
Dense organic molecules/graphene network anodes with superior volumetric and areal performance for asymmetric supercapacitors†
Lina
Zhang‡
ab,
Daliang
Han‡
ab,
Ying
Tao
ab,
Changjun
Cui
ab,
Yaqian
Deng
c,
Ximan
Dong
ab,
Wei
Lv
 c,
Zifeng
Lin
d,
Shichao
Wu
ab,
Zhe
Weng
*ab and
Quan-Hong
Yang
*ab
c,
Zifeng
Lin
d,
Shichao
Wu
ab,
Zhe
Weng
*ab and
Quan-Hong
Yang
*ab
aNanoyang Group, State Key Laboratory of Chemical Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300350, China. E-mail: zweng@tju.edu.cn; qhyangcn@tju.edu.cn
bJoint School of National University of Singapore, Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
cShenzhen Key Laboratory for Graphene-based Materials, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, China
dCollege of Materials Science and Engineering, Sichuan University, Chengdu 610065, China
First published on 22nd November 2019
Abstract
Volumetric and areal capacitance are as important as gravimetric capacitance for small energy storage devices. However, achieving both a high volumetric and a high areal capacitance is still a big challenge. Here we report a dense redox organic molecules/graphene network, in which highly redox active sodium anthraquinone-2-sulfonate (AQS) molecules are anchored on interconnected and highly conductive graphene sheets by noncovalent π–π interactions to form high-performance supercapacitors (SCs). The AQS/graphene (AQS/G) has a high volumetric specific capacitance of up to 650 F cm−3 and an excellent rate capability (422 F cm−3 even at 30 A g−1), as well as a good cycling stability. A maximum areal specific capacitance of 13.3 F cm−2 is achieved at a high mass loading of 32 mg cm−2 (200 μm in thickness), which is amongst the highest values recorded for organic-based materials for SCs. An asymmetric SC constructed with AQS/G and RuO2/graphene delivers a maximum volumetric energy density of 44 W h L−1. This outstanding performance is attributed to the excellent electron conduction and ion transport provided by the dense but interconnected graphene network. This work suggests a new way for organic-based high-performance electrode materials to be used in electrochemical energy storage devices.
Introduction
Supercapacitors (SCs), one of the most important electrochemical energy storage (EES) devices, have received much attention because they have a higher energy density than traditional electrolytic capacitors, a high power performance and a remarkable cycling stability.1,2 The rapid development of electric vehicles and portable electronic devices has made miniature energy storage devices essential, thus making the volumetric energy density more important than the gravimetric energy density for practical applications.3 To achieve high-volumetric-performance EES devices, we must take into account not only the volumetric capacitance but also the areal capacitance.4–6 This means that high-gravimetric-capacitance materials and thicker electrodes with high areal capacitance are desirable, which is still challenging.Based on the energy-storage mechanism, SCs are classified into electrical double-layer capacitors (EDLCs) and pseudocapacitors.7 The latter typically have higher volumetric energy densities than the former, as a result of not only storing more charge from faradaic redox reactions compared with physical ion adsorption/desorption, but also their higher density than porous electrical double-layer (EDL) materials.8 Therefore, pseudocapacitive electrode materials are promising for building SCs with a high volumetric energy density. However, pseudocapacitive materials such as transition metal oxides and conductive polymers often suffer from the high cost and complexity of their synthesis, hence hindering their large-scale manufacture.9 Due to their high reversible redox reactivity, good sustainability and low cost, redox-active organic molecules, including quinone and its derivatives, have become one of the most attractive pseudocapacitive materials.10,11 For example, sodium anthraquinone-2-sulfonate (AQS) is suitable for use as an active material in aqueous SCs. On the one hand, it has a remarkable theoretical specific capacitance due to the two-electron reaction mechanism between H+ and carbonyl groups.12,13 On the other hand, the –SO3− functional groups not only offer excellent surface wettability with aqueous electrolytes but also act as proton donors and carriers to facilitate proton transport.14 Together with its high density (1.44 g cm−3), AQS has great potential in realizing high volumetric and areal energy densities. However, its intrinsic electrical resistivity significantly hinders the full utilization of the intrinsic redox-active centers, resulting in a low specific capacitance and inadequate rate performance, and the expected high volumetric and areal capacitance is not achieved.
Introducing a conductive substrate is the most widely used and effective strategy to address this problem. Graphene, the basic structural unit of sp2 carbon materials, has excellent electrical conductivity, a large specific surface area (SSA), wonderful flexibility and remarkable compatibility with other materials, and has been widely used as an active material and conductive substrate in EES systems.15–18 It can be easily assembled into 1D fibers, 2D films and 3D monoliths, which can be densified by mechanical pressure and capillary drying methods to achieve a high volumetric capacitance.19–22 An AQS/graphene xerogel has been used in SCs and has delivered a high gravimetric capacitance and good rate capability.14 However, the low density of the xerogel prevents the realization of electrodes with a high volumetric and areal capacitance. Furthermore, the exact electrochemical behavior of AQS in aqueous systems is still unclear. Last but not least, most of the examined high-performance pseudocapacitive materials can only be produced as thin electrodes with a sub-micrometer thickness. However, for practical applications, much thicker sub-millimeter electrodes with a high areal capacitance are needed, and these require unimpeded electron and ion transport to retain the performance of the thin electrodes. Numerous strategies have been used to improve the kinetics of ion transport in thicker electrodes. Integrating a nonvolatile liquid electrolyte with the electrodes and constructing a continuous ion transport network by using a sacrificial template or pore former can increase the accessible surface area between the electrolyte and electrodes.23–25 Changing the ion transport direction from parallel-to to perpendicular-to the current collector of two-dimensional flakes (for example, MXene and graphene)26,27 shortens the ion transport path, thus realizing thickness-independent electrochemical performance. However, these methods are often too difficult and/or costly to take advantage of, and usually decrease the electrode density and electrochemical performance. Therefore, achieving both a high volumetric capacitance and a high areal capacitance is still a challenge.
