
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Epoxide and oxetane based liquid crystals for advanced functional materials
Davey C.
Hoekstra
 ab,
Albert P. H. J.
Schenning
*ab and
Michael G.
Debije
a
ab,
Albert P. H. J.
Schenning
*ab and
Michael G.
Debije
a
aLaboratory of Stimuli-responsive Functional Materials and Devices, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, Groene Loper 5, 5612 AE Eindhoven, The Netherlands. E-mail: a.p.h.j.schenning@tue.nl
bInstitute for Complex Molecular Systems, Eindhoven University of Technology, Groene Loper 5, 5612 AE Eindhoven, The Netherlands
First published on 21st May 2020
Abstract
Liquid crystalline elastomers (LCEs) and liquid crystalline networks (LCNs) are classes of polymers very suitable for fabricating advanced functional materials. Two main pathways to obtain LCEs and LCNs have gained the most attention in the literature, namely the two-step crosslinking of LC side-chain polymers and the photoinitiated free-radical polymerisation of acrylate LC monomers. These liquid crystal polymers have demonstrated remarkable properties resulting from their anisotropic shapes, being used in soft robotics, responsive surfaces and as photonic materials. In this review, we will show that LCs with cyclic ethers as polymerisable groups can be an attractive alternative to the aforementioned reactive acrylate mesogens. These epoxide and oxetane based reactive mesogens could offer a number of advantages over their acrylate-based counterparts, including oxygen insensitivity, reduced polymerisation shrinkage, improved alignment, lower processing viscosity and potentially extended resistivity. In this review, we summarise the research on these materials from the past 30 years and offer a glimpse into the potential of these cyclic ether mesogens.
 Davey C. Hoekstra | Davey Hoekstra received his Master's degree in Chemical Engineering in 2016 after having studied mechanoluminescent polymers under supervision of Prof. Dr Rint Sijbesma. Afterwards, he joined the group of ‘Stimuli-responsive Functional Materials and Devices’ at the same University for his PhD studies. He is currently working on advanced functional materials from oxetane liquid crystal monomers under supervision of Prof. Dr Albert Schenning and Dr Michael Debije. |
 Albert P. H. J. Schenning | Albert Schenning studied chemistry at Radboud University Nijmegen, where he obtained his master's degree in 1992 and his doctorate in 1996. His PhD thesis on supramolecular architectures based on porphyrin and receptor molecules was supervised by Dr Martin Feiters and Prof. Roeland Nolte. Between June and December 1996, Schenning was a post-doctoral fellow in the group of Prof. Bert Meijer at Eindhoven University of Technology (TU/e), working on dendrimers. In 1997, he investigated pi-conjugated oligomers and polymers based on triacetylenes with Prof. François Diederich at the ETH in Zurich. From 1998 until 2003, Schenning was a fellow of the Royal Netherlands Academy of Arts and Sciences (KNAW) at the TU/e, active in the field of supramolecular organization of pi-conjugated systems. In 2003 he was appointed assistant professor, in 2005 associate professor and in 2014 full professor of Stimuli-responsive Functional Materials and Devices at TU/e. |
 Michael G. Debije | Michael G. Debije received a MSc in High Energy Physics from Iowa State University, Ames, Iowa in 1994 and a PhD in Biophysics from the University of Rochester in Rochester, NY, USA in 2000. As an Assistant Professor he is currently responsible for the energy cluster within the ‘Stimuli-responsive Functional Materials and Devices’ (SFD) group at Eindhoven University of Technology (TU/e). His interests include the control of light in urban and horticultural settings using responsive materials. |
Introduction
Advanced functional materials are interesting from both scientific and industrial points of view. These materials can have several properties, including, but not limited to, shape memory,1,2 reversible adhesion,3 self-healing,4 energy conservation,5–7 soft robotics8,9 and photonic characteristics.10Many organic building blocks can be used to construct advanced functional materials. Liquid crystalline networks (LCNs) and liquid crystalline elastomers (LCEs) are particularly interesting and suitable classes of materials; their fabrication is often straightforward, and a wide variety of LC ordered phases and orientations can be employed, allowing the resulting polymers to be used in a range of applications, including actuators,11–13 switching surfaces,14 sensors,15,16 shape memory17 and anti-counterfeit coatings.18
LCNs and LCEs can be constructed from a variety of starting materials and polymerisation methods. Over time, however, two distinct approaches have gained the most attention in the literature. In the first, LC side-chain linear polymers are crosslinked in a two-step process.19 Initially, the material is only slightly crosslinked, after which the mesogenic groups in the polymer chains are aligned by mechanical stretching. The monodomain alignment obtained is then fixed with a second crosslinking step to obtain the final materials. Usually, LC materials prepared in this way have the mechanical properties of elastomers. These LCEs have mainly found applications as actuators and sensors.20
The second approach uses reactive LC monomers consisting of a mesogenic group with one or more polymerisable groups, typically acrylates, attached to the core by a flexible spacer.21,22 The low molar mass of these reactive mesogens allows for their alignment in the monomeric state. The aligned monomers are then immobilised by (photo)polymerisation resulting in moderately-to-highly crosslinked LCNs that have been applied as advanced functional materials. Examples include as birefringent films for optical applications,23 responsive surface topographies,24 energy saving reflecting films25 and soft robotics.26–28 Recent efforts have led to the development of less crosslinked LCEs based on reactive mesogens by first forming a linear prepolymer through step-growth polymerisation that then is crosslinked at a later stage.29
In addition to side-chain polymers and acrylate-based reactive mesogens, alternative building blocks for LCE or LCN-based stimuli responsive materials have been developed. While they may not have attracted as much attention in the literature, LC monomers that contain epoxide or oxetane groups offer potentially intriguing advantages to the classic acrylic monomers. As an example, epoxy monomers can be used in the synthesis of vitrimer-based LCEs, opening the pathway to self-healing, easier processing, soft actuators, with possibilities for recyclability. In free-radical (photo)polymerisation, oxygen acts as a polymerisation inhibitor by reacting with the generated free-radicals, resulting in the need of an inert atmosphere, something that is often difficult to achieve on a large scale. Although a variety of solutions have been industrially employed to prevent this inhibition,30 these solutions are not always suitable for LC systems. Additionally, acrylates suffer from polymerisation shrinkage. In contrast, epoxide and oxetane LC monomers that link through a cationic ring-opening mechanism are insensitive to the presence of oxygen and suffer significantly less from polymerisation shrinkage.31 Therefore, cationic ring-opening polymerisation may have significant advantages over free-radical polymerisations of LC monomers. When epoxide LC monomers are used as a step-growth monomer in the production of LC thermosets, they may prove to have interesting physical properties, including better alignment,32 extended lifetimes or resistivity,33 and lower viscosity during processing.34
In this work, we describe the current catalogue of available reactive cyclic ether LC monomers and, when available, describe their physical characteristics to provide a type of ‘cookbook’ for the researcher interested in this class of materials. We will discuss materials prepared by step growth and cationic polymerisation, respectively. We offer suggested pathways towards future research that will lead to advanced materials with intriguing functional properties.
Advanced functional materials through step-growth polymerisation
LC monomers containing cyclic ethers as polymerisable groups can be cured through step-growth polymerisation, resulting in both highly crosslinked LC thermosets (i.e. LCNs) as well as more flexible LCEs.LCNs from liquid crystalline epoxy resins
Conventional thermosets are polymers that irreversibly harden by curing and have been abundant in industrial applications. In thermoset resins containing cyclic ethers, epoxide-functionalised monomers (i.e. epoxy resins) are the most utilised and most studied. The first reports on LC epoxy resins date back to the 1990s and were mainly targeted to improve the mechanical and thermal stabilities of thermosets.32–34The introduction of LC behaviour in the monomer gives rise to anisotropic properties in the resulting polymer networks. As is the case for all LC systems, the properties of thermosets based on LC epoxy resins are highly dependent on the molecular structure of the mesogenic group. For this reason, LC epoxy resins based on a wide variety of mesogenic units have been reported. The most common structures among them are biphenyl (BP),35–37 methylstilbene (DOMS),36,38–41 phenyl benzoate (PHBHQ),31,32,42,43 binaphthyl (EPBN)33,44 and azomethine (EF3).45–47 An overview of these mesogenic epoxides is given in Table 1.
| Name | Abbreviation | Chemical structure | T m (°C) | T N–I (°C) | Ref. |
|---|---|---|---|---|---|
| a T m: melting temperature. b T N–I: nematic to isotropic transition temperature. | |||||
| Biphenyl | BP |

|
155 | No LC phase | 35–37 |
| Methylstilbene | DOMS |
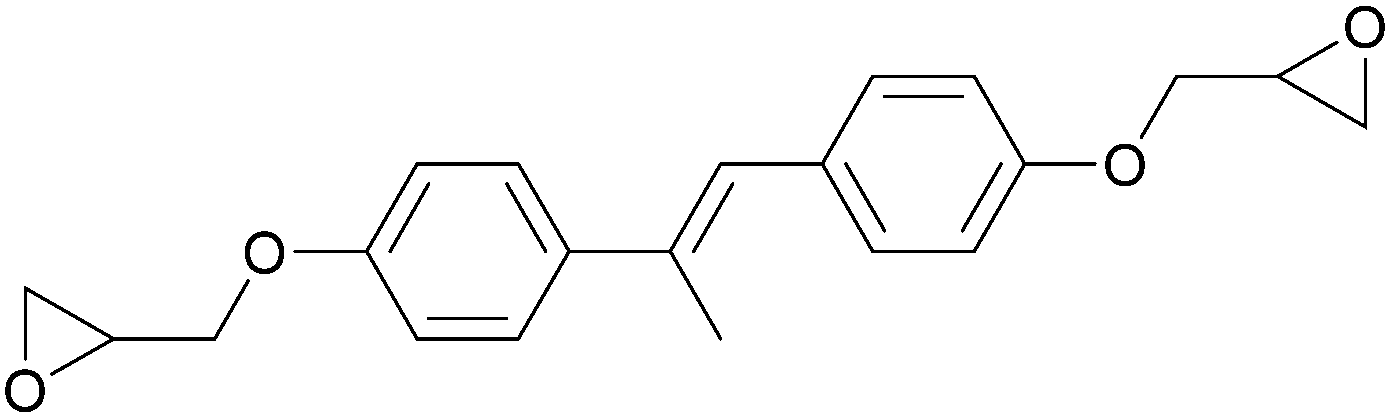
|
130 | 115 (monotropic) | 36 and 38–41 |
| Phenyl benzoate | PBHQ |
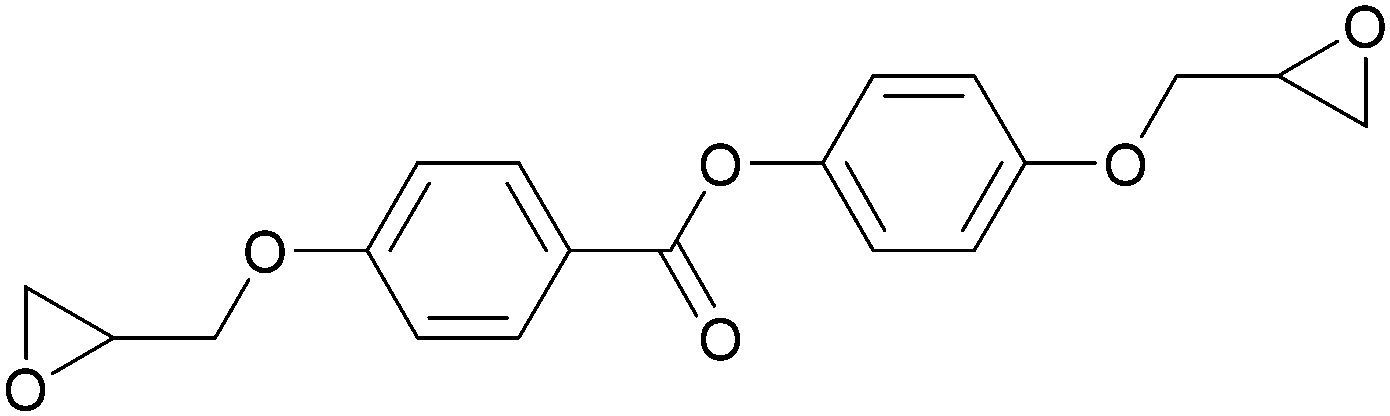
|
113 | 97 (monotropic) | 31, 32, 42 and 43 |
| Binaphthyl | EPBN |
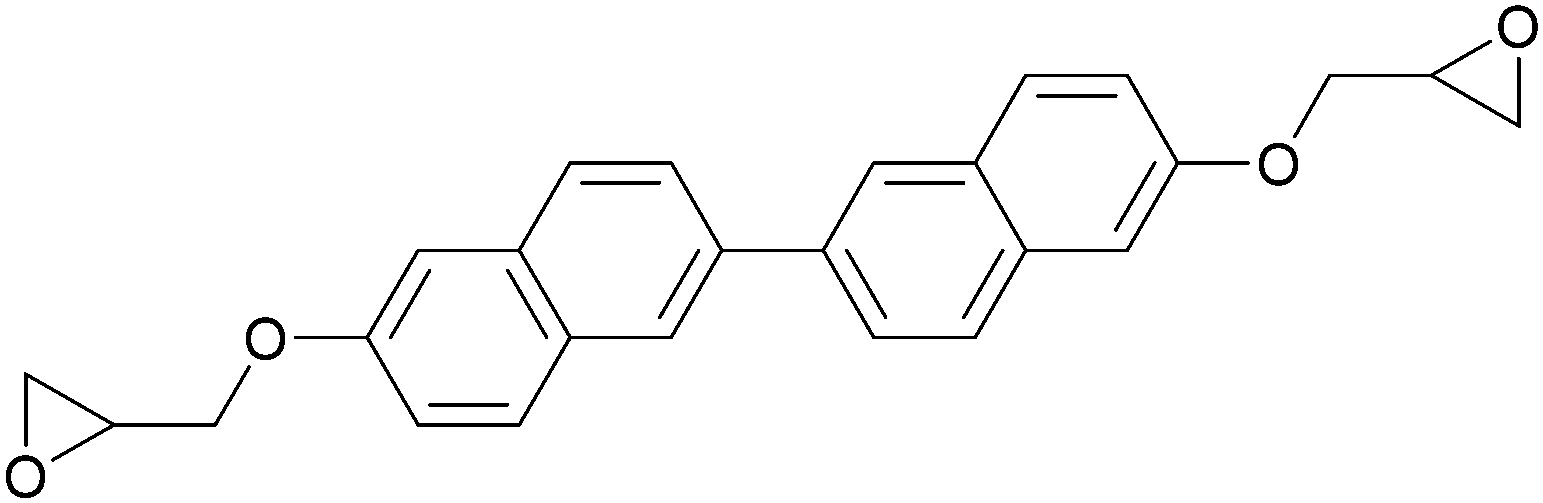
|
240 | 270 | 33 and 44 |
| Azomethine | EF3 |
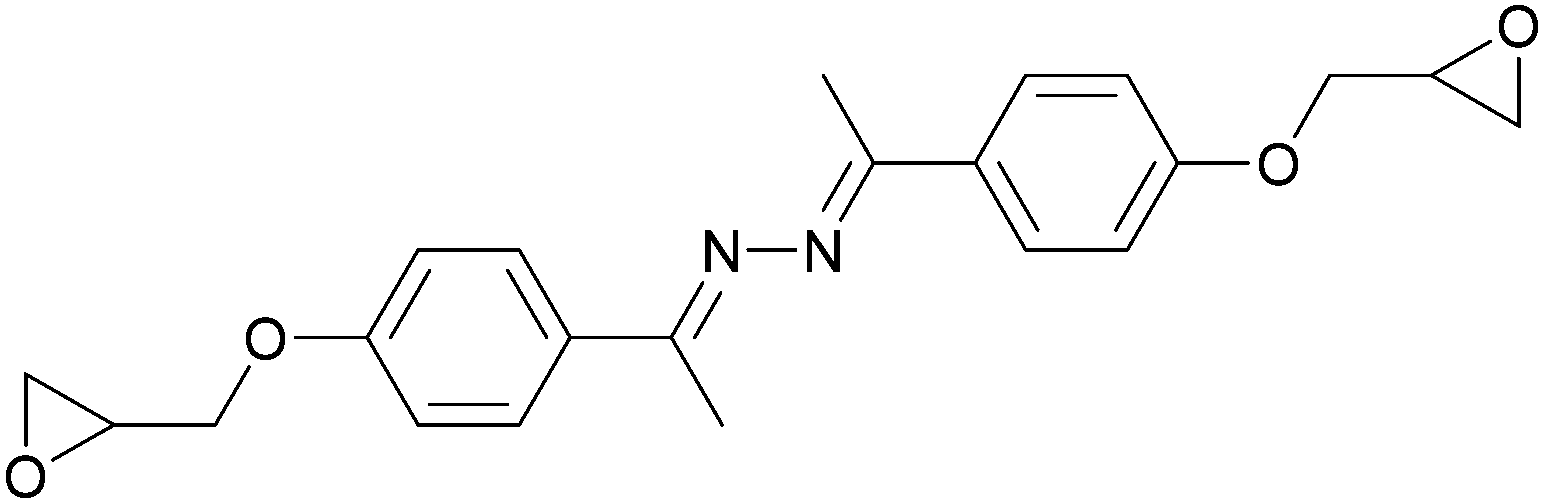
|
176 | No LC phase | 45–47 |
Although it is common to refer to these monomers as LC epoxy resins, on their own they do not necessarily exhibit LC phase behaviour. In fact, most of the common monomers either have no LC phase, or only a monotropic LC phase (Table 1). To obtain a crosslinked LCE, they may be cured with curing agents or hardeners. Diamines are the most popular class of curing agents for epoxy based LCEs. A few examples of common curing agents are shown in Chart 1. Generally, the LC phase of the LCE is formed during the curing reaction. Consequently, to a large extent the conditions during curing determine the final properties of the LCE obtained. As an example, curing methylstilbene epoxy together with sulphanilamide (SAA) gives a smectic LC thermoset,39 while curing the same monomer with diaminotoluene (DAT, Chart 1) results in a nematic LCE.40 The reason for this is the two amines in sulphanilamide have different reactivities, so in the curing process linear chains are initially formed, before the second amine group reacts to give crosslinks, resulting in longer chain formation and a smectic phase.39 In diaminotoluene, the amine groups have similar reactivities, leading to shorter chain lengths and hence a nematic phase in the methylstilbene epoxy-diaminotoluene cured LCE. As another example, a rigid aromatic diamine such as diaminodiphenyl sulphone (DDS) results in a stiffer polymer network, and thus a higher glass transition temperature (Tg), than a more flexible curing agent such as diaminodiphenylmethane (DDM), trans-1,4-diaminocyclohexane (t-DAC), or even diaminobutane (DB) (Chart 1).48 In summary, the vast number of combinations of LC epoxy monomers and curing agents allows for the development of epoxy-based LCEs with a large variety of thermal, mechanical and LC properties.
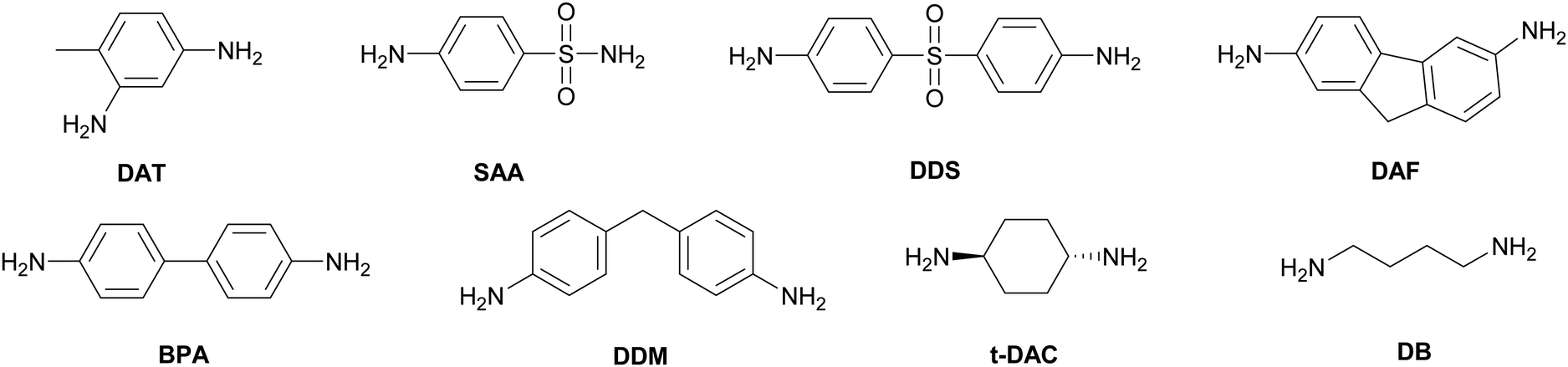 | ||
| Chart 1 Chemical structures of common aromatic diamine curing agents for LCERs. | ||
Several other factors in the curing reaction play a role in determining the structure of the final network. For example, the temperature at which the curing reaction is performed determines which LC phase is present in the final network: when a mixture of methylstilbene epoxy and diaminotoluene is cured below 180 °C, a nematic phase is observed in the polymer network. However, curing at temperatures above 190 °C leads to an isotropic network.40 Another study showed that a mixture of methylstilbene epoxy and diaminodiphenylmethane can be cured from the isotropic melt to a nematic network, and from a nematic monomeric phase to a smectic network.48 Hence, not only does the choice of monomer determine the properties of the LC thermosets, but also the conditions at which they are cured. An extensive overview of these temperature and kinetic effects is given in a review article by Carfagna et al.49
As mentioned earlier, LC epoxy resins were initially developed to improve the properties of conventional epoxy resins. The LC domains in the LC epoxy resins lead to an increase in fracture toughness and resistance to brittle crack propagation.50–52 Furthermore, the presence of an LC phase provides a higher resistance to permanent creep deformation.53 Other interesting aspects of LC epoxy resins include increased thermal stability,33,54 increased thermal conductivity55,56 and moisture barrier properties.57
Epoxide-based LCEs as shape memory polymers
After the development of LC epoxy resins as improvements to classical epoxy resins, researchers progressed to developing soft actuators from these resins. LC epoxy resins have found to be very useful in the fabrication of shape memory polymers (SMPs). SMPs are a class of polymer that can be programmed into a temporary shape through deformation and return to their original (permanent) shape upon exposure to an external stimulus. Multiple classes of SMPs exist, with one-way and two-way SMPs being the most important. A one-way SMP can be programmed in a temporary shape and irreversibly recover to its original shape upon increasing the temperature.58 Usually, a phase transition taking place at this temperature triggers the recovery of the permanent shape. Reprogramming of the one-way SMP is required to adopt the temporary shape again (Fig. 1a). Two-way SMPs can reversibly contract upon heating and extend on cooling.58 LCEs are particularly suitable for the fabrication of SMPs, as they exhibit a reversible isotropic-LC transition that can be tuned relatively easily.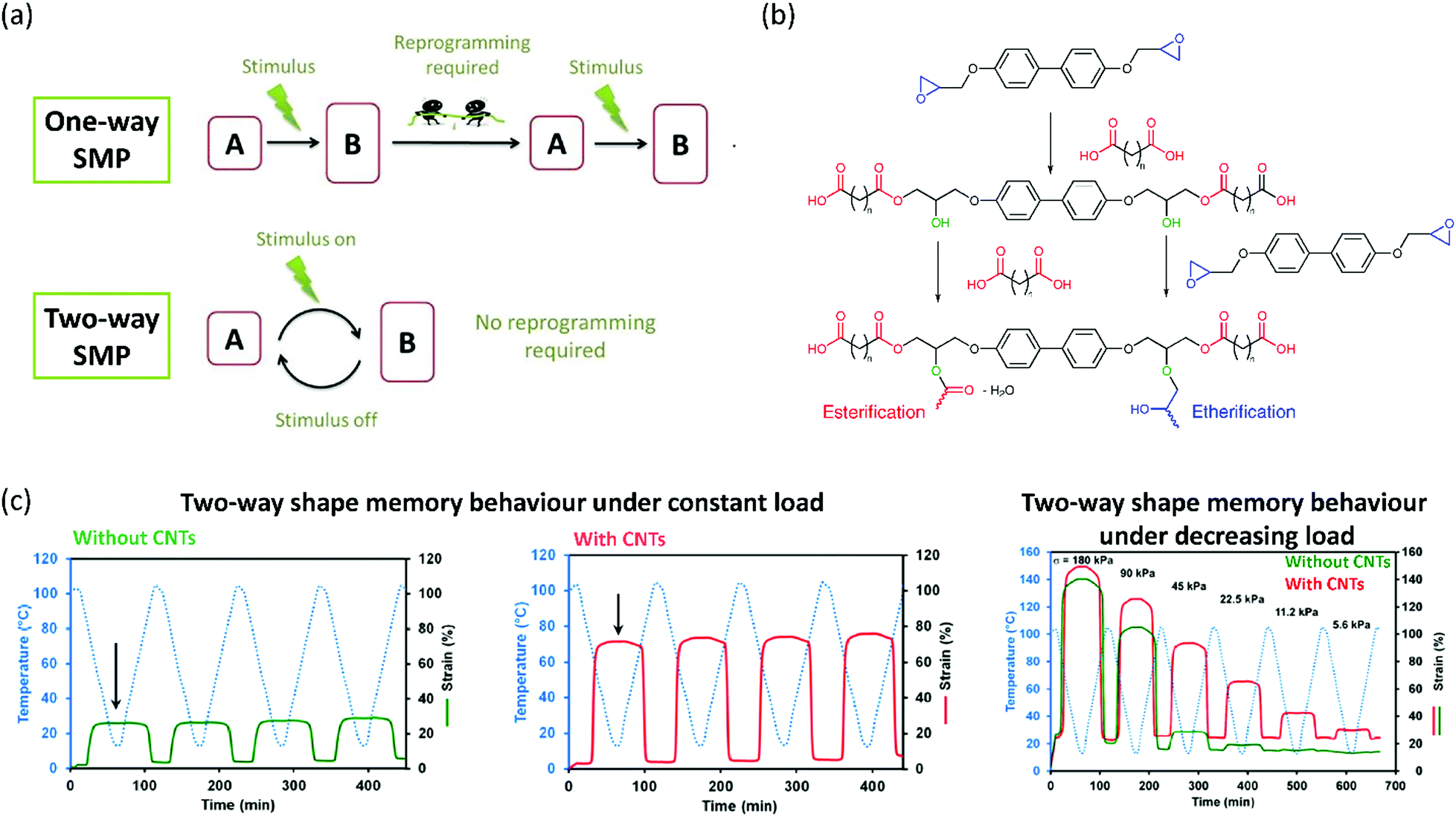 | ||
| Fig. 1 (a) Definitions of one-way, and two-way SMPs. A one-way SMP can irreversibly revert to its permanent shape after a stimulus, after which reprogramming is required. A two-way SMP shows reversible actuation. Reproduced from ref. 58 with permission from Elsevier, copyright 2015. (b) Mechanism of the step-growth polymerisation of a diepoxide monomer and a dicarboxylic acid to obtain LCEs. Reproduced (adapted) from ref. 59 with permission from American Chemical Society, copyright 2015. (c) Two-way shape memory behaviour of a typical diepoxide–dicarboxylic acid LCE (left), a similar LCE with CNTs grafted on the epoxide monomer (middle) and the behaviour of both LCEs under decreasing tensile loadings, revealing a lower threshold for the CNT–epoxy LCE composite. Reproduced from ref. 60 with permission from American Chemical Society, copyright 2015. | ||
To increase the stimuli-responsivity of LC epoxy resins by reducing the crosslink density, Li et al. have used a combination of biphenyl epoxy and a dicarboxylic acid curing agent, sebacic acid, to produce a smectic main-chain LCE.59 The step-growth polymerisation mechanism is shown in Fig. 1b. In this process, two parallel reactions are taking place. In a first step, the epoxide is ring-opened by the carboxylic acid, giving a linear oligomer with additional hydroxy groups. These hydroxy groups can then, in a second step, react with another carboxylic acid in an esterification reaction, or with an epoxy monomer in an etherification reaction. The authors assumed that the nature of the chemical bonds formed in this second step had a significant influence on the liquid crystallinity and network structure of the LCE. Therefore, by varying the ratio between the biphenyl epoxy monomer and the curing agent sebacic acid, they were able to tune, to a large extent, both the LC transition and thermomechanical properties of the generated LCE. The authors concluded that their approach could help in the practical application of LCEs as two-way SMPs, as the actuation properties can be easily adjusted by varying the stoichiometric ratio between epoxide monomer and dicarboxylic acid curing agent.
Inspired by this work, other researchers have found new ways to tune the thermomechanical response of shape memory materials based on smectic epoxy networks. Lama et al. have incorporated carbon nanotubes into a similar smectic main-chain epoxy LCE to tune the actuation properties of the obtained elastomer.60 They achieved this by grafting methylstilbene epoxy with multiwalled carbon nanotubes (CNTs) and subsequently curing the grafted epoxy monomer with curing agent sebacic acid, which led to the formation of homogeneous composites. It turned out that the incorporation of multiwalled CNTs led to a significant enhancement of the thermal actuation behaviour of the elastomer alongside a decreased stress threshold required to achieve actuation (Fig. 1c). The authors attributed these effects to the better alignment of the LC domains when nanotubes are present in the elastomer. Hence, it was possible to tune the transition temperatures and stress thresholds required for actuation by varying the amount of multiwalled CNTs dispersed into the LCE. Another publication by the same group studied the effect of varying the aliphatic chain length in the dicarboxylic acid curing agent.61 By increasing the chain length, the isotropisation temperature (Ti) of the LC domains in the elastomer increased as well. Effectively, this means that the temperature at which actuation will occur can be adjusted. The authors also showed that it is possible to tune the amount of strain actuation in this way. Hence, the shape changing properties can be easily tuned in several ways. However, in all the actuators described in this section, reversible shape memory behaviour was only achieved when a constant load was applied in the actuation direction to revert the polymer to its original shape.61
Belmonte et al. claim that autonomous two-way SMPs can be obtained by preparing a bilayer of an epoxide-based LCE with a glassy thermoset.62 In this system, the glassy thermoset was composed of an excess of bisphenol A-based epoxy crosslinker and a tri-functional thiol. This mixture was first cured at a relatively low temperature to retain the excess of epoxy and obtain a sticky intermediate network. A programmed LCN actuator based on biphenyl epoxy and sebacic acid was attached, and the bilayer was heated to 90 °C to further cure the glassy thermoset and connect it to the LCN actuator. The resulting bilayer was now able to bend to a temporary shape upon heating above the LCN's Ti, and back to its original shape upon cooling due to the stress induced in the glassy thermoset, making this material a true two-way SMP.
Vitrimer LCEs
All the materials we have described so far are thermosets: polymers with high mechanical stability. This mechanical stability, however, comes at a price. The crosslinks in these polymer networks are permanent, making it virtually impossible to remould, reprocess, or recycle them. In 2011, Leibler and co-workers published their pioneering article on vitrimers: polymer networks based on epoxy and carboxylic acid or anhydride monomers, to which they have added a transesterification catalyst.63 This resulted in a crosslinked epoxy thermoset in which transesterification reactions occur at higher temperatures (Fig. 2a). Effectively, this means that the crosslinks in the material become dynamic at elevated temperatures (Fig. 2b). This phenomenon introduces an additional viscoelastic transition temperature, the “topology freezing temperature”, Tv. Above this temperature, the timescale of the transesterification reactions becomes shorter than the timescale of material deformation, which effectively means that the material can flow. Hence, vitrimers have the unique property of the mechanical stability of a classical thermoset at room temperature, yet can be processed or reprocessed at elevated temperatures. Therefore, the invention of vitrimers has opened the way to the development of thermosetting polymers that can be remoulded, repaired and recycled in a similar manner to glasses and metals.64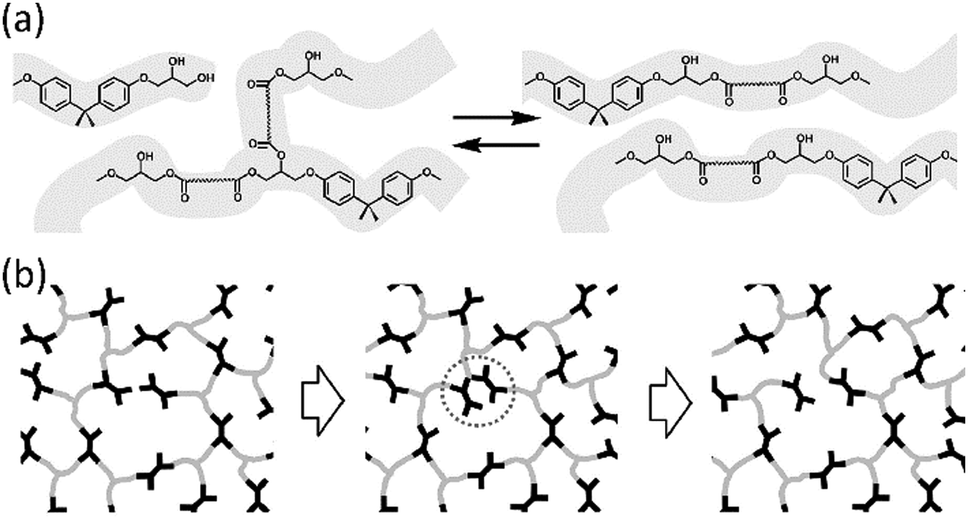 | ||
| Fig. 2 (a) Transesterification reactions in thermosets based on epoxy resins and carboxylic acids. (b) In these thermosets, crosslinks become dynamic at elevated temperature. Figures reproduced from ref. 63 with permission from AAAS, copyright 2011. | ||
Initially, vitrimers were based on conventional epoxy resins. A few years after their invention, Ji and co-workers realised that the same approach should also be possible for LC epoxy resins, since the earlier approach of curing LC epoxy resins with dicarboxylic acids for applications such as shape memory polymers gives an LCE with ester bonds that are susceptible towards transesterification. In 2014, Pei et al. reported the development of liquid crystal elastomers with exchangeable links (xLCEs).65 The epoxy LCE network they used consisted of a combination of the biphenyl LC epoxy monomer and curing agent sebacic acid (Fig. 3a). A transesterification catalyst was added to this monomer mixture, which was subsequently cured. This resulted in a polydomain nematic LCE. Similar to LCEs based on these monomers, the xLCE has a Tg at about 55 °C and an isotropic transition at 100 °C. However, upon further heating, the transesterification catalyst introduced dynamic crosslinks into the xLCE, giving it a Tv of 160 °C. This made the xLCE re-shapeable and re-processable, a feature that had not been seen in LCEs before. The authors demonstrated that they could use the dynamic crosslinks to re-join two pieces of xLCE film by compression moulding. Not only did this work with two pieces of the same xLCE, but also with a conventional epoxy elastomer cured with a diacid. This allowed for the fabrication of bi- or multilayers of elastomers with different properties. The authors also demonstrated that the xLCEs showed two-way shape memory behaviour under a constant load, and that this behaviour was reversible without any signs of creep (Fig. 3b). In contrast, a non-LC analogue of the xLCE only showed creep upon repeated exposure to high temperatures. Furthermore, a remoulded-xLCE sample showed almost identical thermal actuation behaviour to a virgin sample (Fig. 3c).
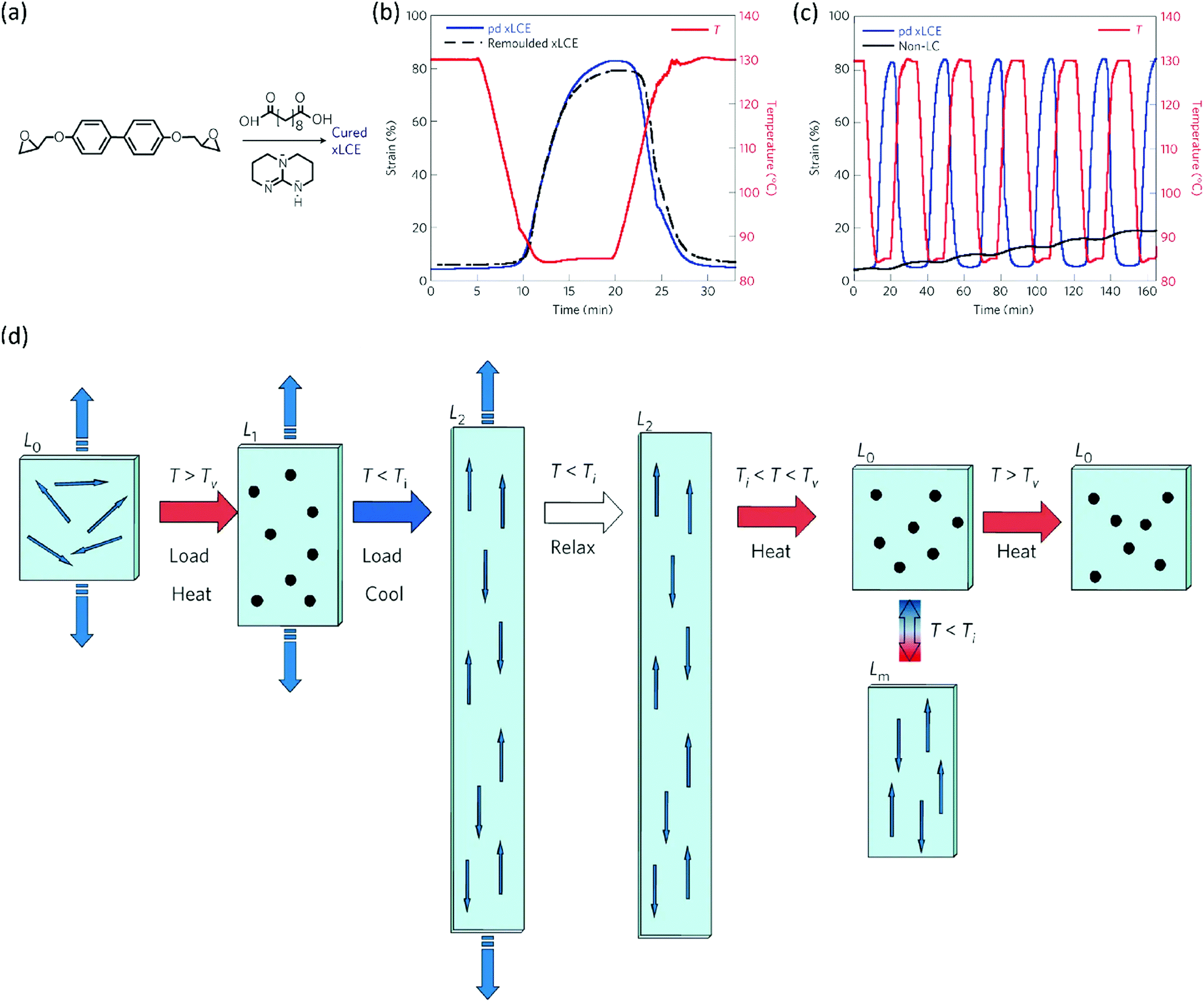 | ||
| Fig. 3 (a) Composition of the xLCE: biphenyl epoxy monomer, curing agent sebacic acid and transesterification catalyst triazabicyclodecene are cured to obtain a polydomain xLCE. (b) Thermal actuation behaviour under constant load of a polydomain xLCE compared to a non-LC analogue containing exchangeable crosslinks. The xLCE shows reversible actuation without any creep, whereas the non-LC analogue only shows repeated creep. (c) Reversible thermal actuation behaviour under constant load of a virgin polydomain xLCE compared to a remoulded sample. Both samples show near-identical actuation behaviour. (d) Programming a polydomain xLCE. From left to right: a polydomain sample (L0) is heated above Tv under load, giving isotropic sample L1 which is subsequently cooled under load to <Ti to obtain monodomain alignment (L2). This xLCE can be actuated between L0 and Lm by heating above and cooling below Ti. Reprogramming the alignment is possible by heating above Tv again. Figures reproduced from ref. 65 with permission from Springer Nature, copyright 2014. | ||
A remarkable aspect of xLCEs is that polydomain elastomers that are obtained after curing can be processed into a monodomain alignment, and that even this is reversible.65 One of the main challenges with LCEs is obtaining monodomain alignment, which is generally achieved with external fields in LC monomers, or mechanical stretching in a two-step crosslinking process. Both methods, however, have their disadvantages. Alignment using an external field generally only works for thin films. In the two-step crosslinking method, it is difficult to obtain three-dimensional shapes. The xLCEs are a promising solution to these challenges, as the presence of a Tv allows monodomain alignment by stretching a film at a temperature above Tv, shown schematically in Fig. 3d. From left to right: when the polydomain sample is loaded above Tv, the LC domains will become isotropic, and the crosslinks reversible. Subsequent cooling of the sample then results in reforming the smectic LC domains that are now aligned (L2). Effectively, the sample is now an actuator that will shrink to L0 upon heating above Ti and expand back to Lm upon cooling. The unique feature of the xLCE is the monodomain alignment can be reprogrammed by again heating to above Tv and loading the sample. Furthermore, the authors showed that xLCEs could be formed into virtually any shape by heating them in a mould. In summary, the development of xLCEs has led to responsive polymers that can be more easily processed than conventional LCEs and can be repaired, reprogrammed, and remoulded.
Since the initial report of xLCEs by Ji and co-workers, researchers have improved on the concept with alternatives for transesterification reactions. Li et al. have shown that xLCEs can also be obtained by adding disulphide chemistry into the LCE.66 They found it was possible to make use of disulphide–disulphide exchange reactions to reprocess the LCE. Furthermore, they showed that the material can be fully recycled by adding small thiol compounds to break down the network structure.
Another strategy to improve the functionality of xLCEs is to make nanocomposites. Yang et al. showed that by incorporating carbon nanotubes (CNTs) into the xLCE, a mould was no longer required to program or reprogram the shape of the xLCE.67 The CNTs made the LCE responsive to light, as illumination resulted in photothermal heating, a concept that has been previously employed in conventional LCEs to achieve actuation68 and transesterification in regular epoxy vitrimers.69 Here, the authors used infrared (IR) light to heat the elastomer above its Tv and activate the transesterification reactions.67 Therefore, a film could be processed into a dynamic 3D shape by stretching it into the desired shape and subsequently illuminating it (Fig. 4a). Again, this process could be repeated many times, as was demonstrated by the repeated reconfiguring of a six-petal flower into more than 20 dynamic 3D structures (Fig. 4b). Furthermore, segments of different chemical compositions could be welded together to obtain even more complex actuators.67 This welding principle was employed in another study by Pei et al. to increase the number of shapes that can be ‘remembered’ in an SMP.70 To this end, vitrimer films consisting of sebacic acid and different epoxy monomers (bisphenol A, methylstilbene, biphenyl) were prepared. A multi-SMP was then prepared by welding pieces of these films together by hot pressing, and then giving the combined film its temporary shape. As each part had a different transition temperature, they would recover their original shapes at different temperatures. This can, for instance, be applied to engineer a quadruple SMP that can fold from a cross into a cube by strategically placing the different LCE compositions in the cross (Fig. 4c). Furthermore, CNTs can be incorporated in parts of the multi-SMP to make those parts responsive to IR light. The approach of welding pieces of LCE vitrimers with different chemical compositions allows for partial reprogramming of a strip (Fig. 4d) and reuse of strips by cutting and reassembling them into new structures (Fig. 4e).
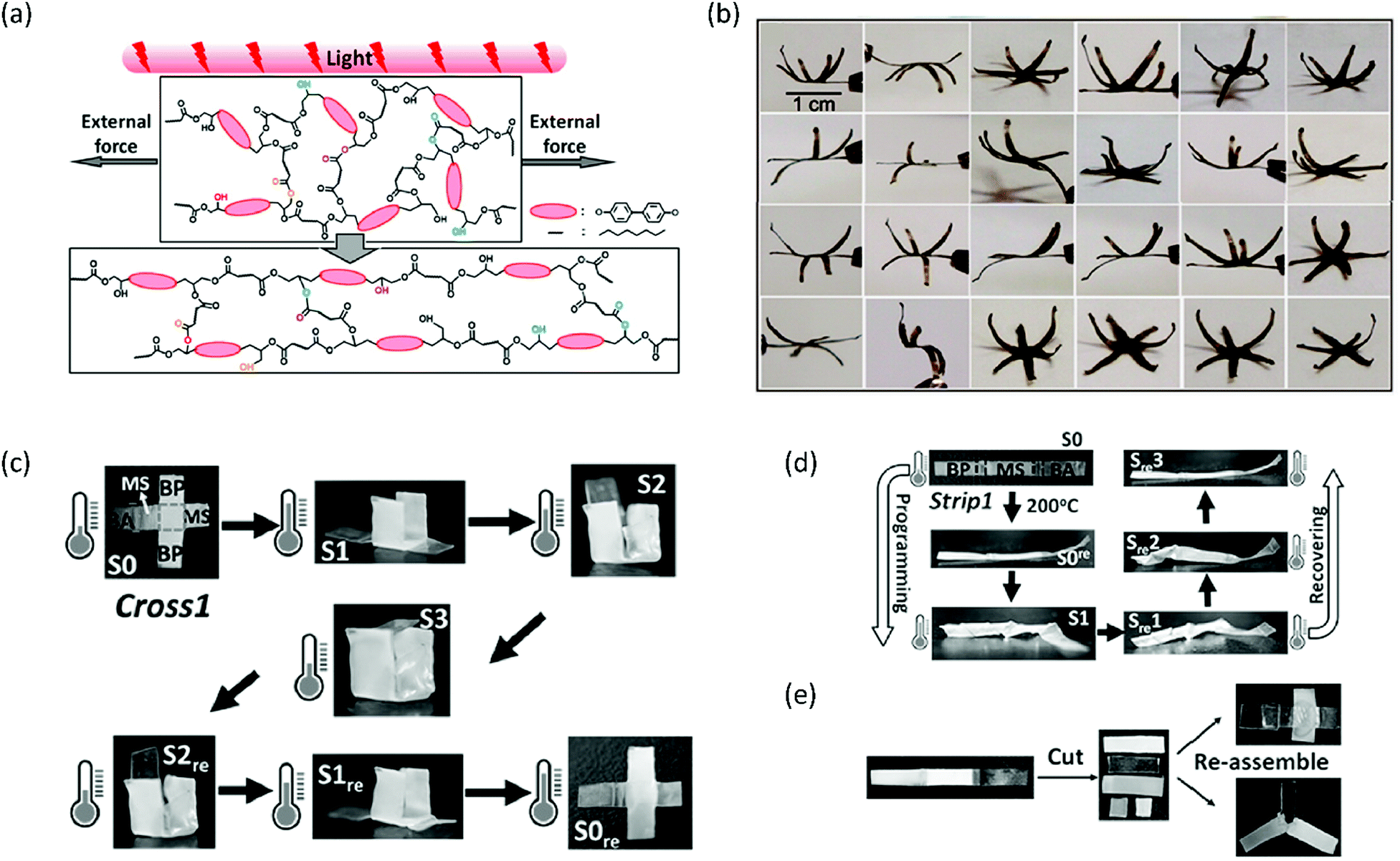 | ||
| Fig. 4 (a) Programming of a xLCE–CNT composite through generation of heat by shining light whilst keeping the film under load. Reproduced from ref. 67 with permission from American Chemical Society, copyright 2016. (b) Processing and reprocessing of a six-petal flower into various dynamic 3D shapes. Reproduced from ref. 67 with permission from American Chemical Society, copyright 2016. (c) Quadruple shape memory behaviour of a cross consisting of pieces of LCE vitrimers with different chemical compositions that are welded together. Reproduced from ref. 70 with permission from Wiley-VCH, copyright 2016. (d) Partial reprogramming of a welded strip by only twisting the BA part of the film, and the related shape memory behaviour. Reproduced from ref. 70 with permission from Wiley-VCH, copyright 2016. (e) Cutting and reassembling welded strips to obtain new structures. Reproduced from ref. 70 with permission from Wiley-VCH, copyright 2016. | ||
The possibility of making LCE vitrimers responsive to light of other wavelengths has also been investigated. In conventional LCEs and LCNs, chromophores are commonly used to convert light energy into mechanical work. A classic example is the azobenzene moiety that changes its geometry from trans to cis upon illumination with ultraviolet (UV) light. This effect has been used extensively to achieve shape changes in LCEs and LCNs, such as bending, twisting and oscillating.70 Li et al. incorporated azobenzene into an LCE vitrimer by curing an azobenzene epoxy monomer with sebacic acid and adding a transesterification catalyst (Fig. 5a).71 This led to the formation of a multifunctional LCE that had the shape memory behaviour of an LCER, the (re)processability of an LCE vitrimer and the light-responsivity of an azobenzene LCN. The authors have demonstrated this by programming a cross-shaped sample into a cube (Fig. 5b). Heating of this cube above Tg led to unfolding, whilst further heating above Ti led to refolding. The same shape memory behaviour could be achieved by illuminating the cube with UV light. The isomerisation of the azobenzene moieties heated the cube photothermally to unfold above Tg and further heating to above Ti caused refolding (Fig. 5b). The authors also showed that the material could be reprocessed or repaired by heating above Tv, and that this could also be locally achieved by UV illumination. Furthermore, in a follow-up publication, they showed that the material could also be prepared with exchangeable disulfide bonds as the dynamic crosslinks.72
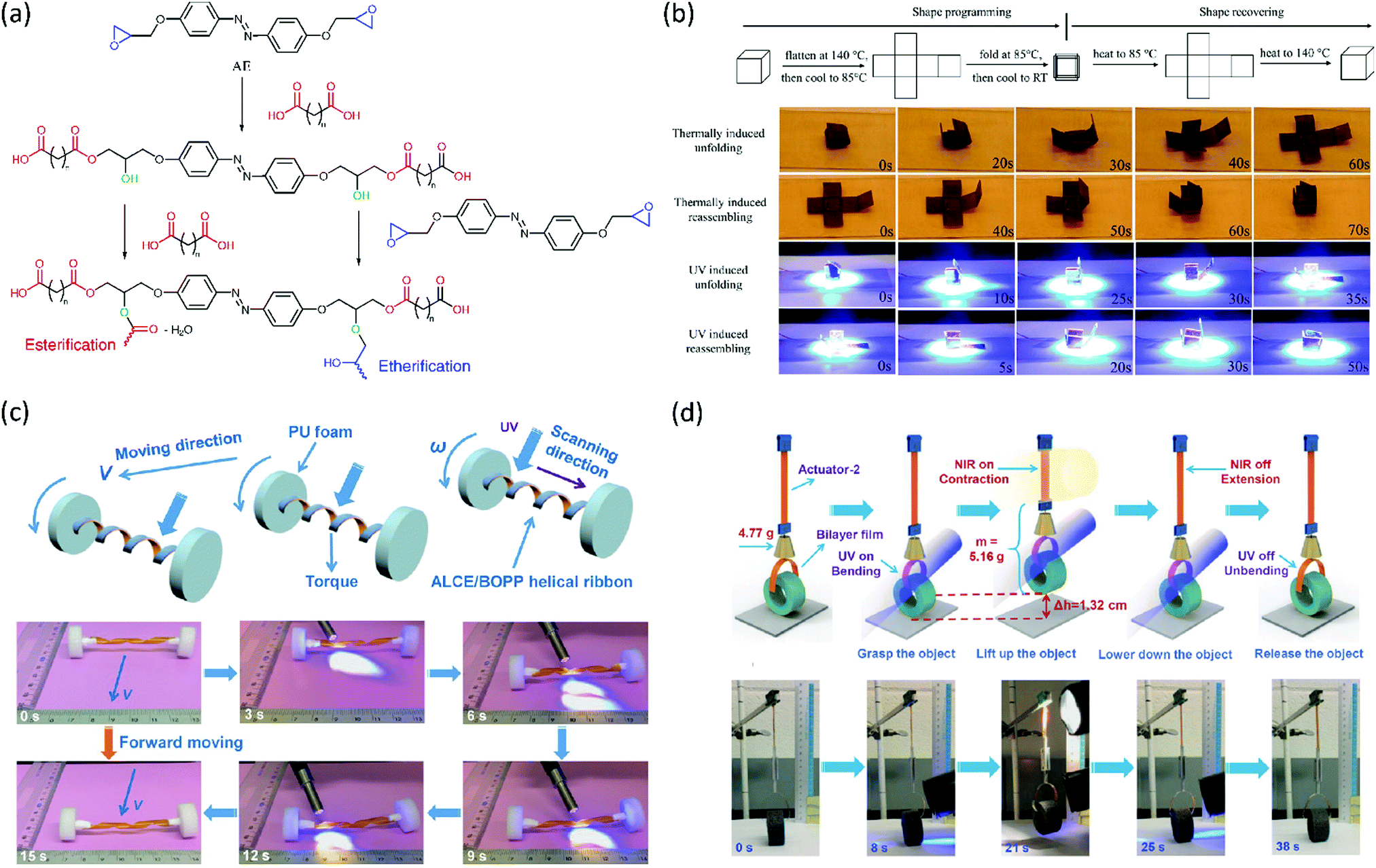 | ||
| Fig. 5 (a) Synthesis of a photoresponsive LCE vitrimer by step-growth polymerisation of an azobenzene epoxide monomer (AE) and a dicarboxylic acid, and with adding a transesterification catalyst. Reproduced (adapted) from ref. 71 with permission from American Chemical Society, copyright 2016. (b) Programming of a photoresponsive LCE vitrimer cross into a cube, and its thermal and UV induced folding and unfolding behaviour. Reproduced from ref. 71 with permission from American Chemical Society, copyright 2016. (c) Light-driven forward motion of an azobenzene LCE vitrimer ‘vehicle’ with a spring-like ‘motor’. Reproduced from ref. 73 with permission from Wiley-VCH, copyright 2017. (d) ‘Polymer crane’ constructed from a gold nanorod – azobenzene LCE vitrimer nanocomposite that is capable of grasping, lifting, lowering, and releasing objects upon illumination with UV and NIR light. Reproduced from ref. 74 with permission from Wiley-VCH, copyright 2018. | ||
Lu et al. have developed the system further by synthesising a light responsive LCE vitrimer from the azobenzene epoxy monomer and dodecanedioic acid, which is a slightly longer dicarboxylic acid than sebacic acid.73 They showed the UV-induced contraction force generated in the LCE vitrimer was higher when the film strain was higher, due to enhanced orientation of the azobenzene mesogens. They also demonstrated the contraction force generated originates from both mechanical action by trans–cis isomerisation of the azobenzene mesogens, as well as a photothermal effect of heating the film above a transition temperature. By making bilayers of the LCE vitrimer with transparent polypropylene, they were able to construct wheels that could move away from or towards a light source via a photoinduced shifting of its centre of mass. Taking this a step further by twisting the bilayer into a spring and attaching it to wheels of polyurethane foam (Fig. 5c), it was possible to construct a ‘vehicle’ that is light driven by UV-induced winding/unwinding of the spring.73
To further increase the potential of this system, the same group doped the azobenzene LCE vitrimer with gold nanorods to expand responsivity to near-infrared light (NIR).74 This resulted in a nanocomposite capable of two light-triggered molecular changes: a transition from the LC to isotropic state by absorption of NIR and subsequent heat release by the gold nanorods, as well as photoisomerisation of the azobenzene moieties. The authors demonstrated the full potential of this system by creating a polymer ‘crane’ capable of performing ‘light-controlled sophisticated, concerted robot-like, macroscopic motions to produce useful work’ (Fig. 5d).74 To this end, they attached a bilayer film acting as a gripper upon UV illumination to a telescopic arm capable of contracting upon NIR illumination. The resulting polymer crane could grasp and lift objects, move them, and then lower and release the cargo, demonstrating the potential applications of LCE actuators in soft robots.
Stability of LCE vitrimers
Dynamic crosslinks in LCE thermosets have been shown to have many advantages in the processing, reprocessing, and repairing of LCE thermosets. However, they also limit the stability and durability of these materials, as operation of the functional material above its Tv will activate the dynamic crosslinks and remove the monodomain alignment. To improve the stability of the LCE vitrimer, a balance must be found for the Tv: it should be sufficiently high to operate the functional material at high temperatures, yet not too high to allow for easy (re)processability. One strategy is to optimise the amount of transesterification catalyst in the material. Chen et al. have published a systematic study where they reduced the amount of catalyst and monitored the effects on processability and actuation stability of an LCE vitrimer actuator.75 They found that a lower catalyst concentration indeed led to a higher Tv and thus a higher actuation stability. Processing and reprogramming of the actuator, however, remained possible. Another strategy pursued by researchers is to make a re-processable actuator without incorporation of any transesterification catalyst. Yang et al. have demonstrated that, unlike the common perception that a catalyst is required to trigger the transesterification reactions, they still occur in LC epoxy thermosets that do not contain a catalyst, albeit at a much lower rate.76 These LC epoxy thermosets could be processed, reprogrammed and welded, similarly to the xLCEs described earlier, with the difference that these processes take longer in the LC epoxy thermosets. However, due to the slower transesterification reactions at high temperatures, actuators made from these LC epoxy thermosets have a significantly higher stability than the xLCEs.Advanced functional materials through cationic (photo)polymerisation of LC epoxides and oxetanes
So far, we have mainly discussed the step-growth polymerisation of LC epoxy resins with curing agents. The second major pathway towards LCEs and LCNs is the photoinitiated polymerisation of LC monomers. Low molar mass reactive mesogens can be photopolymerised into a crosslinked LCE or LCN. The advantage of this approach is that the temperature at which the photopolymerisation is initiated can be, within limits, freely chosen. Therefore, it is relatively easy to bring the reactive mesogens in the desired phase (e.g. smectic, nematic) by controlling the temperature and subsequently photopolymerising it. Furthermore, the mesogens can be aligned (prior to polymerisation) by a variety of alignment techniques, such as surface alignment, shear force, electric fields, or magnetic fields. This allows for a wide variety of possible alignments (e.g. planar, homeotropic) and orientations (i.e. homogeneous, splay, twisted). A possible disadvantage, though, is the use of photopolymerisation is limited to relatively thin films.The development of acrylate reactive mesogens at Philips Research in the 1980s opened the way to moderately to highly crosslinked LCNs that have proven useful in many practical applications.23 Use of multifunctional monomers will result in a high crosslink density, which can be reduced by adding mono-functional monomers. The relatively straightforward synthetic methods to obtain reactive mesogens have led to a large molecular toolbox of different monomers that can be used to tune the properties of the resulting LCN. They do, however, suffer from the aforementioned drawbacks of oxygen inhibition and polymerisation shrinkage. Cationic (photo)polymerisation of cyclic ether LC monomers such as epoxides and oxetanes can be used to solve these issues. These monomers can polymerise through a ring-opening mechanism that is shown in Scheme 1. Upon illumination with UV light, the photoinitiator (typically arylsulfonium or iodonium salts) will decompose, generating a radical cation (Scheme 1a). This radical cation will abstract a hydrogen atom from its surroundings. Subsequently the counter anion (e.g. PF6−) can attack this hydrogen atom to obtain a strong acid (e.g. HPF6). For this reason, cationic photoinitiators are sometimes referred to as photoacid generators. Only after generation of the acid can the actual polymerisation mechanism start, shown in Scheme 1b. One of the lone pairs on a monomer can attack the acid, turning the monomer into a cation that is susceptible to being attacked by the next monomer, which happens many times in the propagation phase to obtain the polymer chain. Eventually, this growing chain will be terminated by, for instance, an alcohol.
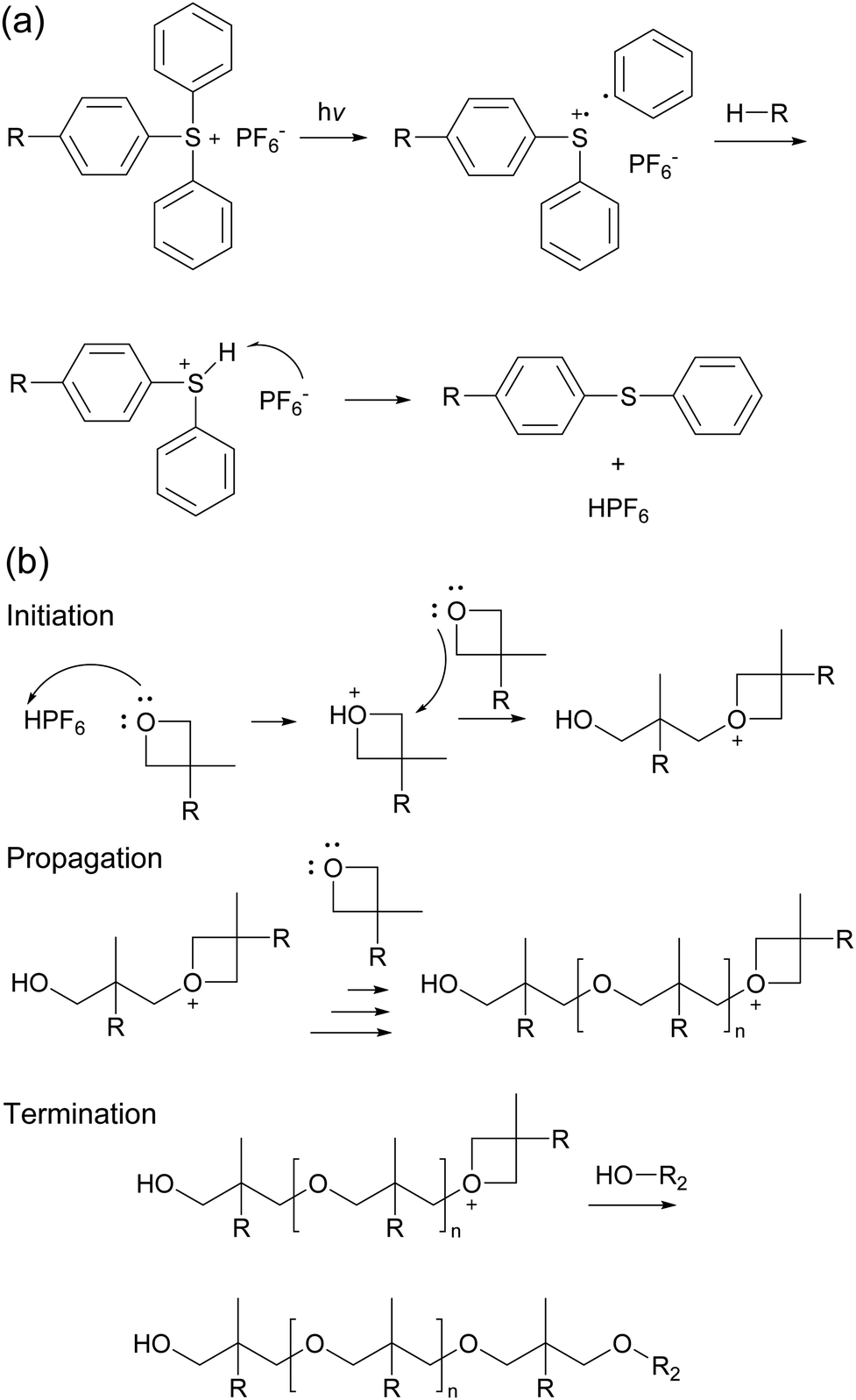 | ||
| Scheme 1 Cationic ring-opening polymerisation of cyclic ether (LC) monomers. (a) Mechanism of photoacid generation. (b) Initiation of polymerisation by the photogenerated acid, followed by propagation to generate the polymer chain. Termination occurs by reaction with a monofunctional alcohol. | ||
In 1994, two publications from Philips Research reported the synthesis of diepoxide monomers with a similar mesogenic unit as the conventional acrylate reactive mesogens.31,32 They found that it was possible to obtain LCNs by cationic photopolymerisation of these monomers. The resulting LCNs had a higher degree of order than similar networks obtained from acrylate reactive mesogens, showed lower polymerisation shrinkage, and had a higher adhesion to various substrates.31 Drawbacks, however, were a relatively slow rate of polymerisation and the occurrence of side reactions, resulting in yellow, scattering films.32
To overcome these issues, liquid crystalline oxetanes were developed in 2004 by Lub et al. (Fig. 6a).77 The polymerisation kinetics of these oxetane LCs were found to be much faster than their corresponding epoxides. Moreover, yellowing due to side-reactions during polymerisation did not occur, which makes the oxetanes more suitable for application as optical films. Van der Zande et al. have used the oxetane LCs for the fabrication of patterned retarders for transflective displays.78 These patterned retarders consisted of alternating birefringent (i.e. nematic) and isotropic domains and could be prepared by photolithographic polymerisation. First, a coated film was brought in the nematic phase and subsequently polymerised through a photomask, freezing the birefringent state in the illuminated areas. The coating was then heated above Ti and a second illumination polymerised the rest of the coating. The use of oxetane LCs instead of acrylate reactive mesogens was found to be beneficial in two ways: the possibility of photopolymerising the coating in air allowed for upscaling in future industrial applications, and the resolution of the obtained patterns turned out to be very high, higher than could previously be achieved using acrylate reactive mesogens (Fig. 6b and c).79 The authors attributed this to the absence of surface corrugation (due to reduced polymerisation shrinkage) and a lower UV sensitivity of the cationic photopolymerisation: scattered UV light in the non-illuminated areas did not result in substantial crosslinking.
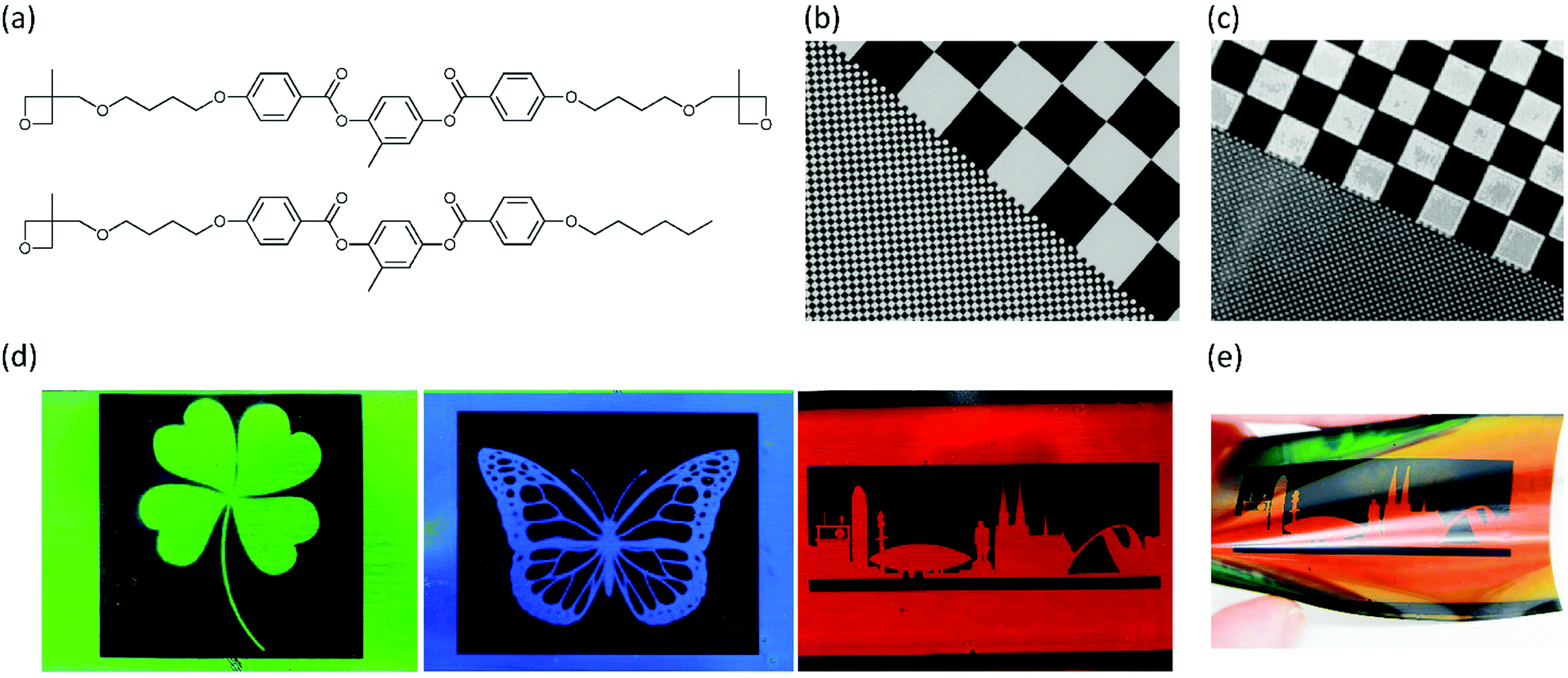 | ||
| Fig. 6 (a) Chemical structures of oxetane LC monomers. (b) Patterned birefringent-isotropic retarder for transflective displays based on oxetane LCs. Reproduced from ref. 78 with permission from Wiley-VCH, copyright 2005. (c) Patterned birefringent-isotropic retarder for transflective displays based on acrylate LCs. Reproduced from ref. 79 with permission from AIP Publishing, copyright 2006. (d) Patterned Ch-LC-isotropic coatings based on Ch-LC oxetane LCs. The colour can be tuned by changing the amount of the chiral oxetane monomer. Reproduced from ref. 80 with permission from American Chemical Society, copyright 2019. (e) Changing the viewing of a Ch-LC coating changes the observed colour. Reproduced from ref. 80 with permission from American Chemical Society, copyright 2019. | ||
Inspired by this work, in a recent publication the oxetane monomer toolbox has been extended with a chiral dioxetane monomer.80 This allowed for the fabrication of photonic coatings that could reflect light, as the addition of the chiral oxetane to a nematic oxetane LC would result in a twist of the molecular director, and consequently in a periodic refractive index variation. Therefore, the resulting chiral nematic LC (Ch-LC) phase resembled a 1D photonic crystal that could selectively reflect light.15 These Ch-LC oxetanes were used to fabricate photonic coatings on a relatively large scale using flexographic printing on flexible substrates. The photonic oxetane LC coatings could be patterned lithographically at a high resolution, and in a wide variety of patterns (Fig. 6d and e), giving these coatings potential for application in both visually pleasing coatings as well as anticounterfeit labels.
Along with oxetane and epoxide LC monomers that are based on the phenyl benzoate mesogenic structure, it is also possible to polymerise the LCERs described earlier in this review via a cationic ring-opening polymerisation. Lv et al. have demonstrated that light responsive LCN films can be obtained via the cationic photopolymerisation of azobenzene diepoxides.81 They found that simultaneous illumination with unpolarised blue light during UV polymerisation of a planar aligned monomer mixture, induced reorientation of the azobenzene moieties at the illuminated side, resulting in a splay alignment. This way, they were able to control the bending behaviour of the films, either away from or towards the light source by illuminating it from either the planar or the homeotropic sides of the film, respectively.
Earlier, we mentioned the increased thermal conductivity of LC epoxy resins over conventional epoxy resins. Islam et al. have found that the cationic ring-opening polymerisation of biphenyl epoxy increased the thermal conductivity of the resulting network.82 They attributed this to weaving of the pre-aligned epoxy groups in a linear fashion via cationic polymerisation, whereas curing with a diamine hardener disturbed the order of the LC monomer. Therefore, cationic polymerisation of biphenyl epoxy resulted in a higher molecular order and reduced phonon scattering, which substantially enhanced the thermal conductivity.
Combining free-radical and cationic ring-opening polymerisations
Free-radical and cationic polymerisations are orthogonal mechanisms. This means that, under the right conditions, both polymerisation mechanisms can be performed independently, either simultaneously or sequentially. As an example, this could be done by using a radical and a cationic photoinitiator that have different activation wavelengths. Combining cationic and free-radical polymerisations could prove useful in producing chiral nematic LC systems. Generally, the reflection band of a Ch-LC is quite narrow, limiting the effectiveness of a Ch-LC coating designed to reflect IR light for energy saving. To solve this, researchers have set out to broaden the reflection band of Ch-LCs by photopolymerisation-induced diffusion.83,84 By combining diacrylate with monoacrylate LCs, and illuminating at a low UV intensity during polymerisation, the highly reactive diacrylates diffuse towards the top of the film, leading to a gradient in the concentration of the chiral LC through the film thickness, and hence to a pitch gradient and a broadened reflection band. Zhang et al. have shown that the same principle can be applied to a Ch-LC mixture of epoxide and acrylate LCs.85 In their approach, a free-radical initiator was activated first by illumination with 365 nm UV light, inducing diffusion of the acrylate LCs to the top of the film, which induced a pitch gradient in a similar way to the diacrylate–monoacrylate approach (Fig. 7a). In a second polymerisation step, the cationic photoinitiator was activated by 254 nm UV light, freezing the pitch distribution in the Ch-LC, and resulting in broadening of the reflection band (Fig. 7b). Since the difference in reactivity between the acrylate and epoxide LC in the free-radical polymerisation step is essentially infinite, obtaining a pitch gradient is potentially more effective then when a mixture of mono- and diacrylate LCs is used.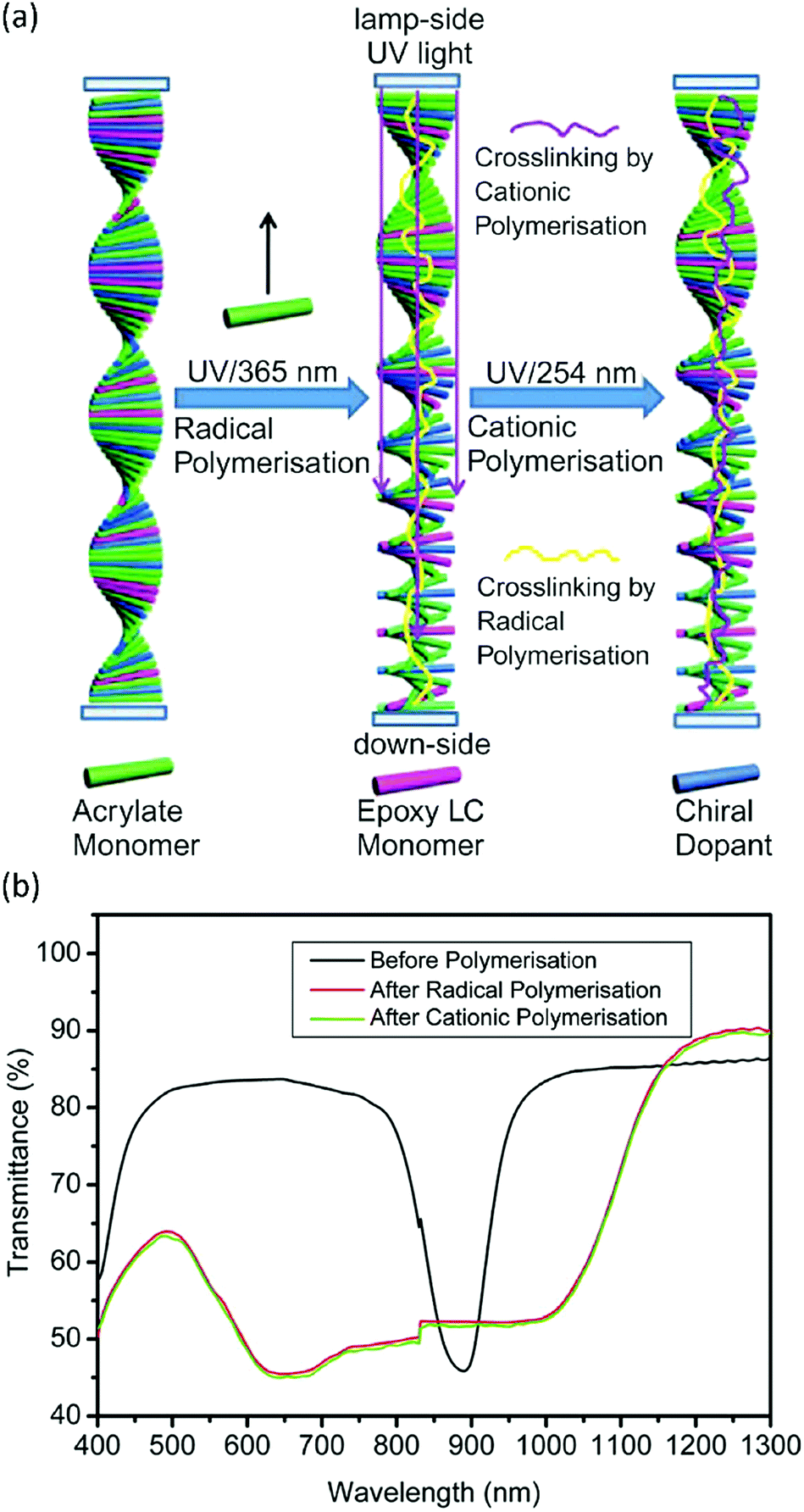 | ||
| Fig. 7 (a) Mechanism of photoinduced diffusion by sequential polymerisation of acrylate and epoxide LC monomers to obtain a broadband reflecting Ch-LC film. (b) Transmission spectra of the Ch-LC film before and after the sequential polymerisation process. Reproduced from ref. 85 with permission from Taylor & Francis Ltd, copyright 2011. | ||
Another area where cationic ring-opening polymerisations could be of importance is additive manufacturing. Since epoxy and oxetane LC monomers can be polymerised in oxygen-rich environments, obtaining complex form factors or easier fabrication protocols become possible where oxygen inhibition of acrylate monomers poses an issue. Recently, McCracken et al. published a study where they used epoxy LCs with a mesogenic structure analogous to common acrylate reactive mesogens to obtain LCE micro actuators.86 By using two-photon direct laser writing, they were able to prepare microstructured LCEs from aligned epoxy LC monomers in an oxygen-rich environment. An extensive study of the thermal- and mechanical properties of bulk films of these epoxy monomers revealed that the thermal response of epoxy-based LCEs is complementary to acrylate-based LCEs: the epoxies actuated at higher temperatures than the acrylates. This, in combination with the orthogonality of the polymerisation mechanisms of the epoxy and acrylate LCs, allowed them to prepare hybrid microactuators that were effectively composites of epoxy and acrylate LCEs. To this end, they first prepared the microstructure by using two-photon direct laser writing of the epoxy LCE, where the oxygen-insensitivity of the cationic polymerisation aided substantially in obtaining complex structures at a high resolution. This microstructure was then filled with the acrylate LC monomer, which was subsequently polymerised through a free-radical polymerisation. The resulting hybrid microactuator could be actuated at a wide temperature range, owing to the thermal properties of both components.
Conclusions and future perspectives
Advanced functional materials based on cyclic ether liquid crystals have great potential of being applied in fields such as soft actuators, self-healing, and optical materials. Liquid crystalline epoxy resins that polymerise through step-growth were initially developed in the 1990s to improve the properties of conventional epoxy resins. They were found to exhibit an increased fracture toughness and resistance to creep, higher thermal stability, increased thermal conductivity and better moisture barrier properties. Later, LC epoxy resins have been applied in smart, responsive materials by curing them with dicarboxylic acids, resulting in LCEs that can act as actuators and shape memory materials. By making nanocomposites of these LCEs with carbon nanotubes, for instance, may further increase the responsivity and functionality.A major breakthrough in the development of advanced functional materials from LC epoxy resins came with the development of LCE vitrimers. These materials have the mechanical and thermal stability of conventional LCEs, but the dynamic crosslinking at higher temperatures allows for easily processability, re-processability and repairability. Moreover, the alignment of the LC domains can be easily reprogrammed, making the resulting actuators very versatile in use. In a world where people will have to move to a circular economy, recyclable network polymers will become increasingly important. LCE vitrimers are therefore very suitable candidates as durable materials in soft robotics and other micromechanical systems.
In addition to curing LC epoxy resins with hardeners such as diamines and dicarboxylic acids in a step-growth manner, cyclic ether LC monomers such as epoxies and oxetanes can also be cured through cationic ring-opening (photo)polymerisation. Benefits of this approach include insensitivity to oxygen, allowing for polymerisation in air, and reduced polymerisation shrinkage. In particular, for applications such as coatings and additive manufacturing, where relatively thin layers need to be polymerised, the possibility of photopolymerisation in air is a major benefit over free-radical polymerisation of LC acrylates. Moreover, free-radical polymerisation and cationic ring-opening polymerisation are orthogonal mechanisms. Combining acrylate and cyclic ether LC monomers and independently addressing their polymerisation gives rise to new smart materials with unique properties. We expect that this will continue to promote the development of novel smart materials in the near future.
With this review, we show the potential of LC monomers with cyclic ethers as polymerisable groups in the fabrication of advanced functional materials. It is foreseeable that these materials will continue to expand at a rapid pace.
Abbreviations
| LC | Liquid crystalline |
| LCN | Liquid crystalline network |
| LCE | Liquid crystalline elastomer |
| T g | Glass transition temperature |
| T i | Isotropisation temperature |
| T v | Topology freezing temperature |
| SMP | Shape memory polymer |
| CNT | Carbon nanotube |
| xLCE | Liquid crystalline elastomer with exchangeable cross-links |
| UV | Ultraviolet |
| IR | Infrared |
| NIR | Near-infrared |
| Ch-LC | Chiral nematic liquid crystal |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge the inspiring discussions with and contributions of all current and former colleagues. This work was financially supported by the Netherlands Organization for Scientific Research (TOP-PUNT 718.016.003).References
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- A. Lendlein and O. E. C. Gould, Nat. Rev. Mater., 2019, 4, 116–133 CrossRef.
- L. F. Boesel, C. Cremer, E. Arzt and A. Del Campo, Adv. Mater., 2010, 22, 2125–2137 CrossRef CAS PubMed.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- J. Kim, G. K. Ong, Y. Wang, G. LeBlanc, T. E. Williams, T. M. Mattox, B. A. Helms and D. J. Milliron, Nano Lett., 2015, 15, 5574–5579 CrossRef CAS PubMed.
- A. Ghosh, B. Norton and A. Duffy, Appl. Energy, 2015, 159, 362–369 CrossRef.
- J. Shintake, V. Cacucciolo, D. Floreano and H. Shea, Adv. Mater., 2018, 30, 1707035 CrossRef PubMed.
- S. Nocentini, C. Parmeggiani, D. Martella and D. S. Wiersma, Adv. Opt. Mater., 2018, 6, 1800207 CrossRef.
- J. E. Stumpel, D. J. Broer and A. P. H. J. Schenning, Chem. Commun., 2014, 50, 15839–15848 RSC.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- A. H. Gelebart, D. Jan Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS PubMed.
- M. Pilz da Cunha, Y. Foelen, R. J. H. van Raak, J. N. Murphy, T. A. P. Engels, M. G. Debije and A. P. H. J. Schenning, Adv. Opt. Mater., 2019, 7, 1801643 CrossRef.
- D. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Macromolecules, 2012, 45, 8005–8012 CrossRef CAS.
- D. J. Mulder, A. P. H. J. Schenning and C. W. M. Bastiaansen, J. Mater. Chem. C, 2014, 2, 6695–6705 RSC.
- E. P. A. Van Heeswijk, A. J. J. Kragt, N. Grossiord and A. P. H. J. Schenning, Chem. Commun., 2019, 55, 2880–2891 RSC.
- K. Nickmans, D. A. C. van der Heijden and A. P. H. J. Schenning, Adv. Opt. Mater., 2019, 7, 1900592 CrossRef CAS.
- M. Moirangthem and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2018, 10, 4168–4172 CrossRef CAS PubMed.
- J. Küpfer and H. Finkelmann, Die Makromol. Chem., Rapid Commun., 1991, 12, 717–726 CrossRef.
- C. Ohm, M. Brehmer and R. Zentel, Adv. Mater., 2010, 22, 3366–3387 CrossRef CAS.
- D. Liu and D. J. Broer, Langmuir, 2014, 30, 13499–13509 CrossRef CAS.
- D. L. Thomsen, P. Keller, J. Naciri, R. Pink, H. Jeon, D. Shenoy and B. R. Ratna, Macromolecules, 2001, 34, 5868–5875 CrossRef CAS.
- J. Lub and D. J. Broer, in Cross-Linked Liquid Crystalline Systems: From Rigid Polymer Networks to Elastomers, ed. D. J. Broer, G. P. Crawford and S. Zumer, CRC Press, Boca Raton, United States, 1st edn, 2011, pp. 4–47 Search PubMed.
- F. L. L. Visschers, M. Hendrikx, Y. Zhan and D. Liu, Soft Matter, 2018, 14, 4898–4912 RSC.
- H. Khandelwal, A. P. H. J. Schenning and M. G. Debije, Adv. Energy Mater., 2017, 7, 1602209 CrossRef.
- O. M. Wani, H. Zeng and A. Priimagi, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- M. Pilz da Cunha, S. Ambergen, M. G. Debije, E. F. G. A. Homburg, J. M. J. den Toonder and A. P. H. J. Schenning, Adv. Sci., 2020, 7, 1902842 CrossRef CAS PubMed.
- A. Buguin, M. H. Li, P. Silberzan, B. Ladoux and P. Keller, J. Am. Chem. Soc., 2006, 128, 1088–1089 CrossRef CAS PubMed.
- T. H. Ware, Z. P. Perry, C. M. Middleton, S. T. Iacono and T. J. White, ACS Macro Lett., 2015, 4, 942–946 CrossRef CAS.
- S. C. Ligon, B. Husár, H. Wutzel, R. Holman and R. Liska, Chem. Rev., 2014, 114, 577–589 CrossRef PubMed.
- D. J. Broer, J. Lub and G. N. Mol, Macromolecules, 1993, 26, 1244–1247 CrossRef CAS.
- S. Jahromi, J. Lub and G. N. Mol, Polymer, 1994, 35, 622–629 CrossRef CAS.
- C. Carfagna, E. Amendola and M. Giamberini, Macromol. Chem. Phys., 1994, 195, 2307–2315 CrossRef CAS.
- M. Giamberini, E. Amendola and C. Carfagna, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1995, 266, 9–22 CrossRef CAS.
- W. A. Su, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 3251–3256 CrossRef CAS.
- W. Mormann and M. Brocher, Macromol. Chem. Phys., 1996, 197, 1841–1851 CrossRef CAS.
- Y. L. Jun, J. Jyongsik, S. H. Seung, M. H. Soon and U. K. Kwang, Polymer, 1998, 39, 6121–6126 CrossRef.
- G. G. Barclay, C. K. Ober, K. I. Papathomas and D. W. Wang, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1831–1843 CrossRef CAS.
- Q. Lin, A. F. Yee, J. D. Earls, R. E. Hefner and H. J. Sue, Polymer, 1994, 35, 2679–2682 CrossRef CAS.
- E. Amendola, C. Carfagna, M. Giamberini and G. Pisaniello, Macromol. Chem. Phys., 1995, 196, 1577–1591 CrossRef CAS.
- E. Amendola, C. Carfagna, M. Giamberini and L. Komitov, Liq. Cryst., 1996, 21, 317–325 CrossRef CAS.
- W. Mormann and M. Bröcher, Polymer, 1998, 39, 4905–4910 CrossRef CAS.
- J. Y. Lee and J. Jang, Polymer, 2006, 47, 3036–3042 CrossRef CAS.
- J. Y. Lee, J. Jang, S. S. Hwang, S. M. Hong and K. U. Kim, Polymer, 1998, 39, 6121–6126 CrossRef CAS.
- W. Mormann, M. Brochec, P. Schwarz, U. Siegen and M. Chemie, Macromol. Chem. Phys., 1997, 198, 3615–3624 CrossRef CAS.
- E. J. Choi, H. K. Ahn, J. K. Lee and J. Il Jin, Polymer, 2000, 41, 7617–7625 CrossRef CAS.
- Z. Gao, Y. Yu, Y. Xu and S. Li, J. Appl. Polym. Sci., 2007, 105, 1861–1868 CrossRef CAS.
- P. Xie and R. Zhang, in Thermotropic Liquid Crystal Polymers, ed. T.-S. Chung, Technomic Publishing Company, Lancaster, USA, 2001, pp. 281–302 Search PubMed.
- C. Carfagna, E. Amendola and M. Giamberini, Prog. Polym. Sci., 1997, 22, 1607–1647 CrossRef CAS.
- H. J. Sue, J. D. Earls and R. E. Hefner, J. Mater. Sci., 1997, 32, 4031–4037 CrossRef CAS.
- C. Ortiz, R. Kim, E. Rodighiero, C. K. Ober and E. J. Kramer, Macromolecules, 1998, 31, 4074–4088 CrossRef CAS.
- C. Ortiz, L. Belenky, C. K. Ober and E. J. Kramer, J. Mater. Sci., 2000, 35, 2079–2086 CrossRef CAS.
- Y. Li and M. R. Kessler, Polymer, 2014, 55, 2021–2027 CrossRef CAS.
- W. F. A. Su, K. C. Chen and S. Y. Tseng, J. Appl. Polym. Sci., 2000, 78, 446–451 CrossRef CAS.
- M. Akatsuka and Y. Takezawa, J. Appl. Polym. Sci., 2003, 89, 2464–2467 CrossRef CAS.
- M. Harada, M. Ochi, M. Tobita, T. Kimura, T. Ishigaki, N. Shimoyama and H. Aoki, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 1739–1743 CrossRef CAS.
- L. Nie, A. Burgess and A. Ryan, Macromol. Chem. Phys., 2013, 214, 225–235 CrossRef CAS.
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 49–50, 3–33 CrossRef CAS.
- Y. Li, C. Pruitt, O. Rios, L. Wei, M. Rock, J. K. Keum, A. G. McDonald and M. R. Kessler, Macromolecules, 2015, 48, 2864–2874 CrossRef CAS.
- G. C. Lama, P. Cerruti, M. Lavorgna, C. Carfagna, V. Ambrogi and G. Gentile, J. Phys. Chem. C, 2016, 120, 24417–24426 CrossRef CAS.
- A. Belmonte, G. C. Lama, G. Gentile, X. Fernández-Francos, S. De La Flor, P. Cerruti and V. Ambrogi, J. Phys. Chem. C, 2017, 121, 22403–22414 CrossRef CAS.
- A. Belmonte, G. C. Lama, G. Gentile, P. Cerruti, V. Ambrogi, X. Fernández-Francos and S. De la Flor, Eur. Polym. J., 2017, 97, 241–252 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, O. Rios, J. K. Keum and M. R. Kessler, Soft Matter, 2017, 13, 5021–5027 RSC.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, J. Am. Chem. Soc., 2016, 138, 2118–2121 CrossRef CAS PubMed.
- R. R. Kohlmeyer and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9234–9237 CrossRef CAS PubMed.
- Y. Yang, Z. Pei, X. Zhang, L. Tao, Y. Wei and Y. Ji, Chem. Sci., 2014, 5, 3486–3492 RSC.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- Y. Li, O. Rios, J. K. Keum, J. Chen and M. R. Kessler, ACS Appl. Mater. Interfaces, 2016, 8, 15750–15757 CrossRef CAS PubMed.
- Y. Li, Y. Zhang, O. Rios, J. K. Keum and M. R. Kessler, RSC Adv., 2017, 7, 37248–37254 RSC.
- X. Lu, S. Guo, X. Tong, H. Xia and Y. Zhao, Adv. Mater., 2017, 29, 1606467 CrossRef PubMed.
- X. Lu, H. Zhang, G. Fei, B. Yu, X. Tong, H. Xia and Y. Zhao, Adv. Mater., 2018, 30, 1706597 CrossRef.
- Q. Chen, Y. Li, Y. Yang, Y. Xu, X. Qian, Y. Wei and Y. Ji, Chem. Sci., 2019, 10, 3025–3030 RSC.
- Y. Yang, E. M. Terentjev, Y. Zhang, Q. Chen, Y. Zhao, Y. Wei and Y. Ji, Angew. Chem., Int. Ed., 2019, 58, 17474–17479 CrossRef CAS PubMed.
- J. Lub, V. Recaj, L. Puig, P. Forcén and C. Luengo, Liq. Cryst., 2004, 31, 1627–1637 CrossRef CAS.
- B. M. I. Van Der Zande, S. J. Roosendaal, C. Doornkamp, J. Steenbakkers and J. Lub, Adv. Funct. Mater., 2006, 16, 791–798 CrossRef CAS.
- B. M. I. van der Zande, J. Steenbakkers, J. Lub, C. M. Leewis and D. J. Broer, J. Appl. Phys., 2005, 97, 123519 CrossRef.
- D. C. Hoekstra, K. Nickmans, J. Lub, M. G. Debije and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2019, 11, 7423–7430 CrossRef CAS.
- J. A. Lv, W. Wang, J. Xu, T. Ikeda and Y. Yu, Macromol. Rapid Commun., 2014, 35, 1266–1272 CrossRef CAS PubMed.
- A. M. Islam, H. Lim, N. H. You, S. Ahn, M. Goh, J. R. Hahn, H. Yeo and S. G. Jang, ACS Macro Lett., 2018, 7, 1180–1185 CrossRef CAS.
- D. J. Broer, G. N. Mol, J. A. M. M. Van Haaren and J. Lub, Adv. Mater., 1999, 11, 573–578 CrossRef CAS.
- H. Khandelwal, R. C. G. M. Loonen, J. L. M. Hensen, A. P. H. J. Schenning and M. G. Debije, J. Mater. Chem. A, 2014, 2, 14622–14627 RSC.
- L. Zhang, K. Li, W. Hu, H. Cao, Z. Cheng, W. He, J. Xiao and H. Yang, Liq. Cryst., 2011, 38, 673–677 CrossRef CAS.
- J. M. McCracken, V. P. Tondiglia, A. D. Auguste, N. P. Godman, B. R. Donovan, B. N. Bagnall, H. E. Fowler, C. M. Baxter, V. Matavulj, J. D. Berrigan and T. J. White, Adv. Funct. Mater., 2019, 29, 1903761 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |