
A novel single-atom catalyst for CO oxidation in humid environmental conditions: Ni-embedded divacancy graphene†
Quanguo
Jiang
 a,
Jianfeng
Zhang
*a,
Huajie
Huang
a,
Yuping
Wu
a and
Zhimin
Ao
*b
a,
Jianfeng
Zhang
*a,
Huajie
Huang
a,
Yuping
Wu
a and
Zhimin
Ao
*b
aCollege of Mechanics and Materials, Hohai University, Nanjing 210098, China. E-mail: jfzhang@hhu.edu.cn
bGuangzhou Key Laboratory Environmental Catalysis and Pollution Control, Guangdong Key Laboratory of Environmental Catalysis and Health Risk Control, Institute of Environmental Health and Pollution Control, School of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou, 510006, China. E-mail: zhimin.ao@gdut.edu.cn
First published on 7th November 2019
Abstract
The degradation of catalysts for CO oxidation in humid air is a common issue owing to the blocking of the active site by the adsorption and dissociation of water molecules. In order to evaluate the effect of humidity on the CO oxidation, the adsorption and dissociation of the common gas molecules in air, namely CO, O2, H2O, and N2, on single Ni-embedded divacancy graphene (Ni-DG) have been investigated using first-principles calculations. It was found that all the molecules keep their molecular state and the adsorption energy of CO is much larger than those of the other molecules. In addition, the CO molecules can sufficiently substitute the pre-adsorbed O2, H2O, and N2 molecules with small energy barriers, indicating that the active site of the Ni-DG will not be blocked by water molecules in humid environments. At most two CO molecules can be chemically adsorbed on the Ni-DG, indicating that a new termolecular Eley–Rideal (TER) mechanism is preferred, and the energy barrier for the rate-limiting step (2CO + O2 → OCOOCO) is only 0.34 eV. Hirshfeld charge analysis shows that the charge transfer from the O2-2π* orbital to the CO-2π* orbital plays an important role in the CO oxidation via the TER mechanism. Overall, our results show that the low-cost Ni-DG is an efficient catalyst for CO oxidation, even in humid air at low temperature.
1. Introduction
With environmental pollution getting worse, the transformation and elimination of waste gases has become particularly important.1 Among these waste gases, CO is very harmful to human health, even in small quantities, thus CO conversion plays an important role in solving the environmental issues, where the most common method is the oxidation of CO into CO2.1–3 Some noble metals, such as Au,4 Pt,5,6 Pd7 and Rh,8 have been studied as catalysts for CO oxidation. However, these noble metal catalysts are costly and usually require high reaction temperatures for efficient operation owing to the high energy barriers, which is energy-consuming and may trigger explosions due to the high temperatures. Therefore, reducing or replacing the use of noble metals in the catalysts is needed. Non-noble metal oxide catalysts, such as Co3O4, show high catalytic activity at low temperature.9–11 However, the Co3O4 surface is very sensitive to the reaction conditions and water concentration,9,10 where the performance towards CO oxidation may severely deteriorate in the presence of trace amounts of moisture owing to the blocking of active sites by the adsorption and dissociation of water molecules. As a result, developing efficient non-noble metal catalysts for CO oxidation in humid air is desirable.In recent decades, carbon nanomaterials (such as fullerene, carbon nanotubes, graphene and graphdiyne) have been studied as either catalyst supports or catalysts for various applications. As a novel form of carbon, graphene12 is a promising matrix to support metal atoms to realize new catalysts owing to its outstanding electrical,13 mechanical14 and thermal properties.15 Moreover, the large surface-to-volume ratio also benefits the use of graphene as a support for heterogeneous catalysts. Decorating metal atoms into graphene is usually used to enhance the interactions between the inert graphene surface and gas molecules. Theoretically, single metal atoms supported on graphene with a single carbon vacancy (SV), such as Au-,16 Fe-,17 Cu-,18 Pt-,19 Al-,20 Co-,21 Ni,22,23 and Pd-embedded24 SV graphenes, exhibit high reactivity toward CO oxidation. The energy barriers for the CO oxidation reaction on the above catalysts can be reduced to 0.31–0.58 eV compared with the metal catalysts. In addition, single metal atoms supported on graphene with double carbon vacancies (DV), such as Mn-,25 and Co-embedded26 DV graphenes, also exhibit high reactivity toward CO oxidation. However, water vapor is inevitable in practical applications and the catalysts always deactivate rapidly in the presence of a small amount of water vapor, which was not considered in the previous DFT studies.16–26 On the other hand, recent experimental studies have revealed that oxide supports, such as FeOx,27 MgO,28 and TiO2,29 or graphitic layers30–32 can anchor various single metal atoms and hence allow the synthesis of single-atom catalysts (SAC).
In general, CO oxidation on catalysts may occur via either the Eley–Rideal (ER) mechanism or the Langmuir–Hinshelwood (LH) mechanism.16–26 In the ER mechanism, the gas-phase CO molecule directly reacts with the pre-adsorbed and activated O2 to form a carbonate-like intermediate (CO3) or the final product of CO2. The LH mechanism involves the co-adsorption of O2 and CO molecules, the formation and dissociation of a peroxide-like (OCOO) complex intermediate, and finally desorption of CO2. The LH mechanism is preferred for CO oxidation on most metal-decorated graphenes because the adsorption energy of an O2 molecule is larger than that of a CO molecule and the co-adsorption energy of CO and O2 molecules on these metal-embedded graphenes is larger than the individual adsorption energy of both CO and O2 molecules. More interestingly, a recently reported termolecular Eley–Rideal (TER) mechanism with the O2 molecule being directly activated by the two pre-adsorbed CO molecules has been identified to act as a possible or even preferable mechanism for CO oxidation.33–36
Nickel has Earth-abundant reserves and is environmentally benign. In addition, Ni-decorated single vacancy graphene has shown good performance for CO oxidation with a rate-limiting energy barrier of 0.59 eV (ref. 22) (0.63 eV (ref. 23)). Note that the single carbon vacancies have a low migration barrier (about 1.3 eV),37,38 which means that the single vacancies can merge into divacancies. On the other hand, the carbon divacancies need to overcome a larger energy barrier (about 7 eV) to migrate,37,38 which makes the divacancies much more stable than the single vacancies, thus Ni-decorated divacancy graphene can be synthesized more easily. Generally, the adsorption energy of O2 is larger than that of CO, where the O2 molecule has the priority to adsorb on the active site of the single-atom catalyst.17–26 If the adsorption energy of the CO molecule is larger than that of the O2 molecule, the catalyst will easily capture the CO molecule and then promote the CO oxidation process. Fortunately, the adsorption energy of CO is larger than that of O2 on Ni-DG, as reported in previous literature.25,26 However, further study of CO oxidation on Ni-DG is lacking.
In this work, we will study the reaction mechanism of CO oxidation on Ni-embedded graphene via the TER mechanism through first-principles calculations. The effects of H2O and N2 molecules on the energy barriers for the reaction are also discussed and the corresponding mechanisms are analyzed through understanding the charge transfer and electronic orbitals.
2. Calculation methods
All density functional theory (DFT) calculations with spin-unrestricted in this work are done by using the Dmol3 module.39 Generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)40 functional has been adopted as the exchange–correlation functional. To take into consideration the effect of van der Waals forces, the Grimme scheme41 for DFT-D correction is used in all the calculations. DFT semicore pseudopotentials (DSPPs) core treatment is implemented for relativistic effects, which replaces core electrons with a single effective potential. The convergence tolerance of energy of 10−5 Hartree is taken (1 Hartree = 27.21 eV), and the maximal allowed force and displacement are 0.002 Hartree Å−1 and 0.005 Å, respectively. It was reported that the selected exchange–correlation functional has an evidential effect on the adsorption energy results. However, the effect on the calculated reaction energy barriers is much smaller.42 To investigate the minimum energy pathway for CO oxidation on graphene, the linear synchronous transit/quadratic synchronous transit (LST/QST)43 and nudged elastic band (NEB)44 tools in the Dmol3 module are used, which have been well validated to determine the structure of the transition state and the minimum energy reaction pathway. In the simulation, three-dimensional periodic boundary conditions are taken. The simulation cell consists of a 4 × 4 graphene supercell with a vacuum width of 20 Å above the graphene layer to minimize the interlayer interaction. The k-point is set to 5 × 5 × 1, and all atoms are allowed to relax according to our convergence test. After structure relaxations, the density of states (DOS) is calculated with a finer k-point grid of 15 × 15 × 1. The electron orbits of the molecule/graphene systems are calculated with CASTEP code,45 where the ultrasoft pseudopotentials, the GGA-PBE functional, an energy cutoff of 340 eV and 5 × 5 × 1 k-point meshes are used.For the molecules (CO or O2) adsorbed on graphene, the adsorption energy Ead is determined by:
| Ead = Emolecule/graphene − (Egraphene + Emolecule) | (1) |
3. Results and discussion
Based on our previous reports,25 the adsorption energies of CO and O2 molecules on divacancy graphene embedded with transition metals (from Sc to Zn) are systemically studied, where the adsorption energies of CO and O2 on Ni-DG are −1.21 eV and −0.41 eV, respectively. These moderate adsorption energies indicate that the Ni-decorated divacancy graphene (Ni-DG) may possess good catalytic properties for CO oxidation. To carefully find the most stable structure for decorating Ni atom on the divacancy graphene, the possible adsorption sites for Ni on DG are studied, as shown in Fig. S1.† Based on the DFT calculations, the Ni atom at the T1 site simultaneously diffused to the B1 site (see Fig. S2a†), while the Ni atom on the T2, T3, T4, B2, B3, B4, B5, and Hol2 sites simultaneously diffused to the Hol1 site (see Fig. S2b†) after geometry optimization. Finally, the Ni atom can adsorb on the B1, Hol1, Hol3 and Hol4 sites with adsorption energies of −1.96 eV, −7.93 eV, −2.42 eV, and −2.43 eV, respectively (see Fig. S2†). Therefore, the configuration with the Ni atom adsorbed on the Hol1 site has the largest adsorption energy. To check the mobility of the adsorbed Ni atom at different sites (B1, Hol1, Hol3 and Hol4) on the divacancy graphene, the diffusion pathways of the Ni atom from the B1, Hol3 and Hol4 sites to the Hol1 site are also shown in Fig. S2,† where the diffusion barriers are 0.21 eV, 0.20 eV and 0.22 eV, respectively, which further confirms that the divacancy graphene with the Ni atom adsorbed at Hol1 is the most stable configuration, as shown in Fig. 1a.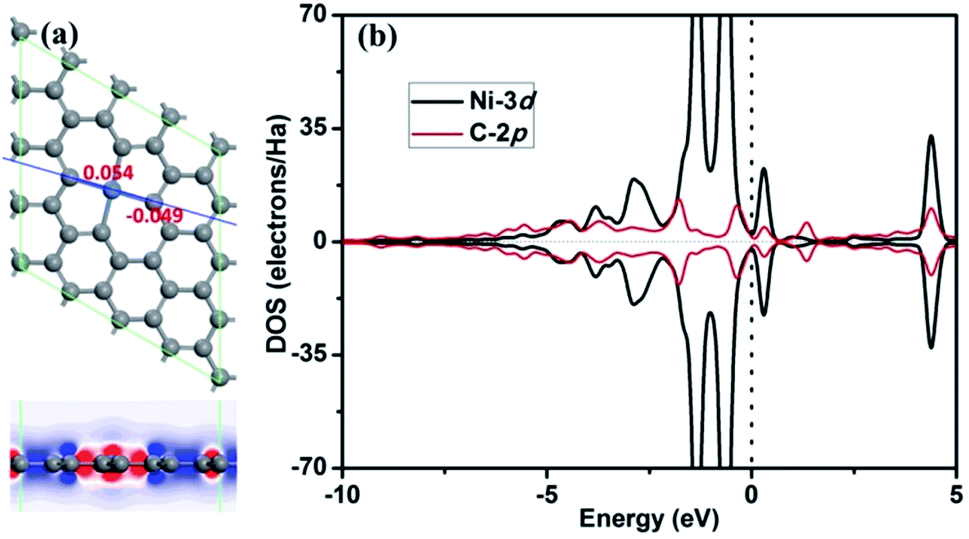 | ||
| Fig. 1 (a) The most stable structure of Ni-embedded divacancy graphene, where the grey and blue atoms are C and Ni atoms in this and the following figures. The atomic charge obtained by Hirshfeld analysis near the dopant is also given. The differential charge density along C–Ni–C bonds for Ni-DG is shown, where the blue and red isosurfaces correspond to the increase in the number of electrons and the depletion zone, respectively. (b) PDOS of Ni atom and C atom on the Ni-embedded divacancy graphene. The vertical line indicates the Fermi level. | ||
The electronic and atomic structures for Ni-DG are first studied in the following. After embedding Ni atom into the divacancy graphene, where two C atoms are substituted by one Ni atom, the structure of graphene is reconstructed and the relaxed structure is shown in Fig. 1a. Single chemical Ni–C bonds are formed in graphene by calculating the Mayer bond order of 0.838. The Ni atom is in the same plane with the C atoms, which is different from single vacancy graphene where the Ni atom moves out of the plane to get more space due to its relatively large atomic radius.22,23 The C–Ni bond length lC–Ni is 1.89 Å, which is in good agreement with the reported result of 1.86 Å.26 The binding energy of the Ni atom in graphene is −7.93 eV, which is larger than that of −7.43 eV (−6.98 eV (ref. 22) or −7.57 eV (ref. 23)) for the Ni atom in single vacancy graphene. The atomic charges obtained by the Hirshfeld method near the dopant are also given in the atomic configuration in Fig. 1a, where the Ni atom forms an electron-depletion position by losing electrons (0.054e) from graphene. The strong interaction between the Ni atom and the C atoms can be further confirmed by the partial density of states (PDOS) (Fig. 1b), where significant overlap of the bands between the embedded Ni atom and the nearby C atom is found. In addition, the differential charge density along Ni–C bonds for Ni-G is shown in Fig. 1a, where the blue and red isosurfaces correspond to the increase in the number of electrons and the depletion zone, respectively. It shows that electrons are depleted near the doped Ni atom, indicating the high chemical active area. The remaining unsaturated d orbital of the Ni atom is reactive, which can adsorb small molecules and promote the subsequent reactions.
The aggregation problems for the adsorbed metal atoms on the substrate are significant for the catalytic performance, especially when the concentration of metal atoms is high.20,25 To determine the possibility of aggregation for Ni atoms on divacancy graphene, the diffusion pathway of the Ni atom to its neighbouring positions is investigated based on DFT calculations (see Fig. 2a), where the corresponding diffusion energy barrier for the decorated Ni atom is 6.23 eV. It is claimed that a surface reaction will occur when the reaction barrier is smaller than the critical value of Ecbar = 0.75 eV,46 thus the Ni-decorated divacancy graphene is quite stable without aggregation problems. The phonon dispersion of Ni-DG has been calculated as shown in Fig. 2b, where there are no soft modes for the Ni-DG, indicating that the studied structure is stable at room temperature. In addition, the adsorption energy of the Ni atom on divacancy graphene is −7.93 eV. Such strong binding indicates that Ni-DG is rather stable and the Ni atom cannot be removed thermally or by irradiation with sub-MeV electrons, similar to W-DG (W-doped divacancy graphene).47,48
 | ||
| Fig. 2 (a) The pathway for the diffusion of the embedded Ni atom on divacancy graphene, where IS, TS and FS represent the initial, transition and final structures, respectively, in this and the following figures. (b) The phonon structure of Ni-embedded divacancy graphene. | ||
O2, CO, H2O, and N2 are the common gas molecules in humid atmospheres, which may affect the oxidation reaction of CO on the supported single-atom catalyst. Before studying the CO oxidation reaction, the adsorption properties of the above gas molecules on Ni-DG are first discussed. In addition, the performance of CO oxidation catalysts usually falls in moist environments owing to the dissociation of water molecule and its subsequent reactions with the catalyst.49 Therefore, the interactions between the mentioned molecules and the Ni-DG are important, which will determine the final performance of the catalyst and are not considered in the previous literature using the DFT method.16–26 The most stable configurations for the adsorption of O2, CO, H2O and N2 on Ni-DG are shown in Fig. 3 (see the IS structure). It can be seen that the O2, CO and H2O molecules are chemically adsorbed on the Ni-DG, while the N2 molecule is physically adsorbed on the Ni-DG. The adsorption energies on Ni-DG in Table 1, where the corresponding adsorption energies are −0.41, −1.24, −0.43 and −0.17 eV for O2, CO, H2O and N2, respectively. To evaluate the stability of Ni-DG after the adsorption of the molecules, the adsorption energy of the Ni–M species (M is the adsorbed molecules) on divacancy graphene is calculated to estimate the strength of the Ni–C bond as reported in previous literature,33 where the adsorption energies of Ni–CO, Ni–O2, Ni–H2O and Ni–N2 species on divacancy graphene are −6.57 eV, −7.10 eV, −7.55 eV and −7.91 eV, respectively, which are similar to that of Ni atom on divacancy graphene (−7.93 eV). Therefore, the Ni-DG is also thermally stable after adsorption of the above molecules. The adsorbed molecules may dissociate on the surface, as discussed in the previous literature.20,25,50,51 To assess the stability of the adsorbed molecules, the dissociative reactions of individual O2, CO, H2O and N2 molecules on Ni-DG are studied in Fig. 3.
 | ||
| Fig. 3 The pathway for the dissociation of O2 (a), CO (b), H2O (c), and N2 (d) molecules on the Ni-embedded graphene. IS represents the most stable structure for each adsorbed molecule on Ni-embedded divacancy graphene. | ||
| E ad (eV) | Q (e) | l ad (Å) | l free (Å) | |
|---|---|---|---|---|
| O2 | −0.41 | −0.066 | 1.26 | 1.22 |
| CO | −1.21 | 0.058 | 1.15 | 1.14 |
| H2O | −0.43 | 0.125 | 0.98 | 0.98 |
| N2 | −0.17 | 0.012 | 1.11 | 1.11 |
After NEB calculations, the dissociation reaction barrier for an O2 molecule on the Ni-DG is 1.83 eV > Ecbar = 0.75 eV,46 which indicates that the adsorbed O2 molecule prefers to stay on the Ni-DG in the molecular state at low temperature. The binding energy Eb of a C–O bond is 11.57 eV, which is much larger than Eb = 6.36 eV for an O–O bond based on DFT calculations.25 Thus CO should be more difficult to be dissociated, which is confirmed by the fact that the dissociative energy barrier for the CO molecule on Ni-DG is 4.02 eV, as shown in Fig. 3b. The dissociation barriers of H2O and N2 molecules on Ni-DG are 1.15 eV and 5.86 eV, as shown in Fig. 3c and d, which indicates that the adsorbed H2O and N2 molecules also prefer to stay on Ni-DG in the molecular state at low temperatures. Based on the above discussions, the catalytic performance of Ni-DG will not be degenerated by water molecules and nitrogen gas. The reaction mechanism for CO oxidation is then studied in the following.
The adsorption energy for a CO molecule on Ni-DG is much larger than those for O2, H2O, and N2 molecules, as shown in Table 1. We thus consider the possibility of the substituted adsorption of CO for the other pre-adsorbed molecules, as shown in Fig. 4. For the pre-adsorbed O2 molecule in Fig. 4a, the free CO molecule will adsorb on the Ni atom after overcoming a small barrier of 0.25 eV, and the pre-adsorbed O2 molecule will become physically adsorbed on the graphene surface, as shown in the FS structure in Fig. 4a. Therefore, the CO molecule will automatically adsorb on the Ni-embedded graphene even with the large amount of O2 molecules in the atmosphere. In the presence of moisture, the H2O molecule may adsorb on the active site and prevent the subsequent CO oxidation reactions. The substituted adsorption process of CO to H2O molecules is shown in Fig. 4b, where only a small energy barrier of 0.12 eV needs to be overcome and heat of −0.83 eV is released, indicating that the CO molecule will automatically adsorb on the Ni-DG in a humid environment. A similar reaction path for the substituted adsorption of CO for the N2 molecules is shown in Fig. 4c, where only a small energy barrier of 0.22 eV needs to be overcome. Therefore, the CO molecule will automatically adsorb on the Ni-DG in an ambient atmosphere with and without humidity. With sufficient CO molecules in the atmosphere, it is possible for two or more CO molecules to adsorb on the surface of the catalyst.
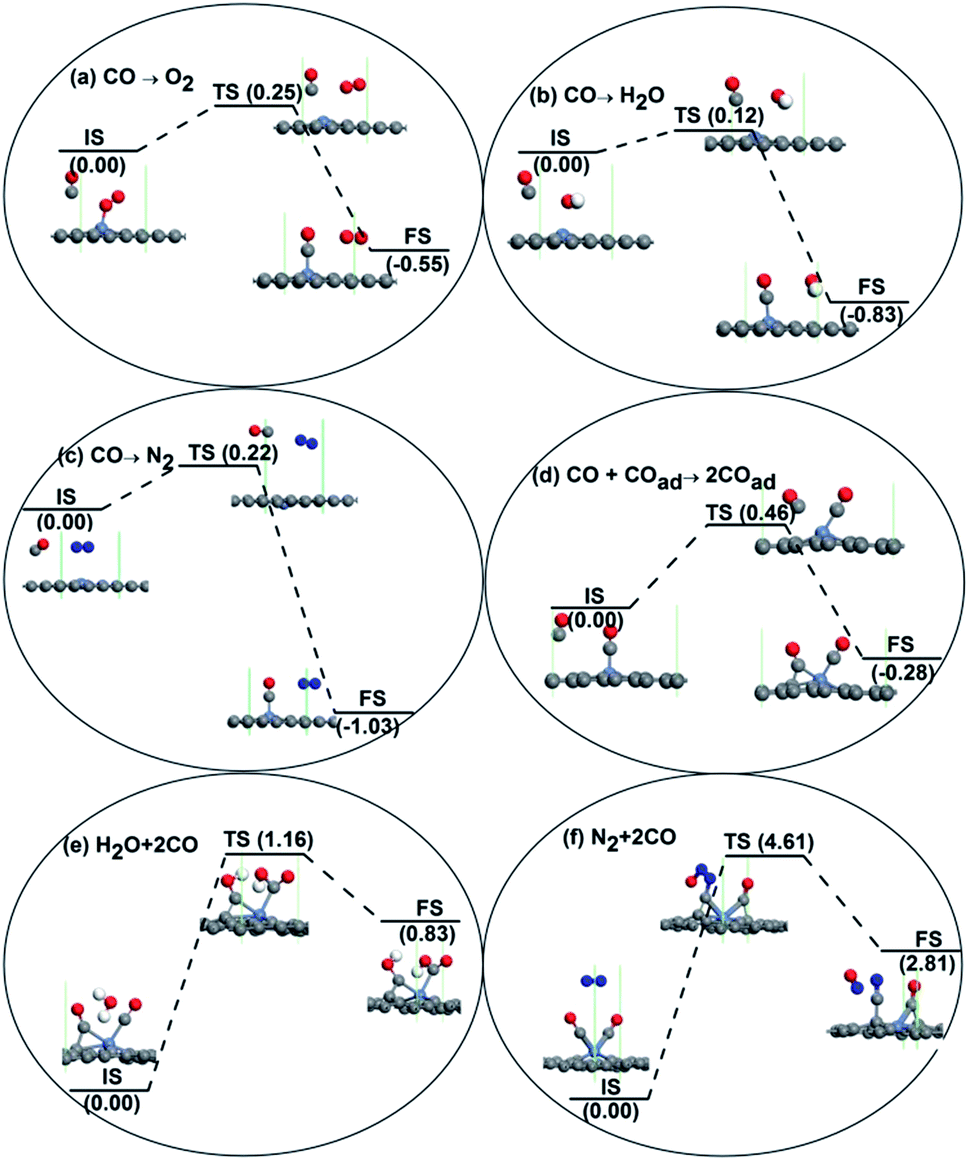 | ||
| Fig. 4 The reaction pathway for the substituted adsorption of CO on the Ni-embedded graphene with pre-adsorbed O2 (a), H2O (b), and N2 (c) molecules. (d) The reaction pathway for the co-adsorption of two CO molecules is also shown. The possible reaction between H2O and the two CO molecules is shown in (e), while the possible reaction between N2 and the two CO molecules is shown in (f). | ||
To check whether the O2 and H2O molecules will block the second CO adsorption, the co-adsorption of H2O and CO, as well as the co-adsorption of O2 and CO, is then studied. When the CO and O2 were manually put on the Ni atom with a co-adsorption configuration on Ni-DG, the O2 molecule simultaneously desorbed from the Ni atom after geometry optimization, leaving the CO molecule solely chemically adsorbed on the Ni atom. In the final adsorption configuration, the CO chemically adsorbs on the Ni atom, while the O2 molecule physically adsorbs on the graphene surface with a Ni–O distance of 4.755 Å, as shown in Fig. 4a. Similar to the O2 molecule, after geometry optimization, the manual co-adsorption configuration for the CO and H2O on the Ni atom will be broken with the H2O molecule simultaneously desorbing from the Ni atom. In the final adsorption configuration, the CO molecule chemically adsorbs on the Ni atom while the O2 molecule physically adsorbs on the graphene surface with a Ni–O distance of 4.146 Å, as shown in Fig. 4b. Therefore, the co-adsorption of O2 and CO, as well as H2O and CO, cannot occur, indicating that the O2 and H2O molecules will not block the adsorption of the second CO on Ni-DG.