Here we report a densification strategy to make maximum use of the high volumetric and areal capacitance of AQS. An AQS/G material was produced for use in high-volumetric-performance SCs. The graphene network not only has a bulk density of 1.38 g cm−3 but also provides paths for the easy migration of electrons and protons. As a result, AQS/G shows a high volumetric capacitance up to 650 F cm−3 together with a good rate capability. As the mass loading increases from 3 to 32 mg cm−2 (20 to 200 μm in thickness), its volumetric specific capacitance remains 84% of its initial value. Moreover, it delivers a remarkable areal capacitance of 13.3 F cm−2 when the mass loading reaches 32 mg cm−2 (200 μm in thickness), which is among the best results of redox-active organic molecule-based materials for SCs. After coupling with a RuO2/graphene (RuO2/G) cathode, the asymmetric SC (ASC) delivers a maximum volumetric energy density of 44 W h L−1, among the best for aqueous systems.
Experimental
Preparation of AQS/G composites
Graphite oxide was prepared by a modified Hummers method. First, a homogeneous graphene oxide (GO) colloidal dispersion (2 mg mL−1) was prepared from graphite oxide treated by ultrasonication in deionized (DI) water for 2 h. Then, sodium anthraquinone-2-sulfonate (AQS) and the GO colloidal dispersion were mixed (with the mass ratios of AQS to GO being 0.5, 1, and 2) and stirred at ambient temperature for 6 h to obtain a homogeneous solution. The mixture was then transferred to a Teflon-lined autoclave and heated at 180 °C for 12 h to assemble the black AQS/graphene (AQS/G) hydrogel, which was then washed three times with DI water to remove impurities. A compact AQS/G monolith was finally obtained by capillary drying at 70 °C. Pure AQS and a high-density porous graphene monolith (HPGM) as control samples were obtained by similar methods. As a control, the mixture (labeled AQS/SP-1) was prepared by mixing AQS and conductive carbon black (Super P) with the same ratio as that in AQS/G-1.Preparation of RuO2/G
The RuO2/G composite was synthesized by a method similar to that reported in a previous paper.28 First, a cylindrical graphene hydrogel was prepared by a hydrothermal process and then dispersed by vigorous magnetic stirring to form a slurry of microscale 3D networks and soaked in a 20 mL RuCl3 aqueous solution (0.075 M) for 12 h to ensure the complete adsorption of Ru3+. A 1 M NaOH aqueous solution was slowly added to the above solution until the pH was adjusted to about 7 to obtain a Ru(OH)3/graphene composite. After repeated washing, the composite obtained was annealed at 150 °C for 2 h in air to obtain the RuO2/graphene (RuO2/G) composite.Materials characterization
The microscopic morphology of the samples was examined using a scanning electron microscope (SEM) (Hitachi S4800, Japan) at 5.0 kV. Transmission electron microscopy (TEM) characterization was performed on a JEM 2100F (JEOL, Japan), operated at 200 kV. Raman spectroscopy was performed using a multi-wavelength micro-Raman spectrometer (JY HR800) with 532.05 nm incident radiation. X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI 5000 Versa Probe II with Al Kα radiation. Fourier transform infrared (FTIR) spectroscopy was conducted on a FTIR Spectrometer (NICOLET 380). X-ray diffraction (XRD) (X'Pert Pro) was used to investigate the structure and phase composition. UV-vis spectroscopy was performed on a HITACHI U-3900 UV/vis spectrophotometer. The electrical conductivities were measured using a standard four-point-probe resistivity measurement system (FT-341, Ningbo, China).Electrochemical measurements
A three-electrode system was used to analyze the electrochemical performance of AQS/G and RuO2/G, with Pt foil as the counter electrode and Ag/AgCl as the reference electrode. The AQS/G and RuO2/G composites were first pulverized into powders and mixed with conductive carbon black powder and a polytetrafluoroethylene (PTFE) binder in a ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in ethanol. The mixture was evaporated to a dried slurry and then kneaded into a wafer with a diameter of 10 mm. The wafer was then pressed onto a stainless-steel sheet current collector under a pressure of 10 MPa and dried in a vacuum oven at 80 °C. For AQS/G, the operating potential was from −0.2 V to 0.4 V vs. Ag/AgCl, which changed to 0–1 V vs. Ag/AgCl for RuO2/G. The electrochemical performance of the asymmetric supercapacitors was examined using 2032-type coin cells with various mass ratios of AQS/G-1 to RuO2/G from 1 to 1.6, and 1.12 is the optimal one. The electrolyte for both the three-electrode and the two-electrode system is a 1 M H2SO4 aqueous solution. Electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were conducted on an Eco Chemie Autolab 128N instrument (Metrohm, Switzerland). Typically, in the three-electrode system, current densities are based on the weight of the active material only in the individual electrodes. For the ASCs, current densities are based on the total weight of the active materials both in the anode and in the cathode.
:1:1 in ethanol. The mixture was evaporated to a dried slurry and then kneaded into a wafer with a diameter of 10 mm. The wafer was then pressed onto a stainless-steel sheet current collector under a pressure of 10 MPa and dried in a vacuum oven at 80 °C. For AQS/G, the operating potential was from −0.2 V to 0.4 V vs. Ag/AgCl, which changed to 0–1 V vs. Ag/AgCl for RuO2/G. The electrochemical performance of the asymmetric supercapacitors was examined using 2032-type coin cells with various mass ratios of AQS/G-1 to RuO2/G from 1 to 1.6, and 1.12 is the optimal one. The electrolyte for both the three-electrode and the two-electrode system is a 1 M H2SO4 aqueous solution. Electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements were conducted on an Eco Chemie Autolab 128N instrument (Metrohm, Switzerland). Typically, in the three-electrode system, current densities are based on the weight of the active material only in the individual electrodes. For the ASCs, current densities are based on the total weight of the active materials both in the anode and in the cathode.
The gravimetric specific capacitance (Cg, F g−1) based on the active materials was calculated from the GCD curves using the following equation:

The volumetric specific capacitance (CV, F cm−3) was calculated from
| CV = Cg × ρ |
The volumetric energy density (EV, W h L−1) and power density (PV, W L−1) of the AQS/G-1//RuO2/G asymmetric supercapacitors were calculated from the GCD curves using the following equations:


where V (cm−3) is the total volume of both the anode and cathode, I (A) is the discharge current, t (s) is the corresponding discharge time, and U (V) is the voltage of the ASC device.

The utilization rate of AQS is calculated from the following equation:

Results and discussion
The typical synthesis of AQS/G composites is illustrated in Fig. 1. AQS powder was added to a graphene oxide (GO) dispersion to obtain a homogeneous AQS and GO mixture under intense stirring. It should be noted that the –SO3− functional groups in AQS endow it with excellent solubility in water29,30 compared with the insoluble –SO3−-free anthraquinone (AQ) (Fig. S1†) and facilitate the uniform noncovalent modification of AQS on GO sheets at a molecular level. Hydrothermal treatment was then used to reduce the insulating GO sheets to conductive graphene sheets, which were then assembled into a 3D interlinked AQS/G hydrogel. After washing and capillary drying, a shrunken AQS/G monolith was obtained. The as-prepared AQS/G composites were labeled AQS/G-x, where x is the initial mass ratio of AQS to GO in the mixture. In this composite, the 3D graphene network guarantees unimpeded electron conduction from the graphene sheets to the redox-active AQS molecules. | ||
| Fig. 1 Schematic of the synthesis of AQS/G composites. | ||
The electrochemical performance of the AQS/G composites was investigated using a three-electrode system in an aqueous electrolyte (1 M H2SO4). A well-defined pair of redox peaks at around −0.07/−0.01 V versus Ag/AgCl (at 5 mV s−1) was detected in the cyclic voltammograms (CVs) of AQS and AQS/G composites (Fig. 2a and S2†). These correspond to the reversible two-electron reaction between H+ and carbonyl groups, namely the transformation between AQS and AQSH2.31 In comparison, the CV curve of the high-density porous graphene monolith (HPGM) without the AQS has an almost rectangular shape, indicating an EDL capacitive behaviour. The AQS/G-x composites have a higher gravimetric specific capacitance than the HPGM and pure AQS deduced from the larger area of the CV curves, because of the combined faradaic and EDL capacitive behaviours, and AQS/G-1 shows the largest gravimetric specific capacitance (Fig. 