
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetecting structural transformation of cobalt phosphonate to active bifunctional catalysts for electrochemical water-splitting†
Arindam
Indra
 ab,
Prashanth W.
Menezes
*b,
Ivelina
Zaharieva
c,
Holger
Dau
*c and
Matthias
Driess
*b
ab,
Prashanth W.
Menezes
*b,
Ivelina
Zaharieva
c,
Holger
Dau
*c and
Matthias
Driess
*b
aDepartment of Chemistry, Indian Institute of Technology (BHU), Varanasi, Uttar Pradesh-221005, India
bDepartment of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Straße des 17 Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: prashanth.menezes@mailbox.tu-berlin.de; matthias.driess@tu-berlin.de
cFachbereich Physik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany. E-mail: holger.dau@fu-berlin.de
First published on 7th January 2020
Abstract
In recent years, several cobalt-based catalysts have been developed for water splitting because of their promising activity, stability and structural motifs. Here, we report that cobalt phosphonate represents a novel class of bifunctional single-source precursors for highly efficient alkaline electrochemical O2 evolution (OER) and H2 evolution reaction (HER). Inspired by its favorable catalytic OER and HER activity, an overall water-splitting device has been constructed from this precursor, showing very low cell voltage (1.62 V @ 10 mA cm−2) and excellent long-term stability. Depending on the applied oxidation and reduction potential on cobalt phosphonate, two distinct modified structures at the anode and cathode have been uncovered employing the quasi in situ X-ray absorption spectroscopy and ex situ methods. During OER, the phosphonate precursor reorganized itself to layered CoOx(OH)y structure with defects and disorders, while the contribution of the metallic Co along with Co3O4 spinel and Co(OH)2 is evident to drive the HER. The presented work demonstrates the advantage of using the ‘all-in-one’ precursor approach to realize bifunctional water-splitting electrocatalysts through the evolution of different species with self-supporting interfacial structural features at the anode and cathode during electrochemical water splitting.
Introduction
Electrochemical energy conversion by splitting water into O2 and H2 may become pivotal in providing clean and sustainable energy replacing fossil fuels.1–3 The water-splitting process comprises of two half-cell reactions-oxygen evolution reaction (OER) at the anode and hydrogen evolution reaction (HER) at the cathode.4–6 Water splitting is a thermodynamically uphill reaction and electrolysis of water requires a huge amount of energy in terms of potential.5 Moreover, the O–O bond formation during OER requires the removal of four electrons and four protons from two water molecules involving high-energy intermediates.7 Therefore, special attention has been provided to develop an efficient catalyst system for the overall water splitting reactions.8Although noble metal-based RuO2 and IrO2 catalysts produce high water oxidation activity and Pt is known for the excellent hydrogen evolution, scarcity and cost of these materials limit their use for large-scale applications.9,10 Recently, first-row transition metal-based catalysts have been widely explored for the OER and HER.11–13 In this respect, cobalt-based materials are of particular importance because of their promising water-oxidation activity at reasonable overpotentials and regarding the understanding of structural and functional properties during water oxidation.14–17 After the first report of in situ generated cobalt (oxide) phosphate (CoPi) catalyst for the efficient oxygen evolution in the neutral medium by Nocera and co-workers,18,19 several crystalline and amorphous cobalt phosphates and oxides have extensively been investigated for electrocatalytic OER.16,20–24 Most of the cobalt-based catalysts produce high electrochemical water oxidation activity in the alkaline medium through the activation of the catalyst into amorphous cobalt hydroxide–oxyhydroxide.16,25–27 In many cases, the transformation is limited to the surface of the particles to form an amorphous shell while complete amorphization of the particles has been observed for the ultra-small nanoparticles.24,26 Reversible amorphization of the catalytic species in cobalt oxide has also been demonstrated by some of us.15 Moreover, the coordination environment around the cobalt center and the local structure has also shown to control the OER activity.22,28
On the other hand, the structural transformation of the cobalt-based catalyst during electrochemical HER has mostly been investigated only by ex situ methods.16,29–31 The ex situ methods have various drawbacks including the oxidation of the precatalysts in alkaline conditions and the possible surface oxidation of the active phase in aerial conditions.5 Therefore, it is highly desirable to investigate the active catalyst structure through in situ methods.
Though a series of cobalt (oxide) phosphate-based catalysts have been explored for the OER16,20–24,26,27 and HER,16,29,30,32–34 their use for the overall water splitting is fostered in the last few years. Cobo et al. have developed a Janus cobalt-based catalyst for the OER and HER at neutral pH.35 The catalyst showed the formation of metallic Co coated with a cobalt-oxo/hydroxo-phosphate during HER in neutral medium.35 Recently, we reported helical cobalt borophosphates for the electrochemical water splitting in the alkaline medium where the structural transformation of the catalyst at the anode and cathode was observed to attain two different active catalyst structures.36 Therefore, understanding the structural transformation and realizing the structure of such active catalysts for both HER and OER is of fundamental interest to establish the structure–activity correlation.
