
An ultrasmall Ru2P nanoparticles–reduced graphene oxide hybrid: an efficient electrocatalyst for NH3 synthesis under ambient conditions†
Runbo
Zhao‡
 a,
Chuangwei
Liu‡
b,
Xiaoxue
Zhang
c,
Xiaojuan
Zhu
c,
Peipei
Wei
a,
Lei
Ji
a,
Yanbao
Guo
b,
Shuyan
Gao
d,
Yonglan
Luo
*c,
Zhiming
Wang
*a and
Xuping
Sun
*a
a,
Chuangwei
Liu‡
b,
Xiaoxue
Zhang
c,
Xiaojuan
Zhu
c,
Peipei
Wei
a,
Lei
Ji
a,
Yanbao
Guo
b,
Shuyan
Gao
d,
Yonglan
Luo
*c,
Zhiming
Wang
*a and
Xuping
Sun
*a
aInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China. E-mail: zhmwang@gmail.com; xpsun@uestc.edu.cn
bCollege of Mechanical and Transportation Engineering, China University of Petroleum, Beijing 102249, China
cChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, School of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637002, Sichuan, China. E-mail: luoylcwnu@hotmail.com
dSchool of Materials Science and Engineering, Henan Normal University, Xinxiang 453007, Henan, China
First published on 27th November 2019
Abstract
Industrial NH3 synthesis highly relies on the Haber–Bosch process which consumes a large amount of energy and emits a massive amount of CO2. Electrochemical N2 reduction is an eco-friendly and sustainable approach to realize NH3 synthesis under ambient conditions, but its implementation requires efficient electrocatalysts for the N2 reduction reaction. In this work, a hybrid of Ru2P nanoparticles and reduced graphene oxide is proposed as an efficient electrocatalyst for artificial N2-to-NH3 fixation with excellent selectivity under ambient conditions. Electrochemical tests in 0.1 M HCl show that such a hybrid achieves a large NH3 yield of 32.8 μg h−1 mgcat.−1 and a high faradaic efficiency of 13.04% at −0.05 V vs. the reversible hydrogen electrode. Furthermore, it also exhibits remarkable electrochemical and structural stability. Theoretical calculations reveal that Ru2P–rGO can efficiently catalyze NH3 synthesis with a low energy barrier.
As one of the most produced chemicals, NH3 is widely employed as a nitrogen source to produce fertilizers, medicaments, plastics, and so on.1 Furthermore, with a considerable hydrogen content, high energy density and zero carbon emission, NH3 is also considered an ideal candidate for energy storage and clean fuel.2 Nowadays, industrial NH3 synthesis is still predominantly performed by the Haber–Bosch process, which is conducted under harsh conditions with massive energy consumption and greenhouse gas emission.3 Therefore, a sustainable and economical approach is needed for artificial NH3 synthesis under mild reaction conditions.
Electrochemical N2 reduction is an energy-saving and environmentally friendly approach which can replace the Haber–Bosch process for NH3 synthesis under ambient conditions; however, efficient electrocatalysts for the N2 reduction reaction (NRR) are needed to break the inert N2 molecule.4–28 Ru performs efficiently in the traditional Haber–Bosch process.29 Ru is also theoretically predicted to be a good NRR electrocatalyst due to its appropriate overpotential and energy for N2 adsorption and activation, which are much lower than those of Pt and Pd.30–32 A recent experimental study showed that Ru nanoparticles catalyze the NRR under ambient conditions with an NH3 yield of 0.55 μg h−1 cm−2 and a faradaic efficiency (FE) of 5.4%.33 Theoretical investigation further revealed that *N2Hx species (0 ≤ x ≤ 2) could accumulate on the surface of Ru due to the strong binding strength between them, resulting in a decreased performance toward the NRR.34 Obviously, to improve the NRR activity, the binding strength should be weakened. Bonding metal atoms with electronegative nonmetals is an effective way to weaken the binding strength through controlling the electron transfer between them.4 Our recent study verified that the NRR performance of a B catalyst is greatly improved by alloying with P due to further weakened N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds.35 Hence, enhanced N2 reduction electrocatalysis is expected for the Ru catalyst after alloying with P, which however has not been explored before.
N bonds.35 Hence, enhanced N2 reduction electrocatalysis is expected for the Ru catalyst after alloying with P, which however has not been explored before.
Herein, we report the development of an ultrasmall Ru2P nanoparticles–reduced graphene oxide (Ru2P–rGO) hybrid as an efficient NRR electrocatalyst for ambient N2-to-NH3 conversion with excellent selectivity. In 0.1 M HCl, this catalyst is capable of achieving a large NH3 yield of 32.8 μg h−1 mgcat.−1 and a high FE of 13.04% at −0.05 V vs. the reversible hydrogen electrode (RHE), superior to those of its Ru–rGO counterpart (6.4 μg h−1 mgcat.−1; 1.86%). Furthermore, it also exhibits remarkable electrochemical and structural stability. Density functional theory (DFT) calculations reveal that Ru2P–rGO can efficiently catalyze NH3 synthesis with a low energy barrier of 0.68 eV.
The X-ray diffraction (XRD) pattern of Ru2P–rGO is shown in Fig. 1a. The characteristic diffraction peaks at 30.5°, 38.1°, 40.6°, 42.2°, 47.1°, and 76.7° are indexed to the (111), (112), (211), (103), (020), and (403) planes of Ru2P (JCPDS no. 65-2382). Transmission electron microscopy (TEM) analysis indicates that Ru2P nanoparticles with ultrasmall diameters (2–4 nm) are uniformly anchored on the surface of rGO nanosheets (Fig. 1b). As shown in Fig. 1c, the high-resolution TEM (HRTEM) image depicts clear crystallographic fringes of Ru2P–rGO with an average interplanar distance of 0.357 nm, which corresponds to the (112) lattice plane of Ru2P. Furthermore, the selected area electron diffraction (SAED) pattern (Fig. 1d) shows three diffraction rings, which can be separately assigned to the (112), (103), and (403) planes of Ru2P. Fig. 1e shows the scanning TEM (STEM) image and energy-dispersive X-ray (EDX) elemental mapping images of Ru2P–rGO, which confirm the uniform distribution of Ru and P species on rGO nanosheet.
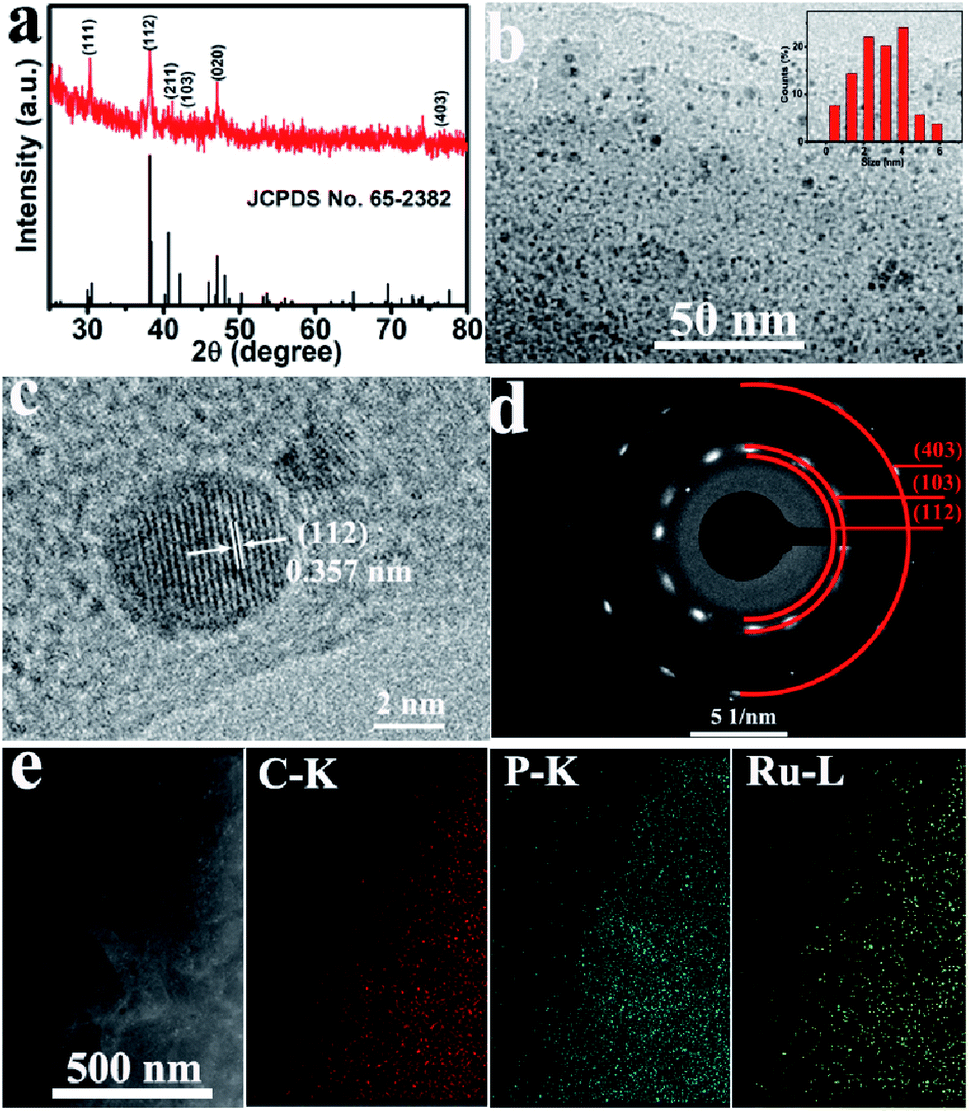 | ||