2a). Higher and lower AQS loadings show a worse performance, because less AQS provides a lower specific capacitance and more AQS results in a worse rate performance because of the reduced electronic conductivity (Fig. S3†). The CV results are consistent with the corresponding galvanostatic charge–discharge (GCD) curves (Fig. 2d), which show the highest gravimetric specific capacitance (473 F g−1) and the best rate performance for AQS/G-1 (Fig. S4a†). After taking electrode density into account, AQS/G-1 also shows the highest volumetric specific capacitance of 650 F cm−3, which is almost twice that of the HPGM (328 F cm−3) and almost 70 times greater than that of pure AQS (9 F cm−3) (Fig. 2b). This results from not only the high density of AQS/G-1 but also the improved expression of its pseudocapacitance. However, for the AQS composite without the graphene network, only 0.67% of the active sites in AQS molecules participate in the redox reaction due to its inherent insulating properties. The specific capacitance of AQS/G-1 remains at 77% of the initial value even when the current density is increased to a 100-fold value (10 A g−1) (Fig. 3b). Even when the current density is increased to a 300-fold level (30 A g−1), the volumetric specific capacitance of AQS/G-1 is 376 F cm−3 (58% of the initial value), showing excellent rate capability (Fig. 3b). The well-defined and symmetric CV curves of AQS/G-1 at different scan rates indicate that the redox reaction of the composite is highly reversible (Fig. 3c). The GCD curves of AQS/G-1 show similar shapes and negligible IR drops for different current densities (Fig. 3d and S4b†), also indicating the outstanding reversibility and rate capability of AQS/G-1. In addition, AQS/G-1 delivers a long-term cycling stability of 73% after 20000 cycles at a high current density of 5 A g−1 (Fig. S4c†). This is mainly due to the dissolution of active AQS molecules into the electrolyte during cycling32,33 (Fig. S5†). However, it is much more serious for AQS/SP-1, demonstrating that the strong noncovalent π–π interaction between AQS molecules and graphene sheets can effectively alleviate the dissolution of AQS into the electrolyte.14,32
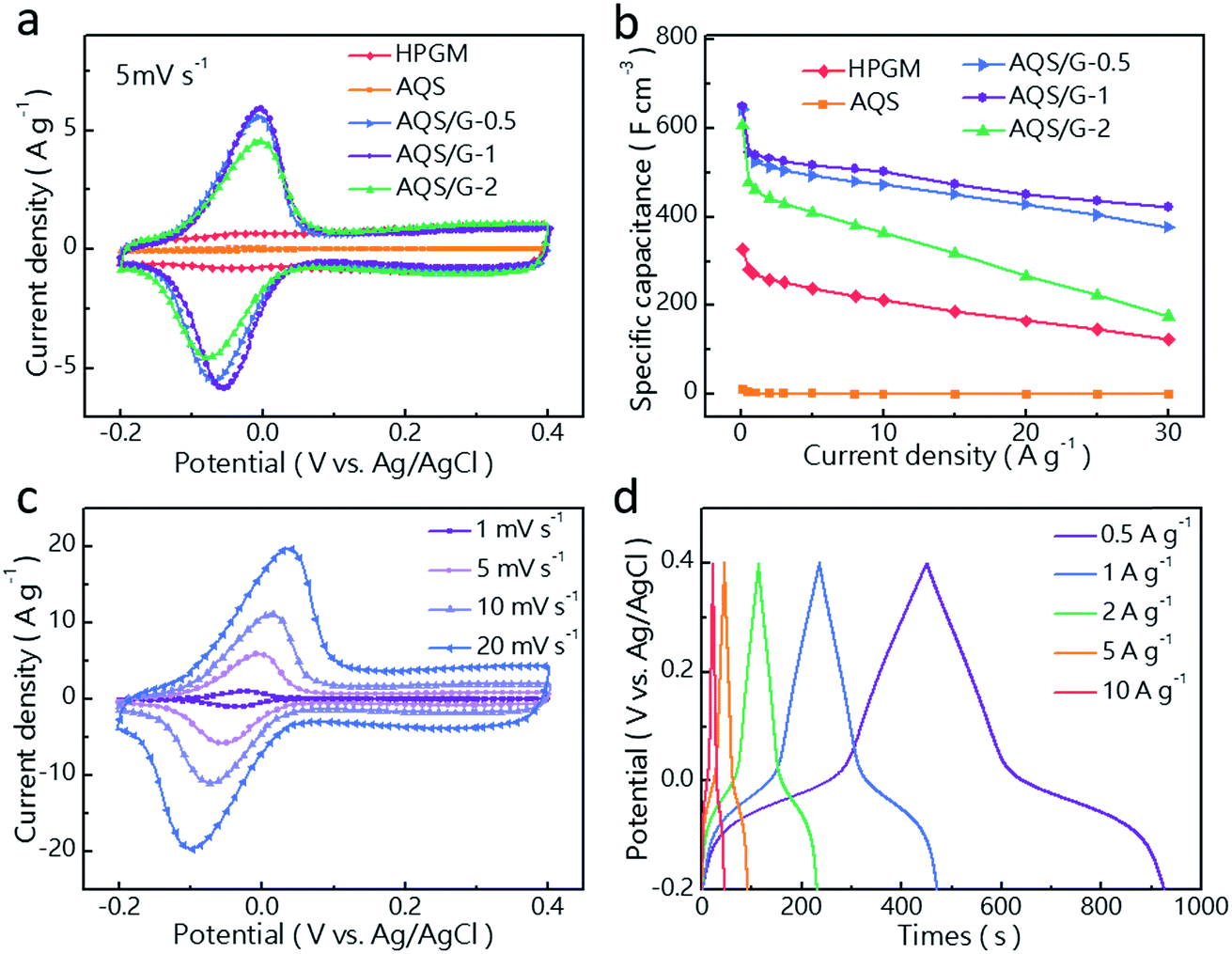 | ||
| Fig. 2 (a) Cyclic voltammetry profiles and (b) specific volumetric capacitances of HPGM, AQS and AQS/G composites with various AQS loadings. (c) Cyclic voltammetry profiles and (d) galvanostatic charge–discharge curves of AQS/G-1. | ||
 | ||
| Fig. 3 (a) SEM image (the inset shows the photograph of volume change after densification), (b) TEM image and (c) corresponding EDS elemental (C, O and S) maps of AQS/G-1. (d) XRD patterns and (e) FTIR spectra of AQS, the HPGM, AQS/G-0.5, AQS/G-1, and AQS/G-2. (f) XPS spectrum of AQS/G-1. | ||
The microstructure of the AQS/G composites was investigated to analyze the reason for the differences in their electrochemical performance. The shrunken AQS/G-1 monolith obtained by capillary drying showed a more compact interconnected nanostructure than the AQS/G-1 xerogel obtained by freeze drying (Fig. 3a and S6†), and shrank to about 2.5% of its original volume (inset in Fig. 3a), resulting in a bulk density as high as 1.38 g cm−3. In contrast, bare AQS powder produced irregular solid blocks without any interconnected nanostructure, quite different from the AQS/G composites (Fig. S8†). No AQS particles are visible in the scanning electron microscope (SEM) images of the AQS/G-1 monolith even at a high magnification (Fig. S9†), indicating the uniform modification of nano-sized AQS on graphene sheets. Such high-density, interlinked AQS/G composites have great potential for constructing thicker electrodes with high volumetric/areal capacitance.
