
Controlling DOPA adsorption via interacting with polyelectrolytes: layer structure and corrosion resistance†
 *a
Jinben
Wang
a
*a
Jinben
Wang
a
Abstract
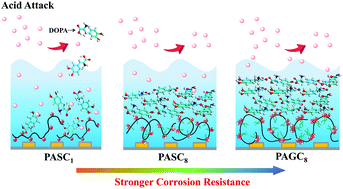
Protein adsorption on polyelectrolyte (PE) surfaces has aroused intensive attraction, but there are still few investigations on tuning the protein adsorption at a solid surface by controllable layer structures and surface properties of PE adlayers. Furthermore, there is a lack of understanding regarding the correlation between molecular conformation and anticorrosion performance of composite materials. With this in mind, we synthesized a series of PEs and constructed 3,4-dihydroxy-L-phenylalanine (L-DOPA) adlayers on the PE surfaces, monitoring the whole adsorption process in situ. A highly charged cationic PE surface exhibits a low adhesion of DOPA molecules, leading to a loose structure, rough surface morphology, and strong solvation effects and, accordingly, this kind of multilayer provides a poor anticorrosion capacity. In comparison, amphiphilic and highly charged cationic PE surfaces are in favor of DOPA adsorption and the formation of compact and smooth multilayers due to cation–π and hydrophobic interactions between DOPA and PEs. Interestingly, one of the multilayers exhibits a remarkable enhancement of inhibition efficiency of about 460-fold compared with that of the bare substrate, which is much higher than that of other anticorrosion coatings reported previously. Our findings reveal the interaction mechanism between DOPA and PE surfaces to achieve the controllable adsorption of biomolecules, providing a promising way to optimize the layer structures to improve the anticorrosion capacity.
 Please wait while we load your content...
Please wait while we load your content...