| Fig. 1 (a) XRD pattern for Ru2P–rGO. (b) TEM (inset: the particle size distribution of Ru2P) and (c) HRTEM image of Ru2P–rGO. (d) SAED pattern of Ru2P–rGO. (e) STEM and EDX elemental mapping images of Ru2P–rGO. | ||
Fig. 2a shows the X-ray photoelectron spectroscopy (XPS) survey spectrum of Ru2P–rGO, which reveals the existence of Ru, P, C and O elements in the hybrid with Ru2P about 38.69%. As shown in Fig. 2b, the peaks at 284.5 and 280.6 eV are assigned to the binding energies (BEs) of Ru 3d3/2 and Ru 3d5/2,36,37 while the peaks at 288.7, 285.8, and 284.9 eV are attributed to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CO, and CC bonds, respectively.38 In the Ru 3p region (Fig. 2c), the peaks at 484.3 and 462.1 eV are assigned to the BEs of Ru 3p1/2 and Ru 3p3/2 peaks.39 Note that the peaks at 486.9 and 464.3 eV can be ascribed to the oxidized RuPx species.39 In the P 2p region (Fig. 2d), the peaks located at 130.7 and 129.7 eV are consistent with the P 2p1/2 and P 2p3/2 peaks, respectively.40 Additionally, the BE at 134.1 eV is ascribed to the P–O bond, which is formed by surface oxidation after exposure to air.39 It is worth noting that the BE of Ru 3d5/2 (280.6 eV) exhibits a positive shift compared with that of metallic Ru (279.8 eV),41 while the BE of P 2p3/2 (129.7 eV) shows a negative shift compared with that of elemental P (130.2 eV).42 These shifts reveal that Ru possesses a partially positive charge, while P has a partially negative charge, implying that the electron density is transferred from Ru to P.38
O, CO, and CC bonds, respectively.38 In the Ru 3p region (Fig. 2c), the peaks at 484.3 and 462.1 eV are assigned to the BEs of Ru 3p1/2 and Ru 3p3/2 peaks.39 Note that the peaks at 486.9 and 464.3 eV can be ascribed to the oxidized RuPx species.39 In the P 2p region (Fig. 2d), the peaks located at 130.7 and 129.7 eV are consistent with the P 2p1/2 and P 2p3/2 peaks, respectively.40 Additionally, the