The uniform distribution of AQS on the graphene sheets was also characterized by transmission electron microscopy (TEM). From the TEM images, wrinkled graphene sheets can be easily observed without any obvious agglomeration of AQS, even at a high magnification (Fig. 3b and S10a, b and d, e†). However, in the energy-dispersive X-ray spectroscopy (EDX) elemental maps of AQS/G-1, C, O and S are distinctly seen, revealing the uniform distribution of oxygen- and sulfur-containing groups in the AQS/G-1 composite (Fig. 3c), which confirms the successful and uniform combination of AQS and graphene sheets. The selected area electron diffraction (SEAD) images of AQS/G-1 and bare AQS show that AQS loses its high crystallinity and becomes amorphous when anchored on the graphene sheets (Fig. S10c–f†), and thus exposes more active sites for the electrochemical reaction. The amorphous structure was confirmed using X-ray diffraction (XRD) patterns, in which the strong characteristic peaks of highly crystalline AQS disappeared for the AQS/G composites (Fig. 3d). Instead, all the AQS/G composites have two similar broad characteristic peaks comparable to those seen for the HPGM, indicating the amorphization of AQS after being combined with graphene sheets (Fig. 3d and S11†). The characteristic peaks of AQS were also identified in the Fourier-transform infrared (FTIR) spectra of AQS/G composites, which confirms the combination of AQS and graphene sheets (Fig. 3e). AQS/G shows bands at 1668, 1223, 1045 and 717 cm−1 which correspond to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, –SO3− stretching and C–H vibration, respectively (Fig. 3e). Moreover, the typical CC vibration band of bare AQS at 1587 cm−1 shows a slight red-shift to 1575 cm−1 in AQS/G composites (Fig. 3e), which can be ascribed to the π–π interaction between AQS and graphene sheets.34 In the Raman spectra, the slight change of ID/IG, the characteristic peak position and the peak shape indicates that the noncovalent interaction between AQS and graphene sheets does not significantly affect the characteristics of the graphene sheets (Fig. S12†).14,35 X-ray photoelectron spectroscopy (XPS) of AQS/G-1 shows three peaks, C 1s, O 1s and S 2p (Fig. S13†), with two S 2p signals (at 167 eV and 162.6 eV) corresponding to the –SO3− group detected (Fig. 3f), which confirms the anchoring of AQS. Overall, all these results confirm the successful and uniform noncovalent modification of AQS molecules on graphene sheets. The noncovalent π–π interaction between AQS and graphene maintains the high conductivity of the graphene and the high redox activity of AQS, thus resulting in the above outstanding electrochemical performance.
O stretching, –SO3− stretching and C–H vibration, respectively (Fig. 3e). Moreover, the typical CC vibration band of bare AQS at 1587 cm−1 shows a slight red-shift to 1575 cm−1 in AQS/G composites (Fig. 3e), which can be ascribed to the π–π interaction between AQS and graphene sheets.34 In the Raman spectra, the slight change of ID/IG, the characteristic peak position and the peak shape indicates that the noncovalent interaction between AQS and graphene sheets does not significantly affect the characteristics of the graphene sheets (Fig. S12†).14,35 X-ray photoelectron spectroscopy (XPS) of AQS/G-1 shows three peaks, C 1s, O 1s and S 2p (Fig. S13†), with two S 2p signals (at 167 eV and 162.6 eV) corresponding to the –SO3− group detected (Fig. 3f), which confirms the anchoring of AQS. Overall, all these results confirm the successful and uniform noncovalent modification of AQS molecules on graphene sheets. The noncovalent π–π interaction between AQS and graphene maintains the high conductivity of the graphene and the high redox activity of AQS, thus resulting in the above outstanding electrochemical performance.
Research on AQS as an electrode material for SCs began in 2018,14 and the exact electrochemical behaviour of AQS is still unclear. Therefore, electrochemical kinetic analysis was conducted to investigate the electrochemical behaviour and capacitance contribution of AQS/G-1. In general, current (i) and scan rates (v) in CV curves obey the relationship shown in eqn (1):36
| i = avb | (1) |
i vs. logv. The b-value can be used to distinguish the type of energy-storage mechanism, with a b-value of 0.5 indicating a total diffusion-controlled process, while 1.0 is representative of capacitive dominance. In order to minimize the influence of the current peak shift due to polarization at high scan rates, a MUSCA technique was used in this work37 with a potential step amplitude (ΔV) of 100 mV. The calculated average currents (ī) were calculated using eqn (2):37 | (2) |
 | (3) |
 | (4) |
 indicate the capacitive and diffusion-controlled contributions to the overall current (i), respectively.38 At relatively low scan rates, a capacitive process contributed the minority of the current, indicating that the two-electron redox reaction of AQS behaves like a fast diffusion-controlled process (Fig. 4c). With the increasing scan rates, the capacitive contributions are dominant (Fig. 4c). When the scan rate was increased to 50 mV s−1, the capacitive process contributed 63% of the total charge and a well-fitted CV curve was obtained (Fig. 4c and S14†). Both volumetric specific capacitance and areal specific capacitance are key metrics for portable and miniature devices. Therefore, the electrochemical performance of AQS/G-1 was measured with different mass loadings. The AQS@G-1 shows a superior volumetric specific capacitance of 650 F cm−3 for 3 mg cm−2 (20 μm in thickness) and as high as 547 F cm−3 even for a high mass loading of 32 mg cm−2 (200 μm in thickness) (Fig. 4d). Even when the current density is increased to 0.5 A g−1, AQS/G-1 delivers a maximum volumetric specific capacitance of 544 F cm−3 and shows little capacitance decay as a result of the increasing mass loading (Fig. 4d). However, pure AQS was completely different. Even at a low current density of 0.1 A g−1, its volumetric specific capacitance sharply decreases to near zero (3.8 F cm−3) when the mass loading is increased to 11 mg cm−2 (Fig. 4d). The areal specific capacitance of a thicker AQS/G-1 electrode can be over 10 F cm−2 with a mass loading of 23 mg cm−2 (Fig. 4e), and by increasing this to 32 mg cm−2 (200 μm in thickness), the areal specific capacitance continues to increase to 13.3 F cm−2 (Fig. 4e), which is amongst the best results for organic molecule-based electrodes as far as we know. The 200 μm AQS/G-1 electrode also shows outstanding rate capability, and even with a mass loading of 23 mg cm−2, the electrode delivers a capacitance retention of 88% when the current density is increased to a 10-fold value (Fig. 4e). It should be noticed that our results are among the best in all the current reports39–46 when taking volumetric capacitance, areal capacitance and electrode thickness into account (Fig. 4f).