The adsorption of two CO molecules on the Ni-DG is also studied and shown in Fig. 4d, where the free CO molecule will co-adsorb on Ni-DG after overcoming a small energy barrier of 0.46 eV. We note that the adsorption energies for the first CO and second CO molecules on Ni-DG are −1.21 eV and −0.39 eV, respectively, indicating that the adsorption for the second CO molecule is preferred in energy. In addition, the desorption energy barrier (the energy difference between FS and TS in Fig. 4d) for the second CO molecule is 0.74 eV, which is nearly the same as the critical barrier of 0.75 eV;46 thus the desorption of the second CO molecule is difficult at low temperature. The adsorption energy for a third CO molecule on Ni-DG is 0.75 eV, which is an endothermic process, indicating that the Ni-DG can adsorb only two CO molecules at most in the 4 × 4 supercell. Based on the above discussions, two CO molecules will adsorb on the Ni-embedded divacancy graphene in the presence of CO molecules in the atmosphere, which is the initial state for the CO oxidation on Ni-DG catalyst. The adsorbed CO molecules are more reactive than the free CO owing to the electron transfer from the graphene substrate. Thus we further consider the possibility for the reaction between the H2O or N2 molecules in the atmosphere and the two adsorbed CO molecules on Ni-DG, and the detailed reaction path is shown in Fig. 4e and f. The reaction barriers are 1.16 eV and 4.61 eV for H2O and N2 molecules, respectively, which are much larger than the critical barrier of 0.75 eV.45 This result confirms that the H2O or N2 molecules in atmosphere will not react with the two adsorbed CO molecules on Ni-DG and the subsequent CO oxidation can proceed smoothly.
Before studying the CO oxidation mechanism on Ni-DG, the electronic structures of the adsorbed O2 and CO on Ni-DG are studied. Fig. 5a shows the most stable configuration for O2 molecule adsorbed on Ni-DG, where the angle between the O–O bond and the graphene sheet is about 45°, and 0.066e is transferred from the Ni-DG to the O2 molecule based on Hirshfeld charge analysis. The 2π* anti-bond orbital of the free O2 molecule is half-filled, as discussed in previous literature.20,25,52 The PDOS of the Ni atom and the adsorbed O2 molecule is shown in Fig. 5b, where all orbitals of the adsorbed O2 molecule are also labelled to understand the interaction between the adsorbed O2 and Ni-DG. About 0.066e is transferred from Ni-DG to the adsorbed O2 molecule based on the Hirshfeld method, which occupies the O2-2π* orbital above the Fermi level and is confirmed by the new peaks for the O2-2π* orbital below the Fermi level (see Fig. 5b). This charge transfer elongates the O–O bond from 1.23 Å in free O2 to 1.26 Å in the adsorbed O2 molecule, which is much shorter than that of 1.43 Å and 1.40 Å on Al-G20 and Mn-DG,25 respectively. Note that the adsorption energies for O2 molecules are −0.41 eV, −1.57 eV (ref. 20) and −1.96 eV (ref. 25) on Ni-DG, Al-G, and Mn-DG, respectively. This result is generally consistent with the fact that the adsorbed O2 molecule on Ni-DG has the smallest adsorption energy and the O2 on Ni-DG is less activated with the shortest O–O bond length compared with that on Al-G and Mn-DG. The interaction between the O2 and the Ni atom is relatively weaker (see Fig. 5b) than that on Al-G20 and Mn-DG,25 and the PDOS of O2 on Ni-DG is similar to that of the free O2 molecule, further confirming the small adsorption energy for O2 on Ni-DG.
 | ||
| Fig. 5 The most stable structure of O2 (a) and CO (c) adsorbed on Ni-DG, where the differential charge density along O2 and CO on Ni-DG is shown. The blue and red isosurfaces correspond to the increase in the number of electrons and the depletion zone, respectively. PDOS of the adsorbed O2 (b) and CO (d) molecules and the Ni atom for the Ni-DG is also shown. The vertical lines indicate the Fermi level. | ||
The adsorption configuration of a CO molecule on Ni-DG is shown in Fig. 5c, where the CO molecule is vertically adsorbed on the top of the decorated Ni atom. CO is chemically adsorbed on Ni-DG, which is confirmed by the chemical bond between the Ni atom and the carbon atom of the CO molecule. The adsorption energy of a CO molecule on Ni-DG is Ead = −1.21 eV, and the CO molecule transfers 0.058e to the Ni-DG. Fig. 5d shows the PDOS of the adsorbed CO molecule on Ni-DG, where the orbitals of the adsorbed CO molecule are labelled and displayed. The 5σ peak of the CO molecule adsorbed on Ni-DG is significantly depressed compared to the free CO molecule owing to the charge transfer. Although the 2π* anti-bond orbital far above the Fermi level for the free CO molecule is fully empty, the Ni atom transfers some electrons to the CO-2π* orbital due to the fact that CO-2π* is close to the Fermi level in the absorbed state, which slightly elongates the C–O bond from 1.14 Å for the free CO molecule to 1.15 Å for the adsorbed CO molecule. The above discussions indicate that O2 and CO have chemical interactions with Ni-DG (corresponding adsorption energies are −0.41 and −1.21 eV, respectively), but the adsorption of CO is much stronger. The CO molecule is more activated on Ni-DG, which is different from how the O2 molecule is usually more activated on metal-decorated graphene.20,25 The Ni-DG may be CO poisoned, as discussed in the previous literature.20,25 However, the CO oxidation along a new TER mechanism may occur and will be discussed in the following.
Two classical reaction mechanisms have been established for the oxidation of the CO molecule: the Langmuir–Hinshelwood (LH) mechanism and the Eley–Rideal (ER) mechanism.16–26 For the ER mechanism, the O2 molecule is first adsorbed and activated by the catalyst, then a free CO molecule approaches the substrate to form an intermediate product. For the LH mechanism, the O2 and CO molecules should first co-adsorb on the Ni atom. However, as discussed above, when the CO and O2 were manually put on the Ni atom with a co-adsorption configuration on Ni-DG, the O2 molecule simultaneously desorbs from the Ni atom after geometry optimization in the software, leaving the CO molecule solely chemically adsorbed on Ni atom. In the final adsorption configuration, the CO chemically adsorbs on the Ni atom, while the O2 molecule physically adsorbs on the graphene surface with a Ni–O distance of 4.755 Å, as shown in Fig. 4a. Therefore, the co-adsorption of O2 and CO cannot occur and CO oxidation via the LH mechanism is not further considered. The ER mechanism for the CO oxidation seems to be possible and is studied in the following.
