
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable liquid crystal elastomer microactuators prepared via thiol–ene dispersion polymerization†
Xiaohong
Liu
ab,
Xinglong
Pan
ab,
Michael G.
Debije
 a,
Johan P. A.
Heuts
bc,
Dirk J.
Mulder
ab and
Albert P. H. J.
Schenning
*ab
a,
Johan P. A.
Heuts
bc,
Dirk J.
Mulder
ab and
Albert P. H. J.
Schenning
*ab
aStimuli-Responsive Functional Materials and Devices, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands. E-mail: a.p.h.j.schenning@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands
cSupramolecular Polymer Chemistry group, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, PO Box 513, 5600 MB, Eindhoven, The Netherlands
First published on 18th May 2020
Abstract
Narrowly dispersed, 10 micron-sized, liquid crystalline elastomer polymer actuators were first prepared via thiol–ene dispersion polymerization and then embedded and stretched in a polyvinyl alcohol film, followed by photopolymerization of the residual acrylate groups. Prolate micro spheroids in which the mesogens are aligned parallel to the long axis were obtained and showed reversible thermally driven actuation owing to nematic to isotropic transition of the liquid crystal molecules. The particles were also compressed to form disk-shaped oblate microactuators in which the mesogens are aligned perpendicular to the short axis, demonstrating that the reported method is a versatile method to fabricate liquid crystal elastomer microactuators with programmable properties.
Liquid crystalline (LC) microactuators have received considerable interest as they exhibit reversible changes in morphology in response to specific stimuli, and thus have potential applications in biomedicine and microfluidics.1–6 Various stimuli have been used to trigger shape changes of these LC polymer particles, including heat and light.7–10 Changes in shape are a result of phase changes of the LC mesogens, leading to changes in molecular order and conformation of polymer chains in the particles.11,12 Most LC microactuators have been prepared via microfluidics by polymerizing spherical LC monomer droplets, and upon disrupting the LC order, the particles undergo a shape change to a non-spherical morphology.1–6,8,9 An alternative method of producing shape changing particles was by deforming shape memory particles. Ho et al. developed a simple approach, in which polymer particles were immobilized in a poly(vinyl alcohol) (PVA) matrix and stretched.13,14 The approach has been widely applied for its simplicity and versatility comparing to other techniques aiming to deform microscopic objects, such as nanoimprint lithography.15–21 However, these non-LC particles only show “one-way” shape recovery; that is, from the temporary shape to the programmed permanent shape. Marshall et al. combined microfluidics and stretching and prepared LC microactuators by stretching spherical LC monomer droplets prior to polymerization; the resulting LC microactuators showed a “reverse” shape change, that is, from ellipsoidal (LC) to spherical (isotropic), although only ellipsoidal particles were prepared.22 Microfluidics, however, face challenges in preparing small-sized particles (with diameters (D) ∼ 10 μm).
Recently, an advanced method has been reported for fabricating centimeter-size liquid crystal elastomer (LCE) actuators with programmable shapes through a two-step polymerization approach.23–30 Briefly, in the first step, LC monomers are partially polymerized via a thiol–ene polymerization reaction and then mechanically aligned. During the second step, a radical polymerization is carried out to polymerize the remaining acrylate end groups. In doing so, the LCE actuators consist of two competing polymer structures: the first thiol–ene structure that favors the initial shape before programming, and the second polyacrylate structure that favors the programmed shape.
To the best of our knowledge, the fabrication of LC microactuators in the 10 μm range with reversible shape changes has not been reported. Bowman et al. described the preparation of micron-size non-LC particles with a narrow particle size distribution without microfluidics based on thiol–ene dispersion polymerization.31,32 By incorporating dynamic covalent functional groups in the network, the permanent shape of the particles can be tuned with specific stimuli.33 However, reversible shape change was not achieved with these non-LC particles. Nevertheless, this thiol–ene dispersion polymerization opens up a route towards the preparation of LC microactuators with reversible shape changes through the two-step polymerization approach (vide supra).