BE at 134.1 eV is ascribed to the P–O bond, which is formed by surface oxidation after exposure to air.39 It is worth noting that the BE of Ru 3d5/2 (280.6 eV) exhibits a positive shift compared with that of metallic Ru (279.8 eV),41 while the BE of P 2p3/2 (129.7 eV) shows a negative shift compared with that of elemental P (130.2 eV).42 These shifts reveal that Ru possesses a partially positive charge, while P has a partially negative charge, implying that the electron density is transferred from Ru to P.38
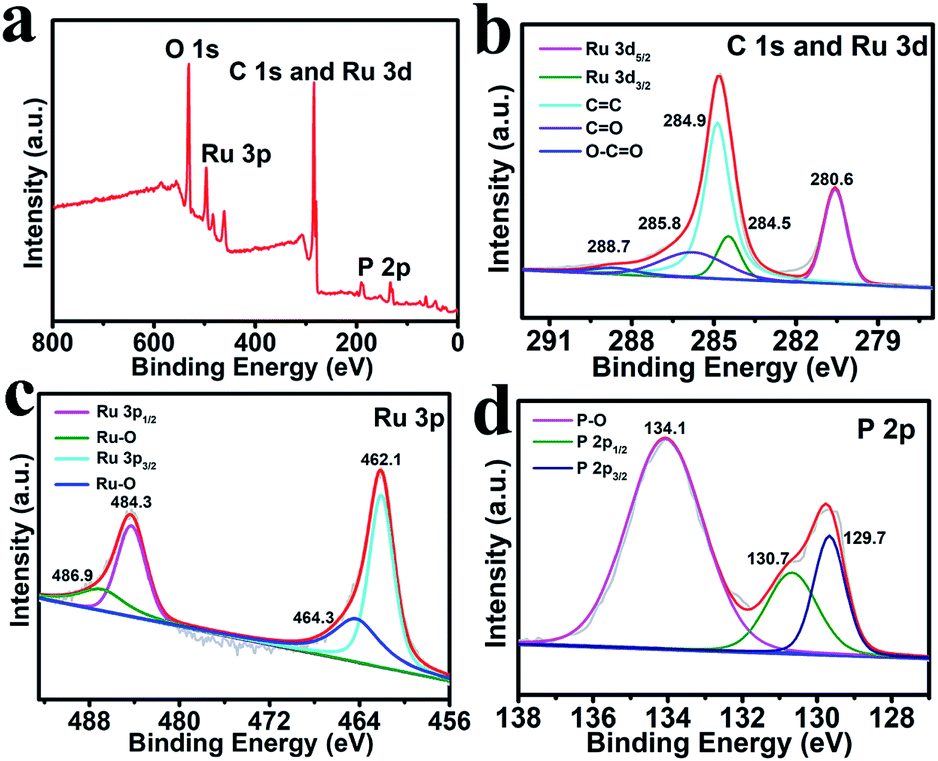 | ||
| Fig. 2 (a) XPS survey spectrum of Ru2P–rGO. XPS spectra of Ru2P–rGO in the (b) C 1s and Ru 3d, (c) Ru 3p, and (d) P 2p regions. | ||
Ru2P–rGO was loaded on carbon paper (Ru2P–rGO/CP) with a loading of 0.1 mg cm−2 as the working electrode to evaluate the electrocatalytic NRR performance of this electrocatalyst in 0.1 M HCl. All potentials in the electrocatalytic NRR process were calibrated to the RHE scale. Chronoamperometry was performed at certain potentials ranging from 0.00 to −0.20 V for 2 h to obtain enough target product (NH3) for quantitative evaluation of the catalytic activity of Ru2P–rGO. As depicted in Fig. 3a, the current density increases with the increasing working potential. After electrolysis, NH3 and the possible by-product (N2H4) were determined by the indophenol blue43 and Watt–Chrisp spectrophotometry methods,44 respectively. The corresponding calibration curves for NH3 and N2H4 are shown in Fig. S1 and S2,† respectively. Notably, the NH4+ calibration solution was prepared in 0.1 M HCl. The UV-vis absorption spectra of the electrolytes dyed with the indophenol indicator after electrolysis for 2 h are displayed in Fig. 3b and employed to quantify the produced NH3. The corresponding NH3 yields and FEs are depicted in Fig. 3c. The largest NH3 yield of 32.8 μg h−1 mgcat.