
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe hierarchical bulk molecular structure of poly(acrylamide) hydrogels: beyond the fishing net†
Yvonne
Gombert
a,
Fabrice
Roncoroni
 a,
Antoni
Sánchez-Ferrer
b and
Nicholas D.
Spencer
*a
a,
Antoni
Sánchez-Ferrer
b and
Nicholas D.
Spencer
*a
aLaboratory for Surface Science and Technology, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: nspencer@ethz.ch
bWood Research Munich, School of Life Sciences, TU Munich, Winzererstrasse 45, 80797 Munich, Germany
First published on 23rd September 2020
Abstract
The polymeric structure of hydrogels is commonly presented in the literature as resembling a fishing net. However, this simple view cannot fully capture all the unique properties of these materials. Crucial for a detailed description of the bulk structure in free-radical polymerized vinylic hydrogels is a thorough understanding of the cross-linker distribution. This work focuses on the precise role of the tetra-functional cross-linker in the hydrogel system: acrylamide-N,N′-methylenebis(acrylamide). Clusters of crosslinker smaller than 4 nm and their agglomerates, as well as polymer domains with sizes from the 100 nm to the μm-range, have been identified by means of both X-ray and visible-light scattering. Placed in the context of the extensive literature on this system, these observations demonstrate the heterogeneous organisation of the polymer within the hydrogel network structure, and can be accounted for by the different polymerization behavior of the monomer and crosslinker. Together with polymer-network chain-length approximations based on swelling experiments and structural observations with scanning electron microscopy, these results indicate a hierarchical structure of the polymer network surrounding pockets of water.
1. Introduction
Hydrogels constitute a class of soft materials consisting of a three-dimensional, crosslinked polymer network with large amounts of water tightly incorporated within their structure. It is this structure combining water with polymer that makes hydrogels unique among synthetic materials. The polymer network provides water, a Newtonian fluid, with form-stability—a property traditionally associated with solids. On the other hand, water provides the polymer, typically a solid, with the possibility of rapid gas and liquid diffusion together with a low modulus that is otherwise atypical for solids. This combination results in a material that is intermediate in its properties between solids and liquids. Typical characteristics are a high diffusion rate for gases and small molecules, transparency, oxygen permeability, flexibility and lubricity.1–4With this unique set of properties, the material exhibits similarities to human body tissue like no other existing synthetic material. Due to this resemblance, the applications of hydrogels have often been targeted towards biomedical areas—a challenging field, in which the benefits of the polymeric network structure in combination with a high water content can be utilized to full advantage. The application of hydrogels in the medical sector is a constantly growing field, with diverse current and future applications ranging from spray bandages and contact lenses to intelligent drug-delivery systems, as well as injectable tissue replacements and implants in all parts of the human body.5–7
However, the demands placed on the material are also growing. Despite their rising importance, fundamental research is still needed in the structural analysis of this class of materials. A thoroughly comprehensive and in-depth understanding is lacking, as to how all the structural elements of the polymer network combine with water to create a unique set of properties.
Free-radical polymerized (FRP) poly(acrylamide) (pAAm) hydrogels have been researched since the 1960s. A large number of studies exist, highlighting the heterogeneous character of the polymer network structure of vinylic hydrogels.8–10 In this study, we build upon existing knowledge of this system by combining it with a focused analysis of the network architecture in relation to the bis-acrylamide (bis-AAm) cross-linker concentration. The distribution of the cross-linker in the polymer network is analyzed on both nanoscopic and microscopic scales by means of small-angle X-ray scattering (SAXS) and light transmittance. These results are compared with calculations on the chain length of these networks. Backed up by the extensive literature on the different polymerization behavior of the monomers and our scanning-electron-microscopy (SEM) observations, we present a revised picture of the polymeric network structure in FRP vinylic hydrogels.
Through the combined analysis of many structural elements of the polymer network, we have been able to create an overall picture of the hierarchical vinylic hydrogel structure, and the mutual influence of its constituent elements. The common view of a fishing-net-like structure has thereby been revised. This may guide future design principles for specific hydrogel applications.
2. Cross-linker distribution in the hydrogel polymer network
Poly(acrylamide) (pAAm) hydrogels consist of water held within a polymer network that is formed by the monomer acrylamide (AAm) and the cross-linker N,N′-methylenebis(acrylamide) (bis-AAm). In this work, we maintained the AAm monomer concentration at a constant 7.5 wt% and varied the bis-AAm cross-linker concentration from a minimum of 0.03 wt% (“pAAm003”) to a maximum of 0.8 wt% (“pAAm08”). Samples are named after their bis-AAm content (pAAm003 to pAAm08) throughout. This approach allowed us to analyze the different, cross-linker-concentration-dependent hydrogel structures that are produced by the free-radical polymerization (FRP) reaction.2.1. X-ray scattering
A typical SAXS intensity profile displays the scattering data as intensity I(q) vs. scattering wave vector q (Fig. 1). We have fitted the scattering spectra using the broad-peak model, as described in Section 6.2. An initial inspection of the data reveals that the hydrogel-network structure contains regular electron-density fluctuations in two size ranges.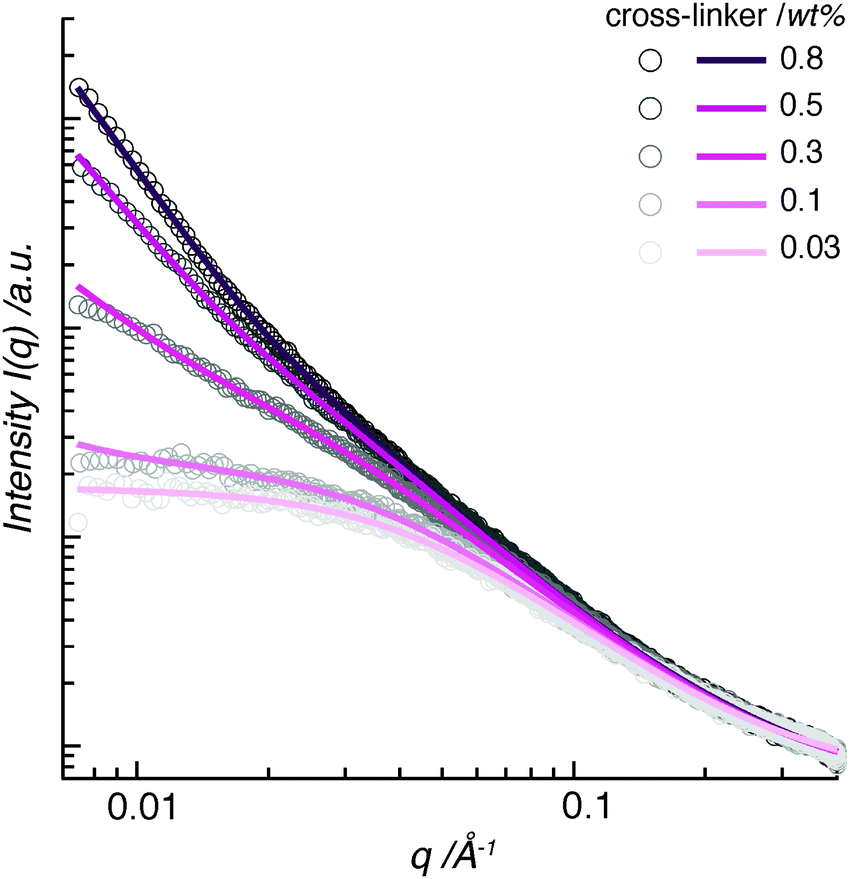 | ||
| Fig. 1 SAXS data of pAAm hydrogels with different amounts of bis-acrylamide cross-linker show two scattering regimes. In the high-q range, similar scattering intensities indicate similarly small scattering features in the samples. In the low-q range, a fanning-out towards stronger scattering intensity I(q) was observed with increasing cross-linker concentration. Dots indicate the raw data and straight lines their corresponding fits. | ||
The high-q range displays scattering from small features, below approximately 6 nm. A Lorentzian function, the second term of the broad-peak model, can fit the scattering spectra in this size range. This term contains ξ, which often is denoted the correlation length in semi-dilute polymer solutions. It provides information on the spatial resolution between polymer chains and can be considered as a solvent–polymer interaction parameter.11 It is commonly applied to give an estimation of the average distance between cross-links, namely the mesh size, in a polymer network.8,12 However, in chemically cross-linked hydrogels, this parameter resembles the size of a polymer cluster rather than the average distance between cross-links13,14—a discrepancy that is often disregarded in the hydrogel-user community. In this work, we rename ξ as ε, in order to avoid confusion. For our data, the fits result in similar diameters of the polymer clusters in the pAAm hydrogels (Fig. 1). With values lying between ε = 2.3–3.7 nm, the size of the clusters increases only little with increasing cross-linker concentration.