indicate the capacitive and diffusion-controlled contributions to the overall current (i), respectively.38 At relatively low scan rates, a capacitive process contributed the minority of the current, indicating that the two-electron redox reaction of AQS behaves like a fast diffusion-controlled process (Fig. 4c). With the increasing scan rates, the capacitive contributions are dominant (Fig. 4c). When the scan rate was increased to 50 mV s−1, the capacitive process contributed 63% of the total charge and a well-fitted CV curve was obtained (Fig. 4c and S14†). Both volumetric specific capacitance and areal specific capacitance are key metrics for portable and miniature devices. Therefore, the electrochemical performance of AQS/G-1 was measured with different mass loadings. The AQS@G-1 shows a superior volumetric specific capacitance of 650 F cm−3 for 3 mg cm−2 (20 μm in thickness) and as high as 547 F cm−3 even for a high mass loading of 32 mg cm−2 (200 μm in thickness) (Fig. 4d). Even when the current density is increased to 0.5 A g−1, AQS/G-1 delivers a maximum volumetric specific capacitance of 544 F cm−3 and shows little capacitance decay as a result of the increasing mass loading (Fig. 4d). However, pure AQS was completely different. Even at a low current density of 0.1 A g−1, its volumetric specific capacitance sharply decreases to near zero (3.8 F cm−3) when the mass loading is increased to 11 mg cm−2 (Fig. 4d). The areal specific capacitance of a thicker AQS/G-1 electrode can be over 10 F cm−2 with a mass loading of 23 mg cm−2 (Fig. 4e), and by increasing this to 32 mg cm−2 (200 μm in thickness), the areal specific capacitance continues to increase to 13.3 F cm−2 (Fig. 4e), which is amongst the best results for organic molecule-based electrodes as far as we know. The 200 μm AQS/G-1 electrode also shows outstanding rate capability, and even with a mass loading of 23 mg cm−2, the electrode delivers a capacitance retention of 88% when the current density is increased to a 10-fold value (Fig. 4e). It should be noticed that our results are among the best in all the current reports39–46 when taking volumetric capacitance, areal capacitance and electrode thickness into account (Fig. 4f).
 | ||
| Fig. 4 (a) Calculated cyclic voltammetry profiles obtained by Multiple Step Chrono Amperometry (MUSCA) measurements with a potential step size of 100 mV. (b) b-Values of AQS/G-1 at various potentials in cathodic scans. The inset shows the b-values determined by using the logi–logv plot. (c) Capacitive contributions at different scan rates. (d) Volumetric specific capacitance of AQS and AQS/G-1 with various mass loadings. The inset shows the illustration of the electron and ion transport in the electrode. (e) Areal specific capacitance of AQS/G-1 at different current densities with various mass loadings. (f) Ragone plot of AQS/G-1 and other reported materials. | ||
The outstanding electrochemical performance of AQS/G-1 can be attributed to three factors. First, the dense graphene network provides not only excellent electron conduction but also easy ion transport for the highly active AQS molecules. Second, the face-to-face modification of AQS molecules on graphene sheets by noncovalent π–π interaction maximizes their contact with the conductive graphene sheets and electrolytes, which significantly facilitates electron transfer between them and the graphene and the two-electron reaction between AQS and protons. Third, the amorphous structure of AQS provides more active sites exposed to the electrolyte and thus enables a high specific capacitance. Last but not least, –SO3− functional groups in AQS not only give AQS/G-1 excellent surface wettability with aqueous electrolytes but also act as proton donors and carriers to facilitate proton transport.
