
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Layered double hydroxide-based antioxidant dispersions with high colloidal and functional stability†
Adél
Szerlauth
,
Szabolcs
Muráth
 and
Istvan
Szilagyi
*
and
Istvan
Szilagyi
*
MTA-SZTE Lendület Biocolloids Research Group, Interdisciplinary Excellence Centre, Department of Physical Chemistry and Materials Science, University of Szeged, 1 Rerrich Béla tér, H-6720 Szeged, Hungary. E-mail: szistvan@chem.u-szeged.hu
First published on 28th September 2020
Abstract
Highly stable antioxidant dispersions were designed on the basis of ring-opened ellagic acid (EA) intercalated into MgAl-layered double hydroxide (LDH) nanoparticles. The morphology of the composite was delicately modified with ethanolic washing to obtain EtOH–EA–LDH with a high specific surface area. The colloidal stability was optimized by surface functionalization with positively charged polyelectrolytes. Polyethyleneimine (PEI), protamine sulfate (PS) and poly(acrylamide-co-diallyl dimethyl ammonium chloride) (PAAm-co-DADMAC) was adsorbed onto the surface of the oppositely charged EtOH–EA–LDH leading to charge neutralization and overcharging at appropriate doses. Formation of adsorbed polyelectrolyte layers provided remarkable colloidal stability for the EtOH–EA–LDH. Modification with PEI and PAAm-co-DADMAC outstandingly improved the resistance of the particles against salt-induced aggregation with a critical coagulation concentration value above 1 M, while only limited stability was achieved by covering the nanoparticles with PS. The high antioxidant activity of EtOH–EA–LDH was greatly preserved upon polyelectrolyte coating, which was proved in the scavenging of radicals in the test reaction applied. Hence, an active antioxidant nanocomposite of high drug dose and remarkable colloidal stability was obtained to combat oxidative stress in systems of high electrolyte concentrations.
1. Introduction
Antioxidants are the cornerstone defence systems against naturally occurring oxidative species, which can induce a variety of illnesses that originate from oxidative stress.1,2 In addition, it is important to prevent the unwanted oxidative degradation of foods,3,4 paints5 or cosmetics,6,7 since it leads to lower quality and rapid expiration of these commercial products. Undoubtedly, antioxidant enzymes (e.g., peroxidases, catalase and superoxide dismutase)8 are the most effective tools to suppress such deterioration. Besides, numerous composites were developed by the combination of inorganic particles used as carriers and immobilized enzymatic or enzyme mimicking compounds to overcome the limitations of the native enzymes.9–11Since the working conditions of enzymes are strictly regulated by multiple factors (temperature, pH, ionic strength, etc.), the use of non-enzymatic, with another term molecular antioxidants is also a popular choice.3,4,12,13 However, the application of molecular antioxidants is often unfavoured due to their limited efficiency compared to the enzymes, low water solubility and considerable chemical reactivity. To deal with these issues, the development of nanocomposites of antioxidant substances with solid supports were the focus of research groups in the past.14 As solid carriers, various compounds have been considered, e.g., titanium dioxide,15,16 graphene oxide,17,18 aluminosilicates19,20 and layered double hydroxides (LDHs).21–23
The latter substances offer a large selection of materials that can be perceived as mixed metal hydroxides containing divalent and trivalent cations.24,25 The positively charged LDH layers consist of hydroxide ions and coordinated metal cations, which most commonly are Mg2+, Ca2+, Ni2+, Cr3+, Al3+ or Fe3+.26 LDHs are excellent anion exchangers with anions situated (i.e., intercalated) in the interlamellar space giving rise to practically countless possibilities of their composition.25,27 In this way, molecular antioxidants were immobilized in the interlamellar space and/or on the outer surface of LDHs. LDH-based composite materials containing chlorogenic acid,22 curcumin,23,28 folic acid,29 gallate30 and phenolic compounds31 of remarkable antioxidant activities were prepared and used to reduce oxidative stress in biomedical and industrial applications. Owing to the interchangeable anions, the surface charge of LDHs can be either negative (with anions of higher negative charge) or positive (with anions of lower charge), which is an important aspect of colloidal stability studies.
Despite the fact that nanoparticle-based antioxidants are usually used in heterogeneous systems, i.e., the particles are dispersed in a liquid, the colloidal stability of these systems have been rarely assessed. In turn, the activity of the materials strongly depends on the surface area, which can be improved by stabilization of the nanocomposites in the dispersions. Synthetic (e.g., polyacrylates,32 polyamines33 and polystyrenes34) and natural (e.g., polypeptides,35 polysaccharides36 and humic substances37) polyelectrolytes have been proven to be excellent stabilizing agents for nano- or colloidal particles in various applications including drug delivery,38 wastewater treatment39 and dye production.40 Comprehensive studies carried out using surface sensitive techniques have revealed that their adsorption on oppositely charged surfaces is rather strong and irreversible.41–43 These phenomena are accompanied by charge neutralization and overcharging in most of the cases.44 The aggregation mechanisms in the dispersions are affected by the surface charge features and rapid particle aggregation was usually reported at doses close to the charge neutralization point, while highly charged particle–polyelectrolyte systems form more stable dispersions, i.e., the rate of particle aggregation is slow or not even detectable due to the high colloidal stability.45,46
Similar observations were also made in LDH–polyelectrolyte systems.47 Accordingly, highly stable polyacrylate–LDH composites were developed by applying the appropriate dose in the dispersions.48 Surface charge properties and subsequent aggregation of LDH particles were extensively studied in the presence of biologically relevant media and the importance of protein corona formation was emphasized.49 The colloidal stability and cellular uptake were improved by coating the LDH particles with albumin macromolecules, making the developed composite promising in drug delivery applications.50 The dispersion stability of LDH–enzyme hybrids was remarkably improved by the formation of polysaccharide layers leading to fine particle dispersions with excellent enzymatic activities.51,52 Polyelectrolyte multilayers were built-up on the outer particle surfaces and used to design delivery systems with high colloidal stability.53,54 Stabilization of LDH–molecular antioxidant composites by polyelectrolyte adsorption was not reported in the past, although this opportunity shows great promise towards the development of highly stable antioxidant dispersions.
In our earlier work,27 we prepared MgAl-LDH intercalated with ellagic acid (EA, see Fig. 1a for the structure), a natural antioxidant with excellent radical scavenging activity.55 The developed product proved to be an efficient antioxidant material of negative surface charge. In the present contribution, we have aimed to functionalize its surface with positively charged polyelectrolyte layers to tune the colloidal stability and thus, to improve the composite's applicability in aqueous systems, which are the most common media in the areas where antioxidant systems are required. Three cationic polyelectrolytes (polyethyleneimine (PEI), protamine sulfate (PS) and poly(acrylamide-co-diallyl dimethyl ammonium) (PAAm-co-DADMAC)) were considered as surface modifying agents. The colloidal behaviour and the antioxidant activity of the bare and coated composites were investigated using light scattering techniques and a spectrophotometric assay, respectively.