The low-q range displays scattering from large-scale features in the polymer network. A power law, the first term in the broad-peak model, accounts for the scattering in this size range. This term contains the Porod exponent, n. It indicates density distributions of large-scale scattering of the polymer network. For polymer samples, this exponent generally covers a range between n = 1 to 4, describing the spatial range from one-dimensional objects (I(q) ∼ q−1) through two-dimensional scattering relations (I(q) ∼ q−2 − q−3), so-called mass fractals, to three-dimensional objects (I(q) ∼ q−4).15
The fitting of our data covers a wide range of n-values between n = 1.5 to 2.93. Hydrogels with lower concentrations of cross-linker, namely pAAm003 (containing 0.03 wt% bis-AAm), pAAm01 (0.1 wt%), and pAAm03 (0.3 wt%), show Porod exponents between n = 1.5 to 1.78. This is in the higher range of polymer mass fractals. These numbers demonstrate that the origin of scattering in these samples lies in swollen polymer chains in close proximity (n = 1.6) with weak physical and chemical cross-linking contributions (n = 2). In contrast, hydrogels pAAm05 (0.5 wt% bis-AAm) and pAAm08 (0.8 wt%) show Porod exponents of n = 2.48 and 2.93, respectively. Such an increase in n-values towards n = 3, which corresponds to a closely packed polymer mesh, shows that the source of X-ray scattering in pAAm hydrogels with large amounts of cross-linker, lies within dense, large polymer domains.
Therefore, the wide span in the Porod exponent in our data marks a clear transition in structural morphology from a loosely cross-linked polymer mesh with swollen polymer chains to a packed polymer network with dense cross-links.16
In the case of pAAm003, the scattering intensity is constant in the low-q range (Fig. 1). Here, the scattering intensity is independent of the wave vector q of the incoming X-ray beam. This indicates that no periodically distributed structural elements larger than approximately 30 nm exist in this hydrogel. As a result, we can consider the polymer network of the pAAm003 hydrogel, besides containing polymer clusters in the low-nm range, to have a random structure similar to a dilute polymer solution.
With an increase in cross-linker concentration from 0.03 wt% upwards, the data fanned out to stronger scattering intensities (Fig. 1). We interpret this change as a further indication of the transition in structural morphology, as already demonstrated by the Porod exponent. The scattering intensity and its dependence on q increased slightly for the pAAm01 hydrogel compared to pAAm003. Here, we observe the trend towards a periodic-structure formation within the hydrogel polymer network. The next increase in cross-linker concentration, pAAm03, led to a further increase in scattering intensity, which is further enhanced with even more cross-linker molecules in the polymer (pAAm05 and pAAm08). This indicates an increase in the number of cross-linker-rich scatterers of a certain size in the hydrogel. Since the data do not peak within the measurement limits of SAXS, the scattering features in the low-q regime could not be fully captured.
In summary, the evaluation of SAXS data identifies the existence of regions of high polymer density in the pAAm polymer network. The high-q range uncovered small polymer clusters with diameters below 4 nm in all hydrogels, independent of the cross-linker concentration. The low-q range demonstrated how the polymer hydrogel morphology transitions with cross-linker concentration from an unstructured polymer mesh, which resembles a dilute polymer solution, to a polymer network with polymer aggregates in a periodic arrangement.
2.2. PAAm polymerization studies in the literature
In order to explain the heterogeneity of the hydrogel polymer structure, we need to gain insight into the polymerization behavior of both AAm monomer and bis-AAm crosslinker. According to Flory, three factors determine the polymerization process of a gel: first, the functionality of the cross-linker, second, the reactivity of the monomer, and third, the relative concentration of the monomer and cross-linker in solution.17 In the pAAm hydrogel system, the cross-linker, bis-AAm, is tetra-functional. The reactivity and concentration of both the chain-monomer, AAm, and the cross-linker, bis-AAm, were the subject of intensive research from Baselga et al. in the late 1980s and others up to the present day.16,18–21Baselga et al. studied the polymerization kinetics of the AAm–bis-AAm system in a series of in situ1H-NMR measurements. They made two important observations on the reactivity of bis-AAm. First, bis-AAm shows higher reactivity than AAm. Therefore, its conversion rate is higher than that of AAm at any point during the initial stages of hydrogel polymerization. Second, bis-AAm preferentially reacts with other bis-AAm molecules, even at very low concentration.16,18 This was confirmed by Okay et al. They found that the reaction of bis-AAm with other bis-AAm molecules, can consume up to 80% of all available crosslinker in the reaction solution. They also observed an acceleration of the polymerization rate with higher bis-AAm concentration.19 This preferential homomonomer reaction of bis-AAm leads to a large number of free vinyl groups in the pAAm oligomers and polymers that form during the early stage of polymerization. Consequently, the free vinyl groups in these polymer strands are prone to reacting with radicals in the on-going polymerization. This leads to cyclization reactions, formation of side chains, branching and intra-molecular cross-linking reactions. Instead of connecting four polymer chains, as is often assumed due to bis-AAm's tetra-functionality, a significant proportion of bis-AAm is consumed in homomonomer reactions. As a result, the overall cross-linking efficiency in the system decreases. Instead of forming a polymer mesh with tetra-functional cross-linking points, the FRP of pAAm produces a network with polymer-dense cross-linking knots of uncontrolled functionality and size. These knots are rich in bis-AAm monomers with reacted and unreacted pendant vinyl groups trapped in this dense polymer cluster. These bis-AAm-rich areas polymerize first in the reaction solution and are not connected in the early polymerization phase. Therefore, this phase is generally denoted as the pre-gel reaction.16,19,22 It is reasonable to assume that the polymer-dense cross-linking knots of this pre-gel reaction correspond to the small polymer clusters observed in our SAXS measurements.
For the AAm reactivity, Baselga et al. found a preferential reaction for heteromonomer polymerization—the reaction of AAm with bis-AAm molecules—and overall lower conversion rates than for bis-AAm.16,18 Consequently, AAm molecules become incorporated in the bis-AAm-rich polymer clusters during the pre-gel reaction. While they also form polymer chains during this first polymerization stage, these chains are too short to connect the cross-linking clusters. AAm monomers polymerize into longer chains only after approximately 50% of all monomer in the reaction solution has reacted and a significant fraction of the bis-AAm has polymerized.19,21,22 In this second phase of polymerization, the so-called gelling phase, the viscosity of the polymerization solution increases while AAm-rich polymer chains join together the insoluble cross-linking clusters from the pre-gelation phase. The polymer gels and forms one gigantic molecule, as was already described by Flory in 1942.17,23
Baselga et al. also described an increase in number and density of cross-linker-rich polymer clusters with increasing amount of bis-AAm in the reaction solution. As a result, more cross-linker in the reaction solution leads to an overall reduction in cross-linking efficiency and network connection.20,24 Therefore, pAAm hydrogels with a higher concentration of cross-linker molecules contain more, but shorter AAm chains.