ASCs were assembled with AQS/G-1 as the anode and RuO2/G as the cathode (Fig. S15†), since the RuO2/G composite has a suitable potential range and good performance in SCs (Fig. S16†). When the voltage was 1.35 V (Fig. S17†) and the mass ratio between the anode and cathode is 1.12:1, the AQS/G-1//RuO2/G ASC shows optimum performance (Fig. S18†). The CV profile for the ASC has an approximately rectangular shape with a pair of pronounced redox peaks at different scan rates from 2 to 50 mV s−1 (Fig. 5a). The GCD curves also show pseudocapacitance with slight plateaus at different current densities corresponding with the peaks in the CV curves (Fig. 5b). The AQS/G-1//RuO2/G ASC delivers a maximum volumetric energy density of 44 W h L−1 at a power density of 2307 W L−1 (Fig. 5c). It retains a high volumetric energy density of 38 W h L−1 when the power density is increased to 8.6 kW L−1, which is quite outstanding compared to that of other reported aqueous ASCs (Fig. 5c).39,46–53 Moreover, the AQS/G-1//RuO2/G ASC retains 76% of its initial capacitance after 10000 cycles at a current density of 5 A g−1 (Fig. S19†). These results clearly indicate that AQS/G-1 can be used as the anode for high-performance ASCs.
 | ||
| Fig. 5 (a) Cyclic voltammetry profiles and (b) galvanostatic charge–discharge curves of the as-assembled AQS/G-1//RuO2/G ASC. (c) Ragone plot of the as-assembled AQS/G-1//RuO2/G ASC and previously reported devices. | ||
Conclusions
A dense organic molecule modified graphene hybrid network (AQS/G) was designed to realize both a high volumetric and a high areal capacitance. The graphene network provides unimpeded electron conduction and ion transport to guarantee a high-performance output, even for a 200 μm thick electrode. Meanwhile, the densification of the AQS/G network by capillary drying makes maximum utilization of the high volumetric and areal performance of AQS. The as-fabricated AQS/G-1 shows a high bulk density and a high volumetric specific capacitance up to 650 F cm−3 together with a 77% capacitance retention even at a 100-fold current density, which is far better than that of pure AQS and the HPGM. After an in-depth analysis of the electrochemical behaviour of AQS/G-1, it is found that the stored charge is derived from both capacitive and diffusion-controlled processes, and the diffusion-controlled process dominates at the redox peak potentials. More importantly, a maximum areal specific capacitance of up to 13.3 F cm−2 can be achieved with a mass loading of 32 mg cm−2 (200 μm in thickness), which as far as we know is the best result for organic molecule-based electrodes in SCs. After coupling with a RuO2/G cathode to assemble an ASC, the device delivers a maximum volumetric energy density of 44 W h L−1, which is also among the best results for aqueous ASCs. We believe that our material-design strategy can be applicable to other organic electrode materials. And the AQS/G hybrid network can also be used to construct other high-performance EES systems, such as zinc-ion batteries and flow batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Thousand Talents Plan for Young Professionals of China, the National Science Fund for Distinguished Young Scholars of China (No. 51525204) and the National Natural Science Foundation of China (No. 51702229 and U1710256).Notes and references
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- S. Wu and Y. Zhu, Sci. China Mater., 2017, 60, 25–38 CrossRef CAS.
- D. Sheberla, J. C. Bachman, J. S. Elias, C. J. Sun, Y. Shao-Horn and M. Dinca, Nat. Mater., 2017, 16, 220–224 CrossRef CAS.
- D. Han, Z. Weng, P. Li, Y. Tao, C. Cui, L. Zhang, W. Lin, Y. Gao, D. Kong and Q.-H. Yang, Energy Storage Materials, 2019, 18, 133–138 CrossRef.
- H. Li, C. Qi, Y. Tao, H. Liu, D. W. Wang, F. Li, Q. H. Yang and H. M. Cheng, Adv. Energy Mater., 2019, 9, 1900079 CrossRef.
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed.
- M. Salanne, B. Rotenberg, K. Naoi, K. Kaneko, P. L. Taberna, C. P. Grey, B. Dunn and P. Simon, Nat. Energy, 2016, 1, 16070 CrossRef CAS.
- H.-P. Cong, X.-C. Ren, P. Wang and S.-H. Yu, Energy Environ. Sci., 2013, 6, 1185–1191 RSC.
- R. Emanuelsson, M. Sterby, M. Stromme and M. Sjodin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC.
- Y. Liao, H. Wang, M. Zhu and A. Thomas, Adv. Mater., 2018, 30, 1705710 CrossRef PubMed.
- L. Xu, R. Shi, H. Li, C. Han, M. Wu, C.-P. Wong, F. Kang and B. Li, Carbon, 2018, 127, 459–468 CrossRef CAS.
- R. Shi, C. Han, H. Duan, L. Xu, D. Zhou, H. Li, J. Li, F. Kang, B. Li and G. Wang, Adv. Energy Mater., 2018, 8, 1802088 CrossRef.
- Y. Q. Sun, Q. O. Wu and G. Q. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- D. Han, J. Zhang, Z. Weng, D. Kong, Y. Tao, F. Ding, D. Ruan and Q.-H. Yang, Materials Today Energy, 2019, 11, 30–45 CrossRef.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC.
- C. Zhao, C. Yu, B. Qiu, S. Zhou, M. Zhang, H. Huang, B. Wang, J. Zhao, X. Sun and J. Qiu, Adv. Mater., 2018, 30, 1702486 CrossRef PubMed.
- D. Yu, K. Goh, H. Wang, L. Wei, W. Jiang, Q. Zhang, L. Dai and Y. Chen, Nat. Nanotechnol., 2014, 9, 555–562 CrossRef CAS PubMed.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- Y. Tao, X. Xie, W. Lv, D. M. Tang, D. Kong, Z. Huang, H. Nishihara, T. Ishii, B. Li, D. Golberg, F. Kang, T. Kyotani and Q. H. Yang, Sci. Rep., 2013, 3, 2975 CrossRef PubMed.
- P. Li, H. Li, D. Han, T. Shang, Y. Deng, Y. Tao, W. Lv and Q. H. Yang, Adv. Sci., 2019, 6, 1970086 CrossRef.
- X. Yang, C. Cheng, Y. Wang, L. Qiu and D. Li, Science, 2013, 341, 534–537 CrossRef CAS PubMed.
- H. Li, Y. Tao, X. Zheng, J. Luo, F. Kang, H.-M. Cheng and Q.-H. Yang, Energy Environ. Sci., 2016, 9, 3135–3142 RSC.
- F. Xu, R. Cai, Q. Zeng, C. Zou, D. Wu, F. Li, X. Lu, Y. Liang and R. Fu, J. Mater. Chem., 2011, 21, 1970–1976 RSC.
- Y. Xia, T. S. Mathis, M. Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- Y. Yoon, K. Lee, S. Kwon, S. Seo, H. Yoo, S. Kim, Y. Shin, Y. Park, D. Kim, J.-Y. Choi and H. Lee, ACS Nano, 2014, 8, 4580–4590 CrossRef CAS PubMed.
- H. Ma, D. Kong, Y. Xu, X. Xie, Y. Tao, Z. Xiao, W. Lv, H. D. Jang, J. Huang and Q. H. Yang, Small, 2017, 13, 1701026 CrossRef PubMed.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- B. G. Choi, J. Hong, W. H. Hong, P. T. Hammond and H. Park, ACS Nano, 2011, 5, 7205–7213 CrossRef CAS PubMed.
- K. Kalinathan, D. P. DesRoches, X. Liu and P. G. Pickup, J. Power Sources, 2008, 181, 182–185 CrossRef CAS.
- S. K. Kim, J. Cho, J. S. Moore, H. S. Park and P. V. Braun, Adv. Funct. Mater., 2016, 26, 903–910 CrossRef CAS.
- Y. Xu, Z. Lin, X. Huang, Y. Wang, Y. Huang and X. Duan, Adv. Mater., 2013, 25, 5779–5784 CrossRef CAS PubMed.
- M. M. Sk and C. Y. Yue, J. Mater. Chem. A, 2014, 2, 2830–2838 RSC.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- D. P. Dubal, N. R. Chodankar and S. Qiao, Small, 2019, 15, 1804104 CrossRef PubMed.
- H. Shao, Z. Lin, K. Xu, P.-L. Taberna and P. Simon, Energy Storage Materials, 2018, 18, 456 CrossRef.
- D. Chao, C. Zhu, P. Yang, X. Xia, J. Liu, J. Wang, X. Fan, S. V. Savilov, J. Lin, H. J. Fan and Z. X. Shen, Nat. Commun., 2016, 7, 12122 CrossRef CAS PubMed.
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. F. Lin, M. Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P. L. Taberna, M. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- Y. Xu, Y. Tao, X. Zheng, H. Ma, J. Luo, F. Kang and Q.-H. Yang, Adv. Mater., 2015, 27, 8082–8087 CrossRef CAS PubMed.
- Z. K. Yang, J. Ma, S. Araby, D. J. Shi, W. F. Dong, T. Tang and M. Q. Chen, J. Power Sources, 2019, 412, 655–663 CrossRef CAS.
- T. T. Gao, Z. Zhou, J. Y. Yu, J. Zhao, G. L. Wang, D. X. Cao, B. Ding and Y. J. Li, Adv. Energy Mater., 2019, 9, 1802578 CrossRef.
- J. S. Zhou, L. Hou, J. Lian, W. B. Cheng, D. Wang, H. Y. Gou and F. M. Gao, J. Mater. Chem. A, 2019, 7, 476–485 RSC.
- S. Zhang, J. Y. Zhu, Y. Qing, L. X. Wang, J. Zhao, J. Li, W. H. Tian, D. Z. Jia and Z. J. Fan, Adv. Funct. Mater., 2018, 28, 1805898 CrossRef.
- H. Tong, S. Yue, L. Lu, F. Jin, Q. Han, X. Zhang and J. Liu, Nanoscale, 2017, 9, 16826–16835 RSC.
- X. Zhang, Y. Liu, S. Dong, J. Yang and X. Liu, Electrochim. Acta, 2019, 294, 233–239 CrossRef CAS.
- Z. Fan, Y. Wang, Z. Xie, D. Wang, Y. Yuan, H. Kang, B. Su, Z. Cheng and Y. Liu, Adv. Sci., 2018, 5, 1800750 CrossRef PubMed.
- L. Sheng, L. Jiang, T. Wei and Z. Fan, Small, 2016, 12, 5217–5227 CrossRef CAS PubMed.
- Y. Xu, Z. Lin, X. Zhong, X. Huang, N. O. Weiss, Y. Huang and X. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- F. G. Zhao, Y. T. Kong, B. Y. G. Pan, C. M. Hu, B. Zuo, X. P. Dong, B. X. Li and W. S. Li, J. Mater. Chem. A, 2019, 7, 3353–3365 RSC.
- A. VahidMohammadi, M. Mojtabavi, N. M. Caffrey, M. Wanunu and M. Beidaghi, Adv. Mater., 2019, 31, 1806931 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta09941g |
| ‡ These two authors are equal major contributors to this work. |
| This journal is © The Royal Society of Chemistry 2020 |