Here we report a new class of material: cobalt phosphonate (CoPn) for electrochemical water splitting in alkaline medium. Phosphonates coordinate to the metal ion through the oxygen atoms and the reaction conditions significantly affect the local environment of the coordination around the metal centers (Fig. S1†).37 Though these inorganic–organic hybrid materials are known for a long time, their applications for the photochemical and electrochemical water oxidation have been explored only recently.38–42 Inspired by the excellent photochemical as well as electrochemical water oxidation of the phosphonates,38–42 we have prepared CoPn and employed for the unification of electrochemical water oxidation, proton reduction, and overall water splitting. The nickel foam (NF) supported CoPn produced excellent OER activity in an alkaline medium to reach the current density of 10 mA cm−2 at only 240 mV overpotential whereas an overpotential of 144 mV was required for HER to attain the same current density. In addition, the designed alkaline overall water-splitting device displayed a cell voltage of only 1.62 V to reach the current density 10 mA cm−2, making it one of the promising materials towards efficient water splitting. Further, the structural insights gathered from quasi in situ and ex situ experiments revealed that with applied potential CoPn transformed into a layered Co oxide structure, related to the crystalline LiCoO2 or crystalline CoO(OH) but with clearly lower degree of order during OER whereas the HER catalyst comprises of metallic cobalt, Co3O4 and Co(OH)2.
Results and discussion
CoPn was synthesized by the solvothermal reaction of Co(NO3)2·6H2O and diethylenetriamine penta(methylenephosphonic acid) [DTPMP] (see the details in Experimental in ESI†). The material was purely amorphous in nature as revealed by the powder X-ray diffraction (Fig. S2†). Infrared spectrum detects the corresponding vibrations related to CoPn, as discussed in detail in the ESI (Fig. S3†).43 The amorphous structure of the particles was clearly visible by the transmission electron microscopic (TEM) studies (Fig. 1). High-resolution TEM could not detect any lattices, whereas the selected area diffraction pattern (SAED) also revealed the amorphous nature of the CoPn particles (Fig. 1). Energy dispersive X-ray (EDX) detects the presence of Co, N, and P in fresh CoPn (Fig. S4†). The elemental analysis reveals that the ratio C/N ∼2.83 in CoPn (close to the theoretical value of C/N = 9/3 = 3). | ||
| Fig. 1 (a) TEM image of CoPn, (b) high-resolution TEM image of CoPn and (c) electron diffraction pattern representing the amorphous nature of the material. | ||
The surface properties, oxidation state and surface composition of CoPn were analyzed by X-ray photoelectron spectroscopic (XPS) studies (Fig. S5†). In Co 2p XPS, two peaks corresponding to Co 2p1/2 and Co 2p3/2 appeared at the binding energies of ∼797 eV and ∼781 eV, respectively (Fig. S5a†). The spin–orbit coupling spacing value (2p3/2–2p1/2) of 15.96 eV indicates CoII as the dominant species.14,15 The O 1s signal of pristine CoPn showed two peaks (531.2 eV and 532.0 eV) corresponding to P–O–Co linkages and oxygen from water (Fig. S5b†).43 The N 1s peak can also be deconvoluted into two peaks at 399.9 eV and 401.8 eV indicating the C–N-containing moiety and quarternary N (Fig. S5c†).43 The P 2p peaks at binding energy 133.4 eV (2p3/2) and 134.3 eV (2p3/2) indicate the presence of POx unit in the phosphonate and clearly matched with the literature reports (Fig. S5d†).43