We now report the facile preparation of 10 micron-sized LC actuators with low dispersity and reversible shape changes based on this two-step approach. Making use of thiol–ene dispersion polymerization reported by Bowman et al., narrowly dispersed, micron-sized LC particles with excess acrylate groups were obtained.31,32 The LC particles were then immobilized in a PVA matrix and stretched. The deformed particles were then irradiated with UV light to photopolymerize the residual acrylate groups in the nematic phase. The LC mesogens were aligned mechanically during stretching, yielding prolate micro spheroids. Subsequent exposure to different temperatures yielded macroscopic, yet reversible shape changes between spheroids (T < TNI; TNI = nematic to isotropic transition temperature) and spheres (T > TNI), due to the change of LC phase. Furthermore, by compressing the PVA/particle composite films, disk-shaped oblate microactuators can also be produced, indicating that this method can prepare particle actuators of different director patterns.10
Previous literature suggests that establishing a delicate balance between the thiol–ene structure and the acrylate structure in the network is crucial to achieving reversible shape changes: the reported optimal molar ratio of thiol groups to acrylate groups is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1, and 25% of the total number of thiol groups should come from the tetrathiol cross-linker.34 The monomer mixture used in the current study is based on these findings and consists of an LC monomer (1), a dithiol chain extender (2), and a tetrathiol cross-linker (3) (Fig. 1(a)).34
:1.1, and 25% of the total number of thiol groups should come from the tetrathiol cross-linker.34 The monomer mixture used in the current study is based on these findings and consists of an LC monomer (1), a dithiol chain extender (2), and a tetrathiol cross-linker (3) (Fig. 1(a)).34
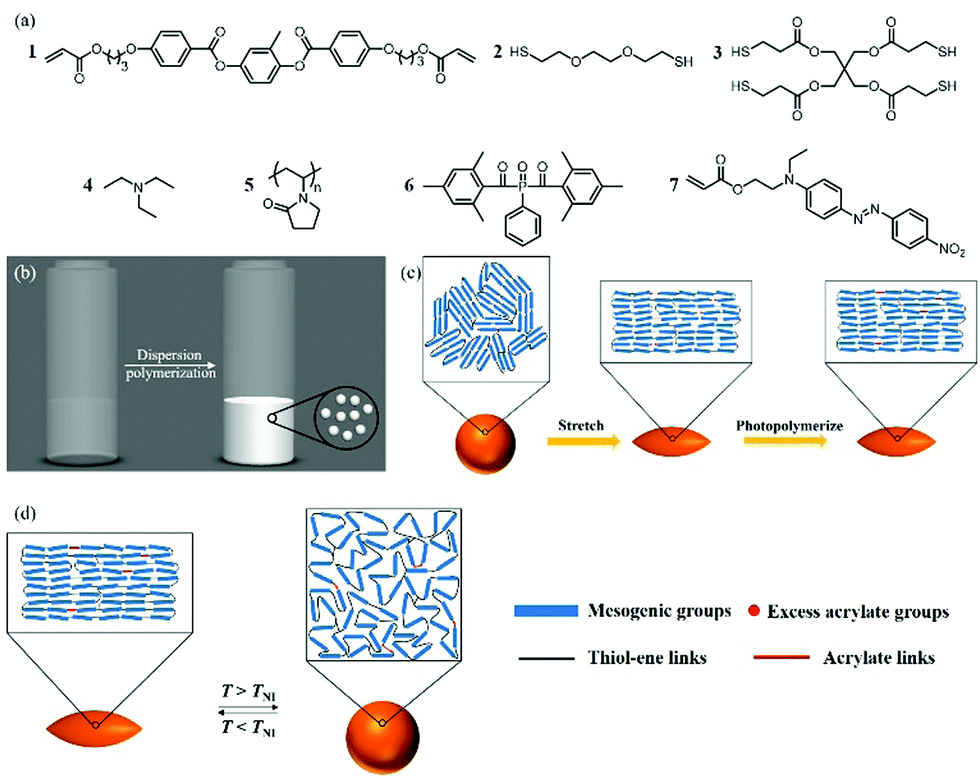 | ||
| Fig. 1 (a) Chemical structures of the particle components. Schematic representation of dispersion polymerization (b), stretching of the particles (c), and thermally driven actuation of the particles (d). | ||
The first polymer structure in the LC particles was prepared via thiol–ene dispersion polymerization. The monomer mixture and the steric stabilizer, polyvinylpyrrolidone (5), were dissolved in the mixed solvent of 2-methoxy ethanol and ethanol. Upon addition of triethylamine (4), the catalyst, the solution quickly turned hazy, and the polymerization was carried out at room temperature overnight. By carefully controlling the composition of the solvent mixture, narrowly distributed particles with an average particle diameter of 10.5 μm and a coefficient of variation of 9% determined by polarized optical microscopy (POM, Fig. 2(a) and Fig. S1, ESI†) were prepared. The particles appeared to be polydomain based on the birefringence observed between cross polarizers (Fig. 2(b)).
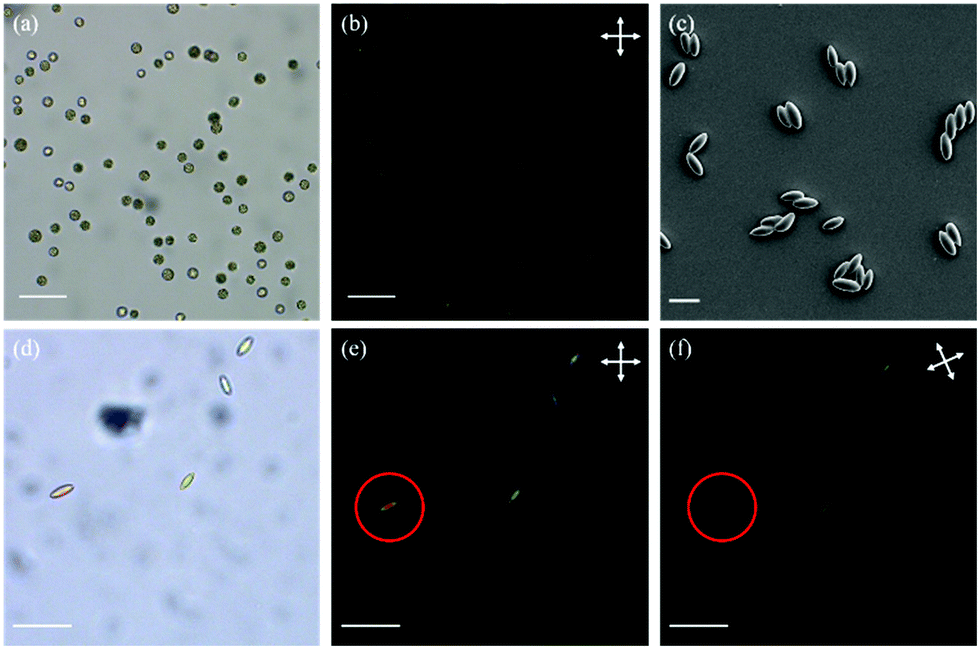 | ||
| Fig. 2 POM images of particles before stretching and photopolymerization at room temperature without (a) and with cross polarizers (b). SEM image (c), POM image without (d) and with (e) cross polarizers of the particles after stretching and photopolymerization. (f) The cross polarizers were rotated such that the long axis of a particle (circled) was parallel to one of the polarizers. (Scale bar = 20 μm for (c); scale bar = 50 μm for other images.) | ||
In order to photopolymerize the excess acrylate groups in the polymer particles, a photoinitiator, Irgacure 819 (6), and a polymerizable dye, disperse red 1 acrylate (7), used as an indicator to verify the photopolymerization, were dissolved in CHCl3 and emulsified in an aqueous sodium dodecyl sulfate (0.2% w/v in water) solution. This emulsion was mixed with the polymer particles and agitated overnight. Upon complete absorption of the CHCl3 droplets, the suspension was added to the PVA solution (composition per 100 mL water: 10 g PVA, 1 g urea, and 1 g formamide as plasticizers) and dried to form the PVA/particle film. The film was then cut into strips and stretched (draw ratio = 3) at 60 °C to deform the spherical particles. The stretched strips were cooled to room temperature, followed by photopolymerization of the remaining acrylate groups. After photopolymerization, the PVA strips were dissolved in water to yield the ellipsoidal particles (Fig. 2(c) and (d)). The lengths along the long and the short axes were determined as 15.5 μm and 7.5 μm, respectively. Most likely the LC moieties align along the long particle axis, as the particles appeared dark if the cross polarizers were parallel or perpendicular to the long axis (Fig. 2(e) and (f)). This indicates that prolate micro spheroids are formed in which the LC mesogens are aligned in the stretching direction along the long axis. Since the number of acrylate groups in the particles is very low, the characteristic signals of acrylate groups were too weak for an accurate conversion measurement with infrared spectroscopy (Fig. S2(a), ESI†). Hence, to verify the success of photopolymerization, the particles were extracted with THF to determine whether the dispersed red 1 acrylate dye was incorporated into the network. The dye was extracted from the particles before photopolymerization but not from the particles after photopolymerization, indicating that the photopolymerization was indeed successful and the dye was grafted to the particles (Fig. S2(b) and (c), ESI†).
For the thermal actuation study, the micro spheroids were dispersed in glycerol, and a drop of the dispersion was added to a glass cell and analyzed with POM. The dispersion was first equilibrated at 50 °C, and then heated to 70 °C at 3 °C min−1. The micro spheroids showed a shape change from ellipsoidal to spherical during heating, and upon cooling to 50 °C, the spheres became ellipsoidal again (Fig. 3). The heating–cooling cycle was repeated and the shape change was similar to the first cycle, which indicates that the process was reversible (Fig. S3, ESI†). The birefringence of the particles was observed with POM at 50 °C and disappeared at 70 °C, suggesting that the actuation was a result of the LC-isotropic phase transition (Fig. 3).
 | ||
| Fig. 3 Thermally driven actuation of the stretched particles without (top) and with (bottom) cross polarizers (scale bar = 10 μm). | ||
To demonstrate that the initial shape and director of the LC microactuators can be programmed, the PVA/particles were also compressed to produce disk-shaped oblate microactuators in which the mesogens are aligned perpendicular to the short axis. The particles were slightly elongated because the PVA matrix flowed outwards during compression and created a shear field (Fig. 4(a)). The average lengths along the long and short axes were determined as 13 μm and 11 μm, respectively. By tracking a particle that rotated in glycerol, the shape was confirmed as a disk (Fig. 4(b)). To study the temperature-responsive shape changes, the disk-shaped microactuator dispersion was first equilibrated at 60 °C and then heated to 75 °C at 3 °C min−1. The shape of the particles changed from ellipsoidal to spherical, corresponding to the appearance of birefringence under POM (Fig. 4(c)), and upon cooling to 60 °C, the shape returns to ellipsoidal again.
 | ||
| Fig. 4 (a) SEM images of the particles after compression and photopolymerization (scale bar = 20 μm); (b) POM images and schematic representation of a particle when facing upwards (top) and to the side (bottom) (scale bar = 20 μm). (c) Thermally driven actuation of the particles without (top) and with (bottom) cross polarizers (scale bar = 10 μm). | ||
Conclusions
We report the preparation of thermally driven microactuators in the 10 μm range with reversible shape changes. The particles were prepared by thiol–ene dispersion polymerization and the excess acrylate groups were photopolymerized after the particles were stretched in a PVA matrix. By changing the temperature of the environment, the LC phase of the prolate microactuators can be altered between the nematic and isotropic phases, and as a result, the shape of the microactuators can be tuned between spherical and ellipsoidal. By compressing the PVA/particle film, disk-shaped oblate microactuators can also be produced. This work can lead to a new, versatile method for preparing micron-sized prolate and oblate shape changing particles with programmable properties, making these actuators appealing for applications ranging from biomedicine to microfluidic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Alberto Belmonte Parra and Sterre Bakker for helpful discussions. We are grateful for the financial support by The Netherlands Organization for Scientific Research (TOP-PUNT-718.016.003).Notes and references
- C. Ohm, N. Kapernaum, D. Nonnenmacher, F. Giesselmann, C. Serra and R. Zentel, J. Am. Chem. Soc., 2011, 133, 5305–5311 CrossRef CAS PubMed.
- E. K. Fleischmann, H. L. Liang, N. Kapernaum, F. Giesselmann, J. Lagerwall and R. Zentel, Nat. Commun., 2012, 3, 1178 CrossRef PubMed.
- C. Ohm, E. K. Fleischmann, I. Kraus, C. Serra and R. Zentel, Adv. Funct. Mater., 2010, 20, 4314–4322 CrossRef CAS.
- C. Ohm, C. Serra and R. Zentel, Adv. Mater., 2009, 21, 4859–4862 CrossRef CAS PubMed.
- T. Hessberger, L. Braun and R. Zentel, Polymers, 2016, 8, 410 CrossRef PubMed.
- E. K. Fleischmann, F. R. Forst, K. Köder, N. Kapernaum and R. Zentel, J. Mater. Chem. C, 2013, 1, 5885–5891 RSC.
- H. Yu, H. Liu and T. Kobayashi, ACS Appl. Mater. Interfaces, 2011, 3, 1333–1340 CrossRef CAS PubMed.
- L. B. Braun, T. Hessberger and R. Zentel, J. Mater. Chem. C, 2016, 4, 8670–8678 RSC.
- V. S. R. Jampani, D. J. Mulder, K. R. De Sousa, A. H. Gélébart, J. P. F. Lagerwall and A. P. H. J. Schenning, Adv. Funct. Mater., 2018, 28, 1801209 CrossRef.
- V. S. R. Jampani, R. H. Volpe, K. Reguengo de Sousa, J. Ferreira Machado, C. M. Yakacki and J. P. F. Lagerwall, Sci. Adv., 2019, 5, eaaw2476 CrossRef CAS PubMed.
- E. M. Terentjev, J. Phys.: Condens. Matter, 1999, 11, R239–R257 CrossRef CAS.
- C. Ohm, M. Brehmer and R. Zentel, Adv. Mater., 2010, 22, 3366–3387 CrossRef CAS PubMed.
- C. C. Ho, A. Keller, J. A. Odell and R. H. Ottewill, Colloid Polym. Sci., 1993, 271, 469–479 CrossRef CAS.
- C. C. Ho, R. H. Ottewill, A. Keller and J. A. Odell, Polym. Int., 1993, 30, 207–211 CrossRef CAS.
- R. A. Meyer, M. P. Mathew, E. Ben-Akiva, J. C. Sunshine, R. B. Shmueli, Q. Ren, K. J. Yarema and J. J. Green, Acta Biomater., 2018, 72, 228–238 CrossRef CAS PubMed.
- C. Wischke and A. Lendlein, Langmuir, 2014, 30, 2820–2827 CrossRef CAS PubMed.
- M. Kohri, Y. Tamai, A. Kawamura, K. Jido, M. Yamamoto, T. Taniguchi, K. Kishikawa, S. Fujii, N. Teramoto, H. Ishii and D. Nagao, Langmuir, 2019, 35, 5574–5580 CrossRef CAS PubMed.
- Y. C. Lo, H. F. Tseng, Y. J. Chiu, B. H. Wu, J. W. Li and J. T. Chen, Langmuir, 2018, 34, 8326–8332 CrossRef CAS PubMed.
- Y. C. Lo, Y. J. Chiu, H. F. Tseng and J. T. Chen, Langmuir, 2017, 33, 12300–12305 CrossRef CAS PubMed.
- X. Liu, H. Zheng, G. Li, H. Li, P. Zhang, W. Tong and C. Gao, Colloids Surf., B, 2017, 158, 675–681 CrossRef CAS PubMed.
- S. Coertjens, R. De Dier, P. Moldenaers, L. Isa and J. Vermant, Langmuir, 2017, 33, 2689–2697 CrossRef CAS PubMed.
- J. E. Marshall, S. Gallagher, E. M. Terentjev and S. K. Smoukov, J. Am. Chem. Soc., 2014, 136, 474–479 CrossRef CAS PubMed.
- C. M. Yakacki, M. Saed, D. P. Nair, T. Gong, S. M. Reed and C. N. Bowman, RSC Adv., 2015, 5, 18997–19001 RSC.
- M. K. McBride, A. M. Martinez, L. Cox, M. Alim, K. Childress, M. Beiswinger, M. Podgorski, B. T. Worrell, J. Killgore and C. N. Bowman, Sci. Adv., 2018, 4, eaat4634 CrossRef CAS PubMed.
- L. Yu, H. Shahsavan, G. Rivers, C. Zhang, P. Si and B. Zhao, Adv. Funct. Mater., 2018, 28, 1–8 Search PubMed.
- D. R. Merkel, N. A. Traugutt, R. Visvanathan, C. M. Yakacki and C. P. Frick, Soft Matter, 2018, 14, 6024–6036 RSC.
- M. Barnes and R. Verduzco, Soft Matter, 2019, 15, 870–879 RSC.
- L. Wang, W. Liu, L. X. Guo, B. P. Lin, X. Q. Zhang, Y. Sun and H. Yang, Polym. Chem., 2017, 8, 1364–1370 RSC.
- E. C. Davidson, A. Kotikian, S. Li, J. Aizenberg and J. A. Lewis, Adv. Mater., 2019, 32, 1905682 CrossRef PubMed.
- N. Torras, K. E. Zinoviev, J. Esteve and A. Sánchez-Ferrer, J. Mater. Chem. C, 2013, 1, 5183–5190 RSC.
- C. Wang, M. Podgórski and C. N. Bowman, Mater. Horiz., 2014, 1, 535–539 RSC.
- C. Wang, X. Zhang, M. Podgórski, W. Xi, P. Shah, J. Stansbury and C. N. Bowman, Macromolecules, 2015, 48, 8461–8470 CrossRef CAS.
- L. M. Cox, X. Sun, C. Wang, N. Sowan, J. P. Killgore, R. Long, H. A. Wu, C. N. Bowman and Y. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 14422–14428 CrossRef CAS PubMed.
- M. Barnes and R. Verduzco, Soft Matter, 2019, 15, 870–879 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm00817f |
| This journal is © The Royal Society of Chemistry 2020 |