The energy profile for the oxidation of the first CO molecule via the ER mechanism is shown in Fig. 6a. The configuration with physisorbed CO perpendicular to the graphene surface was selected as the initial state (IS) after considering all possible adsorption positions. When one CO molecule approaches the activated O2, after overcoming an energy barrier of 3.36 eV, the O–O bond is broken, the O with the dangling bond would form a covalent bond with the C atom in the CO molecule (see TS in Fig. 6a). Then the C atom of the CO binds with the free O atom and a CO2 molecule is formed over the Ni atom (see FS in Fig. 6a). This process is exothermic and releases energy of 4.90 eV. The subsequent CO molecule will react with the remaining O atom to produce the second CO2 molecule after surmounting an energy barrier of 0.07 eV (see Fig. 6b). The reaction profile for the production of the two CO2 molecules is shown in Fig. 6, where the rate-limiting step is the formation of the first CO2 molecule. Because the energy barrier of 3.36 eV for this step is much larger than Ecbar = 0.75 eV,46 the ER mechanism is not preferred for the CO oxidation on Ni-DG at room temperature. The micro-kinetics method is used to calculate the maximum reaction rate for the CO oxidation via different reaction pathways by applying the thermodynamic condition of T = 298 K, PCO = 0.01 bar, PO2 = 0.21 bar as applied in the previous literature53 (see the Calculation details in the ESI†). The maximum reaction rate is 9.60 × 10−60 s−1 for the ER pathway, and the formation of the first CO2 molecule (step 1) is the rate-determining step for the ER pathway based on Campbell's degree of rate control analysis.54 The maximum reaction rate is too small, which again confirms that the CO oxidation along the ER mechanism is difficult to happen at room temperature.
 | ||
| Fig. 6 The reaction pathway for the oxidation of the first CO molecule (a) and the second CO molecule (b) on the Ni-DG with a pre-adsorbed O2 molecule via the ER mechanism. | ||
The adsorption energy of the CO molecule is much larger than that of other molecules. It seems that the Ni-embedded divacancy graphene may be poisoned by the CO molecule owing to the larger adsorption energy for CO than that for O2, as discussed in previous literature.20,25 However, the reaction via a new termolecular ER (TER) mechanism may happen for this graphene system, where the two pre-adsorbed CO molecules will react with the free O2 molecule, which has not been reported for the Ni-DG in the literature. For oxidation of CO on the Ni-DG via the TER mechanism, several steps and also intermediate products (MS) for the oxidation procedure exist.33–36 For each step, e.g. from the initial state to the intermediate state in Fig. 7, a transition state also exists. The configuration of the two co-adsorbed CO molecules and the physically adsorbed O2 molecules on Ni-DG is taken as the reactant (IS in Fig. 7) based on the above discussions. After overcoming an energy barrier of 0.34 eV, an OCOOCO intermediate (MS in Fig. 7) is formed. Then the two CO2 molecules (FS) are formed on Ni-DG after overcoming an energy barrier of 0.29 eV (TS2 in Fig. 7), where the CO2 is physically adsorbed on Ni-DG with an adsorption energy of −0.45 eV. The O–O bond for the OOCOOO configuration in the MS is broken at TS2, and two CO2 molecules with a bond angle of 122.3° are formed. The reaction for this step can release a heat of 5.52 eV, which can sufficiently overcome the adsorption energy of CO2, and the produced CO2 molecules would desorb from the Ni-DG efficiently. The maximum reaction rate is 1.06 × 107 s−1 for the TER pathway by using the micro-kinetics method (see the Calculation details in the ESI†), and the formation of the OCOOCO intermediate (step 1) is the rate-determining step for the TER pathway based on Campbell's degree of rate control analysis.54 Therefore, the TER mechanism is more preferable than the ER mechanism, and the CO oxidation on Ni-DG via the TER mechanism has fast kinetics.
 | ||
| Fig. 7 The reaction pathway for the CO molecule on Ni-DG via the TER mechanism. The unit of E is eV, where E is the energy barrier. The Hirshfeld charge near the adsorbate is also shown. | ||
To comprehensively understand Ni-DG's superior catalytic performance towards CO oxidation, the difference between the single Ni atom on single vacancy graphene (Ni-SG) and that on divacancy graphene (Ni-DG) is then discussed. The binding energy of the Ni atom in Ni-DG is −7.93 eV, which is larger than that of −7.43 eV in Ni-SG (−6.98 eV (ref. 22) or −7.57 eV (ref. 23)), which indicates that the interaction between the Ni atom and the divacancy is stronger than that between the Ni atom and the single vacancy. It is reasonable owing to the fact that the Ni atom is four-coordinated in Ni-DG, while it is three-coordinated in Ni-SG, and the Ni atom saturates more dangling bonds of the divacancy in Ni-DG. Note that the adsorption energies for O2 and CO on Ni-SG are −1.50 eV and −0.68 eV, respectively, while the adsorption energies for O2 and CO on Ni-DG are −0.41 eV and −1.21 eV, respectively. Therefore, the Ni-DG can capture the CO molecules more easily that the Ni-SG. In addition, the energy barrier for the rate-limiting step is 0.59 eV (ref. 22) (0.63 eV (ref. 23)) on Ni-SG via the LH path, while the energy barrier for the rate-limiting step is 0.34 eV on Ni-DG via the TER path. Therefore, the Ni atom on divacancy graphene possesses better catalytic performance than that on single vacancy graphene, which provides guidance for tuning the catalytic properties through controlling the defects of the supporting substrate.
Hirshfeld charge and electronic orbital analyses are performed to further reveal the origin of the high efficiency for the CO oxidation on Ni-DG via the TER mechanism. The Hirshfeld charge for IS, TS and MS is shown in Fig. 7, where the atomic charge transfer of the O2 molecule is −0.129e, −0.098e, and −0.057e in the IS, TS and MS structures, respectively, indicating that the O2 molecule loses charge from the IS to the MS. While the atomic charge transfer of the two adsorbed CO molecules is 0.228e, 0.099e, and −0.118e in the IS, TS and MS structures, respectively, indicating that the CO molecules obtain charge from the IS to the MS. Therefore, the O2 molecule transfers electrons to the two adsorbed CO molecules from the IS structure to the MS structure during the reaction progress. The PDOS of the Ni atom and O2 and CO is shown in Fig. 8, where the HOMO and LUMO orbitals are also shown to understand the charge transfer from the O2 to the adsorbed CO molecules. Fig. 8a presents the HOMO and LUMO orbitals of the IS, where the HOMO level dominantly locates on the O2 molecule and the LUMO level mainly locates on the CO molecule and graphene, indicating that the adsorbed CO molecule allows the incoming electron to occupy this state upon the adsorption of the O2 molecules. Furthermore, the adsorption of O2 is weak with an adsorption energy of 0.25 eV in the IS, and rather strong in the MS with an adsorption energy of 1.13 eV, which indicates that the two carbon atoms will provide the anchor for the O2 molecule to form the OCONiOCO intermediate. Fig. 8b presents the HOMO and LUMO orbitals of the TS, where the HOMO level mainly locates on the O2 molecule while part of the HOMO level locates on the CO, indicating the charge transfer from the O2-HOMO to the CO-LUMO, which is confirmed by the occupation of the LUMO state on the O2 molecule in the upper panel of Fig. 8b. The HOMO and LUMO orbitals of the MS are shown in Fig. 8c, where the HOMO level is mainly located on the OC part, which confirms the charge transfer from the O2-HOMO to the CO-LUMO. For comparison purposes, Fig. 8d gives the orbitals of free CO and O2 molecules, where the energy difference between the O2-HOMO and the CO-LUMO is about 7.0 eV, which indicates that it is difficult for charge transfer from the O2-HOMO orbital to the CO-LUMO orbital to happen. When the two CO molecules are adsorbed on Ni-DG, the CO-LUMO orbital is significantly downshifted to the Fermi level, where the energy difference between the O2-HOMO orbital and the CO-LUMO orbital is about 1.0 eV, which can promote the charge transfer from the O2-HOMO orbital to the CO-LUMO orbital.
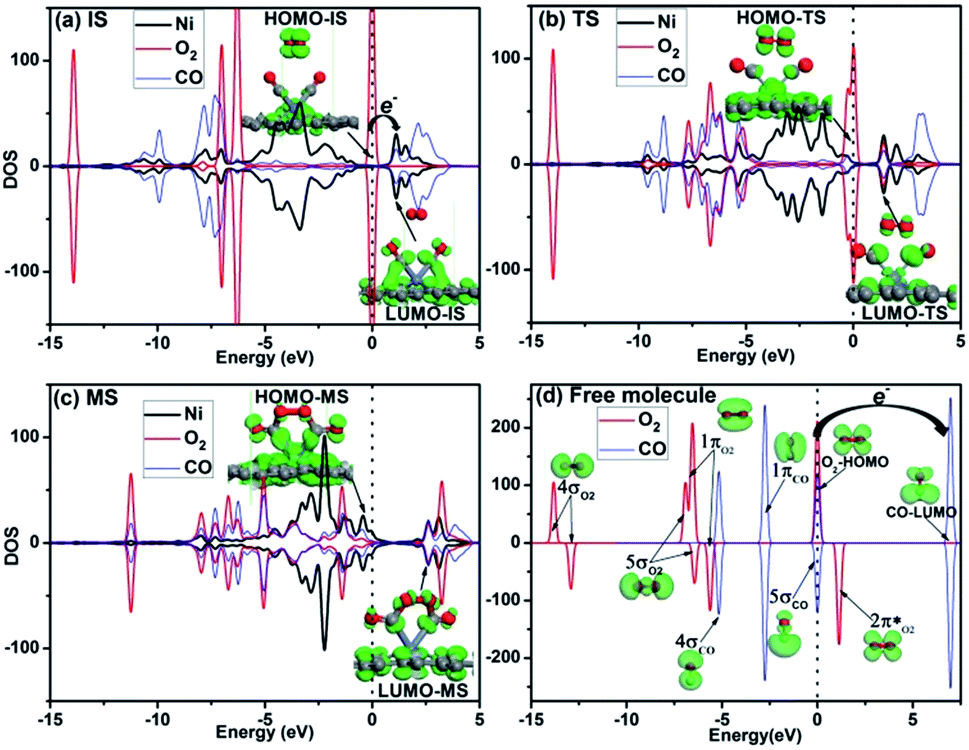 | ||
| Fig. 8 PDOS of the adsorbed O2 and CO and the Ni atom for the IS (a), TS1 (b) and MS (c) configurations via the TER mechanism. The HOMO and LUMO orbitals are also shown. The PDOS of the free O2 and CO molecules is also shown in panel (d). | ||
4. Conclusions
In summary, the oxidation of a CO molecule on Ni-decorated divacancy graphene (Ni-DG) has been studied by using DFT calculations. The diffusion barrier for the single Ni atom on the divacancy site is 6.23 eV, which shows high stability for the Ni-DG. The LH and ER mechanisms are not preferred for the CO oxidation on Ni-DG owing to the much larger adsorption energy of CO than that of O2 molecules. Further study indicates that the TER mechanism is preferred for the CO oxidation on Ni-DG, where the O2 molecule is activated by the two pre-adsorbed CO molecules and a pentagonal OCONiOCO intermediate is formed after overcoming a small energy barrier of 0.34 eV. Then the intermediate dissociates into two CO2 molecules with an energy barrier of 0.29 eV. Therefore, the rate-limiting energy barrier is only 0.34 eV, indicating an efficient oxidation process. The charge transfer from the O2 molecule to CO through the Ni atom via the TER mechanism plays a key role in depressing the energy barrier of the CO oxidation. The adsorption energy of CO is much larger than those of O2, H2O, and N2 molecules, indicating that Ni-DG can be a noble-metal-free and efficient catalyst for CO oxidation, even in humid environments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support by the Fundamental Research Funds for National Natural Science Foundation of China (Grant No. 21703052, 21607029, 21777033), the Central Universities (Grant No. 2017B12914 and 2015B01914), the China Postdoctoral Science Foundation (2015M571652), the Natural Science Foundation of Jiangsu Province (BK20161506), the National 973 Plan Project (2015CB057803), the Science and Technology Planning Project of Guangdong Province (2017B020216003), the Science and Technology Program of Guangzhou City (201707010359), and the “1000 Plan” for Young Professionals Program of China.Notes and references
- J. Neugebohren, D. Borodin, H. W. Hahn, J. Altschäffel, A. Kandratsenka, D. J. Auerbach, C. T. Campbell, D. Schwarzer, D. J. Harding and A. M. Wodtke, Nature, 2018, 558, 280–283 CrossRef CAS PubMed.
- W. Zhan, J. Wang, H. Wang, J. Zhang, X. Liu, P. Zhang, M. Chi, Y. Guo, Y. Guo and G. Lu, J. Am. Chem. Soc., 2017, 139, 8846–8854 CrossRef CAS PubMed.
- Y. Wang, H. Yuan, Y. Li and Z. Chen, Nanoscale, 2015, 7, 11633–11641 RSC.
- Z. P. Liu, P. Hu and A. Alavi, J. Am. Chem. Soc., 2002, 124, 14770–14779 CrossRef CAS PubMed.
- K. Bleakley and P. Hu, J. Am. Chem. Soc., 1999, 121, 7644–7652 CrossRef CAS.
- C. Dupont, Y. Jugnet and D. Loffreda, J. Am. Chem. Soc., 2006, 128, 9129–9136 CrossRef CAS PubMed.
- C. J. Zhang and P. Hu, J. Am. Chem. Soc., 2001, 123, 1166–1172 CrossRef CAS PubMed.
- A. H. Zhang, J. Zhu and W. H. Duan, J. Chem. Phys., 2006, 124, 234703 CrossRef CAS PubMed.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Y. Lou, L. Wang, Z. Y. Zhao, Y. H. Zhang, Z. G. Zhang, G. Z. Lu, Y. Guo and Y. L. Guo, Appl. Catal., B, 2014, 146, 43–49 CrossRef CAS.
- C. L. Wang, S. Liu, D. D. Wang and Q. W. Chen, J. Mater. Chem. A, 2018, 6, 11037–11403 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- C. G. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- Y. H. Lu, M. Zhou, C. Zhang and Y. P. Feng, J. Phys. Chem. C, 2009, 113, 20156–20160 CrossRef CAS.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CrossRef CAS.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678–3683 CrossRef CAS.
- Y. N. Tang, Z. X. Yang and X. Q. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566–16572 RSC.
- Q. G. Jiang, Z. M. Ao, S. Li and Z. Wen, RSC Adv., 2014, 4, 20290–20296 RSC.
- Y. Tang, D. Ma, W. Chen and X. Dai, Sens. Actuators, B, 2015, 211, 227–234 CrossRef CAS.
- Y. Tang, X. Dai, Z. Yang, Z. Liu, L. Pan, D. Ma and Z. Lu, Carbon, 2014, 71, 139–149 CrossRef CAS.
- X. Y. Xu, J. Li, H. Y. Xu, X. F. Xu and C. Y. Zhao, New J. Chem., 2016, 40, 9361–9369 RSC.
- G. Xu, R. Wang, F. Yang, D. Ma, Z. Yang and Z. Lu, Carbon, 2017, 118, 35–42 CrossRef CAS.
- Q. G. Jiang, J. F. Zhang, Z. M. Ao, H. J. Huang, H. Y. He and Y. P. Wu, Front. Chem., 2018, 6, 187 CrossRef PubMed.
- Y. N. Tang, W. G. Chen, Z. G. Shen, C. G. Li, D. W. Ma and X. Q. Dai, Phys. Chem. Chem. Phys., 2018, 20, 2284–2295 RSC.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- A. Uzun, V. Ortalan, N. D. Browning and B. C. Gates, J. Catal., 2010, 269, 318–328 CrossRef CAS.
- P. Liu, et al. , Science, 2016, 352, 797–800 CrossRef CAS.
- D. Deng, et al. , Sci. Adv., 2015, 1, e1500462 CrossRef PubMed.
- X. Zhang, et al. , Nat. Commun., 2013, 4, 1924 CrossRef PubMed.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. L. Wang, J. J. Li, S. Q. Wei and J. L. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- K. K. Mao, L. Li, W. H. Zhang, Y. Pei, X. C. Zeng, X. J. Wu and J. L. Yang, Sci. Rep., 2014, 4, 5441 CrossRef CAS.
- G. L. Xu, R. Wang, F. Yang, D. W. Ma, Z. X. Yang and Z. S. Lu, Carbon, 2017, 118, 35–42 CrossRef CAS.
- Z. S. Lu, P. Lv, Y. L. Liang, D. W. Ma, Y. Zhang, W. J. Zhang, X. W. Yang and Z. X. Yang, Phys. Chem. Chem. Phys., 2016, 18, 21865–21870 RSC.
- Z. S. Lu, P. Lv, J. Xue, H. H. Wang, Y. Z. Wang, Y. Huang, C. Z. He, D. W. Ma and Z. X. Yang, RSC Adv., 2015, 5, 84381–84388 RSC.
- A. A. El-Barbary, R. H. Telling, C. P. Ewels, M. I. Heggie and P. R. Briddon, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 144107 CrossRef.
- A. V. Krasheninnikov, P. O. Lehtinen, A. S. Fostera and R. M. Nieminen, Chem. Phys. Lett., 2006, 418, 132–136 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1789 CrossRef CAS PubMed.
- A. Roldán, J. M. Ricart and F. Illas, Theor. Chem. Acc., 2009, 123, 119–126 Search PubMed.
- T. A. Halgren and W. N. Lipscomb, Chem. Phys. Lett., 1977, 49, 225 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978 CrossRef CAS.
- M. D. Segall, P. L. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- C. Shang and Z. P. Liu, J. Am. Chem. Soc., 2011, 133, 9938–9947 CrossRef CAS PubMed.
- O. Cretu, A. V. Krasheninnikov, J. A. Rodríguez-Manzo, L. Sun, R. M. Nieminen and F. Banhart, Phys. Rev. Lett., 2010, 105, 196102 CrossRef PubMed.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- L. N. Cao, et al. , Nature, 2019, 565, 631–635 CrossRef CAS PubMed.
- Q. G. Jiang, Z. M. Ao and Q. Jiang, Phys. Chem. Chem. Phys., 2013, 15, 10859–10865 RSC.
- Q. G. Jiang, J. F. Zhang, H. J. Huang, Y. P. Wu and Z. M. Ao, J. Phys. Chem. C, 2019, 123, 11591–11601 CrossRef CAS.
- K. Honkala and K. Laasonen, Phys. Rev. Lett., 2000, 84, 705 CrossRef CAS PubMed.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- C. Stegeimann, A. Andreasen and C. T. Campbell, J. Am. Chem. Soc., 2009, 131, 8077–8082 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information and figures. See DOI: 10.1039/c9ta08525d |
| This journal is © The Royal Society of Chemistry 2020 |