The as-prepared CoPn was first deposited on nickel foam (NF) by electrophoretic deposition (EPD) and electrochemical performances were investigated in 1 M aqueous KOH solution by a typical three-electrode system. For comparison, the OER properties of bare NF and commercially available benchmark electrocatalysts were also studied under the same condition. As presented in Fig. 2a, CoPn/NF exhibited an excellent OER activity with an overpotential of 240 mV to drive geometrical catalytic current density of 10 mA cm−2 (330 mV at 100 mA cm−2). The potential is significantly lower than noble metal catalysts RuO2/NF, IrO2/NF, and NF. Interestingly, the achieved current density and overpotentials for CoPn was also found to be comparable to recently reported NF supported transition metal catalysts (Table S1†).44–46
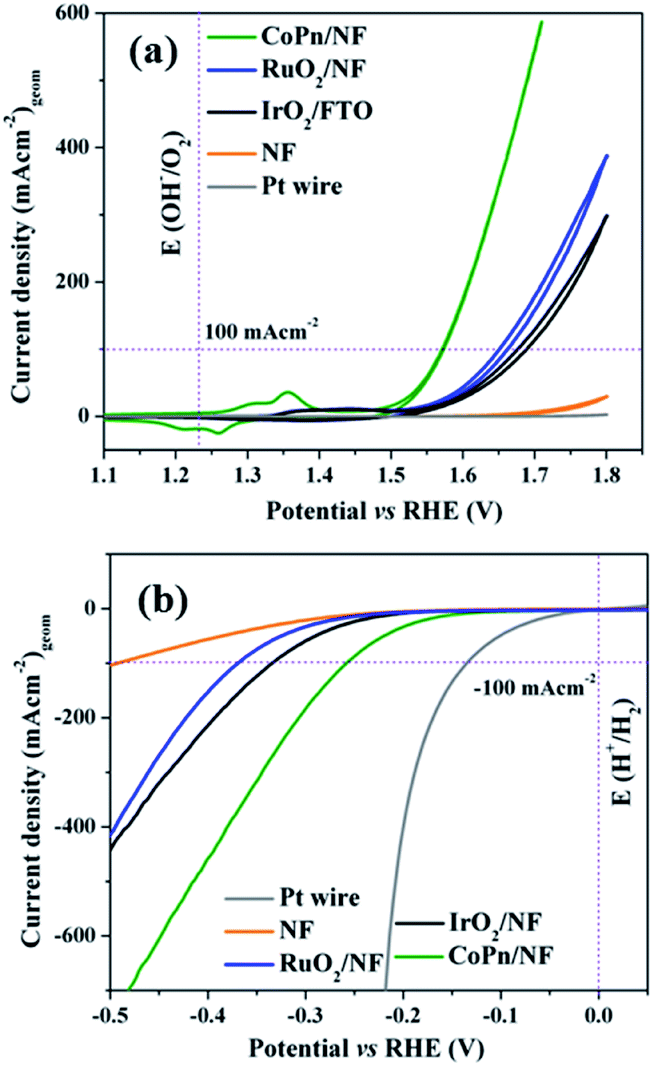 | ||
| Fig. 2 (a) CV of OER and (b) LSV of HER for CoPn/NF and commercial noble metal-based catalysts in 1 M aqueous KOH at a scan rate of 1 mV s−1. | ||
To further illustrate the superior catalytic performance, the electrochemical impedance spectroscopy (EIS) was conducted where the Nyquist plots revealed very low charge-transfer resistance (Rct) for CoPn/NF suggesting faster electron transfer rate between the interface of the catalyst that contributes to the higher OER activity (Fig. S6†).47 The electrochemical double-layer capacitance (Cdl) was determined by cyclic voltammetry (CV) in the non-faradic reaction range (Fig. S9†). A Cdl of 2.14 mF cm−2 was achieved for CoPn/NF that was almost thrice larger compared to bare NF (0.8 mF cm−2).48 The Cdl was then converted into ECSA using the specific capacitance (Cs) of the material per unit area (Cs = 1.7 mF cm−2 for NF),47 and a value of 1.25 cm2 (for details see Fig. S7† and Experimental section) was estimated for CoPn/NF demonstrating the presence of rich catalytically active sites than that of NF (0.47 cm2).49
Two redox peaks were detected while cycling CoPn/NF between the potential values of 1.15 V and 1.45 V vs. RHE (Fig. S8†). These two peaks represent the oxidation of CoII to CoIII and CoIV during the electrochemical process to form active OER catalyst CoOx(OH)y.50 The confirmation of the increase in the oxidation state was further proved by the ex situ XPS and quasi in situ X-ray absorption spectroscopic (XAS) studies (see later). Tafel plots assessed the catalytic reaction kinetics of CoPn/NF, and a slope of close to 60 mV (ca. 68 mV dec−1) indicates that the OER mechanism proceeds through reversible electron transfer before reaching the turnover-limiting step (Fig. S9†).51 Moreover, the excellent stability of the system has been achieved to show constant current production for more than 25 h under chronoamperometric (CA) conditions (Fig. S10†). A careful investigation of the CA profile reveals that there was a significant increment in the current density within the first 12 h. The initial increase in the current density can be explained by the activation of the precatalyst (CoPn) to form the active water oxidation catalyst. As the transformation of the precatalyst to active catalyst was completed, a slight drop in current density was observed, indicating slow deactivation of the catalyst on longer run.
