li, S. Ouyang, H. Xu, D. Lu, M. Zhao, X. Zhang and J. Ye, Synergistic effect of Au and Rh on SrTiO3 in significantly promoting visible-lightdriven syngas production from CO2 and H2O, Chem. Commun., 2016, 52, 5989–5992 RSC
- H. Yoshida, L. Zhang, M. Sato, T. Morikawa, T. Kajino, T. Sekito, S. Matsumoto and H. Hirata, Calcium titanate photocatalyst prepared by a flux method for reduction of carbon dioxide with water, Catal. Today, 2015, 251, 132–139 CrossRef CAS
- M. F. Ehsan, M. N. Ashiq, F. Bi, Y. Bi, S. Palanisamy and T. He, Preparation and characterization of SrTiO3–ZnTe nanocomposites for the visible light photoconversion of carbon dioxide to methane, RSC Adv., 2014, 4, 48411–48418 RSC
- K. Xie, N. Umezawa, N. Zhang, P. Reunchan, Y. Zhang and J. Ye, Selfdoped SrTiO3-δ photocatalyst with enhanced activity for artificial photosynthesis under visible light, Energy Environ. Sci., 2011, 4, 4211–4219 RSC
- H. Nakanishi, K. Iizuka, T. Takayama, A. Iwase and A. Kudo, Highly active NaTaO3-based photocatalysts for CO2 reduction to form CO using water as the electron donor, ChemSusChem, 2017, 10, 112–118 CrossRef CAS PubMed
- Y. Ji and Y. Luo, Theoretical Study on the Mechanism of Photoreduction of CO2 to CH4 on the Anatase TiO2 (101) Surface, ACS Catal., 2016, 6, 2018–2025 CrossRef CAS
- H. He, P. Zapol and L. A. Curtiss, Computational screening of dopants for photocatalytic two-electron reduction of CO2 on anatase (101) surfaces, Energy Environ. Sci., 2012, 5, 6196–6205 RSC
- Q. Wu, J. Cen, K. R. Goodman, M. G. White, G. Ramakrishnan and A. Orlov, Understanding the interactions of CO2 with doped and undoped SrTiO3, ChemSusChem, 2016, 9, 1889–1897 CrossRef CAS PubMed
- H.-C. Chen, C.-W. Huang, J. C. Wu and S.-T. Lin, Theoretical investigation of the metal-doped SrTiO3 photocatalysts for water splitting, J. Phys. Chem. C, 2012, 116, 7897–7903 CrossRef CAS
- J. Baniecki, M. Ishii, K. Kurihara, K. Yamanaka, T. Yano, K. Shinozaki, T. Imada, K. Nozaki and N. Kin, Photoemission and quantum chemical study of SrTiO3 (001) surfaces and their interaction with CO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 195415 CrossRef
- J. Baniecki, M. Ishii, K. Kurihara, K. Yamanaka, T. Yano, K. Shinozaki, T. Imada and Y. Kobayashi, Chemisorption of water and carbon dioxide on nanostructured BaTiO3–SrTiO3 (001) surfaces, J. Appl. Phys., 2009, 106, 054109 CrossRef
- B. Modak and S. K. Ghosh, Origin of enhanced visible light driven water splitting by (Rh, Sb)-SrTiO3, Phys. Chem. Chem. Phys., 2015, 17, 15274–15283 RSC
- S. Woo, H. Jeong, S. A. Lee, H. Seo, M. Lacotte, A. David, H. Y. Kim, W. Prellier, Y. Kim and W. S. Choi, Surface properties of atomically at poly-crystalline SrTiO3, Sci. Rep., 2015, 5, 8822 CrossRef PubMed
- D. Halwidl, B. Stöger, W. Mayr-Schmölzer, J. Pavelec, D. Fobes, J. Peng, Z. Mao, G. S. Parkinson, M. Schmid and F. Mittendorfer,
et al., Adsorption of water at the SrO surface of ruthenates, Nat. Mater., 2016, 15, 450 CrossRef CAS PubMed
- M. Burriel, S. Wilkins, J. P. Hill, M. A. Muñoz-Márquez, H. H. Brongersma, J. A. Kilner, M. P. Ryan and S. J. Skinner, Absence of Ni on the outer surface of Sr doped La2NiO4 single crystals, Energy Environ. Sci., 2014, 7, 311–316 RSC
- K. Momma and F. Izumi, VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- K. Reuter and M. Scheffer, Composition, structure, and stability of RuO2 (110) as a function of oxygen pressure, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 35406 CrossRef
- U. Terranova and D. Bowler, Effect of hydration of the TiO2 anatase (101) substrate on the atomic layer deposition of alumina films, J. Mater. Chem., 2011, 21, 4197–4203 RSC
- https://janaf.nist.gov/janaf4pdf.html .
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS
- C. Kunkel, F. Viñes and F. Illas, Transition metal carbides as novel materials for CO2 capture, storage, and activation, Energy Environ. Sci., 2016, 9, 141–144 RSC
- K. V. Sopiha, O. I. Malyi, C. Persson and P. Wu, Band gap modulation of SrTiO3 upon CO2 adsorption, Phys. Chem. Chem. Phys., 2017, 19, 16629–16637 RSC
- H. Guhl, W. Miller and K. Reuter, Water adsorption and dissociation on SrTiO3 (001) revisited: A density functional theory study, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155455 CrossRef
- W.-J. Yin, B. Wen, S. Bandaru, M. Krack, M. Lau and L.-M. Liu, The effect of excess electron and hole on CO2 adsorption and activation on rutile (110) surface, Sci. Rep., 2016, 6, 23298 CrossRef CAS PubMed
- K. C. Ko, S. T. Bromley, J. Y. Lee and F. Illas, Size-dependent level alignment between rutile and anatase TiO2 nanoparticles: implications for photocatalysis, J. Phys. Chem. Lett., 2017, 8, 5593–5598 CrossRef CAS PubMed
- B. S. Kwak and M. Kang, Photocatalytic reduction of CO2 with H2O using perovskite CaxTiyO3, Appl. Surf. Sci., 2015, 337, 138–144 CrossRef CAS
- S. Carlotto, M. M. Natile, A. Glisenti and A. Vittadini, Adsorption of small molecules at the cobalt-doped SrTiO3 (001) surface: A first-principles investigation, Surf. Sci., 2015, 633, 68–76 CrossRef CAS
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
| This journal is © The Royal Society of Chemistry 2020 |