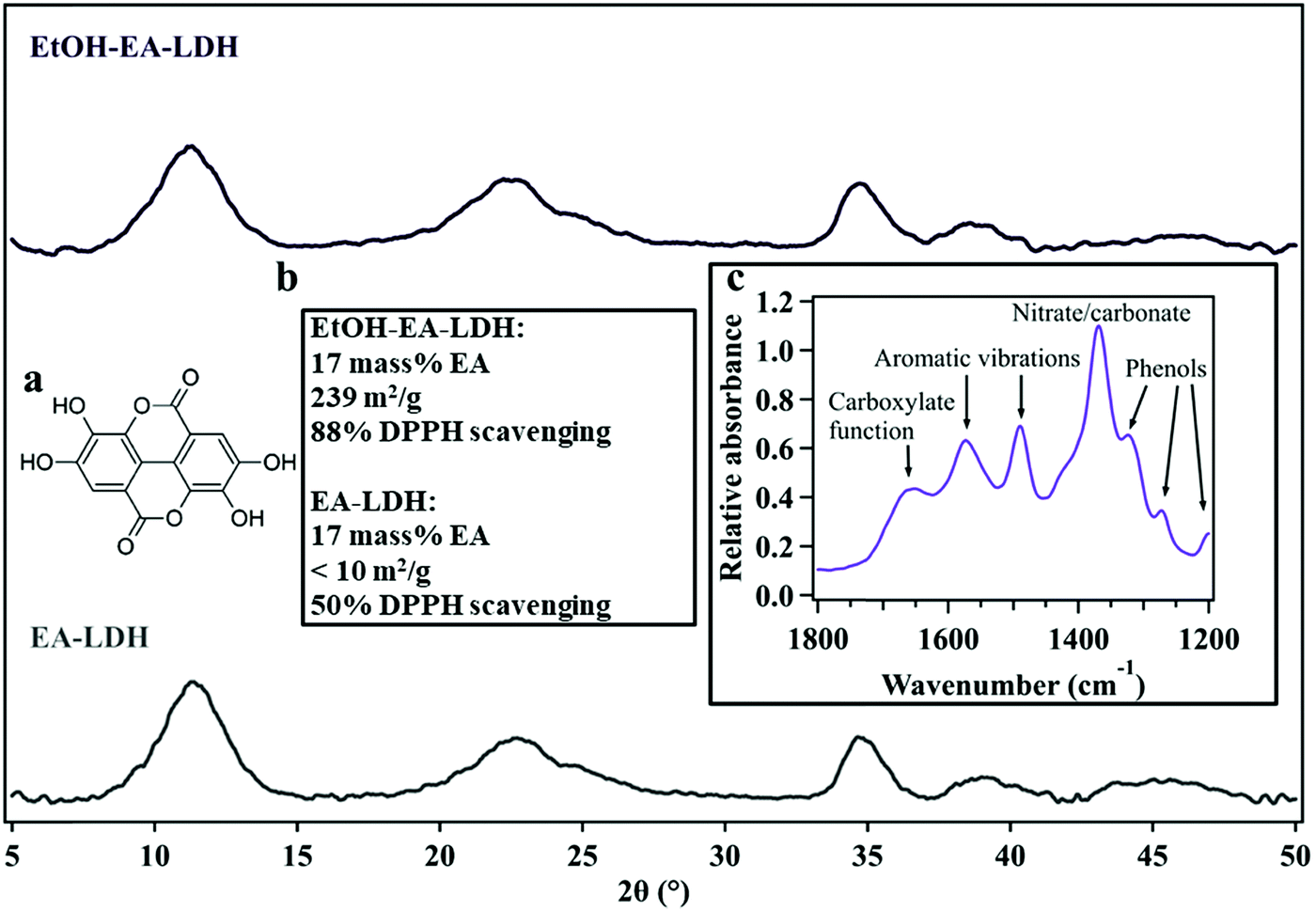 | ||
| Fig. 1 The powder X-ray diffraction patterns of EtOH–EA–LDH (upper) and EA-LDH (lower). Insets show (a) the molecular structure of EA, (b) composition, BET specific surface area and DPPH scavenging activity of the LDHs and (c) the snippet of a typical IR spectrum of EA-LDHs indicating the presence of ring-opened EA in the materials. The data are taken from ref. 27. | ||
2. Experimental
2.1. Materials
For the synthesis of particles, analytical grade Mg(NO3)2·6H2O, Al(NO3)3·9H2O, 4 M NaOH solution, anhydrous EtOH and ellagic acid (EA) were purchased from VWR International. For surface functionalization, PEI (ca. 30% w/v solution, branched, molecular weight of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, catalogue no. 40529 from VWR International), PS (powdered, ca. 22.5% nitrogen content, from salmon, catalogue no. ICN19472905 from Fisher Scientific), and PAAm-co-DADMAC (ca. 10 wt% solution, molecular weight of 250000 g mol−1, ca. 55% acrylamide content, catalogue no. 409081 from Sigma-Aldrich) were used. The molecular structure of the polyelectrolytes is shown in Fig. S1 (ESI†). The ionic strength was adjusted with NaCl (min. 99.8%, from VWR International). For the antioxidant assays, anhydrous MeOH and 2,2-diphenyl-1-picrylhydrazyl free radicals (DPPH) were acquired from VWR International. Water was purified with a Puranity TU 3+ UV/UF system equipped with a UV irradiation unit (VWR). For the light scattering experiments, water was filtered through a hydrophilic syringe filter with 100 nm pore size (Millex). The pH of all solutions and dispersions used was adjusted to 9 with dilute NaOH solutions.
000 g mol−1, catalogue no. 40529 from VWR International), PS (powdered, ca. 22.5% nitrogen content, from salmon, catalogue no. ICN19472905 from Fisher Scientific), and PAAm-co-DADMAC (ca. 10 wt% solution, molecular weight of 250000 g mol−1, ca. 55% acrylamide content, catalogue no. 409081 from Sigma-Aldrich) were used. The molecular structure of the polyelectrolytes is shown in Fig. S1 (ESI†). The ionic strength was adjusted with NaCl (min. 99.8%, from VWR International). For the antioxidant assays, anhydrous MeOH and 2,2-diphenyl-1-picrylhydrazyl free radicals (DPPH) were acquired from VWR International. Water was purified with a Puranity TU 3+ UV/UF system equipped with a UV irradiation unit (VWR). For the light scattering experiments, water was filtered through a hydrophilic syringe filter with 100 nm pore size (Millex). The pH of all solutions and dispersions used was adjusted to 9 with dilute NaOH solutions.
2.2. LDH synthesis
The preparation of the EA-loaded Mg2Al-LDH (simply EA-LDH) and its organic solvent modified variant (EtOH–EA–LDH) is described elsewhere.27 Briefly, 25 mL solution of Mg(NO3)2 and Al(NO3)3 was added to the solution of EA in 4 M NaOH under vigorous stirring. The EA-to-Mg-to-Al molar ratio was 1:8:4 and a final pH value of 13 was reached. The precipitate was separated by centrifugation after 24 h at 4200 rpm (2090 rcf for 10 min in an Orto Alresa Unicen 21 centrifuge), followed by washing with water (3 × 15 mL) and drying at 50 °C overnight. Furthermore, EtOH treatment was applied to modify the surface properties of the material. For this, EA-LDH was prepared as detailed above. After the aqueous washing step, the wet product was stirred in 25–25 mL fresh EtOH for 120 and 60 min, respectively. The material was centrifuged between the two EtOH treatments and at the end of the synthetic procedure (4200 rpm (2090 rcf) for 20 min). The final drying was performed at 50 °C overnight.
2.3. Characterization methods
Electrophoretic mobility was measured on a LiteSizer 500 (Anton Paar) instrument equipped with a 40 mW laser source (λ = 658 nm). The measurements were executed in Ω-shaped plastic cuvettes (Anton Paar) with 400 μL total volume. During sample preparation, calculated amounts of water, polyelectrolyte and NaCl solutions were mixed with the LDH stock suspensions to achieve the appropriate polyelectrolyte doses and ionic strengths. The samples were prepared 1 day before the electrophoretic measurements. The final particle concentration was set to 10 mg L−1 in each experiment.Dynamic light scattering (DLS) was used to determine the size of the dispersed LDH particles. The measurements were carried out in a LiteSizer 500 device in backscattering mode at a 175° angle. The cumulant method was used to fit the correlation functions, which were collected for 20 seconds to obtain the apparent hydrodynamic radius of the particles using the Stokes–Einstein equation (eqn (S1), see the ESI†).56 The measurements were carried out using the same method as that used for electrophoretic mobility determination with the exception that analyses were run immediately after mixing the components. Colloidal stability was expressed by means of stability ratio (eqn (S2), ESI†).57,58 This value is unity if particle aggregation is diffusion controlled and greater than one if aggregation is slower, and thus the dispersion is more stable. We remark that the stability ratios of the EtOH–EA–LDH based samples at each ionic strength were obtained as the proportion of the appropriate aggregation rate and the fast aggregation rate of EtOH–EA–LDH, while for EA-LDH, the same principle was applied using the fast aggregation rate of EA-LDH for the normalization.