Similarly, the NF supported CoPn has been tested for HER in 1 M aqueous KOH solution. Only 144 mV of overpotential was required to achieve a current density of 10 mA cm−2 (Fig. 2b). Similar to OER catalysis, the catalytic HER performance of CoPn is better than the noble metal-based catalysts and bare NF but inferior to Pt. The activity of CoPn is also comparable to recently reported transition metal-based catalysts (Table S2†). A constant current was recorded to produce H2 over 25 h under chronoamperometric conditions (Fig. S11†).
As most of the cobalt-based transition metal catalysts are transformed into CoOx(OH)y during OER in alkaline medium,26,31,36,50 we have extensively carried out studies on the structural transformation of CoPn. First, the catalyst was characterized by ex situ spectroscopic, microscopic and analytical methods after 25 h CA measurements. Destruction of the structure of the CoPn was detected by TEM studies (Fig. 3). A layered structure with overlapping plates was formed after the OER (Fig. 3a and b). The rings of the SAED could be attributed to the (110) and (130) planes of the Co(O)OH (JCPDF: 26-480). Similarly, CoPn also underwent complete transformation after HER-CA displaying rings of (220) (311) and (511) corresponding to the spinel Co3O4 phase (JCPDF: 42-1467) (Fig. 3c and d).
 | ||
| Fig. 3 TEM and HR-TEM images of CoPn after chronoamperometric OER (a and b) and HER (c and d) studies in 1 M alkaline KOH solution. The corresponding electron diffraction pattern is shown in the inset. | ||
The XPS studies revealed that the oxidation state of Co was increased after the oxygen evolution (Fig. 4 and S12†). From the literature, it is well known that CoII and CoIII ions have almost similar 2p binding energies (BE) but can be differentiated by the Co 2p1/2–2p3/2 spin–orbit coupling value, which is 16 eV for high-spin CoII and 15 eV for low spin CoIII.17 An energy spacing of 15.5 eV was attained after OER CV that indicated the presence of mixed-valent CoII–CoIII species. Moreover, after OER CA, the energy spacing was further reduced to 15.5 eV, which clearly confirmed the dominant amount of CoIII species.17 The deconvolution of the Co 2p peaks also indicates the oxidation of CoII to CoIII during the electrochemical process.36 After the electrochemical measurements (both CV and CA), the O 1s peak was deconvoluted into three peaks. As anticipated, the peak corresponding to P–O–Co species was disappeared with the formation of the new peak for Co–O bonds (529.5 eV) from Co-oxide/hydroxide species (Fig. S13†).26,50 The other two deconvoluted peaks of O 1s indicate substantial hydration (532.7–533.6 eV) and hydroxylation (531.4 eV) of the catalyst surface during electrochemical OER.26,50 The signals from N and P in pristine CoPn were also disappeared after alkaline water oxidation (Fig. S14 and S15†). More detailed descriptions of the deconvoluted spectra are given in Fig. S12 to S15 of ESI.†
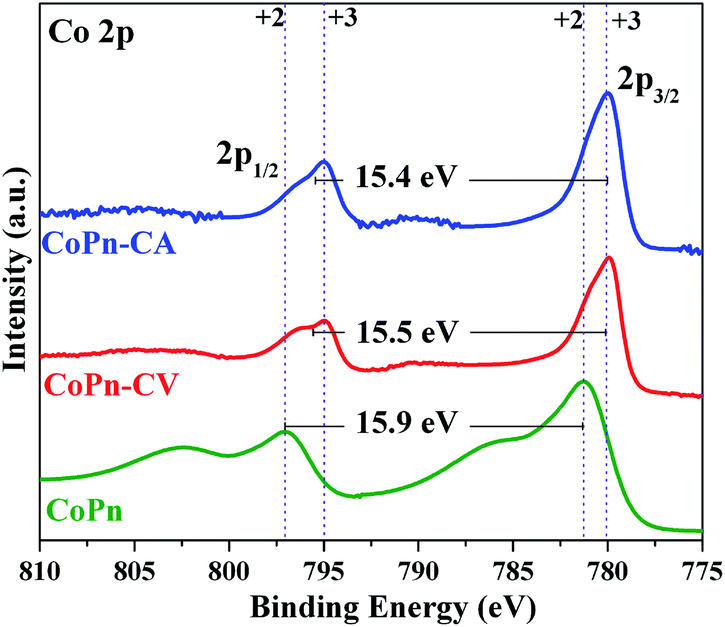 | ||
| Fig. 4 Comparison of the Co 2p XPS spectra in pristine form and after OER-CV and OER-CA. As the binding energies of CoII and CoIII sites are not unambiguous, they can be further differentiated by the Co 2p1/2–2p3/2 spin–orbit level energy spacing. The difference of 16.0 eV represents high-spin CoII whereas 15.0 eV comprises low-spin CoIII. The oxidation of CoII to CoII during OER is reflected in the decreased spin–orbit coupling values. | ||
Similarly, the catalyst at the cathode was also characterized after HER CV and CA by XPS. The Co 2p1/2–2p3/2 spin–orbit coupling value was calculated to be 15.5 eV after HER CV and 15.4 eV after CA, showing the formation of mixed valent CoII–CoIII species (Fig. S16†).36 The structural transformation of CoPn during HER was further confirmed from the O 1s spectra where the P–O–Co bridge was not detected. Instead, a new peak appeared at 529.5 eV for Co–O bonds (Fig. S17†).36 Like in the case of OER, a significant amount of hydration (532.5 eV and 533.6 eV) of the catalyst was detected after HER.14,24,31 The signal from P was also absent after HER confirming severe structural transformation at the cathode (Fig. S18†).24 From XPS, it was clear that CoPn acts as the precatalyst and completely transformed into a combination of distinct active structures at the cathode and anode during electrochemical water splitting. Moreover, the transformation occurred within the first CV measurement where a loss of over 90% phosphorous was attained, which was confirmed by the ICP-AES studies.
To have a more conclusive understanding of the structural transformation of CoPn during water oxidation with varying potential (CV) and at a constant potential (CA) quasi in situ Co K edge, X-ray absorption near edge spectroscopic (XANES) studies were carried out (Fig. 5). The XANES edge shape clearly revealed the structure of as-prepared CoPn remains intact even after EPD on the electrode, which is also consistent with the EXAFS spectra (Fig. S19 and Table S3†). After cycling (CV) and at CA relevant to OER, pronounced structural transformations were attained. The transformation was fast, and the structure of the catalyst after CV or CA at OER potentials was virtually the same (Fig. 4). The edge shape after OER resembles the one from amorphous layered oxides (CoCat) electrodeposited by Nocera's group (Fig. S20†).27,52 This converted structure can be derived from the LiCoO2 crystal structure, but in the present case, as well as in the catalysts from the group of Nocera, the edge was rather smooth and non-structured, a clear indication of the disordered structure of the catalyst. The simulation of the edge shape as a linear combination of spectra was difficult due to the amorphous character of the CoCat (lack of a well-defined crystal structure) and due to the known redox-activity of the Co ions in the structure, which does affect the edge position (the edge position will depend on the history of the applied potential).
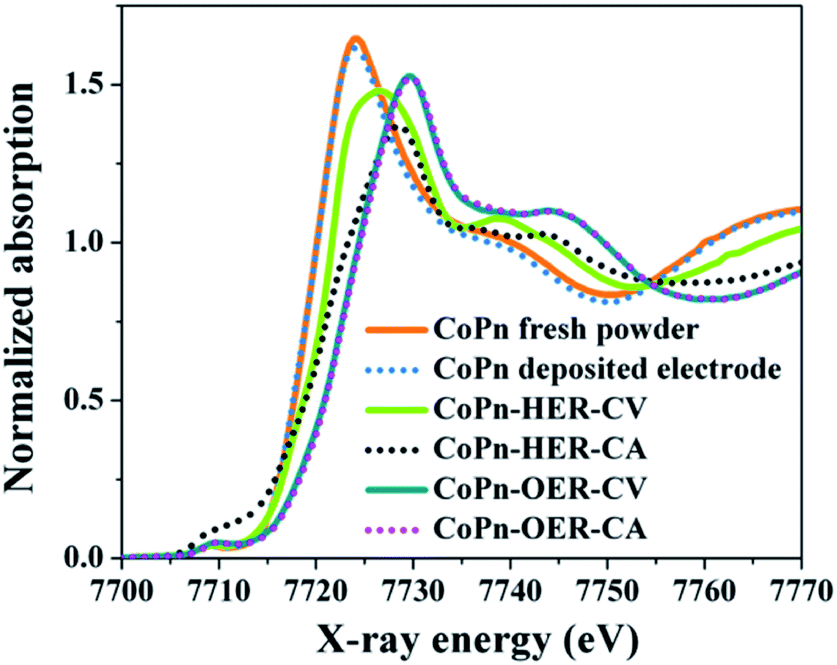 | ||
| Fig. 5 In situ XANES Co K-edge spectra are showing the change in the oxidation state of Co from fresh CoPn during alkaline OER and HER at a constant potential (CA) as well as varying potential (CV). | ||
To draw a further conclusion on the structure of the catalyst at OER potentials, we performed EXAFS simulations (Fig. 6 and S21†). First, we used an unbiased simulation approach to identify possible structures besides the layered CoCat of Nocera type. Evidence for the presence of other structures was ascertained by the appearance of the shoulder at the right side of the second FT peak (Fig. 6). This first simulation approach (Table S4†) indicated the possible presence of a spinel structure in the catalyst. To determine the percentage of the spinel structure, we repeated the simulation assuming a combination of ideal, crystalline spinel and ideal layered Co oxide (simulation approach 2 in Table S4†). The results suggested that there is about 20% of spinel structure in the OER catalysts.