−1 and FE of 13.04% are achieved at −0.05 V under ambient conditions and are comparable to those of most noble-metal electrocatalysts under ambient conditions in aqueous electrolytes (Table S1†). Notably, when the applied potential is below −0.05 V, both the NH3 yield and FE decrease, which is probably caused by the gradually enhanced competitive hydrogen evolution reaction.45
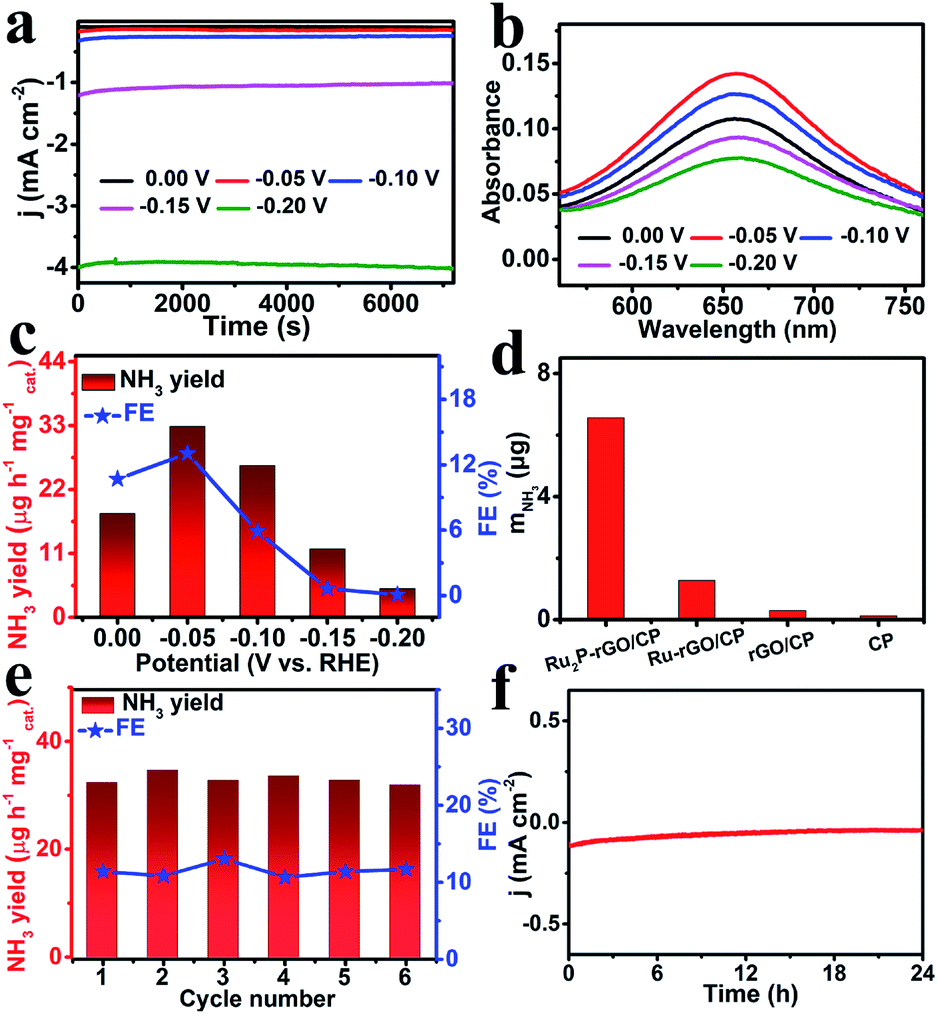 | ||
| Fig. 3 (a) Chronoamperometry curves for Ru2P–rGO/CP at different given potentials in 0.1 M HCl. (b) Corresponding UV-vis absorption spectra of the electrolytes stained with the indophenol indicator after NRR electrolysis at different potentials for 2 h. (c) NH3 yields and FEs of Ru2P–rGO/CP. (d) Amount of NH3 produced using different electrodes at −0.05 V after 2 h of electrolysis under ambient conditions. (e) Recycling tests of Ru2P–rGO/CP at −0.05 V. (f) Chronoamperometry curve for Ru2P–rGO/CP at −0.05 V in a N2-saturated electrolyte for 24 h. | ||
To further verify the superior catalytic performance of Ru2P–rGO, a series of control experiments with catalysts such as Ru–rGO/CP, rGO/CP and blank CP were performed at −0.05 V for 2 h. As depicted in Fig. 3d, CP and rGO/CP exhibit low catalytic activity toward NH3 synthesis. The better catalytic activity of Ru2P–rGO/CP than of Ru–rGO/CP may be caused by the critical function of P in boosting the performance toward the NRR.35 To verify the origin of the generated NH3, NRR experiments using Ru2P–rGO/CP under a N2 atmosphere at open circuit and in Ar gas at −0.05 V were carried out. As shown in Fig. S3,† the weak UV-vis spectra indicate that the produced NH3 stems from N2 electroreduction. Notably, N2H4 was not detected in the final product (Fig. S4†), revealing that Ru2P–rGO/CP possesses good selectivity for NH3 synthesis by the NRR process.