The morphology of the samples with saturated polyelectrolyte layers was examined in 10 mg L−1 dispersions using transmission electron microscopy (TEM). The particles were fixed on a copper-coated carbon mesh TEM grid and were observed using a FEI TECNAI G2 20 X-TWIN instrument with 200 kV accelerating voltage.
2.4. Antioxidant capacity assays
The standard DPPH-based test reaction59 was used to estimate the antioxidant activity of the materials. The general reaction route of the test is shown in Scheme S1 (ESI†). The DPPH-type activity was obtained after mixing 3500 μL of 60 μM methanolic DPPH solution with the dispersion containing the antioxidant material setting a range of DPPH-to-EA ratios. The decrease of absorbance at λ = 517 nm corresponds to the reduction of DPPH radicals, which was observed for 120 min for the polyelectrolyte-coated composites on a Thermo Fisher Genesys 10S dual beam spectrophotometer. The remainder of the DPPH radicals was calculated by Lambert–Beer's law (eqn (S3), ESI†). The absorbance values at each concentration ratio were corrected by the absorbance of the dispersed particles, as they had a slight contribution due to light scattering phenomena. The efficient concentration (EC50) value is the antioxidant concentration at which half of the initial DPPH radicals decompose and was calculated from the fits performed on the remaining DPPH percentage data at different antioxidant concentrations.3. Results and discussion
3.1. General remarks
The solid state characterization of the bare (uncoated) LDHs investigated in this study (EA-LDH and EtOH–EA–LDH) was carried out earlier, including their structural features and antioxidant content.27 Briefly, the XRD patterns showed single phase materials bearing close resemblance to the other LDHs (Fig. 1)24 indicating that no structural damage was observed after EA immobilization and high loading of the antioxidant.Furthermore, the radical scavenging activity of these materials was also assessed. Both materials had a high antioxidant effect in the DPPH test with EtOH–EA–LDH having superior properties (Fig. 1b). It was correlated with the higher specific surface area of the EtOH-modified LDH that made it more facile for the solute DPPH compound to react with the immobilized EA. The underlying mechanism of these observations is a modified hydrogen-bonding system after ethanolic washing and probable low-degree exfoliation, which is a consequence of the partial replacement of structural water molecules by alcohols. The infrared spectra of EA-loaded LDHs attested the presence of organic moieties, although it was found that not EA, but its opened form (4,4′,5,5′,6,6′-hexahydroxydiphenic acid) was intercalated (Fig. 1c). For simplicity reasons, EA-LDH is used to abbreviate these composites.
Negligible release was detected from the carrier and the ethanolic treatment reduced the particle size (Table 1), which is beneficial for potential future applications. Combining the results, we chose EtOH–EA–LDH as a starting material for surface tuning to achieve high colloidal stability. Note that other organic solvents were also applied and EtOH was the most effective as a surface and textural modifier.
| Sample | R h (nm) | σ (mC m−2) | IEPc (mg g−1) | ASPc (mg g−1) | CCCd (mM) | EC50e (10−5 M) |
|---|---|---|---|---|---|---|
| a R h is the apparent hydrodynamic radius of the particles determined by DLS in stable dispersions. The measurement error was ±5 nm. b Surface charge density (σ) was calculated at the slip plane using eqn (1). c Isoelectric point (IEP) and adsorption saturation point (ASP) were determined from the zeta potential versus polyelectrolyte dose graphs. The dimension mg g−1 corresponds to the mass ratio between the polyelectrolyte and the solid particle. These values were determined on the basis of electrokinetic measurements, in which the average error was 5%. d The critical coagulation concentration (CCC) was calculated using eqn (2). e The effective concentration (EC50) values were determined in the DPPH test with an average error of 3%. f These data were taken from ref. 27. | ||||||
| EA-LDH | 317 | −5 | — | — | 21 | 8.73f |
| EtOH–EA–LDH | 120 | −20 | — | — | 65 | 1.98f |
| EtOH–EA–LDH/PEI | 162 | 16 | 9 | 150 | >1000 | 4.46 |
| EtOH–EA–LDH/PS | 142 | 6 | 30 | 200 | 10 | — |
| EtOH–EA–LDH/PAAm-co-DADMAC | 191 | — | 330 | 700 | >1000 | 5.06 |
3.2. Functionalization of EtOH–EA–LDH with polyelectrolytes
As lamellar compounds consisting of positively charged layers, LDHs with simple anions bearing one negative charge generally possess positive overall surface charge.24 However, intercalated EA has multiple negative charges that result in a gross negative surface charge for the EtOH–EA–LDHs. Hence, positively charged PEI, PS and PAAm-co-DADMAC were used as cationic polyelectrolytes to improve the colloidal stability of the EtOH–EA–LDH for the efficient decomposition of harmful radicals. Branched PEI has a high line charge density owing to its frequently abundant primary and secondary amino groups, which are partially protonated under the experimental conditions applied. PS is a small protein with numerous basic side chains of arginine amino acids. PAAm-co-DADMAC is a quaternary polyammonium salt and it also contains non-charged PAAm blocks in its chain possessing, therefore, the lowest line charge density. These polyelectrolytes have been proven as effective stabilizing or aggregating agents for colloidal particles;33,35,46,60,61 however, no systematic colloidal stability studies were reported for LDHs. To assess the charging features at the slip plane, the electrophoretic mobilities were converted to zeta potentials using eqn (S4) ESI.†Initially, the effect of polyelectrolyte dose on the zeta potential was considered (Fig. 2a). Particle concentration (10 mg L−1), pH (9) and the background electrolyte level (1 mM NaCl) was kept constant, i.e., only the polyelectrolyte dose was changed in the samples. At low polyelectrolyte doses, in general, the original zeta potential of EtOH–EA–LDH had a value of about −20 mV. Increasing the polyelectrolyte concentration led to higher potentials with a steep increase into the positive regime of the recorded curves. This sharp change indicated the adsorption of polyelectrolytes onto the EtOH–EA–LDH particles. Eventually, the negative charge of the support LDH was neutralized by the adsorbed polyelectrolytes, which is indicated as IEP (isoelectric point) in Fig. 2a. Further adsorption of the polyelectrolytes led to the formation of coated particles with gross positive charge. At high doses, the curves reached a plateau, which corresponds to a saturated polyelectrolyte layer on the EtOH–EA–LDH surface. This is indicated as ASP (adsorption saturation point) in Fig. 2a. Addition of further polyelectrolyte only increases its concentration in the bulk, as adsorption does not occur after ASP. Note that added polyelectrolytes below the ASP were strongly adsorbed on the oppositely charged particles, i.e., no free polyelectrolyte could be found in the solution phase.46 Similar charging behaviour has been reported earlier for oppositely charged particle–polyelectrolyte systems,32,35,45 including LDH particles.10,47,48,51,52
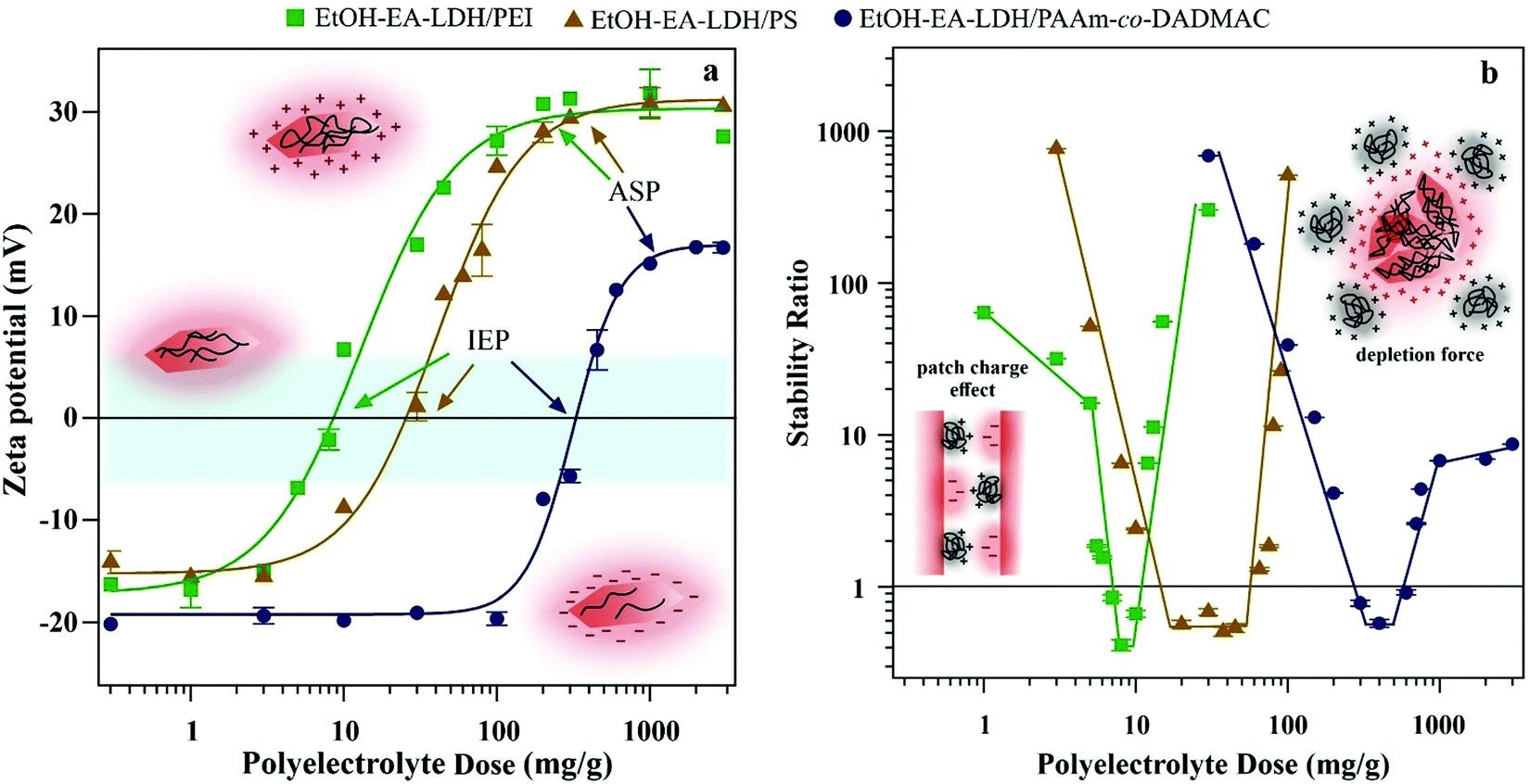 | ||
| Fig. 2 (a) Zeta potential and (b) stability ratio of EtOH–EA–LDHs as a function of the polyelectrolyte dose. Insets show (a) the charging properties of the LDH nanoparticles in different polyelectrolyte concentration regimes and (b) the schematic model of patch–charge effect and depletion force. Particle concentration was 10 mg L−1 at pH 9 and 1 mM ionic strength adjusted with NaCl. The mg g−1 unit means mg of polyelectrolyte per one gram of EtOH–EA–LDH. The solid lines are just a guide to the eye. | ||
Beside these generic findings in the tendency of the zeta potentials, system specific effects were also observed. Both IEP and ASP values increased in the PEI < PS < PAAm-co-DADMAC order (Table 1). Upon careful inspection it is observed that these numbers are not random, as polyelectrolytes with higher line charge density have lower IEP and ASP values. Indeed, it was reported earlier46,60 that highly charged polyelectrolytes adsorb in smaller amounts due to the electrostatic repulsion between the adsorbed chains leading to the formation of polyelectrolyte islands on the surface. However, such a repulsion is much smaller for weakly charged polyelectrolytes giving rise to higher adsorbed amounts. Note that no experimental evidence was found for the influence of the structure of the polyelectrolytes (branched or linear) on the adsorption mechanism.
The aggregation of the polyelectrolyte-modified EtOH–EA–LDHs was followed at different doses by DLS. The time-dependent data of EtOH–EA–LDH coated with polyelectrolytes at three distinct doses (a small and high dose that resulted in slow or no aggregation and one intermediate that induced fast aggregation) are shown in Fig. S2 (ESI†). At the lowest dosages, the particle size is constant within experimental error throughout the course of measurements. Increasing the dose leads to aggregation, which was detected from 5 mg g−1 (PEI), 10 mg g−1 (PS) and 200 mg g−1 (PAAm-co-DADMAC). This slower aggregation turned into a fast aggregation regime close to IEP values, where the dispersions were unstable. Increasing the dose resulted in stable suspensions through the overcharging characteristics of the polyelectrolytes; however, its extent was the most moderate for PAAm-co-DADMAC, which has the lowest line charge density.
Such behaviours lead to classical U-shaped curves of the stability ratios (see eqn (S2) in the ESI†), which were observed for the three polyelectrolytes (Fig. 2b). The DLVO theory (by Derjaguin, Landau, Verwey and Overbeek)44,46,62 served as a qualitative tool to describe such systems. Before and after IEP, where surface charge is significant, repulsive electrical double layer forces prevent the aggregation of the particles and thus, stable dispersions are formed. However, at IEP, van der Waals attractive forces dominate, since repulsive forces of electrostatic origin do not exist at charge neutralization.
Certain deviations from DLVO theory were observed in our measurements. Under a 5 mg g−1 PEI dose, the slope of the stability curve is lower than expected by DLVO theory, which is due to the patch–charge effect (see Fig. 2b inset for schematic representation).44,46,61 Accordingly, the absorbed PEI forms positive islands on the surface of the LDHs that can attract the bare surface of other partially covered LDH particles. Overall, these forces have a slight negative effect on colloidal stability since it acts as an additional attraction among van der Waals interactions. A similar patch–charge effect can explain that in the fast aggregation regime, the stability ratio is less than unity. Accordingly, aggregation is not merely diffusion limited under these conditions, since patch–charge attractive forces accelerate dimer formation. Note that this non-DLVO force is present only at partial surface coverage. The assumption of the presence of patch–charge attraction is based on the tendency in the stability ratio data and on the comparison of results from other particle–polyelectrolyte systems. These forces were directly detected by the atomic force microscopy-based colloidal probe techniques earlier;44 however, our system is not suitable for such measurements.
A non-DLVO destabilizing force appears also at a high dosage of PAAm-co-DADMAC. This is the so-called depletion force (see Fig. 2b inset for schematic representation), which can usually be observed at high polymer concentrations irrespective of the charge of the macromolecules and the particles.62 These forces originate from the difference in the osmotic pressure within the gap between two approaching particles and the bulk solution. Once the particle separation distance is smaller than the size of the polymer, an attractive interparticle force will appear.46
The potential effect of polyelectrolyte adsorption at ASP on the particle morphology was investigated by TEM. All EtOH–EA–LDH particles shared the same motifs, independent of the presence or absence of polyelectrolytes. The materials consisted of particles with irregular shape that aggregated into a mass on the TEM grid. The example of EtOH–EA–LDH and EtOH–EA–LDH/PAAm-co-DADMAC is shown in Fig. 3, while the TEM micrographs from all materials are collected in Fig. S3 (ESI†).
 | ||
| Fig. 3 TEM micrograph of EtOH–EA–LDH (left) and EtOH–EA–LDH/PAAm-co-DADMAC with 700 mg polyelectrolyte per 1 g of LDH dosage (right). Scale bars represent 100 nm. | ||
3.3. Salt-induced aggregation of the polyelectrolyte-coated EtOH–EA–LDH
Since antioxidant efficiency is typically assessed in systems with dissolved electrolytes (e.g., biofluids and industrial liquors), the resistance of the polyelectrolyte-functionalized particles against salt-induced aggregation was assessed. Polyelectrolyte doses were set to ASP equivalent values. First, we explored the surface charge characteristics of the composites at different ionic strengths (Fig. 4a).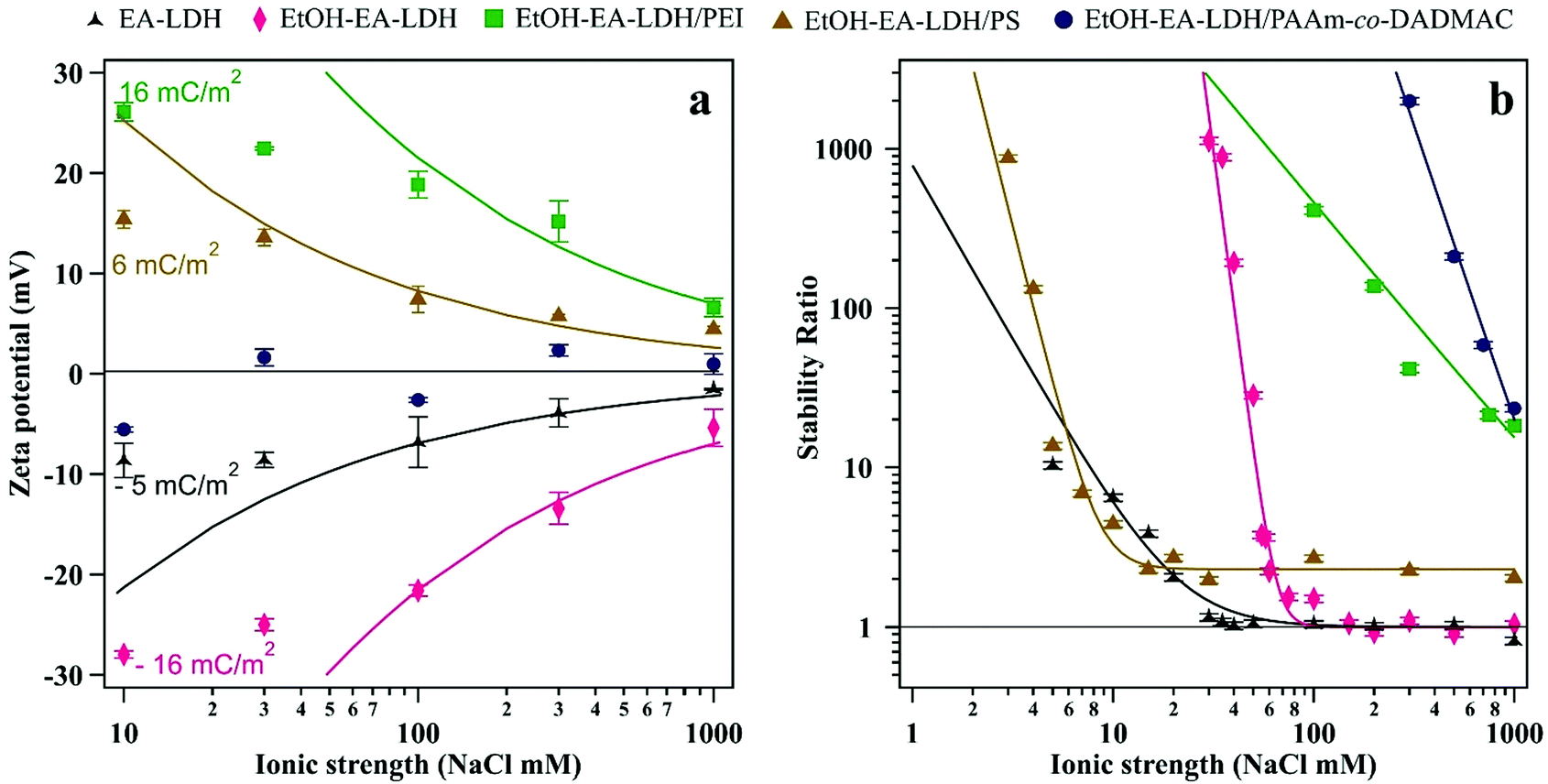 | ||
| Fig. 4 (a) Zeta potential and (b) stability ratio of the bare and polyelectrolyte-coated LDH particles. Particle concentration was maintained at 10 mg L−1. Particles carried polyelectrolyte at doses corresponding to the ASP (shown in Table 1). The solid lines were calculated using eqn (1) in (a) and eqn (2) in (b). | ||
One can see that the absolute value of zeta potentials decreased with the ionic strength, as the corresponding counterions from NaCl screen the charge of the dispersed particles, without causing charge inversion. The only exception was EtOH–EA–LDH/PAAm-co-DADMAC, which possessed close to zero charge at the slip plane in the regime of ionic strength screened. This is due to the abundant neutral acrylamide monomer units in the polymer chain. Note that these results imply that the surface charge is compensated at the slip plane by the neutral part of the copolymer, but the inner surface charge, which is not assessed in the electrokinetic measurements, can still be considerably high. The ionic strength–zeta potential correlation was fitted with the Gouy–Chapman equation to obtain the surface charge density (σ) at the slip plane of the electrical double layer as46
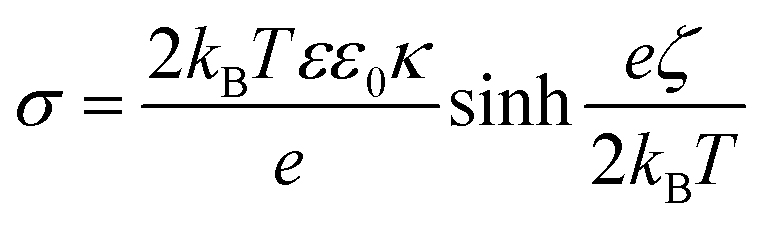 | (1) |
Both bare LDHs are negatively charged and possess low charge (σ is −5 and −16 mC m−2 for EA-LDH and EtOH–EA–LDH, respectively), which is typical for natural inorganic particles.35,47,60 Similarly, relatively low magnitude of charge was determined for the EtOH–EA–LDH/PEI (+16 mC m−2) and EtOH–EA–LDH/PS (+6 mC m−2), while the former one is slightly higher due to the higher abundance of the protonated primary amino groups. Note that these charges were determined at the slip plane and that counterion condensation contributed to this low charge.63,64 As detailed earlier, functionalization with PAAm-co-DADMAC yielded a material of a net charge close to neutral irrespective of the ionic strength applied.
The colloidal stability of the particles was studied and it was found that the velocity of aggregation increases with the NaCl concentration leading to a progressive decrease in the stability ratio values (Fig. 4b). The initial size of the particles was constant at low ionic strengths, meanwhile, high NaCl resulted in rapidly aggregating particles. Such a trend follows the predictions of the DLVO theory. The critical coagulation concentration (CCC), i.e., the NaCl concentration, where electrical double layer forces disappear and van der Waals forces start dominating was defined as58
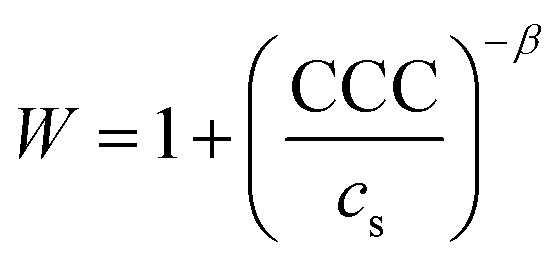 | (2) |
 | (3) |
In general, the different surface charge density of the materials translated into significantly different CCC values (Table 1). Regarding the uncoated LDHs, it was observed that EtOH–EA–LDH (65 mM) had a higher CCC than EA-LDH (21 mM) due to its higher surface charge density. In addition, the slope of the slow aggregation regime of EA-LDH is smaller, which is correlated with its less pronounced negative surface charge and thus, weaker repulsion between the particles. However, these features are typical for bare LDH materials dispersed in monovalent electrolyte solutions.47,65,66
Out of the polyelectrolytes, PS lowered the CCC value of EtOH–EA–LDH to 10 mM. One can explain this using the structural property of PS being a flat-like molecule that forms a thin and weakly charged layer on the surface. The magnitude of the surface charge and hence, the strength of the double layer repulsion is smaller than for the bare particles. These facts give rise to a lower CCC value.
On the other hand, coating EtOH–EA–LDH with PEI and PAAm-co-DADMAC provided dispersions with superior resistance against salt-induced aggregation. The fast aggregation regime of these polyelectrolytes were not reached before 1 M NaCl concentration. The outstanding value of EtOH–EA–LDH/PEI was the consequence of joint repulsive steric and electrostatic forces. The former originates from the overlapping of the adsorbed polyelectrolyte chains and subsequent increase of osmotic pressure.67 The latter is the double layer repulsion, discussed above. This so-called electrosteric stabilization provided extremely high colloid stability for the EtOH–EA–LDH/PEI. Although similar stabilization mechanisms were reported in other particle–polyelectrolyte systems,32,37,40 such a high CCC was rarely reported. Concerning the EtOH–EA–LDH/PAAm-co-DADMAC particles, the fact that the zeta potentials were close to zero at all ionic strengths indicated the absence of double layer forces. They were reported as long-ranged forces in direct force measurements,44,46 and therefore, considerable charge should be detected in the electrokinetic measurements, if these forces are present. Considering this fact, it was assumed that the extremely high CCC of the EtOH–EA–LDH/PAAm-co-DADMAC is the consequence of strong steric repulsion between the coated particles. Such steric forces are usually more pronounced at higher ionic strengths, at which the adsorbed polyelectrolyte layer is swollen giving rise to the formation of polymeric tails and loops on the surface.68
The different modes of stabilization are represented in the apparent hydrodynamic radii (Table 1) data. Accordingly, among the ethanol-treated LDHs, the uncoated material possessed the lowest particle size (120 nm), while the radius was increased to 142 nm after PS adsorption, to 162 nm after PEI adsorption and to 191 nm after PAAm-co-DADMAC adsorption. These values indicate a thicker polyelectrolyte layer on the particles in this order, which enhances the steric repulsion due to the formation of polyelectrolyte tails and loops on the surface. As mentioned above, this effect can be even enhanced by increasing the ionic strength. Although the tendency in the apparent hydrodynamic radii clearly support the above assumption, note that the increment value in the radii is higher than the ones usually reported for thicknesses of adsorbed polyelectrolyte layers.46,68 This is due to the fact that upon formation of the polyelectrolyte layer, the particles pass through the IEP for a very short time period, under which some aggregation may occur leading to higher apparent hydrodynamic radii for the coated particles and hence, to higher layer thickness values. This effect was earlier reported in other particle–polyelectrolyte systems, too.68
3.4. Radical-scavenging activity of the polyelectrolyte-coated LDHs
After assessing the colloidal properties of the materials, their DPPH scavenging activity was determined. The bare EA-LDH and EtOH–EA–LDH had 50 and 88% total scavenging activity with 8.73 × 10−5 and 1.98 × 10−5 M EC50 values determined in the assay, respectively. The EC50 (effective concentration) is the initial concentration of intercalated EA in the composites to decompose 50% of the DPPH radicals and is a common tool to quantify the material's antioxidant efficiency. In a typical test, the decrease in DPPH concentration was observed until steady state at various EA concentrations. These concentrations were calculated from the EA-content of the LDHs. The results of the antioxidant assays are shown in Fig. 5.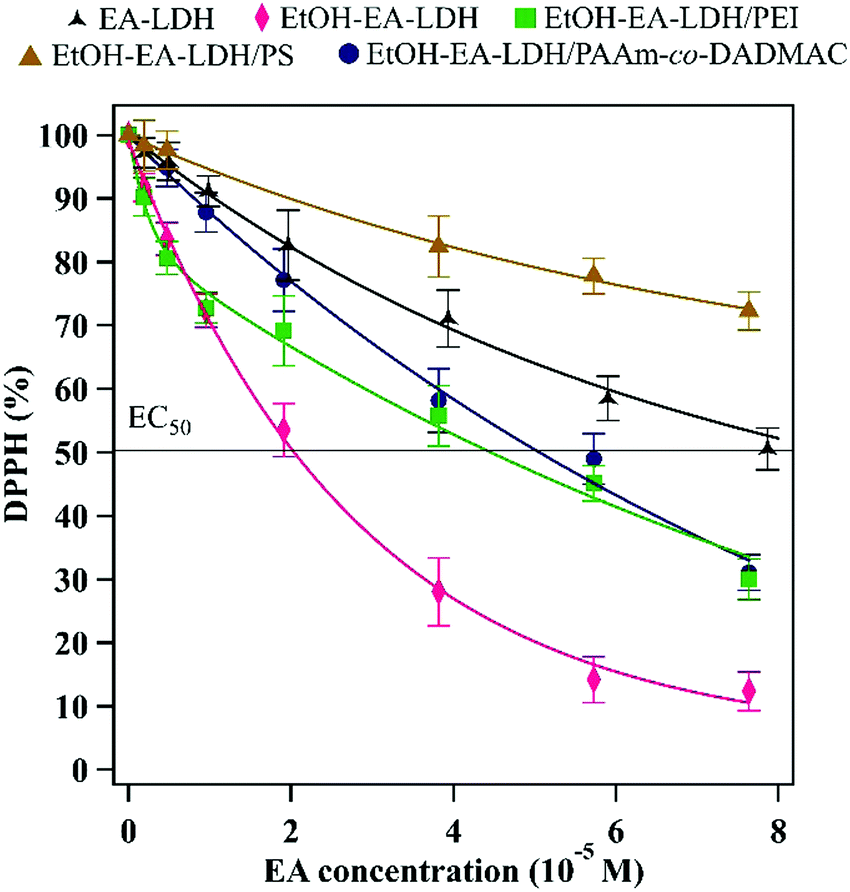 | ||
| Fig. 5 DPPH scavenging activity of the bare and polyelectrolyte-coated LDH materials. | ||
It was observed that in contrast to the uncoated LDHs that expressed their potential in 60 min (Fig. S4A, ESI†), i.e., the steady state of the reaction was reached, the scavenging of DPPH radicals was not finished in this time period and hence, 120 min were allowed for the polyelectrolyte-functionalized EtOH–EA–LDHs to decompose a considerable amount of DPPH (Fig. S4B, ESI†) in the assays. A plausible explanation for this might be the hindrance of the intercalated EA molecules due to the polyelectrolyte layers on the surface of the particles that results in their reduced accessibility for the reacting DPPH radicals. However, this phenomenon provides a long-term radical scavenging activity for the material.
Besides the reaction time, the polyelectrolyte coatings also altered the DPPH scavenging effectiveness of the EtOH–EA–LDH (Table 1). Similar activity was assessed for EtOH–EA–LDH/PEI and EtOH–EA–LDH/PAAm-co-DADMAC with 70 and 69% total scavenging and 4.46 × 10−5 M and 5.06 × 10−5 M EC50 values, respectively. Although, the overall activity of the materials was below that of EtOH–EA–LDH, they showed high activity compared to the literature values of antioxidant-modified LDHs.69–71 EtOH–EA–LDH/PS proved to be the least effective, underachieving untreated EA-LDH with less than 50% scavenging activity, thus no EC50 value could be determined. For reference, we assessed the DPPH scavenging activity of the free polyelectrolytes at ASP doses without any LDH added (Fig. S4C, ESI†). It was found that PS and PAAm-co-DADMAC showed negligible activity, thus their corresponding nanocomposites were efficient antioxidants owing to their EA content. On the other hand, PEI had sizeable contribution (ca. 42%) to the overall DPPH scavenging of its coated LDH. This is probably due to the numerous protonated amino groups of PEI. Interestingly, PS did not show the same effect, although possessing likewise protonated guanidine functions. These results show a clear correlation between the colloidal stability of the coated materials, since highly stable EtOH–EA–LDH/PEI and EtOH–EA–LDH/PAAm-co-DADMAC possessed higher radical scavenging activity than the EtOH–EA–LDH/PS of much lower colloidal stability and CCC, although the overall activity of EtOH–EA–LDH/PEI was partly from the adsorbed PEI.
4. Conclusions
The surface functionalization and colloidal tuning of an antioxidant LDH composite was explored. The EtOH–EA–LDH was prepared by the coprecipitation of EA and LDH followed by EtOH treatment to improve its structural properties, i.e., to enlarge the specific surface area. The nanocomposite possessed negative surface charge, and thus, three cationic polyelectrolytes were chosen for surface functionalization, namely PEI, PS and PAAm-co-DADMAC. To optimize the experimental conditions for particle coating, the charging and aggregation of EtOH–EA–LDH were investigated by varying the polyelectrolyte dose. Stable dispersions were obtained at high doses, where the polyelectrolytes fully cover the surface of the LDH particles to form positively charged composites, meaning that overcharging occurred upon adsorption. However, a more important aspect is that colloidal stability of the particles is low without polyelectrolyte coating, in contrast to those with adsorbed polyelectrolyte layers. Thus, the salt-induced aggregation behaviour of the functionalized particles was also unravelled. While bare EA-LDH and EtOH–EA–LDH possessed lower CCC values, with PEI and PAAm-co-DADMAC on the surface, the dispersions were stable up to 1 M ionic strength. Interestingly, PS lowered the CCC of EtOH–EA–LDH, which was attributed to the flat-type adsorption and low line charge density of PS. On the other hand, it was shown that PAAm-co-DADMAC of neutral acrylamide units in the chain had significant sterical effects on the colloidal stability, while joint steric and electrostatic repulsive forces were responsible for the excellent resistance against salt-induced aggregation for the PEI-coated particles. The results of the radical scavenging activity assay revealed that the stable dispersions containing EtOH–EA–LDH/PEI (aided by the intrinsic scavenging property of PEI) and EtOH–EA–LDH/PAAm-co-DADMAC preserved the majority of the activity of EtOH–EA–LDH, scavenging 70%, 69% and 88% of the DPPH radicals, respectively. The polyelectrolyte layers on the surface somewhat reduced the availability of the intercalated antioxidants and the overall antioxidant capacity of the materials. Such a hindrance, however, provided a long-term activity for the hybrid materials. The long-term radical scavenging abilities, together with the extremely high colloidal stability, make EtOH–EA–LDH/PEI and EtOH–EA–LDH/PAAm-co-DADMAC promising candidates in applications, where antioxidant materials are used in liquid media containing high levels of electrolytes or their mixtures, e.g., suppressing harmful oxidative agents under in vivo conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Ministry of Human Capacities of Hungary (20391-3/2018/FEKUSTRAT), the Hungarian Academy of Sciences (96130) and by the National Research, Development and Innovation Office (SNN131558). The support from the University of Szeged Open Access Fund (4998) is gratefully acknowledged.References
- R. S. Sohal and R. Weindruch, Science, 1996, 273, 59–63 CrossRef CAS.
- S. Reuter, S. C. Gupta, M. M. Chaturvedi and B. B. Aggarwal, Free Radical Biol. Med., 2010, 49, 1603–1616 CrossRef CAS.
- J. W. Finley, A. N. Kong, K. J. Hintze, E. H. Jeffery, L. L. Ji and X. G. Lei, J. Agric. Food Chem., 2011, 59, 6837–6846 CrossRef CAS.
- R. Franco, G. Navarro and E. Martinez-Pinilla, Antioxidants, 2019, 8, 542 CrossRef CAS.
- F. Rasti and G. Scott, Eur. Polym. J., 1980, 16, 1153–1158 CrossRef CAS.
- J. Couto, A. Figueirinha, M. T. Batista, A. Paranhos, C. Nunes, L. M. Goncalves, J. Marto, M. Fitas, P. Pinto, H. M. Ribeiro and M. E. Pina, Antioxidants, 2020, 9, 154 CrossRef CAS.
- S. M. Lin, C. Y. Baek, J. H. Jung, W. S. Kim, H. Y. Song, J. H. Lee, H. J. Ji, Y. Zhi, B. S. Kang, Y. S. Bahn, H. S. Seo and S. Lim, Sci. Rep., 2020, 10, 55 CrossRef CAS.
- M. A. El-Missiry, Antioxidant enzyme, InTech, Rijeka, 2012 Search PubMed.
- J. J. Sun, R. Yendluri, K. Liu, Y. Guo, Y. R. Lvov and X. H. Yan, Phys. Chem. Chem. Phys., 2017, 19, 562–567 RSC.
- M. Pavlovic, M. Nafradi, P. Rouster, S. Murath and I. Szilagyi, J. Colloid Interface Sci., 2019, 543, 174–182 CrossRef CAS.
- G. F. D. del Castillo, M. Koenig, M. Muller, K. J. Eichhorn, M. Stamm, P. Uhlmann and A. Dahlin, Langmuir, 2019, 35, 3479–3489 CrossRef.
- O. O. Erejuwa, S. A. Sulaiman and M. S. Ab Wahab, Molecules, 2012, 17, 4400–4423 CrossRef CAS.
- C. Nirmala, M. S. Bisht, H. K. Bajwa and O. Santosh, Trends Food Sci. Technol., 2018, 77, 91–99 CrossRef CAS.
- S. Murath, N. B. Alsharif, S. Saringer, B. Katana, Z. Somosi and I. Szilagyi, Crystals, 2020, 10, 148 CrossRef CAS.
- K. C. Popat, M. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 4880–4888 CrossRef CAS.
- J. Karlsson, S. Atefyekta and M. Andersson, Int. J. Nanomed., 2015, 10, 4425–4436 CrossRef CAS.
- H. Hashemzadeh and H. Raissi, Appl. Surf. Sci., 2020, 500, 144220 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS.
- A. C. Santos, C. Ferreira, F. Veiga, A. J. Ribeiro, A. Panchal, Y. Lvov and A. Agarwal, Adv. Colloid Interface Sci., 2018, 257, 58–70 CrossRef CAS.
- X. Wen, F. Yang, Q. F. Ke, X. T. Xie and Y. P. Guo, J. Mater. Chem. B, 2017, 5, 7866–7875 RSC.
- X. Mei, S. M. Xu, T. Y. Hu, L. Q. Peng, R. Gao, R. Z. Liang, M. Wei, D. Evans and X. Duan, Nano Res., 2018, 11, 195–205 CrossRef CAS.
- F. Barahuie, M. Z. Hussein, P. Arulselvan, S. Fakurazi and Z. Zainal, Sci. Adv. Mater., 2016, 8, 501–513 CrossRef CAS.
- K. Khorsandi, R. Hosseinzadeh and M. Fateh, RSC Adv., 2015, 5, 93987–93994 RSC.
- D. G. Evans and R. C. T. Slade, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, 2006, vol. 119, pp. 1–87 Search PubMed.
- Z. B. Cao, B. Li, L. Y. Sun, L. Li, Z. P. Xu and Z. Gu, Small Methods, 2019, 1900343, DOI:10.1002/smtd.201900343.
- S. J. Mills, A. G. Christy, J. M. R. Genin, T. Kameda and F. Colombo, Mineral. Mag., 2012, 76, 1289–1336 CrossRef CAS.
- S. Murath, S. Szerlauth, D. Sebok and I. Szilagyi, Antioxidants, 2020, 9, 153 CrossRef CAS.
- O. D. Pavel, A. Serban, R. Zavoianu, E. Bacalum and R. Birjega, Crystals, 2020, 10, 244 CrossRef CAS.
- L. L. Qin, W. R. Wang, S. H. You, J. M. Dong, Y. H. Zhou and J. B. Wang, Int. J. Nanomed., 2014, 9, 5701–5710 CrossRef.
- K. M. Ansy, J. H. Lee, H. Piao, G. Choi and J. H. Choy, Solid State Sci., 2018, 80, 65–71 CrossRef CAS.
- S. P. Lonkar, B. Kutlu, A. Leuteritz and G. Heinrich, Appl. Clay Sci., 2013, 71, 8–14 CrossRef CAS.
- A. Hajdu, M. Szekeres, I. Y. Toth, R. A. Bauer, J. Mihaly, I. Zupko and E. Tombacz, Colloids Surf., B, 2012, 94, 242–249 CrossRef CAS.
- E. Koupanou, S. Ahualli, O. Glatter, A. Delgado, F. Krumeich and E. Leontidis, Langmuir, 2010, 26, 16909–16920 CrossRef CAS.
- T. Klacic, A. Sadzak, J. Jukic, T. Preocanin and D. Kovacevic, Colloids Surf., A, 2019, 570, 32–38 CrossRef CAS.
- B. Katana, P. Rouster, G. Varga, S. Muráth, K. Glinel, A. M. Jonas and I. Szilagyi, ACS Appl. Bio Mater., 2020, 3, 522–530 CrossRef CAS.
- M. X. Liu, R. He, J. Yang, Z. R. Long, B. Huang, Y. W. Liu and C. R. Zhou, Clay Miner., 2016, 51, 457–467 CrossRef CAS.
- A. Tiraferri, L. A. S. Hernandez, C. Bianco, T. Tosco and R. Sethi, J. Nanopart. Res., 2017, 19, 107 CrossRef.
- L. L. Zhao, M. Skwarczynski and I. Toth, ACS Biomater. Sci. Eng., 2019, 5, 4937–4950 CrossRef CAS.
- B. Bolto and J. Gregory, Water Res., 2007, 41, 2301–2324 CrossRef CAS.
- S. Farrokhpay, Adv. Colloid Interface Sci., 2009, 151, 24–32 CrossRef CAS.
- F. Xie, T. Nylander, L. Piculell, S. Utsel, L. Wagberg, T. Akesson and J. Forsman, Langmuir, 2013, 29, 12421–12431 CrossRef CAS.
- L. Muresan, P. Maroni, I. Popa, M. Porus, R. Longtin, G. Papastavrou and M. Borkovec, Macromolecules, 2011, 44, 5069–5071 CrossRef CAS.
- R. Meszaros, I. Varga and T. Gilanyi, Langmuir, 2004, 20, 5026–5029 CrossRef CAS.
- M. Borkovec, I. Szilagyi, I. Popa, M. Finessi, P. Sinha, P. Maroni and G. Papastavrou, Adv. Colloid Interface Sci., 2012, 179, 85–98 CrossRef.
- L. L. Feng, M. C. Stuart and Y. Adachi, Adv. Colloid Interface Sci., 2015, 226, 101–114 CrossRef CAS.
- I. Szilagyi, G. Trefalt, A. Tiraferri, P. Maroni and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- M. Pavlovic, P. Rouster, T. Oncsik and I. Szilagyi, ChemPlusChem, 2017, 82, 121–131 CrossRef CAS.
- C. Vasti, A. Borgiallo, C. E. Giacomelli and R. Rojas, Colloids Surf., A, 2017, 533, 316–322 CrossRef CAS.
- C. Vasti, D. A. Bedoya, R. Rojas and C. E. Giacomelli, J. Mater. Chem. B, 2016, 4, 2008–2016 RSC.
- Z. Gu, H. L. Zuo, L. Li, A. H. Wu and Z. P. Xu, J. Mater. Chem. B, 2015, 3, 3331–3339 RSC.
- M. Pavlovic, P. Rouster, Z. Somosi and I. Szilagyi, J. Colloid Interface Sci., 2018, 524, 114–121 CrossRef CAS.
- M. Pavlovic, P. Rouster and I. Szilagyi, Nanoscale, 2017, 9, 369–379 RSC.
- Z. Somosi, M. Pavlovic, I. Palinko and I. Szilagyi, Nanomaterials, 2018, 8, 986 CrossRef.
- J. Carneiro, A. F. Caetano, A. Kuznetsova, F. Maia, A. N. Salak, J. Tedim, N. Scharnagl, M. L. Zheludkevich and M. G. S. Ferreira, RSC Adv., 2015, 5, 39916–39929 RSC.
- N. Vargas-Mendoza, M. Vazquez-Velasco, L. Gonzalez-Torres, J. Benedi, F. J. Sanchez-Muniz, J. A. Morales-Gonzalez, O. A. Jaramillo-Morales, C. Valadez-Vega and M. Bautista, Antioxidants, 2018, 7, 178 CrossRef.
- P. A. Hassan, S. Rana and G. Verma, Langmuir, 2015, 31, 3–12 CrossRef CAS.
- S. H. Behrens, M. Borkovec and P. Schurtenberger, Langmuir, 1998, 14, 1951–1954 CrossRef CAS.
- D. Grolimund, M. Elimelech and M. Borkovec, Colloids Surf., A, 2001, 191, 179–188 CrossRef CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, Food Sci. Technol., 1995, 28, 25–30 CAS.
- P. Rouster, M. Pavlovic and I. Szilagyi, RSC Adv., 2016, 6, 97322–97330 RSC.
- P. Rouster, M. Pavlovic, E. Horvath, L. Forro, S. K. Dey and I. Szilagyi, Langmuir, 2017, 33, 9750–9758 CrossRef CAS.
- J. Israelachvili, Intermolecular and surface forces, Academic Press, London, 3rd edn, 2011 Search PubMed.
- M. Borkovec, G. J. M. Koper and C. Piguet, Curr. Opin. Colloid Interface Sci., 2006, 11, 280–289 CrossRef CAS.
- N. Malikova, A. L. Rollet, S. Cebasek, M. Tomsic and V. Vlachy, Phys. Chem. Chem. Phys., 2015, 17, 5650–5658 RSC.
- M. Pavlovic, P. Rouster, E. Bourgeat-Lami, V. Prevot and I. Szilagyi, Soft Matter, 2017, 13, 842–851 RSC.
- M. Pavlovic, R. Huber, M. Adok-Sipiczki, C. Nardin and I. Szilagyi, Soft Matter, 2016, 12, 4024–4033 RSC.
- G. Fritz, V. Schadler, N. Willenbacher and N. J. Wagner, Langmuir, 2002, 18, 6381–6390 CrossRef CAS.
- J. Hierrezuelo, I. Szilagyi, A. Vaccaro and M. Borkovec, Macromolecules, 2010, 43, 9108–9116 CrossRef CAS.
- L. P. Amaro, F. Cicogna, E. Passaglia, E. Morici, W. Oberhauser, S. Al-Malaika, N. T. Dintcheva and S. Coiai, Polym. Degrad. Stab., 2016, 133, 92–100 CrossRef.
- Q. Zhang, Q. Jiao, F. Leroux, P. G. Tang, D. Q. Li and Y. J. Feng, Polym. Degrad. Stab., 2017, 140, 9–16 CrossRef CAS.
- S. Olivera, C. Hu, G. S. Nagananda, N. Reddy, K. Venkatesh and H. B. Muralidhara, Int. J. Environ. Sci. Technol., 2019, 16, 2017–2030 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01531h |
| This journal is © The Royal Society of Chemistry 2020 |