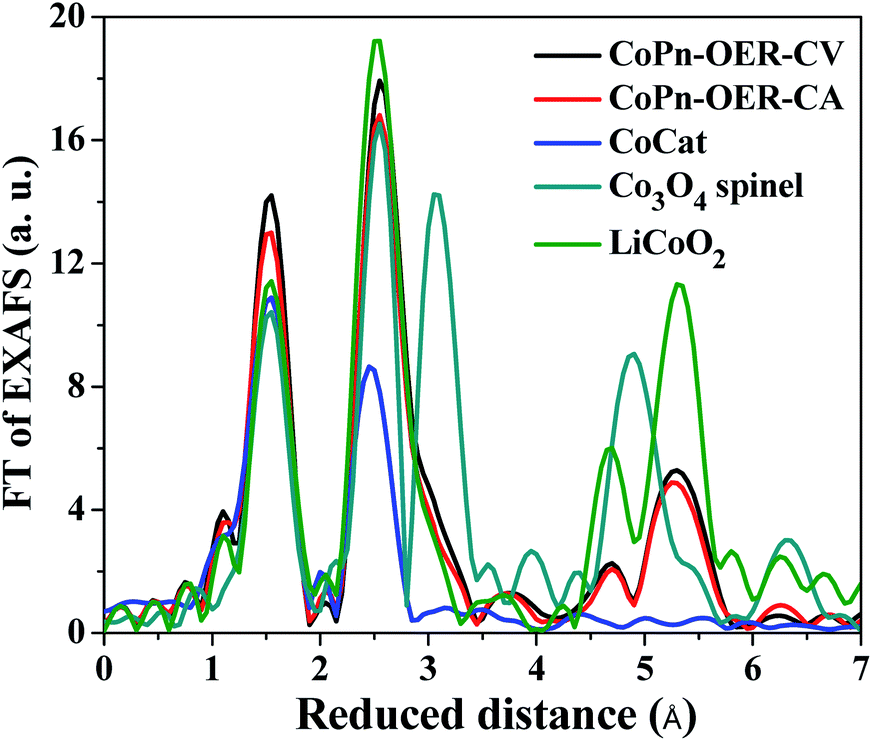 | ||
| Fig. 6 FT of EXAFS of CoPn exposed to OER potentials compared with other catalysts. Two approaches to simulations are shown in Table S4.† Please note that LiCoO2 is isostructural to CoOOH resulting in indistinguishable EXAFS spectra. | ||
In the case of HER, the edge shape significantly changed (Fig. 4) after both CV and CA. The transformation was much slower after CV, and the changes were more pronounced after CA. To understand how these changes related to structural transformation, we simulated the edge spectra after CV and CA as a linear combination of edge spectra of some reference compounds (Fig. S22†). The results show that in both cases, the starting compound is destroyed. The spectrum after CV (Fig. S22a†) can be modeled as a linear combination of 38% hydrated Co2+ ions and 62% Co spinel structure (Co3O4). The spectrum after CA can be described as a linear combination of 4% Co2+ ions, 91% Co spinel structure, and 5% contribution from a metallic Co (Fig. S22b†). In our previous report with cobalt borophosphates,36 we also detected the formation of Co0 during HER, where favorable adsorption of water and proton on cobalt surface as well as the facile desorption of the intermediates from the surface was ascribed to be the reason for the enhancement in HER activity.53,54 In fact, the local environment around the Co0 plays a major role in controlling the Gibbs free energy of adsorption. For example, in the Janus H2-Cat, Co0 species encapsulated in the Co(OH)2 shell provides the active sites for HER, while the N-doped carbon significantly lowers the adsorption free energy in Co@NC.35,53,54 Therefore, in the present case, we propose the participation of Co0 along with Co(OH)2/Co3O4 species are responsible for the efficient alkaline HER.
This edge-shape simulation, especially, in the case of the catalyst after CA is not perfect. The reason can be that the spinel structure in the resulting material is non-crystalline, e.g., the tetrahedral sites in the spinel structure are not 100% occupied and the number of mono-μ-oxo bridges between the Co atoms is lower than predicted. Additional insights into the structure of these materials can be obtained from the simulations of EXAFS spectra (Fig. S23†). In the simulation of the EXAFS spectrum of the catalyst after CV appears an additional Co–Co distance of 3.16 Å (Table S5†) which is not typical for the spinel structure. This distance suggests the presence of structural motifs of a third phase, for example, Co5(OH)4(POH)2 or Co(OH)2.