As one of the important parameters to evaluate the performance of electrocatalysts, the stability should also be considered in the NRR process. Consecutive recycling tests were performed at −0.05 V in a N2-saturated electrolyte 6 times and the corresponding results are shown in Fig. S5.† The well maintained NH3 yields and FEs (Fig. 3e) reveal that Ru2P–rGO/CP is a stable electrocatalyst. In addition, the current density of Ru2P–rGO/CP without obvious variation in 24 h of NRR electrolysis also suggests its excellent long-term stability as shown in Fig. 3f. Furthermore, the photographs of pH test papers for the HCl aqueous solution before and after 24 h of electrolysis are shown in Fig. S6,† which indicate that there is almost no change in pH in our experiment. In addition, we also analyzed the electrocatalytic NRR performance of Ru2P–rGO/CP in 0.1 M Na2SO4. The results indicate that the electrocatalytic NRR performance of Ru2P–rGO in 0.1 M Na2SO4 is inferior to that in 0.1 M HCl (Fig. S7†).
The optimized geometries and Bader charge results before and after nitrogen molecule adsorption on Ru2P–rGO are shown in Fig. 4a and b, respectively. P atoms have negative-mass electrons, which cover the positive charge of Ru, indicating net electron transfer from rGO to Ru2P. Fig. 4b shows the detailed distribution of Bader charges after nitrogen adsorption. Clearly, the net charge on Ru2P is slightly changed, due to donating 0.50 e− to N2. Interestingly, the two N atoms show slightly different charge values. To further understand the NRR catalytic site and mechanism of the Ru2P–rGO, elementary steps associated with distal and alternating possible pathways for four potential N2-adsorption catalytic routes have been studied by DFT calculation (calculation details in the ESI†), including a single Ru site (1), two Ru sites (2), one Ru and one P site (3), and a single P site (4). The (3) and (4) possible catalytic sites are excluded after initial screening in this calculation, due to the fact that the P atom cannot adsorb a N2 molecule. Our calculations show that the dinitrogen molecule would indeed be well captured by the Ru atom, because the adsorption energies are negative on both the (1) and (2) possible catalytic sites. Moreover, the N–N distance increases from 1.10 Å in the free individual nitrogen molecule to 1.14 Å and 1.16 Å on the (1) and (2) possible catalytic sites, respectively, indicating that N2 is activated. The real challenge comes from the first hydrogenation step of 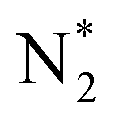 to N2H*, which requires a Gibbs free energy of about 1.30 and 0.68 eV on both possible pathways, respectively. The subsequent reduction steps of route 2 are the most exothermic, but the last desorption step (NH3* to NH3) requires energy input to complete the reaction (0.47 eV) (Fig. 4c and d). But all input energies are smaller than the first hydrogenation step energy, indicating that the first reduction step is the potential-determining step (PDS) for both the distal and alternating mechanisms. However, the reduction steps of route 1 are endothermic reactions before the first NH3-desorption, and all the free energies are lower than that in the first reduction step (Fig. S8†). Therefore, the dinitrogen molecule adsorbed on two Ru atoms (route 2) is the favorable reaction route for the NRR on Ru2P–rGO, and the energy barrier of the NRR is equal to 0.68 eV for both mechanisms.
to N2H*, which requires a Gibbs free energy of about 1.30 and 0.68 eV on both possible pathways, respectively. The subsequent reduction steps of route 2 are the most exothermic, but the last desorption step (NH3* to NH3) requires energy input to complete the reaction (0.47 eV) (Fig. 4c and d). But all input energies are smaller than the first hydrogenation step energy, indicating that the first reduction step is the potential-determining step (PDS) for both the distal and alternating mechanisms. However, the reduction steps of route 1 are endothermic reactions before the first NH3-desorption, and all the free energies are lower than that in the first reduction step (Fig. S8†). Therefore, the dinitrogen molecule adsorbed on two Ru atoms (route 2) is the favorable reaction route for the NRR on Ru2P–rGO, and the energy barrier of the NRR is equal to 0.68 eV for both mechanisms.