2.3. Visible light transmittance
In order to broaden our understanding of the influence of cross-linker concentration on the cross-linker distribution in the hydrogel, we recorded data in the UV/vis range of the electromagnetic spectrum.The optical appearance of our samples changed from transparent through opaque to milky-white—i.e. the turbidity of the hydrogels increased with cross-linker concentration (Fig. 2). We measured the interaction of light with fully polymerized samples under oxygen exclusion with UV/vis spectroscopy, in order to capture this gradual transition. The data were converted from absorbance to transmittance—a measure of sample transparency. Since absorbance of fully polymerized pAAm hydrogels occurs only below 340 nm (see ESI†), all deviations from 100% transmittance in spectra in the visible-light range (660 nm to 440 nm) arise only from scattering processes in the sample.
 | ||
| Fig. 2 The appearance of a fully polymerized pAAm hydrogel strongly depends on its cross-linker concentration: it turns from transparent to milky-white with increasing proportion of cross-linker. Numbers in the photo indicate the wt% of bis-AAm in the sample. | ||
In the previous section we have discussed the polymerization of bis-AAm crosslinkers into clusters. AAm monomers carry only one vinyl group. Thus, they can only polymerize into chains. Consequently, domains in the polymer network can only be composed of agglomerated polymer clusters. In order to enable an estimation of scattering center sizes from UV/vis transmittance spectra, we compared the recorded data to calculated spectra (Section 6.3). Here, all concentration-dependent parameters, namely the particle concentration and the refractive index np of the polymer, were individually adjusted for every sample composition. Therefore, it was possible to calculate the transmittance of pAAm hydrogel systems with different concentrations and polymer-agglomerate radii (for details see ESI†).
The UV/vis transmittance spectrum of a fully polymerized pAAm01 sample revealed its transparency to radiation in the visible-light range. Only a minor shift towards lower transmittance was observed in the low-wavelength region (Fig. 3a). This is in accordance with light-scattering theory, which describes a dependence of the scattering behavior on the wavelength with a power-law λ−4. When we compare the recorded spectrum with the calculated transmittance spectra, we observe an agreement in curve progression between them. This confirms the validity of light-scattering theory to the pAAm hydrogel system in the transparent sample range. Therefore, it is possible to claim that scattering centers in the sample at this cross-linker concentration remain smaller than the wavelength of visible light. As the cross-linker concentration was increased, the transmittance decreased (pAAm03, Fig. 3b). While this sample still appeared macroscopically transparent, the data showed that incident light was scattered more strongly here than it was in pAAm01. A comparison of the recorded spectrum with the theoretical spectra points towards scattering centers that are still smaller than the wavelength of visible light. From this finding, we conclude that a decrease in transmittance in pAAm03 is caused by a combined effect of a small increase in scattering-center size relative to those in pAAm01 and an increase in scattering-center number. This is consistent with the data from SAXS measurements, where an increase in the measured intensity was attributed to an increase in number of scattering centers.
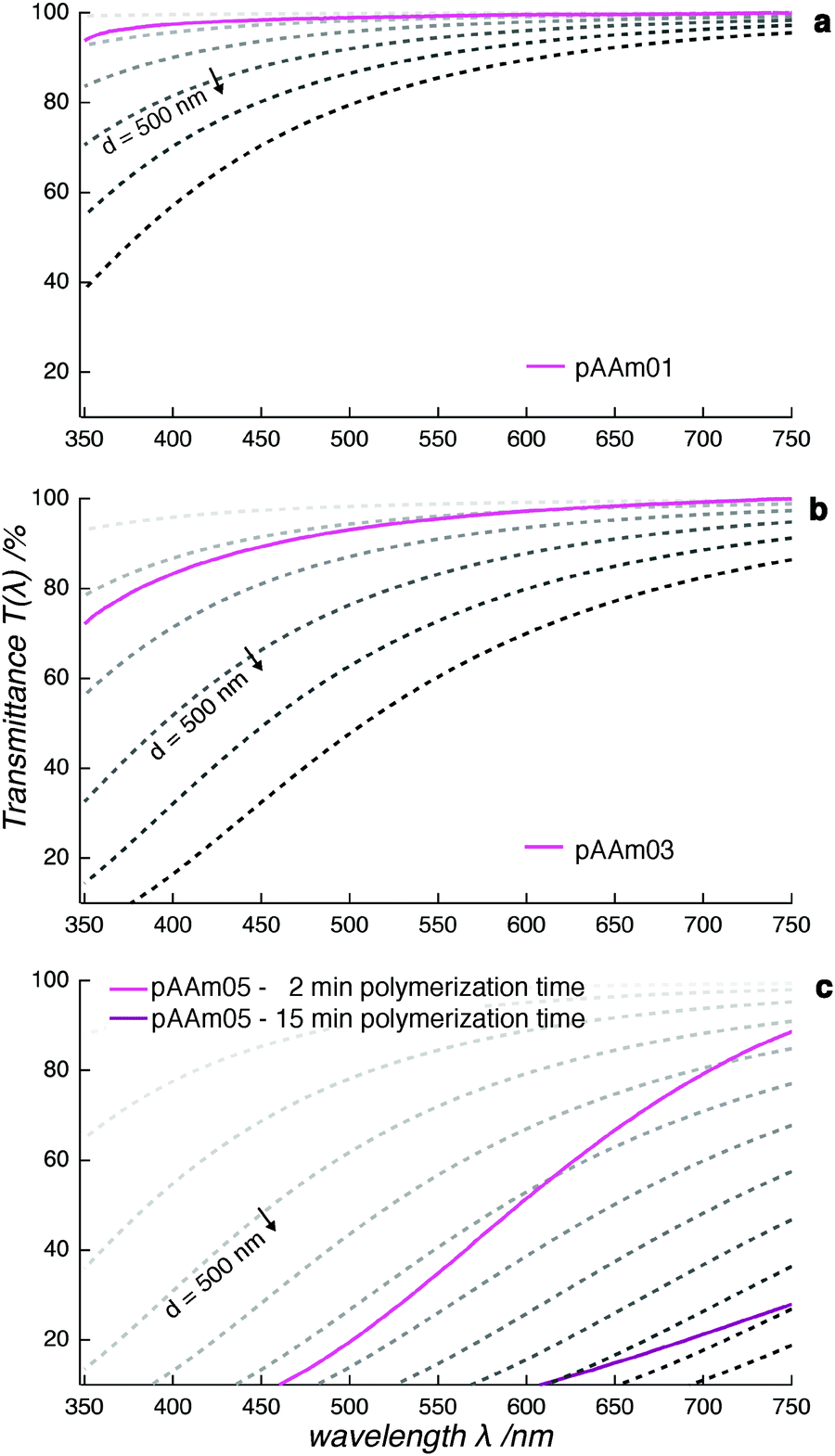 | ||
| Fig. 3 The transmittance spectra of fully polymerized pAAm01 (a), pAAm03 (b), and partially polymerized pAAm05 (c) show decreasing transparency with increasing cross-linker concentration. The data are compared to the calculated transmittance spectra for different polymer domain sizes d. The step size is Δd = +100 nm, with d increasing in the direction indicated by the arrow. | ||
With further increase in the cross-linker concentration, the shape of the recorded spectra changed drastically. pAAm05 already showed a strong reduction in transparency 2 min after the onset of polymerization and turned almost completely opaque after 15 min (Fig. 3c). From comparison with the calculated transmittance spectra, we conclude that the size of scattering centers in this sample is likely to be polydisperse and reached the hundreds-of-nm range, exceeding the wavelength range of visible light. Additionally, the recorded curves deviated from the calculated spectral shapes. Consequently, we cannot expect the polymer system formed in pAAm05 hydrogels to meet the criteria of the light-scattering theory applied here. Instead, we assume that scattering centers in hydrogels with high cross-linker concentration are stretched out towards the μm-range and cause multiple scattering, or that they can no longer be approximated by spheres. The transmittance of pAAm08 samples could not be determined due to its extreme scattering behavior.
In summary, the transmittance of visible light in hydrogels confirmed our findings from X-ray scattering with respect to scattering-center size, which increased in both diameter and number with cross-linker concentration. We interpret the scattering centers as agglomerated polymer clusters and refer to them as “polymer domains”. Their diameter remained smaller than the wavelength range of visible light for transparent pAAm hydrogels with cross-linker concentrations below 0.5 wt% and exceeded this range for higher cross-linker concentrations.
2.4. Electron microscopy
Scanning electron microscopy (SEM) is a widely used analytical method for the characterization of hydrogel samples. We employed it to gain a visual impression of the transition in polymer-network structure of pAAm hydrogels with varying cross-linker concentration.SEM images of bulk samples are shown in Fig. 4. They reveal hollow pore structures for every polymer network composition of the hydrogel. The pore sizes between the samples varied: pAAm003 showed a coarse structure with pore sizes in the μm range (Fig. 4a). The pores grew smaller with more cross-linker in the polymer network and reached the finest pore structure at a cross-linker concentration of 0.3 wt% (Fig. 4c). With further increase in cross-linker concentration, pores appeared to enlarge again (pAAm05, Fig. 4d). A distinctly heterogeneous structure of polymer clusters and domains, as was identified by SAXS analysis and visible light transmittance, was not observed in these images. However, the trend of larger spacing between polymer structures in samples with less cross-linker is consistent with the results from our scattering measurements.
 | ||
| Fig. 4 SEM images of the freeze-dried samples pAAm003 (a), pAAm01 (b), pAAm03 (c), and pAAm05 (d) reveal the hollow pore structures within the bulk hydrogels. The pores decrease in size with an increase in cross-linker concentration and reach a minimum at 0.3 wt% (c). At higher concentration, the pores enlarge (d). The heterogeneous biphasic structure of cross-linker clusters and polymer domains as revealed in SAXS, is not evident in the SEM images. | ||
The reason why the heterogeneous structure of polymer domains was not evident is most likely due to the sample preparation of hydrogels for SEM imaging.25 The water content of many hydrogels lies above 90%. This large amount of water makes them unsuitable for environmental SEM. Hence, the water of hydrogels must be removed, in order to introduce them into the vacuum chamber of an electron microscope. We applied two methods to do this: drying and freeze-drying. In their native state, the optical appearance of our pAAm hydrogels ranged from transparent to milky-white, depending on the concentration of cross-linker (Fig. 2). If the samples were dried in air, their optical appearance changed to fully transparent, independent of their original appearance. Additionally, the samples shrank considerably. If the samples were freeze-dried, their optical appearance changed to dull white, independent of their original appearance. Additionally, they shrank slightly. Therefore, we must assume that water extraction from hydrogel samples has a significant impact on their polymer network structure. The fact that the change in the optical appearance upon drying is independent of the polymer composition but dependent on the drying method indicates that the water-filled polymer structure cannot be satisfactorily preserved in any SEM preparation method. Consequently, SEM images of hydrogels can merely be used to observe substantial changes in polymer structure rather than giving a realistic insight into the polymer distribution in fully hydrated, hydrogel samples. For this reason, it would be rash to draw quantitative conclusions about pore sizes and other polymer-structure features based on SEM imaging alone. Only qualitative comparisons between structural differences of samples that have been prepared in the same way can be obtained from SEM imaging of hydrogels.
3. Polymer chain length of the hydrogel polymer network
Swelling measurements were performed on the pAAm hydrogels, in order to obtain an approximation of the chain length, l, and its dependence on the cross-linker concentration. The following cross-linker distributions established in the previous section were applied for these approximations: pAAm003 solely contains cross-linking clusters with a diameter of 2.3 nm; pAAm01 and pAAm03 comprise domains of agglomerated cross-linking clusters with diameters smaller than the wavelength of visible light; and pAAm05 and pAAm08 incorporate domains of agglomerated cross-linking clusters with diameters in the μm range.To estimate the influence of domain formation on the resulting size distribution in the pore space between cross-link junctions, the calculations were additionally based on the assumption that bis-AAm is tetrafunctional and polymerizes with 100% efficiency.
3.1. PAAm hydrogel swelling
Hydrogel samples of constant AAm concentration and varying cross-linker concentration were swollen from their dry state to equilibrium. As expected, our results show that the swelling behavior of a hydrogel is inversely proportional to the concentration of the cross-linker in the sample (see ESI†). While the volume of the most-cross-linked sample, pAAm08, only expanded from its dry state back to its volume after polymerization, the volume of the least-cross-linked sample, pAAm003, expanded by a further factor of 2.6.3.2. Calculation of the network chain length
The pore spacing between chains and cross-links in a polymer network changes with the amount of solvent in the hydrogel system. However, the length of a polymer chain that connects two cross-links remains constant. Only its extension changes with the amount of water in the hydrogel. Therefore, it is common practice to describe the average chain length in a hydrogel polymer network by the average molecular weight between cross-links,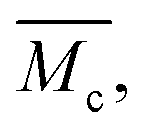 rather than an actual length specification.
rather than an actual length specification.
When we applied the calculation of 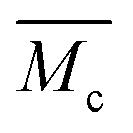 to the pAAm hydrogel system discussed in this work, the trend of decreasing swelling ratio Q with increasing cross-linker repeats for the chain length, independent on the network configuration (Fig. 5). If the calculation is carried out under the assumption that the hydrogel polymerizes in an “ideal” network configuration – each cross-linker molecule connecting four chains, ϕ = 4 – the pre-factor
to the pAAm hydrogel system discussed in this work, the trend of decreasing swelling ratio Q with increasing cross-linker repeats for the chain length, independent on the network configuration (Fig. 5). If the calculation is carried out under the assumption that the hydrogel polymerizes in an “ideal” network configuration – each cross-linker molecule connecting four chains, ϕ = 4 – the pre-factor 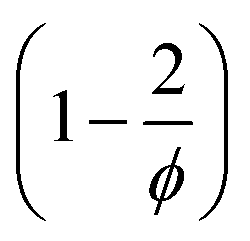 reduces to 0.5 (compare to Section 6.6). In contrast, if the calculation is carried out under the assumption that the hydrogel polymerizes into an inhomogeneous network configuration with dense cross-linking domains, the functionality of single cross-linker molecules reduces, due to their preferred homomonomer reactivity. In this case, ϕ is defined through the number of active bis-AAm molecules on the domain surface, which act as one large cross-linker with a higher functionality. It increases continuously with increasing domain diameter. But since the pre-factor is defined as
reduces to 0.5 (compare to Section 6.6). In contrast, if the calculation is carried out under the assumption that the hydrogel polymerizes into an inhomogeneous network configuration with dense cross-linking domains, the functionality of single cross-linker molecules reduces, due to their preferred homomonomer reactivity. In this case, ϕ is defined through the number of active bis-AAm molecules on the domain surface, which act as one large cross-linker with a higher functionality. It increases continuously with increasing domain diameter. But since the pre-factor is defined as 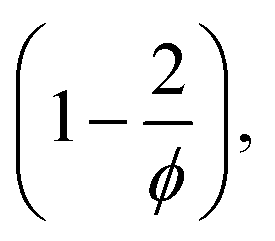 it quickly tends to a value of 1 with increasing domain size. Therefore, while the chain length
it quickly tends to a value of 1 with increasing domain size. Therefore, while the chain length 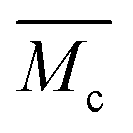 in the inhomogeneous hydrogel polymer network is largely independent of the number of chains originating from a cross-link, it is double that of the “ideal” network configuration.
in the inhomogeneous hydrogel polymer network is largely independent of the number of chains originating from a cross-link, it is double that of the “ideal” network configuration.
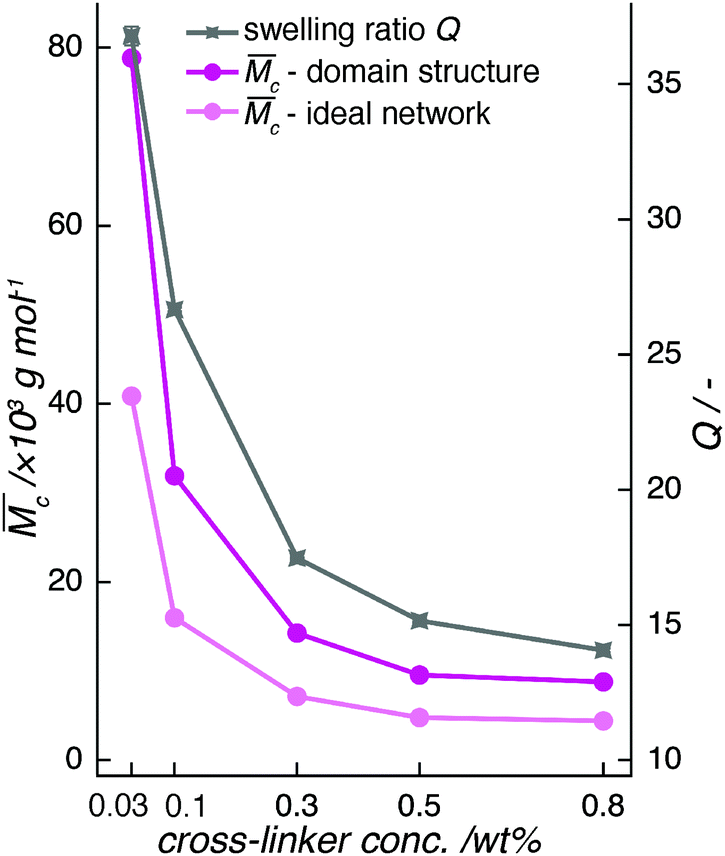 | ||
Fig. 5 The calculated average molecular weight, 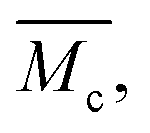 between cross-links in a pAAm hydrogel network, follows the trend of its swelling behavior. It decreases strongly with increasing cross-linker concentration. This trend does not depend on the distribution of the cross-linker in the hydrogel. However, an inhomogeneous organization of the cross-linker in clusters and domains leads to a doubled chain length compared to the organization of cross-links in an “ideal” network formation. between cross-links in a pAAm hydrogel network, follows the trend of its swelling behavior. It decreases strongly with increasing cross-linker concentration. This trend does not depend on the distribution of the cross-linker in the hydrogel. However, an inhomogeneous organization of the cross-linker in clusters and domains leads to a doubled chain length compared to the organization of cross-links in an “ideal” network formation. | ||
4. The overall structure of the hydrogel polymer network
The combination of all the information we have gathered about the distribution of cross-linkers and chains leads us to a comprehensive picture of the hydrogel polymer network. Our results provide an overview of how the cross-linker concentration impacts the distribution of cross-linking knots and chain lengths in the hydrogel and thereby fundamentally determines the polymer network structure.As early as 1971, Richards et al.1 qualitatively identified three different network structures for FRP pAAm, based on the change in turbidity and swelling properties of their samples as a function of cross-linker concentration. According to their theory, so-called “crumpled gels” polymerize when more AAm monomer is available than the amount of tetra-functional bis-AAm monomer is capable to connect. Consequently, long network chains polymerize between cross-links which expand upon swelling, leading to a strong volume change when water is absorbed. An “ideal gel” polymerizes into a balanced distribution of chain- and tetra-functional cross-linker monomers, i.e. a perfect mesh with tetra-functional crosslinking points, much like a (3-D) fishing net. So-called “clustered gels” polymerize when too little AAm monomer is available to connect all cross-linker monomers in the polymerization solution. The cross-linker monomers cluster into dense polymeric structures that are connected by AAm chains. Hydrogels with such inhomogeneous polymer distribution turn turbid. These qualitative predictions are in remarkable agreement with our own observations. When we apply their equation for the prediction of the ideal gel composition to our samples, it yields a cross-linker amount of 0.285 wt% (compare to equations 5 and 6 in Richards et al.1). This value is strikingly close to the composition of pAAm03 (0.3 wt% cross-linker), the sample in our measurement series that marks a transition in optical appearance (from transparent to turbid). While the general predictions of this early study prove to be correct, our measurements provide a more detailed insight into the real polymer-network structure. The fundamental difference between their structural predictions and our findings is our knowledge of the preferred polymerization tendency of bis-AAm over AAm, which was only developed a decade after their publication. Based on our findings, we provide the following refinements to the structural clarification of FRP pAAm hydrogels:
At low cross-linker concentration (corresponding to pAAm003), “crumpled gels” form with long AAm chains between cross-links, as predicted by Richards et al. However, even at this low concentration, these cross-links are not single bis-AAm monomers, but densely polymerized cross-linking clusters as observed with SAXS analysis. With a size in the single-digit nm range they remain small and do not agglomerate, due to the low total concentration in solution. Contrary to Richards’ prediction, a cluster's functionality is not limited to four but connects a multitude of chains due to the large number of bis-AAm monomers in the cluster (Fig. 6, left).
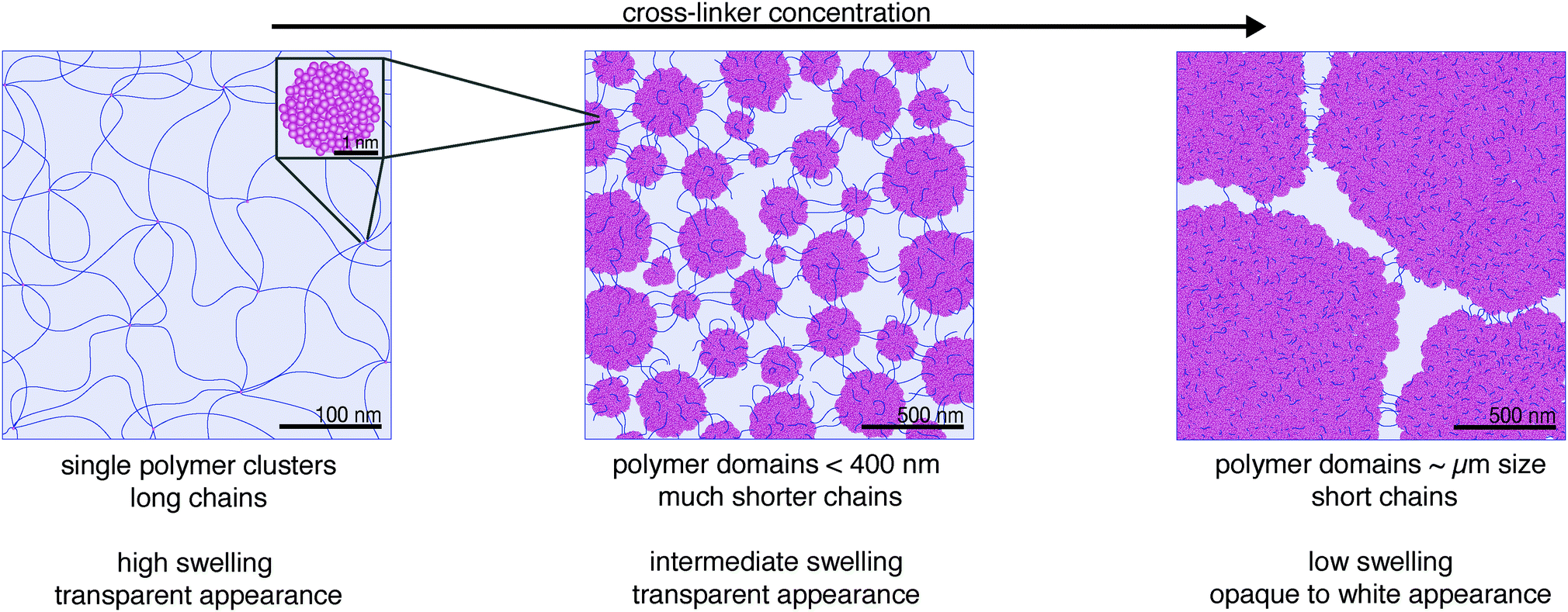 | ||
| Fig. 6 The inhomogeneous polymer network structure of FRP pAAm hydrogels changes significantly as a function of the cross-linker concentration. At low levels of cross-linker (0.03 wt%, left), long polymer chains connect individual cross-linking clusters with a size in the low-nm range. With a ten-fold increase of cross-linker (0.3 wt%, middle), significantly shorter polymer chains connect cross-linking domains (agglomerated clusters) in the low 100-fold nm range. At even higher cross-linker concentration (0.8 wt%, right), domains are in the μm-size and the length of connecting chains reduces further. | ||
With an increase in bis-AAm concentration, our data do not show a distribution of AAm and bis-AAm monomers into the “ideal gel” structure as suggested by Richards. On the contrary, our data reveal an enlargement of the clusters. This increase in cluster volume combines with an increase in the total number of clusters. A rise in cluster density enhances their tendency to agglomerate into cross-linking domains. As a result, the cross-linking knots in the hydrogel occupy more and more volume, and the connecting chains of the polymer network become shorter. Consequently, the transition from a “crumpled gel” to a “clustered gel” is a continuous process. In agreement with Richards et al. and other studies, our measurements demonstrate the existence of an “ideal gel” composition of AAm to bis-AAm ratio around 0.3 wt% (maximum strength in combination with transparency and medium swelling behavior).20,21,26 However, the data strongly suggest that the corresponding polymer network lies in a sweet spot of size distribution of the heterogeneous polymer-dense cross-linking domains with the connecting chains (Fig. 6, right).
We propose an additional structural feature to our direct observations, which adds a level of hierarchy to the hydrogel polymer network: it seems likely that in the FRP process, not only the cross-linker monomers polymerize in an inhomogeneous manner, but also the AAm monomers polymerize with a high dispersity, forming a broad distribution of chain lengths and connections. In other words, there would be no homogeneous network stretching over the macroscopic hydrogel. Instead, the cross-linking knots would be inhomogeneously linked with an irregular number of chains and chain lengths. Many chains would also only be connected by one end. This inhomogeneity of the network would be more pronounced with longer chains in the network. Under this assumption, the swelling of the hydrogels would not be determined by the expansion of the chains between the cross-links. Instead, it would be mainly driven by the swelling of the interstitial spaces, which result from the inhomogeneous network distribution. These pores would also have a broad size distribution and only develop into their equilibrium dimensions as the gel swells. A polymeric structure of inhomogeneous cross-links and chains would encapsulate such water pockets and turn the polymer network into a network that looks similar to the void structure that we observed in the SEM images (Fig. 7, right). Highly cross-linked hydrogels, such as pAAm05 and pAAm08, in which the cross-linker agglomerates into μm-sized domains with only short connecting chains, would hardly form any water pockets. Almost no swelling of the samples happens when they are placed in water. If this picture reflects the true hydrogel structure, an additional hierarchical level in polymer-network organization appears, in addition to the cross-linker distribution: the size and spatial distribution of water pockets, that result from the inhomogeneous chain polymerization.
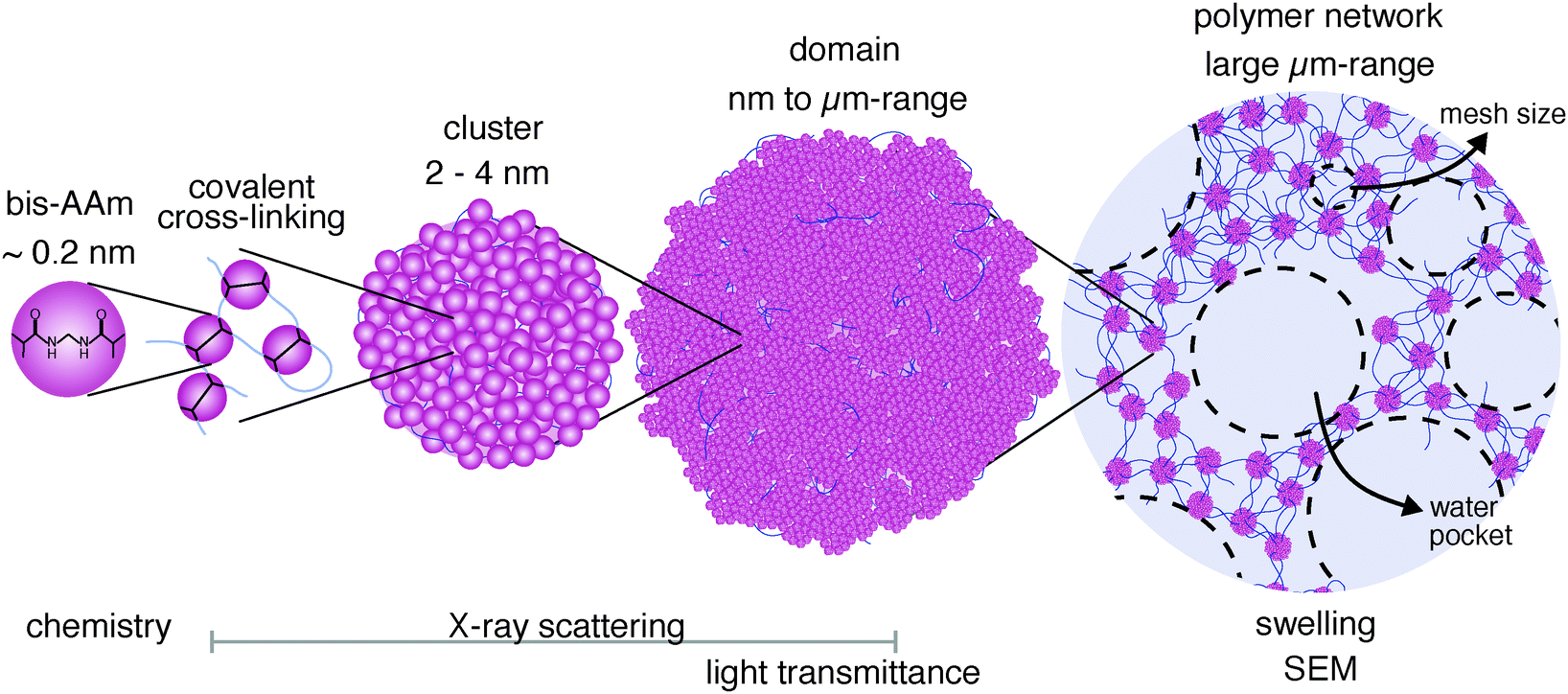 | ||
| Fig. 7 The hierarchical organization of the pAAm hydrogel structure consists of several size levels. The smallest building blocks are the chain monomer AAm and the cross-linker bis-AAm (left). Bis-AAm polymerizes into densely packed polymer clusters. Above a certain total concentration of bis-Aam, clusters agglomerate to domains. Domains connect into a network via AAm chains. During swelling, inhomogeneously interconnected regions of the network fill with water and form water pockets (right). | ||
A study of the distribution of water in the polymer network goes beyond the scope of this work, but would bring our knowledge of the hydrogel structure to completion.
5. Conclusions
In this work, the polymer network structure of FRP pAAm hydrogels has been analyzed, in order to understand the role of cross-linker on network formation and how its distribution influences the structure of the final hydrogel polymer network.A generally heterogeneous network structure for pAAm hydrogels has been identified, which undergoes a structural reorganization with increasing cross-linker concentration. The changes in the structure extend in a hierarchical manner over several organizational levels of the polymer and determine the macroscopic hydrogel properties.
The smallest hierarchical level of the polymer network consists of clusters with high polymer density that are smaller than 4 nm (Fig. 7, left). These clusters consist to a large extent of bis-AAm and polymerize due to the higher polymerization activity of bis-AAm compared to AAm monomers. With increasing concentration, their tendency to agglomerate into domains, the second hierarchical structure level in the inhomogeneously organized network, increases. These domains can grow to sizes in the μm range. Their size and amount determine the transparency of a hydrogel. The domains are connected by polymer chains, which become shorter with increasing cross-linker concentration and domain size. The mesh size, which describes the average distance between cross-linking domains in the hydrogel polymer network, decreases accordingly. Chain length and mesh size have a decisive influence on the mechanical and swelling behavior of a hydrogel.
We predict a third hierarchical level in the inhomogeneous polymer network distribution that plays a dominant role in the type and distribution of water in the network: the broad length distribution of the polymer chains. This heterogeneity increases with increasing chain length and decreasing cross-linking density. Our data indicate that this third hierarchical level causes water pockets in the network to swell in excess amounts of water and drives the strong volume increase in low cross-linked hydrogels (Fig. 7, right).
With the insights gained in this study, we are able to propose a general network heterogeneity with a hierarchical organization of the cross-linker in the polymer network. Combining this with commonly applied calculations for the chain length indicates that existing theories do not adequately describe the polymer network structure of vinylic hydrogels.
6. Experimental
6.1. Hydrogel polymerization
Stock solutions of the monomer acrylamide (AAm, Sigma-Aldrich, ≥99%), the cross-linker N,N′-methylenebis(acrylamide) (bis-AAm, Sigma-Aldrich, ≥99.5%), the initiation system tetramethylethylenediamine (TEMED, Sigma-Aldrich, 99%) and ammonium persulfate (APS, Sigma-Aldrich, ≥98%) were prepared in a deoxygenated environment (concentration O2 <100 ppm). The monomer, cross-linker and deoxygenated ultrapure water were mixed to final concentrations of 7.5 wt% AAm, and 0.03/0.1/0.3/0.5/0.8 wt% bis-AAm, poured into glass molds, and allowed to gel for 24 h. The samples were named according to their cross-linker concentration throughout this work, namely pAAm003, pAAm01, pAAm03, pAAm05 and pAAm08.6.2. X-ray scattering
PAAm polymerization solutions were transferred into thin-walled quartz capillaries, sealed with epoxy glue, and polymerized for 24 h. Small-angle X-ray scattering (SAXS) experiments were performed using a Rigaku MicroMax-002+ equipped with a micro-focused beam (40 W; 45 kV; 0.88 mA) with the λCuKα = 0.15418 nm radiation collimated by three pinhole collimators (0.4, 0.3, and 0.8 mm) to obtain direct information on the scattering patterns. The SAXS intensity was collected by a two-dimensional Triton-200 gas-filled X-ray detector (20 cm diameter; 200 mm resolution). An effective scattering vector range of 0.05 nm−1 < q < 4 nm−1 was obtained, where q is the scattering wave vector. All scattering data were dark-current and transmission corrected and the corrected capillary signal was subtracted. The broad-peak model was used in order to adjust the intensity of the scattering signal in the studied q-range, where the intensity profile was8,13,27Here, n is the Porod exponent, q0 and m the center and the exponent of the peak, ξ is usually described as the correlation length in a polymer network, and bkg is the background scattered intensity. From the fitting of the scattering intensity, the d-spacing, corresponding to the broad peak, is a characteristic distance between the scattering inhomogeneities in the polymer network, and can be calculated following the expression
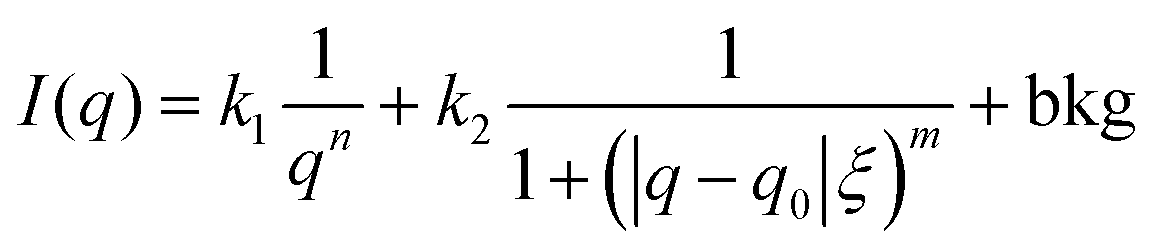
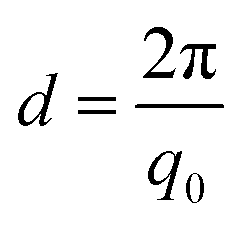
6.3. Visible light transmittance
UV/vis absorbance spectra of fully polymerized samples were recorded and converted to transmittance (3 mL measurement liquid in a cuvette with a path length of 10 mm with Cary 60, Agilent Technologies, United States). The sample absorbance against the baseline of ultrapure water was recorded at room temperature over the entire wavelength range from 190 to 800 nm with a scan rate of 400 nm min−1.The turbidity, τ, of a liquid with particles suspended within it is defined as its transparency as a function of sample thickness, L, and the concentration, N/V, and its scattering cross-section, Csca:
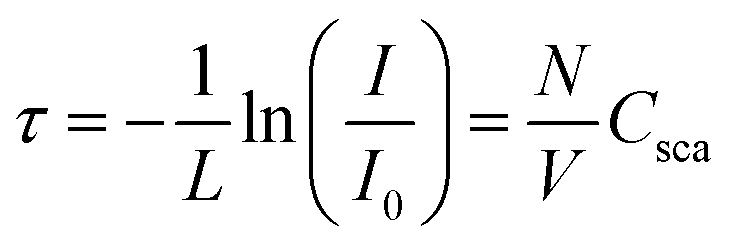
Here, V is the polymer particle volume, λ is the wavelength of light, and np and nm are the refractive indices of particles and medium.

6.4. Scanning electron microscopy
PAAm hydrogels were lyophilized, broken apart, and the broken surfaces sputter-coated with PtPd before SEM imaging. For lyophilization, equilibrium swollen hydrogels were flash frozen. This was carried out in a mixture of liquid ethane and propane, which has a boiling point of −89 °C, instead of liquid nitrogen with a boiling point of −196 °C. The ethane/propane mix was preferred over liquid nitrogen for its higher temperature. It guarantees faster sample freezing, since the Leidenfrost effect is avoided.32 Hydrogel samples were plunged into the ethane/propane mixture for about 10 s, then stored in liquid nitrogen for transportation and subsequently lyophilized. To avoid charging effects during scanning, lyophilized samples were sputtered with a 5 nm PtPd (80/20) layer on a rotating sample holder tilted at 30°.6.5. Swelling
Cylindrical hydrogel samples with a diameter of 10 mm and a height of 9 mm were used. In order to remove uncrosslinked residues from the hydrogel, all samples were allowed to swell in water for one week. After swelling, all samples were lyophilized and their dry weight was determined. Samples were then immersed in deionized water and allowed to swell to equilibrium at room temperature. For weighing, samples were carefully dabbed dry to remove excess water from their surfaces.The swelling ratio, Q, was determined as the ratio of the volume of the equilibrium swollen sample to the volume of the dry sample.1,33 The dry volume of every sample was calculated as Vdry = ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) *mdry with being the partial specific volume of pAAm ( = 0.77 mL g−1; ESI†). For statistics, four samples were evaluated for every cross-linker concentration.
*mdry with being the partial specific volume of pAAm ( = 0.77 mL g−1; ESI†). For statistics, four samples were evaluated for every cross-linker concentration.
6.6. Calculation of the average molecular weight between cross-links, 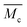
A well-established approach to estimate 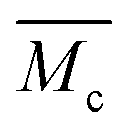
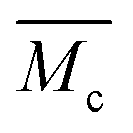 of polymer chains in non-ionic hydrogels comes from Flory's polymer chemistry theory and is based on the volume change upon swelling of hydrogels:34–36
of polymer chains in non-ionic hydrogels comes from Flory's polymer chemistry theory and is based on the volume change upon swelling of hydrogels:34–36Here, V1 is the molar volume of water, ϕ is the number of polymer chains originating from cross-linking junctions in the network, χ is the Flory polymer–solvent interaction parameter,
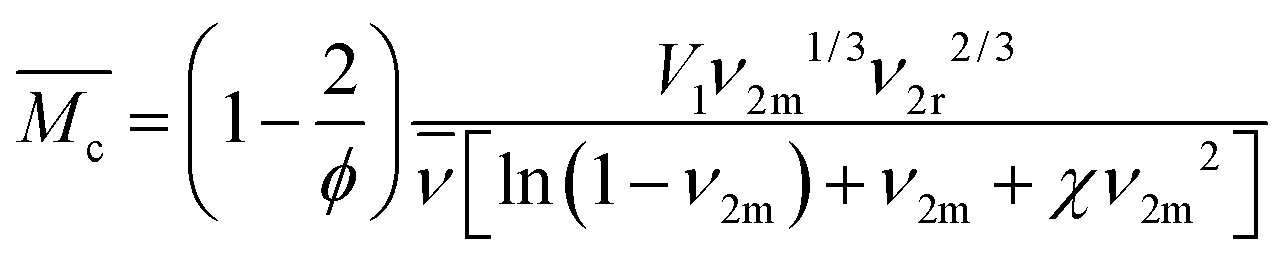 is the specific volume of the polymer, and ν2r and ν2m are the polymer volume fractions in the hydrogel after polymerization and after equilibrium swelling, respectively (see ESI†). Therefore, the parameters with the greatest influence on the chain length in this calculation are the amount of water in the hydrogel and the functionality of cross-linking sites in the polymer network.
is the specific volume of the polymer, and ν2r and ν2m are the polymer volume fractions in the hydrogel after polymerization and after equilibrium swelling, respectively (see ESI†). Therefore, the parameters with the greatest influence on the chain length in this calculation are the amount of water in the hydrogel and the functionality of cross-linking sites in the polymer network.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to Prof. Raffaele Mezzenga for his contributions to this work, which were instrumental in producing the results. Thanks also go to Mr Falk Lucas (ScopeM, ETH Zürich) for his help in SEM sample preparation. Dr Viviane Lutz Bueno is acknowledged for her valuable input on X-ray scattering data interpretation. This work was financed by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement No. 669562).Notes and references
- E. G. Richards and C. J. Temple, Nat. Phys. Sci., 1971, 230, 92 CrossRef CAS.
- Y. Osada and J.-P. Gong, Adv. Mater., 1998, 10, 827–837 CrossRef CAS.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- J. H. Traverse, T. D. Henry, N. Dib, A. N. Patel, C. Pepine, G. L. Schaer, J. A. DeQuach, A. M. Kinsey, P. Chamberlin and K. L. Christman, Basic to Translational Science, 2019, 4, 659 CrossRef.
- H. Yuk, B. Lu, S. Lin, K. Qu, J. Xu, J. Luo and X. Zhao, Nat. Commun., 2020, 11, 1604 CrossRef CAS.
- S. Seiffert, Prog. Polym. Sci., 2017, 66, 1–21 CrossRef CAS.
- S. Seiffert, Polym. Chem., 2017, 8, 4472–4487 RSC.
- M. Lang and T. Müller, Macromolecules, 2020, 53, 498–512 CrossRef CAS.
- P.-G. De Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca, NY, 1979 Search PubMed.
- M. Daoud, J. P. Cotton, B. Farnoux, G. Jannink, G. Sarma, H. Benoit, C. Duplessix, C. Picot and P. G. de Gennes, Macromolecules, 1975, 8, 804–818 CrossRef CAS.
- Z. Yang, Y. Hemar, L. Hilliou, E. P. Gilbert, D. J. McGillivray, M. A. K. Williams and S. Chaieb, Biomacromolecules, 2016, 17, 590–600 CrossRef CAS.
- F. Bode, M. A. da Silva, P. Smith, C. D. Lorenz, S. McCullen, M. M. Stevens and C. A. Dreiss, Soft Matter, 2013, 9, 6986–6999 RSC.
- Center for Neutron Research NIST, https://www.ncnr.nist.gov/staff/hammouda/distance_learning/, 03/20/2020.
- J. L. Nieto, J. Baselga, I. Hernandez-Fuentes, M. A. Llorente and I. F. Piérola, Eur. Polym. J., 1987, 23, 551–555 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- J. Baselga, M. A. Llorente, J. L. Nieto, I. Hernández-Fuentes and I. F. Piérola, Eur. Polym. J., 1988, 24, 161–165 CrossRef CAS.
- H. J. Nachash and O. Okay, J. Appl. Polym. Sci., 1996, 60, 971–979 CrossRef.
- J. Baselga, M. A. Llorente, I. Hernández-Fuentes and I. F. Piérola, Eur. Polym. J., 1989, 25, 471–475 CrossRef CAS.
- D. Calvet, J. Y. Wong and S. Giasson, Macromolecules, 2004, 37, 7762–7771 CrossRef CAS.
- J. Baselga, M. A. Llorente, I. Hernández-Fuentes and I. F. Piérola, Eur. Polym. J., 1989, 25, 477–480 CrossRef CAS.
- P. J. Flory, J. Phys. Chem., 1942, 46, 132–140 CrossRef CAS.
- J. Baselga, I. Hernández-Fuentes, R. M. Masegosa and M. A. Llorente, Polym. J., 1989, 21, 467–474 CrossRef CAS.
- S. M. Paterson, Y. S. Casadio, D. H. Brown, J. A. Shaw, T. V. Chirila and M. V. Baker, J. Appl. Polym. Sci., 2013, 127, 4296–4304 CrossRef CAS.
- W. M. Kulicke and H. Nottelmann, Polymers in Aqueous Media, American Chemical Society, 1989, vol. 2, pp. 15–44 Search PubMed.
- B. Hammouda, D. L. Ho and S. Kline, Macromolecules, 2004, 37, 6932–6937 CrossRef CAS.
- R. J. Nussbaumer, PhD Dissertation, ETH Zürich, 2004.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- H. C. V. D. Hulst, Light scattering by small particles, John Wiley & Sons, New York, 1957 Search PubMed.
- M. Kerker, The scattering of light and other electromagnetic radiation, Academic Press, New York, 1969 Search PubMed.
- J. G. Leidenfrost, Int. J. Heat Mass Transfer, 1966, 9, 1153–1166 CrossRef CAS.
- C.-C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, Ithaca NY, 1953 Search PubMed.
- J. E. Mark and B. Erman, Rubberlike elasticity: a molecular primer, Cambridge University Press, Cambridge, 2007 Search PubMed.
- M. Sen, A. Yakar and O. Guven, Polymer, 1999, 40, 2969–2974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01536a |
| This journal is © The Royal Society of Chemistry 2020 |