To verify the cooperative effect of different true active structures involved in OER and HER catalysis, we synthesized the individual components of active structure present in CoPn during OER (CoOOH, Co3O4, Co(OH)2) and HER (Co3O4, Co and Co(OH)2) and tested in a similar fashion to that of CoPn in 1 M aqueous KOH solution (see Experimental section). When compared, surprisingly, the CoPn exhibited much superior activity in both HER and OER, signifying the importance of the collective components of the active structures over a single constituent (Fig. S24 and S25†).
The excellent performance of CoPn as the bifunctional catalyst led us to construct a two-electrode system by employing NF supported CoPn as the anode and cathode. The overall water splitting reaches a current density of 10 mA cm−2 at only 1.62 V (Fig. S26†). Besides, excellent long-term stability of 25 h was established for CoPn/NF, representing it as one of the most competent catalytic systems for alkaline water electrolysis (Fig. S27†).
Conclusions
For the first time, we have demonstrated that cobalt phosphonate is a new class of highly active pre-catalysts for overall water splitting in alkaline media. Remarkably, it acts as a dual precursor for OER and HER: (i) under OER conditions, pristine cobalt phosphonate is transformed into layered Co oxides (CoCat) similar to crystalline CoO(OH) but with a lower degree of order to facilitate OER, while (ii) under HER conditions, metallic Co is produced as active species for H2 production which is promoted by the presence of spinel Co3O4 and Co(OH)2. The bifunctionality of the (pre)catalyst was further explored for overall water splitting that showed a very low cell voltage with high durability. Our investigation revealed new insights into the evolution and complex nature of interface structures to achieve highly efficient and durable overall water-splitting electrocatalysts, originating from the same metal-phosphorus-containing single-source precursor.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 – 390540038. Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder – EXC 2008/1 – 390540038. A. I. acknowledges the financial support from CSIR [grant no. 01(2977)/19/EMR-II], India. The authors are indebted to Dr Vitaly Gutkin for XPS measurements.Notes and references
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- C. Panda, P. W. Menezes and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 11130–11139 CrossRef CAS PubMed.
- F. Yu, L. Yu, I. K. Mishra, Y. Yu, Z. F. Ren and H. Q. Zhou, Materials Today Physics, 2018, 7, 121–138 CrossRef.
- (a) M.-I. Jamesh, Y. Kuang and X. Sun, ChemCatChem, 2019, 11, 1550–1575 CrossRef CAS; (b) Y. Zhu, H.-C. Chen, C.-S. Hsu, T.-S. Lin, C.-J. Chang, S.-C. Chang, L.-D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS.
- C. Panda, P. W. Menezes, M. Zheng, S. Orthmann and M. Driess, ACS Energy Lett., 2019, 4, 747–754 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- H. M. Xu, S. Q. Ci, Y. C. Ding, G. X. Wang and Z. H. Wen, J. Mater. Chem. A, 2019, 7, 8006–8029 RSC.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. T. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- B. You and Y. J. Sun, Acc. Chem. Res., 2018, 1, 1571–1580 CrossRef PubMed.
- F. L. Lyu, Q. F. Wang, S. M. Choi and Y. D. Yin, Small, 2019, 15, 1804201 CrossRef PubMed.
- K. N. Dinh, Q. H. Liang, C. F. Du, J. Zhao, A. L. Y. Tok, H. Mao and Q. Y. Yan, Nano Today, 2019, 25, 99–121 CrossRef CAS.
- X. H. Deng and H. Tuysuz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araujo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS.
- H. H. Zhong, C. A. Campos-Roldan, Y. Zhao, S. W. Zhang, Y. J. Feng and N. Alonso-Vante, Catalysts, 2018, 8, 559 CrossRef.
- P. W. Menezes, A. Indra, D. Gonzalez-Flores, N. R. Sahraie, I. Zaharieva, M. Schwarze, P. Strasser, H. Dau and M. Driess, ACS Catal., 2015, 5, 2017–2027 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109–114 RSC.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- D. Gonzalez-Flores, I. Sanchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Angew. Chem., Int. Ed., 2015, 54, 2472–2476 CrossRef CAS PubMed.
- Y. Y. Liu, H. T. Wang, D. C. Lin, C. Liu, P. C. Hsu, W. Liu, W. Chen and Y. Cui, Energy Environ. Sci., 2015, 8, 1719–1724 RSC.
- J. Wang and H. C. Zengg, ACS Appl. Mater. Interfaces, 2018, 10, 6288–6298 CrossRef CAS PubMed.
- P. W. Menezes, C. Panda, C. Walter, M. Schwarze and M. Driess, Adv. Funct. Mater., 2019, 1808632 CrossRef.