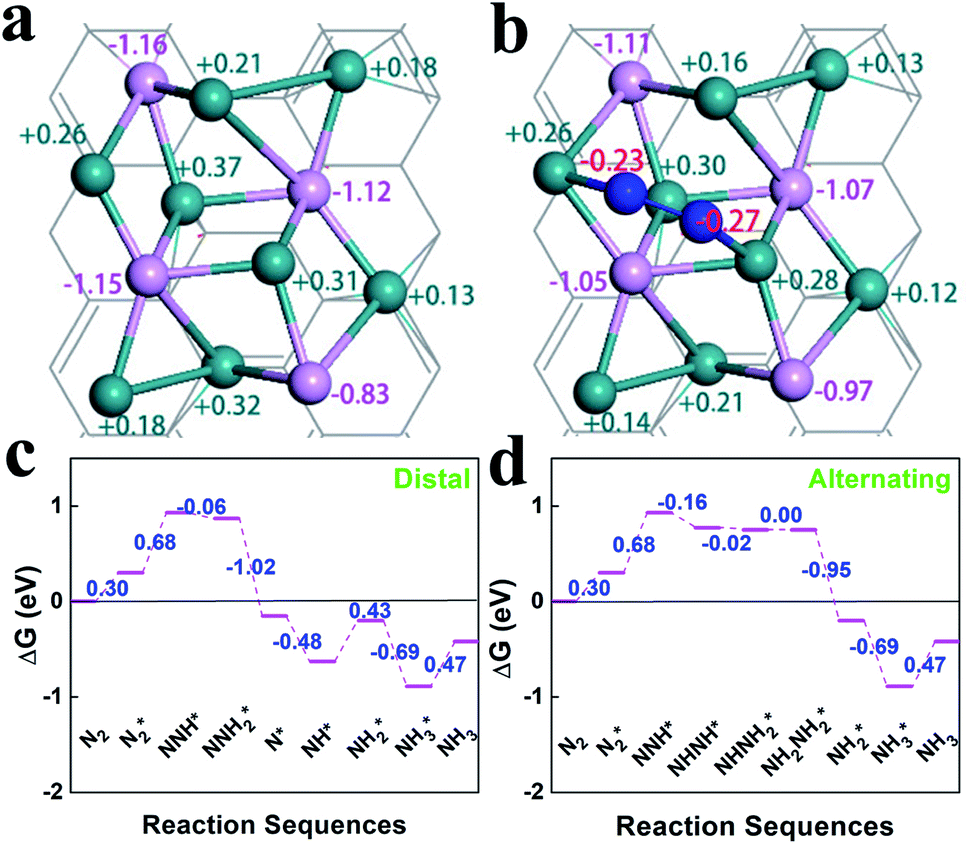 | ||
| Fig. 4 Optimized geometries and Bader charge results (a) before and (b) after N2 adsorption on Ru2P–rGO. Calculated energy profiles for the NRR catalyzed by Ru2P–rGO: (c) distal and (d) alternating mechanisms. | ||
In conclusion, Ru2P–rGO is experimentally and theoretically proved to be an efficient noble-metal electrocatalyst to realize artificial N2-to-NH3 conversion under ambient conditions. In 0.1 M HCl, Ru2P–rGO achieves a large NH3 yield of 32.8 μg h−1 mgcat.−1 and a high FE of 13.04% at −0.05 V. In addition, it also exhibits excellent selectivity toward NH3 synthesis and long-term stability during the electrolysis test process. DFT calculations further reveal that Ru2P–rGO can efficiently catalyze NH3 synthesis with a low energy barrier of 0.68 eV and that the first hydrogenation step is the PDS. This investigation not only provides us with an efficient electrocatalyst toward NH3 synthesis under ambient conditions, but also offers a path to design noble-metal-based phosphides for NRR applications.46
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21575137).References
- R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 2004–2008 CrossRef PubMed.
- R. Lan, J. T. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- I. Dybkjaer, Ammonia Production Processes, ed. A. Nielsen, Springer, Heidelberg, 1995, pp. 199–327 Search PubMed.
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef.
- C. Tang and S. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- G. Chen, S. Ren, L. Zhang, H. Cheng, Y. Luo, K. Zhu, L. Ding and H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- Y. Yang, S. Wang, H. Wen, T. Ye, J. Chen, C. Li and M. Du, Angew. Chem., Int. Ed., 2019, 58, 15362–15366 CrossRef CAS PubMed.
- H. K. Lee, C. S. L. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. Han, C. K. Tsung and X. Ling, Sci. Adv., 2018, 4, eaar3208 CrossRef PubMed.
- J. Zhao, B. Wang, Q. Zhou, H. Wang, X. Li, H. Chen, Q. Wei, D. Wu, Y. Luo, J. You, F. Gong and X. Sun, Chem. Commun., 2019, 55, 4997–5000 RSC.
- Y. Wan, J. Xu and R. Lv, Mater. Today, 2019, 27, 69–90 CrossRef CAS.
- R. Zhang, J. Han, B. Zheng, X. Shi, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2019, 6, 391–395 RSC.