- F. Song, L. C. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. L. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- P. W. Menezes, A. Indra, V. Gutkin and M. Driess, Chem. Commun., 2017, 53, 8018–8021 RSC.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Energy Environ. Sci., 2015, 8, 661–674 RSC.
- H. Kim, J. Park, I. Park, K. Jin, S. E. Jerng, S. H. Kim, K. T. Nam and K. Kang, Nat. Commun., 2015, 6, 8253 CrossRef CAS PubMed.
- L. Su, X. Z. Cui, T. He, L. M. Zeng, H. Tian, Y. L. Song, K. Qi and B. Y. Xia, Chem. Sci., 2019, 10, 2019–2024 RSC.
- G. Q. Zhao, K. Rui, S. X. Dou and W. P. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef.
- P. W. Menezes, C. Panda, S. Garai, C. Walter, A. Guiet and M. Driess, Angew. Chem., Int. Ed., 2018, 57, 15237–15242 CrossRef CAS PubMed.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS.
- Z. F. Dai, H. B. Geng, J. Wang, Y. B. Luo, B. Li, Y. Zong, J. Yang, Y. Y. Guo, Y. Zheng, X. Wang and Q. Y. Yan, ACS Nano, 2017, 11, 11031–11040 CrossRef CAS PubMed.
- M. Caban-Acevedo, M. L. Stone, J. R. Schmidt, J. G. Thomas, Q. Ding, H. C. Chang, M. L. Tsai, J. H. He and S. Jin, Nat. Mater., 2015, 14, 1245–1251 CrossRef CAS.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS.
- P. W. Menezes, A. Indra, I. Zaharieva, C. Walter, S. Loos, S. Hoffmann, R. Schlogl, H. Dau and M. Driess, Energy Environ. Sci., 2019, 12, 988–999 RSC.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS.
- T. H. Zhou, D. P. Wang, S. C. K. Goh, J. D. Hong, J. Y. Han, J. G. Mao and R. Xu, Energy Environ. Sci., 2015, 8, 526–534 RSC.
- D. Shevchenko, M. F. Anderlund, A. Thapper and S. Styring, Energy Environ. Sci., 2011, 4, 1284–1287 RSC.
- R. Zhang, P. A. Russo, A. G. Buzanich, T. Jeon and N. Pinna, Adv. Funct. Mater., 2017, 27, 1703158 CrossRef.
- T. H. Zhou, Y. H. Du, D. P. Wang, S. M. Yin, W. G. Tu, Z. Chen, A. Borgna and R. Xu, ACS Catal., 2017, 7, 6000–6007 CrossRef CAS.
- Z. S. Cai, Y. Shi, S. S. Bao, Y. Shen, X. H. Xia and L. M. Zheng, ACS Catal., 2018, 8, 3895–3902 CrossRef CAS.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, Nanoscale, 2014, 6, 11395–11402 RSC.
- N. K. Chaudhari, H. Jin, B. Kim and K. Lee, Nanoscale, 2017, 9, 12231–12247 RSC.
- F. Lu, M. Zhou, Y. X. Zhou and X. H. Zeng, Small, 2017, 13, 1701931 CrossRef.
- Y. Wang, B. Kong, D. Y. Zhao, H. T. Wang and C. Selomulya, Nano Today, 2017, 15, 26–55 CrossRef CAS.
- Q. Kang, L. Vernisse, R. C. Remsing, A. C. Thenuwara, S. L. Shumlas, I. G. McKendry, M. L. Klein, E. Borguet, M. J. Zdilla and D. R. Strongin, J. Am. Chem. Soc., 2017, 139, 1863–1870 CrossRef CAS PubMed.
- C. C. L. McCrory, S. H. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- L. Yu, H. Q. Zhou, J. Y. Sun, F. Qin, F. Yu, J. M. Bao, Y. Yu, S. Chen and Z. F. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- A. Indra, P. W. Menezes, C. Das, D. Schmeisser and M. Driess, Chem. Commun., 2017, 53, 8641–8644 RSC.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 16501–16509 CrossRef CAS PubMed.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132, 13692–13701 CrossRef CAS PubMed.
- H. Guo, Q. Feng, J. Zhu, J. Xu, Q. Li, S. Liu, K. Xu, C. Zhang and T. Liu, J. Mater. Chem. A, 2019, 7, 3664–3672 RSC.
- H. Fei, Y. Yang, Z. Peng, G. Ruan, Q. Zhong, L. Li, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the catalysts by spectroscopic, microscopic, PXRD and analytical techniques along with the details of the experimental and electrochemical techniques. See DOI: 10.1039/c9ta09775a |
| This journal is © The Royal Society of Chemistry 2020 |