- D. Bao, Q. Zhang, F. Meng, H. Zhong, M. Shi, Y. Zhang, J. Yan, Q. Jiang and X. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- R. Zhang, L. Ji, W. Kong, H. Wang, R. Zhao, H. Chen, T. Li, B. Li, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 5263–5266 RSC.
- L. Zhang, X. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- Z. Xing, W. Kong, T. Wu, H. Xie, T. Wang, Y. Luo, X. Shi, A. M. Asiri, Y. Zhang and X. Sun, ACS Sustainable Chem. Eng., 2019, 7, 12692–12696 CrossRef CAS.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2019, 9, 1803935 CrossRef.
- Y. Zhang, H. Du, Y. Ma, L. Ji, H. Guo, Z. Tian, H. Chen, H. Huang, G. Cui, A. M. Asiri, F. Qu, L. Chen and X. Sun, Nano Res., 2019, 12, 919–924 CrossRef CAS.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- J. Yu, C. Li, B. Li, X. Zhu, R. Zhang, L. Ji, D. Tang, A. M. Asiri, X. Sun, Q. Li, S. Liu and Y. Luo, Chem. Commun., 2019, 55, 6401–6404 RSC.
- R. Zhang, H. Guo, L. Yang, Y. Wang, Z. Niu, H. Huang, H. Chen, L. Xia, T. Li, X. Shi, X. Sun, B. Li and Q. Liu, ChemElectroChem, 2019, 6, 1014–1018 CrossRef CAS.
- S. Zhang, C. Zhao, Y. Liu, W. Li, J. Wang, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Chem. Commun., 2019, 55, 2952–2955 RSC.
- H. Huang, F. Gong, Y. Wang, H. Wang, X. Wu, W. Lu, R. Zhao, H. Chen, X. Shi, A. M. Asiri, T. Li, Q. Liu and X. Sun, Nano Res., 2019, 12, 1093–1098 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- G. Yu, H. Guo, W. Kong, T. Wang, Y. Luo, X. Shi, A. M. Asiri, T. Li and X. Sun, J. Mater. Chem. A, 2019, 7, 19657–19661 RSC.
- T. Wu, X. Zhu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201911153.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- S. Back and Y. Jung, Phys. Chem. Chem. Phys., 2016, 18, 9161–9166 RSC.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Mater. Chem. A, 2019, 7, 4771–4776 RSC.
- D. Wang, L. M. Azofra, M. Harb, L. Cavallo, X. Zhang, B. H. R. Suryanto and D. R. MacFarlane, ChemSusChem, 2018, 11, 3416–3422 CrossRef CAS PubMed.
- Y. Yao, H. Wang, X. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- X. Zhu, T. Wu, L. Ji, C. Li, T. Wang, S. Wen, S. Gao, X. Shi, Y. Luo, Q. Peng and X. Sun, J. Mater. Chem. A, 2019, 7, 16117–16121 RSC.
- Z. Pu, I. S. Amiinu, Z. Kou, W. Li and S. Mu, Angew. Chem., Int. Ed., 2017, 56, 11559–11564 CrossRef CAS PubMed.
- E. Demir, S. Akbayrak, A. M. Önal and S. Özkar, ACS Appl. Mater. Interfaces, 2018, 10, 6299–6308 CrossRef CAS PubMed.
- T. Liu, S. Wang, Q. Zhang, L. Chen, W. Hu and C. Li, Chem. Commun., 2018, 54, 3343–3346 RSC.
- Q. Chang, J. Ma, Y. Zhu, Z. Li, D. Xu, X. Duan, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Sustainable Chem. Eng., 2018, 6, 6388–6394 CrossRef CAS.
- Y. Li, F. Chu, Y. Bu, Y. Kong, Y. Tao, X. Zhou, H. Yu, J. Yu, L. Tang and Y. Qin, Chem. Commun., 2019, 55, 7828–7831 RSC.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- C. Tang, L. Gan, R. Zhang, W. Lu, X. Jiang, A. M. Asir, X. Sun, J. Wang and L. Chen, Nano Lett., 2016, 16, 6617–6621 CrossRef CAS PubMed.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836–841 CrossRef CAS PubMed.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- W. Qiu, X. Xie, J. Qiu, W. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- H. Xie, Q. Geng, X. Zhu, Y. Luo, L. Chang, X. Niu, X. Shi, A. M. Asiri, S. Gao, Z. Wang and X. Sun, J. Mater. Chem. A, 2019, 7, 24760–24764 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c9ta10346e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |