
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of macromolecular structure on the rheology and surface properties of amphiphilic random polystyrene-r-poly(meth)acrylate copolymers prepared by RDRP†
Nicola
Migliore
 ,
Francesco
Picchioni
and
Patrizio
Raffa
*
,
Francesco
Picchioni
and
Patrizio
Raffa
*
Department of Chemical Engineering, University of Groningen, Nijenborgh 4 9747 AG, The Netherlands. E-mail: p.raffa@rug.nl
First published on 17th February 2020
Abstract
In this work rheological and surface properties of various random copolymers of styrene and sodium (meth)acrylate, prepared using reversible deactivation radical polymerization (RDRP), were studied. It is shown that the properties of these polymers in water solution, relevant for several applications, are affected by their chemical structure and molecular weight. Cryo-TEM images of their concentrated water solutions do not show the presence of nano-objects as micelles, however the existence of some aggregates seems to be confirmed by fluorescence measurements using pyrene as a hydrophobic probe and by surface tension measurements. Moreover, interesting results are displayed about the viscosity as well as the surface tension of these water polymer solutions, due probably to different interactions at the molecular level as suggested by fluorescence measurements.
Introduction
Amphiphilic copolymers have received great attention in recent years, due to their interesting rheological and surface properties, and for their ability to self-assemble in stable micelles or other aggregates in selected solvents.1–5 The self-assembly in micelles is known to happen above a certain critical micellar concentration (CMC), which is an important parameter to consider when comparing different amphiphilic copolymers and their solution properties. Indeed, it is known that the CMC is affected by many factors, such as the molecular weight and polydispersity, the chemical structure of the polymers, and the balance between the hydrophilic and hydrophobic moieties in the polymer.1,2,6–8It is easy to understand that the aggregation of polymeric structures in vesicles and micelles represents an important phenomenon to take into account in the design of polymers for applications in specific fields, including (mini)emulsion polymerizations, coatings, biotechnology, nanotechnology, medicine, pharmacology, cosmetics, agriculture, water purification, electronics, optoelectronics, and enhanced oil recovery.3,8–10 Many studies on amphiphilic copolymers have focused on the synthesis and systematic study of their rheology in water solution.7,8,11–15 It is important to point out that the formation of micellar aggregates in water is a typical behavior of amphiphilic block copolymers, where the architecture and the length of the hydrophilic and hydrophobic blocks influence the final morphology and properties.2,8,10,16–18 For example, Colombani et al. reported the synthesis and study of poly(n-butyl acrylate)-block-poly(acrylic acid) diblock copolymers prepared via ATRP. They point out that the micelles’ dimensions in water are affected by the amount of acrylic acid present in the polymer.19 Moreover, several studies carried out in our group on well-defined polystyrene-b-polymethacrylate copolymers report a correlation between rheological properties in solution and polymer structure, in particular the length of the hydrophilic block, that can be used as indirect evidence for the arrangement of the polymers into micelles.2,7 Nevertheless, it is reported in the literature that spherical aggregates formed by block copolymers form often “frozen” micelles, this means that the chains are kinetically trapped. The interesting thing about these frozen micelles is that, in this case, the polymer solution displays a surface tension value similar to water.20,21
Although much less frequent, there are also examples of studies of random or gradient structures. For example, Lee et al. synthesized random, gradient and block copolymers using 2-dimethylaminoethyl methacrylate and n-butyl methacrylate and studied their aggregation properties. They found that thermo-responsive behavior and aggregation properties of the random, gradient and block copolymers significantly depended on the architecture of the copolymers.22 Also relevant are the results reported by Bendejacq et al., who synthesized a diblock copolymer comprising one block of statistical copolymer of polystyrene with polyacrylic acid and a second block of polyacrylic acid ((PS-r-AA)-b-PAA). In this case, the amount of styrene incorporated in the polymer chains is the factor mainly responsible for the self-assembly of the copolymer in solution, where nano-objects with different dimensions are formed. Three different scenarios are possible in this case, depending on the ratio S/AA. Indeed, with a low AA intake (typically ≤25 mol%) the structures found in water are micelle-like objects, whereas, at relatively high AA content (in the order of ≥50 mol%), soluble macromolecules are observed. For a content of AA ∼ 50 mol%, a colloidal dispersion is formed.23 Another recent study was carried out by Neal et al. on random copolymers of n-butyl acrylate and methacrylic acid, synthesized via RAFT. When dissolved in water, these polymers self-assemble into nano-objects, whose size is independent of the molecular weight, but dependent on the copolymer composition.24
As shown by the examples reported above, the structure of the polymers and the synthetic method used to prepare them have crucial roles in the final properties of the polymer solutions. In particular, it is important to understand how the different monomers are incorporated in the growing chains during synthesis. In this respect, particularly interesting is the work reported by Smith et al. about the prediction of the polymer composition when a living polymerization method is used to copolymerize two different monomers.25 The authors point out the fact that using living polymerization (either RAFT, ATRP or NMP), the copolymer composition can be different ranging from statistical to gradient. Of course, relevant parameters determining the final composition are relative molar ratios, number-average degree of polymerization (DP) at 100% conversion, and reactivity ratios. Importantly, the use of living polymerization should avoid the possibility to form homopolymer chains in the batch when a random copolymer is synthesized. In contrast, when a random copolymer is synthesized using free radical polymerization, formation of homopolymer in the batch cannot be completely prevented. This point results in being particularly relevant for the solution properties of the polymers, because these can be greatly affected by the possible presence of homopolymer chains in the synthesized copolymer.
Furthermore, as it is possible to observe from the literature, the solution rheology and surface properties of amphiphilic random copolymers are rarely studied, although several papers report controlled synthesis of such structures.26,27 A systematic study of the rheological and interfacial properties of these systems can be really interesting, considering their many applications in industry. In the literature some studies of amphiphilic “random” copolymers have been reported, but these systems are actually diblock copolymers, where one of the two blocks is random.23,27
In this context, herein we report a study aimed at evaluating in the first instance the effect of various structural parameters, namely molecular weight, polydispersity and polymer composition, on the rheology, surface activity, and aggregation behavior of amphiphilic random copolymers in water solution and then how different synthesis methods can affect the same properties. More specifically, a series of random copolymers styrene–acrylic acid and styrene–methacrylic acid were synthesized via RDRP,28 by varying the amount of acrylic monomer in the polymers. For comparison as the synthetic method can affect the solution properties, a sample prepared via free radical polymerization was also investigated. To the best of our knowledge, the solution rheology of various random copolymers of styrene and sodium (meth)acrylate (PS-r-AA PS-r-MAA) was never studied before, although similar studies were carried out by our group on diblock, triblock and star-block copolymers PS-b-PMAA.2,7,20 Finally, water solutions were characterized by surface tension measurements, cryo-TEM microscopy and fluorescent probe UV-vis spectra, to investigate more in detail the structure dependence of the aggregation behavior of such polymers.
Experimental
Materials
Styrene (S, Sigma-Aldrich, ≥99%), tert-butyl acrylate (tBA, Sigma-Aldrich, 98%) and tert-butyl methacrylate (tBMA, Sigma-Aldrich, 98%) were vacuum-distilled over CaH2 and kept under nitrogen before use. CuBr (Sigma-Aldrich, ≥98%) was stirred in glacial acetic acid for at least 5 h then filtered, washed with acetic acid, ethanol and ethyl acetate and dried under vacuum before use. Anisole (Sigma-Aldrich, anhydrous, 99.7%) was deoxygenated by bubbling with nitrogen for at least 40 min before use. Methanol (MeOH, Sigma-Aldrich 99.8%), THF (Sigma-Aldrich, ≥99.9%), 1,4-dioxane (Sigma-Aldrich, 99.9%), methyl-α-bromoisobutyrate (MBiB, Sigma-Aldrich, ≥99.0%), pentamethyldiethylenetriamine (PMDETA, Aldrich, 99%), 2,2′-azobis(2-methylpropionitrile) (Sigma-Aldrich, 98%), NaOH (Sigma-Aldrich, ≥97.0%), triethylamine (Sigma-Aldrich, ≥97.0%), HCl (Sigma-Aldrich, ACS reagent 37%) and pyrene (Sigma-Aldrich, ≥99.0%) were used as received, without further purifications.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v methanol/water. The polymers that contain a higher amount of styrene, precipitate better in the water solution compared to the polymers that contain a higher amount of acrylic monomers. The resulting polymers were dried overnight at 60 °C and then characterized by 1H-NMR (CDCl3), FT-IR and GPC (respectively Fig. S1, S2 and S3, ESI†).
:1 v/v methanol/water. The polymers that contain a higher amount of styrene, precipitate better in the water solution compared to the polymers that contain a higher amount of acrylic monomers. The resulting polymers were dried overnight at 60 °C and then characterized by 1H-NMR (CDCl3), FT-IR and GPC (respectively Fig. S1, S2 and S3, ESI†).
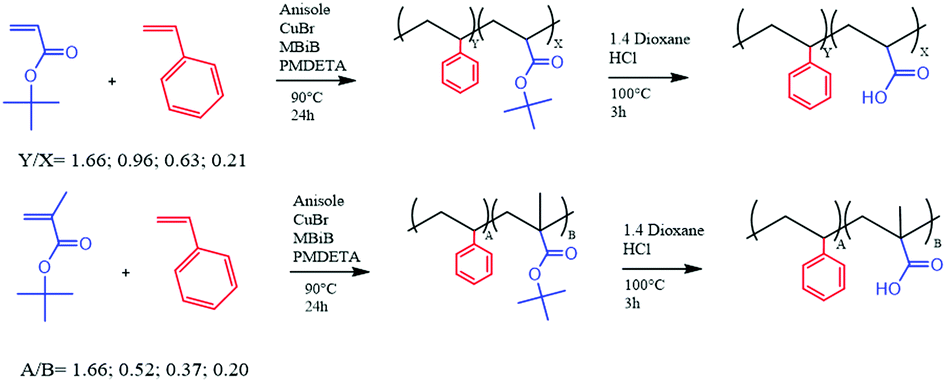 | ||
| Scheme 1 Synthesis of the random co-polymers prepared in this work. | ||
:1 v/v methanol/water. The precipitate was washed with methanol and dried overnight at 60 °C, affording a white solid. The molecular weight was determined by GPC and the effective ratio between the two monomers was determined by 1H-NMR (in CDCl3).
720, 46500, 96000 and 188700 Da) were used for calibration. Rheology experiments were carried out on the random amphiphilic copolymers. Solutions at 1.0, 5.0 and 20.0 wt% concentrations were prepared by dissolving the neutralized polymers in MilliQ water, followed by stirring for at least 10 h before the measurement in order to get homogeneous solutions. All the prepared polymers were soluble in water in their sodium salt form, without need for co-solvents or heating. The rheological measurements were performed with a HAAKE Mars III (Thermo Scientific, Waltham, MA, USA) rheometer equipped with a cone-plate geometry (diameter 60 mm, angle 2°) using 2 mL of solution. Solution viscosity was measured as a function of shear rate (0.1 to 1750 s−1, T = 20 °C), using a trap for the solvent in order to avoid water evaporation during the measurements and be careful to not trap air during the loading of the solutions on the plate, so that no rest time was needed. Surface tension was measured with an OCA 15EC tensiometer from Dataphysics, using the pendant drop method. Fluorescence spectra of water polymer solutions, having different polymer concentrations, were recorded with a Jasco FP-8300 fluorimeter (right angle geometry, 1 cm × 1 cm quartz cell) using the following conditions: excitation at 333 nm, slit width 3 nm for the excitation, and 1.5 nm for the emission. The intensities of the bands I1 at 372 nm and I3 at 383 nm were then evaluated, and their ratio was plotted vs. the polymer concentration. Each sample was prepared in order that the final concentration of pyrene (previously dissolved in MeOH) in the water polymer solution was 2.5 × 10−7 M. The polymer solutions were analyzed by using cryo-TEM in order to evaluated the micelles’ formation. A drop of solution was placed on a glow discharged plain carbon coated 400 mesh copper grid. The samples were examined in an FEI T20 electron microscope operating at 200 keV. Images were recorded on a slow scan CCD camera.
Results and discussion
Synthesis
In order to study the properties of the random copolymers of styrene and acrylic monomers, several polymers were synthesized, via ATRP and via free radical polymerization, changing the relative amounts of monomers in the feed (Scheme 1).The main purpose of this work was to study the influence, if any, of the hydrophobic/hydrophilic balance, as well as the molecular weight, on the micellization behavior and the final solution properties of the prepared random copolymers. Generally, the micellization behavior is better known and understood for amphiphilic block copolymers, but much less for amphiphilic random copolymers. The same can be said about the effect aggregation displays on the interfacial and rheology properties of the corresponding water solutions. In this study, the polymers were synthesized via RDRP using CuBr as catalyst and PMDETA as ligand (Scheme 1), as described in the Experimental section. The use of tert-butyl acrylate (and its methacrylate analogue) as a precursor for acrylic acid is a common procedure used to overcome the infeasibility of ATRP in the direct polymerization of carboxyl-containing monomers.13,18,19,23,27,29 The polymerizations were carried out successfully, even if the GPC analysis (Table 1) does not display an efficient control of the molecular weight (Mn) as portrayed by the corresponding polydispersity values (D).
| Synthesis | Sample | Molar feed ratio | Molar H-NMR ratio | Molar TGA ratio | GPC (kDa) | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | tBA | S | tBA | S | tBA | M n | D | |||
| RDRP | PS-r-tBA 1:1 |
1.00 | 1.03 | 1.00 | 0.60 | 1.00 | 0.30 | 4.19 | 1.29 | 6 |
| PS-r-tBA 1:3 |
1.00 | 2.97 | 1.00 | 1.04 | 1.00 | 0.40 | 23.61 | 1.62 | 50 | |
| PS-r-tBA 1:5 |
1.00 | 4.50 | 1.00 | 1.57 | 1.00 | 0.56 | 3.93 | 1.38 | 15 | |
| PS-r-tBA 1:10 |
1.00 | 9.52 | 1.00 | 4.71 | 1.00 | 0.86 | 17.77 | 1.77 | 33 | |
| Free radical | PS-r-tBA 1:5 |
1.00 | 5.00 | 1.00 | 4.23 | 1.00 | 0.83 | 5.13 | 3.46 | 73 |
| Synthesis | Sample | Molar feed ratio | Molar H-NMR ratio | Molar TGA ratio | GPC (kDa) | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| S | tBMA | S | tBMA | S | tBMA | M n | D | |||
| RDRP | PS-r-tBMA 1:1 |
1.00 | 1.08 | 1.00 | 0.61 | 1.00 | 0.35 | 5.85 | 2.09 | 11 |
| PS-r-tBMA 1:3 |
1.00 | 2.45 | 1.00 | 1.93 | 1.00 | 0.52 | 8.04 | 2.01 | 24 | |
| PS-r-tBMA 1:5 |
1.00 | 4.40 | 1.00 | 2.68 | 1.00 | 0.56 | 7.09 | 1.89 | 8 | |
| PS-r-tBMA 1:10 |
1.00 | 8.21 | 1.00 | 5.04 | 1.00 | 0.73 | 16.34 | 2.63 | 48 | |
The copolymers containing tBA show a narrower MWD, compared to the ones containing tBMA, (1.29 < D < 1.77) and (1.89 < D < 2.63) respectively. The GPC peaks show a monomodal distribution, however the MWD is too large for a typical C/LRP. The yields reported in Table 1 are calculated considering the weight of the polymer obtained over the weight of the monomers used. Again, the low yields obtained and the relative polydispersities of the synthesized polymers suggest that the random copolymerization of the selected monomers in these conditions is not well controlled. However, the polydispersity remains significantly lower than that obtained with the corresponding free radical process (Table 1, compare entries 3 and 5). Moreover, the obtained yields and polydispersities suggest that RDRP is not the most suitable way to synthesize random copolymers of styrene and (meth)acrylates. It is known from the literature that in the traditional ATRP styrene and acrylate grow really slowly on the acrylate macroinitiator because there is poor initiation efficiency in the ATRP of methacrylate monomer from acrylate or styrene-based macroinitiators.19 Nevertheless, the objective of this work did not entail the optimization of the controlled radical polymerization and the polymers described above were then used for further characterization.
1H-NMR and TGA characterization
Molar ratios of the S and tB(M)A moieties were obtained based on 1H-NMR and later compared with the ones obtained from TGA analyses. Indeed, particularly interesting is the alternative way that is proposed in this work to estimate the molar ratio styrene/acrylate incorporated in the polymers based on the TGA data. As it is possible to observe from the thermographs (Fig. 1), the degradation of all copolymers follows two main steps: the first step can be formally attributed to the acrylic monomer degradation30,31 with the loss of the tert-butyl group as isobutene, and the latter can be attributed to the styrene degradation.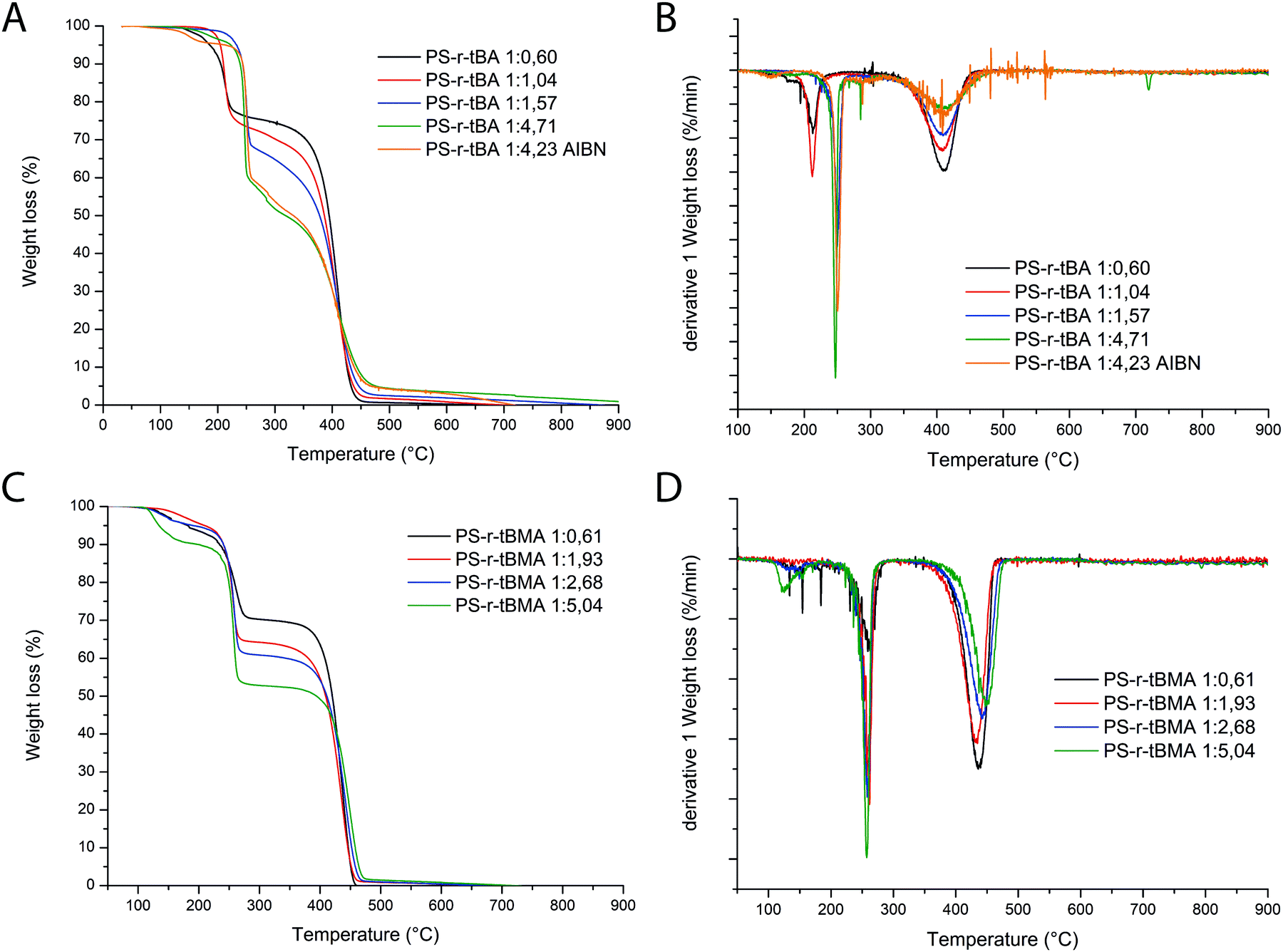 | ||
| Fig. 1 TGA thermographs of PS-r-tBA with different ratios of S:tBA (A) and their first derivatives (B), and TGA thermographs of PS-r-tBMA with different ratios of S:tBMA (C) and their first derivatives (D). | ||
Based on this hypothesis, the ratio between the two monomers in the polymer composition should be proportional to the ratio between the two areas of the first derivative of the thermographs (Fig. 1A and B). The obtained monomer ratios calculated from 1H-NMR and TGA results are reported in Table 1 and plotted in Fig. 1. It is then possible to obtain a polynomial fit that empirically correlates these values (Fig. 2A and B).
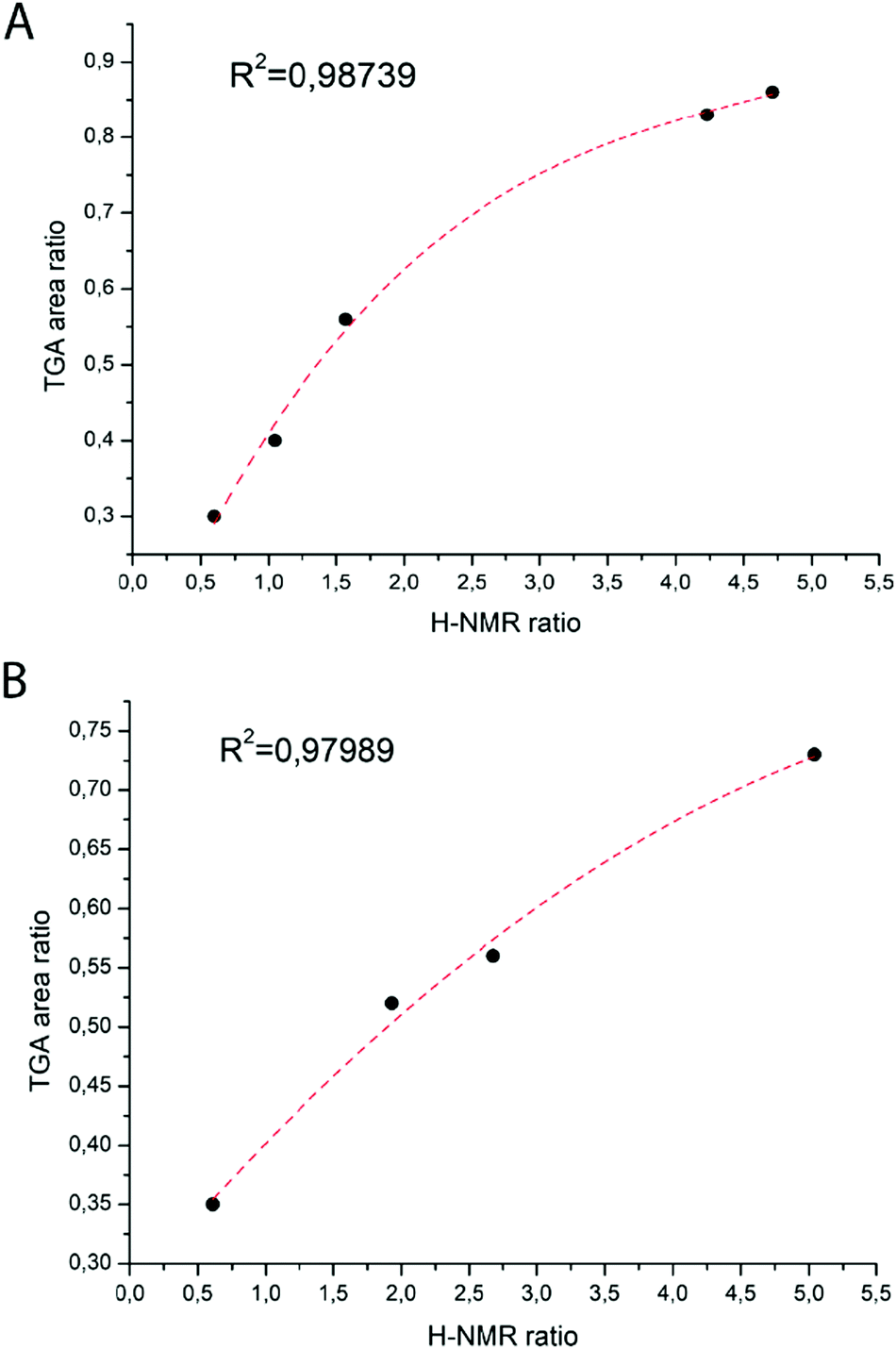 | ||
| Fig. 2 Correlation between the S/tBA (A) and S/tBMA (B) ratios calculated from NMR and TGA. | ||
It is nice to point out that even if the TGA data are affected by an error due to the complexity of the second degradation step, the values are consistent with the 1H-NMR data.
Hydrolysis and solubility
After neutralization and drying, the recovered solids were directly dissolved in water. The obtained solutions were not all clear, but some presented various degrees of turbidity (Fig. 3 and Fig. S4, ESI†), depending on the polymer composition and concentration, as summarized in Table 2, with an apparent trend. | ||
| Fig. 3 Examples of C = clear, T = turbid, M = milky polymer solutions. | ||
| Sample name | Concentration | |||||
|---|---|---|---|---|---|---|
| 0.5 wt% | 1 wt% | 2.5 wt% | 5 wt% | 10 wt% | 20 wt% | |
| PS-r-AA 1:0.60 |
C | C | C | C | C | C |
| PS-r-AA 1:1.04 |
M | M | M | M | M | M |
| PS-r-AA 1:1.57 |
T | T | T | M | T | T |
| PS-r-AA 1:4.71 |
C | C | C | C | C | C |
| PS-r-AA 1:4.23 AIBN |
T | T | M | M | M | M |
| PS-r-MAA 1:0.61 |
T | M | M | M | M | M |
| PS-r-MAA 1:1.93 |
C | C | C | C | C | C |
| PS-r-MAA 1:2.68 |
C | C | C | C | C | C |
| PS-r-MAA 1:5.04 |
C | C | C | C | C | C |
Indeed, except for the PS-r-AA 1:0.60 solution that looks clear even if at relatively high styrene intake, the other polymer solutions are clear and transparent as soon as the acrylate moieties in the polymer composition increase. This is in agreement with what is generally reported for associative polyelectrolytes, where self-assembly to big aggregates becomes more relevant as the hydrophobicity increases.32 The anomalous solubility of PS-r-AA 1:0.60 could be ascribed to its lower molecular weight, compared to most polymers synthesized in this work, but the data do not allow for a clear explanation. Interestingly, the PS-r-AA 1:4.23 AIBN solutions are milky (Fig. S4, ESI†) even if the polymer has a higher relative amount of acrylate. This can possibly be explained considering that in free radical polymerization, contrarily to RDRP, secondary radical reactions and terminations lead to the formation of dead chains and non-linear structures. In this respect, as mentioned previously, it is not to be excluded the possibility of homopolymer chains formation, along with copolymers, during free radical copolymerization.25 In conclusion, the general trend is that the turbidity of the solutions gets higher as the ratio S/(M)AA increases. This is probably due to a combined effect of higher hydrophobicity, causing more aggregations, and the possible presence of polystyrene homo-polymeric chains in the products.
Rheology
Shear viscosity in water solutions at various concentrations in the range 1–20 wt% of the copolymers synthesized in this work has been studied. No measurements were carried out for PS-r-AA 1:4.23 AIBN at 20 wt%, due to its partial insolubility at this concentration.
The main results of the rheology studies are shown in Fig. 4. The solutions at 1 wt% show a Newtonian behavior in a large range of shear rate, with values of viscosity not much above the value of pure water (Fig. 4A and B). The apparent increase in viscosity at high shear is probably due to an artefact. At high shear rate and low viscosity, therefore high Reynold numbers, the solutions are in the turbulent regime. This contributes positively to the flow energy and leads to an apparent increase in viscosity with shear. The Newtonian behavior suggests that at 1 wt% we are still in diluted conditions. Although it is difficult to detect any specific trend, a few observations are possible: the polymers providing the better viscosifying properties are those with simultaneously higher molecular weight and higher relative amounts of hydrophilic moieties. This is compatible with the theory of polyelectrolytes:32 for low hydrophobicity the rheology behavior is that of typical polyelectrolytes, while at higher hydrophobicity (which in our case means higher relative styrene content), the system transitions to a “polysoap” and the viscosity decreases, as a consequence of shrinking of the hydrodynamic volume (Scheme 2). This is visible comparing PS-r-AA 1:4.71 with PS-r-AA 1:1.04 in Fig. 4A. As the concentration of the polymer is increased, the viscosity increases as well (Fig. 4C–F), as expected.
 | ||
| Fig. 4 Viscosity as function of shear for PS-r-AA (A) and PS-r-MAA (B) at 1 wt% concentrations, PS-r-AA (C) and PS-r-MAA (D) at 5 wt% concentrations and PS-r-AA (E) and PS-r-MAA (F) at 20 wt% concentrations at 20 °C. | ||
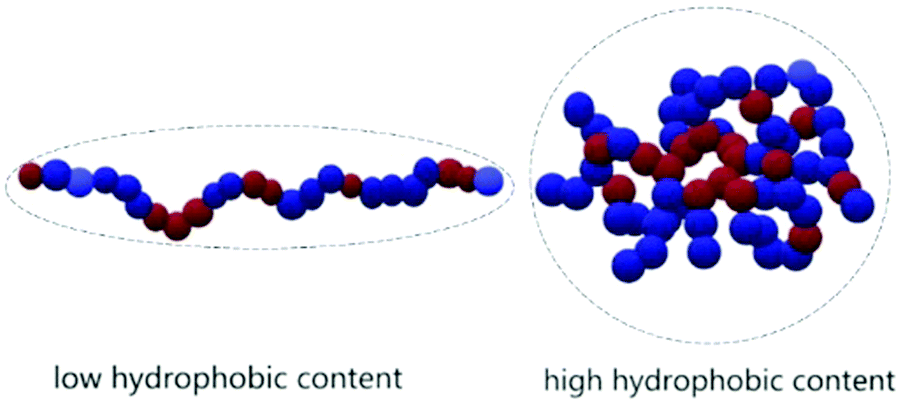 | ||
| Scheme 2 Schematic representation of the hydrodynamic volume of polyelectrolytes and polysoaps due to the lower or higher hydrophobicity. | ||
The solutions at 5 wt% show generally 1 order of magnitude higher viscosities than at 1 wt%, keeping a similar Newtonian behavior. Some solutions seem to display a slightly shear thinning behavior that does not clearly correlate with molecular properties (molecular weight, hydrophobicity). This might represent a transition to an overlapping regime for these systems, where the properties are determined by a non-trivial balance between inter- and intramolecular interactions. Solutions at 20 wt% display significantly higher viscosities, especially the methacrylate series, and in some cases shear thinning behavior. Again, the viscosity seems to correlate well with increase in molecular weight and the solutions have apparent Newtonian or slightly shear thinning behavior. Exceptions to this are PS-r-AA 1:1.157 (Fig. 4E) and PS-r-MAA 1:1.93 (Fig. 4F), which show highly non-Newtonian behavior. Also in this case, this could be ascribed to the high relative amount of styrene. The shear thinning behavior could be again an indication of interaction between aggregated chains being disrupted by the flow.33 Given the non-homogeneity of data of molecular weight, MWD and polymer composition, it is difficult to find extremely exact correlations between these parameters and the solution properties. The general conclusion of this set of measurements is that in most cases, the polymers with higher Mn and lower content of styrene are the ones with higher viscosity. Indeed, the polymers with higher acrylate/styrene ratio have the highest charge density, and as mentioned before, the polymer chains are more stretched in solution, with subsequent higher hydrodynamic values (and thus, viscosity). The polymers with higher styrene/acrylic acid ratio display a lower viscosity due to the easier possibility for the polymer chains to form intermolecular aggregates, which leads to a reduced hydrodynamic volume. As expected, the viscosifying ability of these random copolymers is much less pronounced than analogous block copolymers PS-b-PMAA at the same weight concentration.2 This can be explained by the fact that the block structures can aggregate in giant micelles, with well stretched corona, while random structures are mostly present in solution as single chains, at least in the diluted regime.
Another important aspect to consider are the dynamics of the systems investigated. Hydrophobically associating polymers can give either “frozen” aggregates, especially in the case of block structures, or dynamic associates, especially in the case of “polysoap” structures.8,34,35 In case of non-dynamic systems, the solution properties will be dependent on the sample history, e.g. how the solution is prepared. To help better understand our systems, the surface properties of the prepared polymers in solution have been investigated.
Surface activity
The surface tension as a function of concentration in water was measured for all polymers synthesized in this work. The measured values (Fig. 5) are consistent with what are usually observed for polymeric surfactants.8 | ||
| Fig. 5 Surface tension of PS-r-AA at different concentrations and different amounts of acrylic monomers (A) and surface tension of PS-r-MAA at different concentrations and different amounts of acrylic monomers (B). | ||
Interestingly, this is another clear difference with analogous block copolymers, which are aggregated in “frozen” micelles and do not display any surface activity.20 The PS-r-AA 1:4.23 AIBN is the polymer that shows a more pronounced decrease in the surface tension with concentration, resulting in a significantly lower apparent CMC. We can try to interpret this result based on the hypothesis exposed in the introduction of this paper: the free radical polymerization mechanism produces a large molecular weight distribution, as well as formation of homopolymer chains, altering the apparent surface tension value.
Except for the PS-r-AA synthesized via free radical polymerization, all polymers show very similar surface tension curves, therefore there seem to be no big dependence of the CMC and minimum surface tension value on the molecular structure or hydrophobicity.
The reduced surface tension and the existence of a CMC would suggest that the aggregation behavior of the systems is dynamic. In order to confirm the aggregation behavior and the presence of a CMC, measurements of fluorescence were carried out on the polymer solutions at different concentrations using pyrene as the fluorescent probe. Indeed, it is known in the literature that another way to identify the CMC of a polymer is the use of a fluorescent probe,19,36,37 such as pyrene.
Pyrene is slightly soluble in water and a 2.5 × 10−7 M solution displays a characteristic emission spectrum in the wavelength range between 350 nm and 500 nm (Fig. S5, ESI†). This spectrum is characterized by two emission peaks at 372 nm (I1) and at 383 nm (I3) and their ratio (I1/I3) is an index of the polarity of the pyrene microenvironment.19,36 When the pyrene is in an amphiphilic polymer solution, the emission spectrum changes due to the presence of micelles: above the CMC, the pyrene migrates into the hydrophobic domain of the micellar aggregate. The effect of this phenomenon is clearly displayed in the emission spectrum of pyrene where the I1/I3 intensity ratio changes from ∼1.6 (pyrene in the hydrophilic domain) to ∼0.9 (pyrene in the micelles microdomain). It is possible to express the concentration of the amphiphilic polymer according to the I1/I3 intensity ratio in order to evaluate the presence of the CMC (Fig. 6) in the polymer solution. All the polymers synthesized in this work display a significant decrease in the I1/I3 ratio when they reach a concentration of 2.5 wt%.
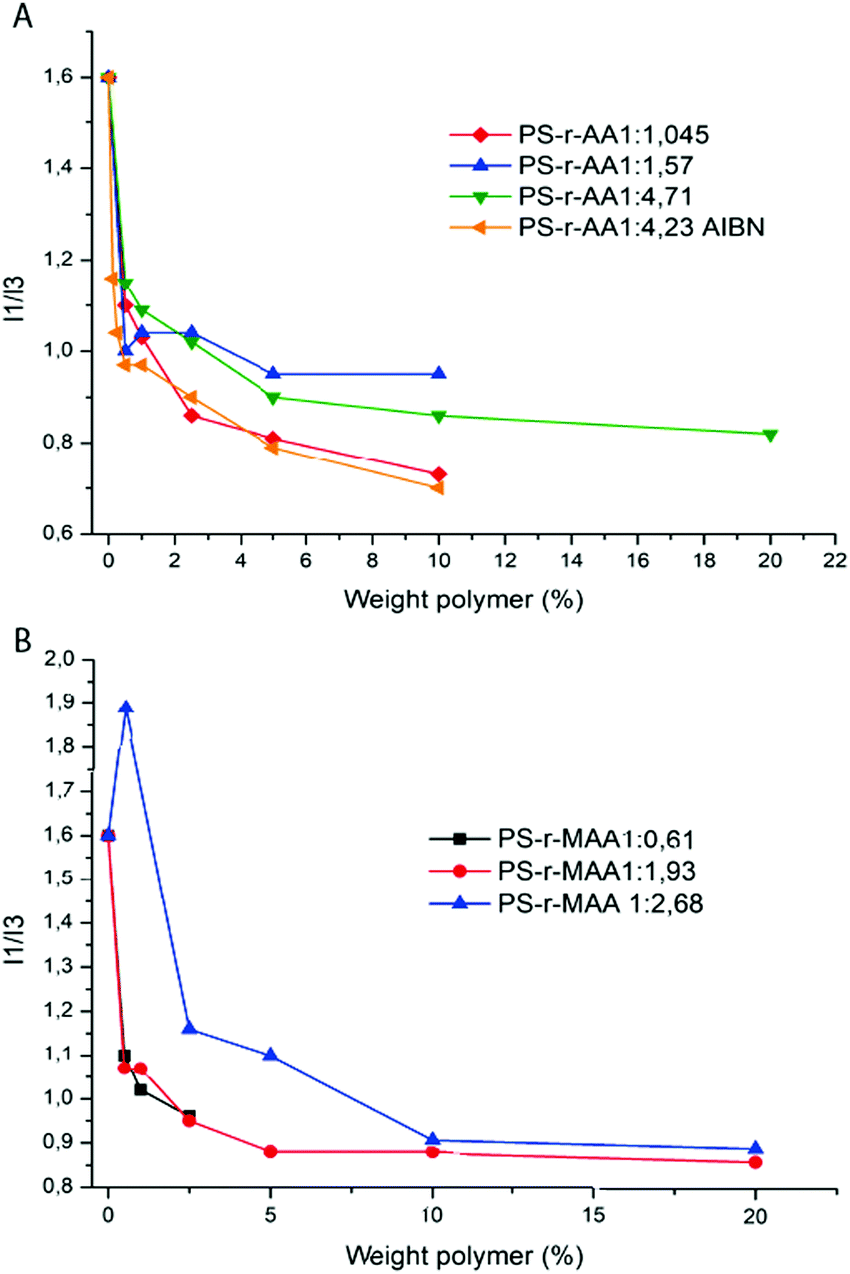 | ||
| Fig. 6 Weight polymer concentration as a function of I1/I3 ratio for PS-r-AA with different amounts of acrylic monomers (A). Weight polymer concentration as a function of I1/I3 ratio for PS-r-MAA at different concentrations and with different amounts of acrylic monomers (B). | ||
Those data are consistent with the surface tension measurements of the same polymer solutions. Indeed, the slight differences that can be noted are probably due to the different physical phenomena used to measure it and a possible effect of the pyrene on the molecular self-assembly.38–40
Moreover, it was observed that the solutions that are milky or turbid display a wide peak in the same wavelengths of emission as pyrene (between ∼370 nm and ∼500 nm) when they are above the 10 wt% of concentration (Fig. S5, ESI†).
Finally, cryo-TEM studies were carried out in order to confirm the presence of aggregates at this concentration. Unfortunately, the TEM images do not show any micellar aggregation (Fig. S7, ESI†).
Comparison between PS-r-AA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1.57 neutralized using NaOH or Et3N
:1.57 neutralized using NaOH or Et3N
After hydrolysis, polymers have to be neutralized to the corresponding polyelectrolyte. Generally, an excess of NaOH is added to the polymer solution/suspension in water, and the excess of base is removed by dialyzing against MilliQ water, changing water at least three times over a period of two days.2 The dialysis step is necessary to remove excess ions present in the solution, which will alter the surface and rheological properties.7 As dialysis is a time-consuming procedure, an alternative way to neutralize the polymers was tested, namely the use of a volatile base such as triethylamine. The excess of amine added to neutralize the polymer solution can then easily be removed by evaporation, eliminating the necessity of a dialysis step. In order to evaluate if this procedure gives a polymer with similar solution properties, one representative polymer was neutralized using both procedures, and measurements of viscosity and surface tension were compared. Remarkably, the resulting ammonium neutralized polymer afforded clear solutions, whereas the Na salt solutions were turbid (Fig. S4, ESI†).
The shear viscosities of solutions at 5 wt% and at 20 wt% (Fig. S6, ESI†) of PS-r-AA 1:1.57 neutralized with NaOH and Et3N show remarkably different behavior. These results suggest that the different neutralization method might provide completely different aggregation behavior in solution, and therefore the final rheological properties of the solution can be affected not only by the polymer molecular characteristics, but also from the method used to solubilize it and the counterion used. Indeed, it is known in the literature that the Hofmeister effect affects amphiphilic polymers’ self-assembly41,42 and their adsorption at the interfaces.43 Anyway, this aspect deserves more future investigation to be fully understood.
On the other hand, slight differences are observed in the surface tension behavior (Fig. 7). This might be explained by the different counter ion.44,45
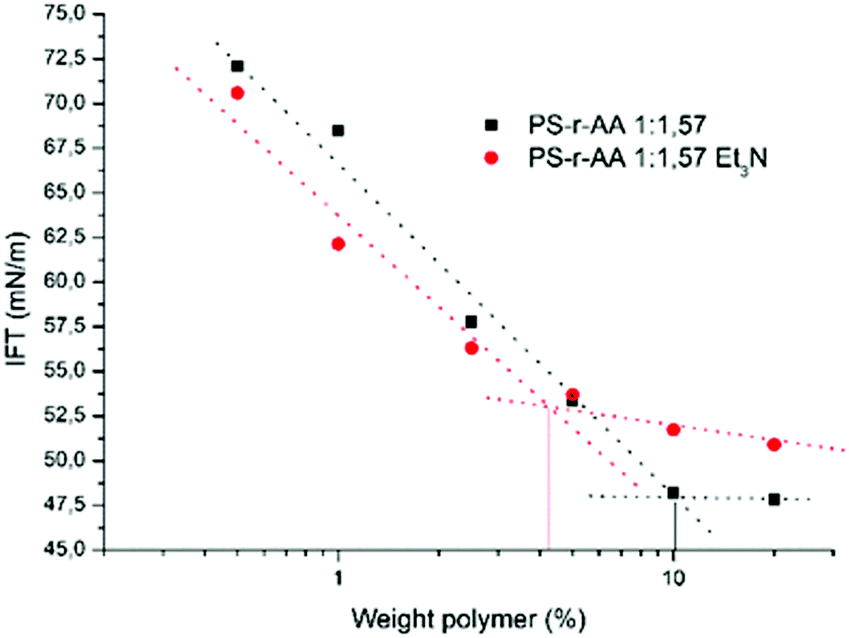 | ||
| Fig. 7 Comparison of surface tension of PS-r-AA 1:57 at different concentrations neutralized with NaOH or with Et3N. | ||
Measurements of fluorescence were carried out on the polymer solutions neutralized in the two different ways in order to estimate the CMC values (Fig. 8) of the polymer solutions.
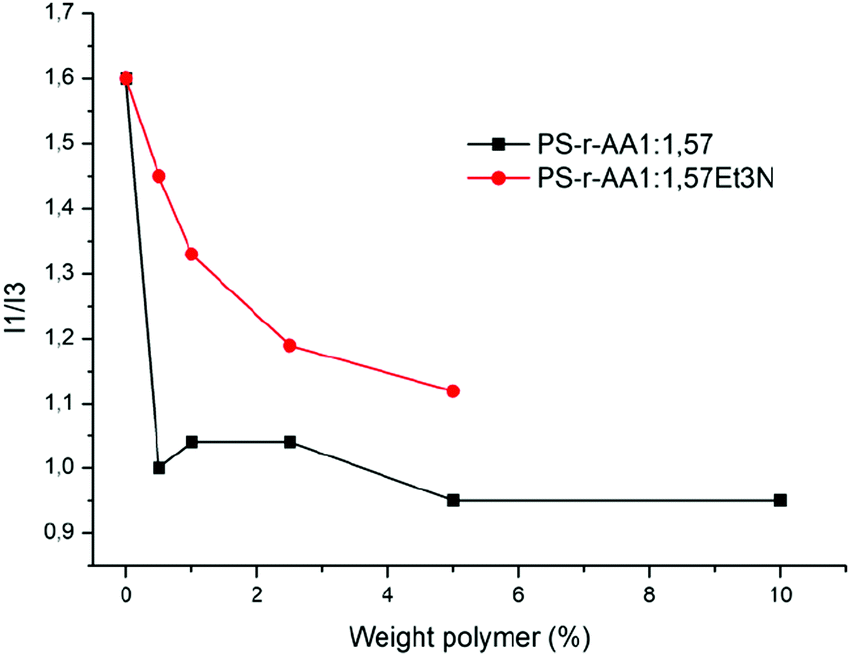 | ||
| Fig. 8 Comparison of the weight polymer concentration as a function of I1/I3 ratio for PS-r-AA 1.57 neutralized with NaOH (black curve) or Et3N (red curve). | ||
Both polymers display a significant decrease in the I1/I3 ratio when they reach a concentration of 2.5 wt%. Also in this case, data are consistent with the surface tension measurements of the same polymer solutions and they do not show big differences.
Conclusions
A series of PS-r-AA and PS-r-MAA random copolymers with various molecular weights and hydrophobic/hydrophilic balance have been synthesized via RDRP28 and via free radical polymerization of styrene and tert-butyl (methacrylate), followed by hydrolysis. The main goal was to study the relevant properties in water solution of such hydrophobically modified polyelectrolytes, as a function of structural parameters and method of synthesis. To the best of our knowledge, the majority of systematic investigations presented in the literature about solution properties of amphiphilic polymers focus on block structures,7,8 whereas random structures are rarely investigated, especially charged ones.One sample was prepared via free radical polymerization for comparison, in order to evaluate the effect of polydispersity and structural control in terms of final solution properties. All the polymers synthesized in this work have a Mn in the range 5.4–43.0 kDa and with a relatively narrow polydispersity (D < 2.63), whereas FRP produced a copolymer with higher polydispersity (D ≈ 3.46).
Also for the polymers prepared via RDRP, the process seems to be not well controlled, probably due to the high difference in reactivities of the two monomers chosen for this study, which would explain the relatively high polydispersity values and the low conversions. Rheological and surface properties have been measured in water solution, in particular shear viscosity and surface tension curves. The aggregation behavior has also been studied via the use of pyrene as a fluorescent probe and cryo-TEM.
The prepared polymers show a rather different behavior in water solution when compared to analogous block copolymers.7 In terms of rheological behavior, the viscosity values are much lower at a concentration comparable to analogous block copolymers (1 wt%), being in most cases not much higher than water, and they are not much dependent on shear. At higher concentration (5 and 20%), the thickening abilities are improved.
The polymers with higher content of acrylate display higher viscosity, possibly due to the higher charge density and low hydrophobicity, that allows the polymer chains to be stretched in solution. At this concentration of polymer, all the solutions are slightly shear thinning in the shear range investigated. The observations are compatible with the theory of polyelectrolytes:32 for low hydrophobicity the rheology behavior is that of typical polyelectrolytes, and generally increases with molecular weight, while at higher hydrophobicity (which in our case means higher relative styrene content), the system transitions to a “polysoap” and the viscosity is lower, as a consequence of shrinking of hydrodynamic volume.
Surface tension curves show that the polymers are surface active and suggest the existence of a CMC for all the prepared systems. The CMC values do not vary significantly, except for the polymer prepared via free radical polymerization, which has a much lower apparent CMC. Remarkably, these polymers are more surface active than corresponding block copolymers. A possible explanation is that the random structure allows a dynamic adsorption of the polymer at the surface/interface, while block copolymers are known to be present in solution as “frozen” aggregates.21 Cryo-electron microscopy did not allow the observation of micellar formation; however, fluorescence measurements using pyrene as the fluorescent probe confirm that some aggregation takes place.
Finally, a new neutralization method used to neutralize the acrylic acid based on the use of Et3N instead of NaOH was tested. Surprisingly, rheological properties are greatly affected by the use of a different neutralization method, while the surface properties remain basically unchanged. A systematic study of the effect of the random polymers structure on the polymer solutions’ properties represents the innovation of this work, and can provide useful tools for the design of new polymeric systems for desired applications in water solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NM gratefully acknowledges the help of Prof. Dr W. Browne and Dr M. C. A. Stuart for their advice respectively on fluorescence measurements and TEM-analysis and Dr A. Postma for the useful talk about the synthesis of statistical copolymer.Notes and references
- L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417–13432 RSC.
- P. Raffa, M. C. A. Stuart, A. A. Broekhuis and F. Picchioni, J. Colloid Interface Sci., 2014, 428, 152–161 CrossRef CAS PubMed.
- P. Alexandridis, Curr. Opin. Colloid Interface Sci., 1996, 1, 490–501 CrossRef CAS.
- P. Guo, W. Guan, L. Liang and P. Yao, J. Colloid Interface Sci., 2008, 323, 229–234 CrossRef CAS.
- S. Imai, Y. Hirai, C. Nagao, M. Sawamoto and T. Terashima, Macromolecules, 2018, 51, 398–409 CrossRef CAS.
- P. Raffa, A. A. Broekhuis and F. Picchioni, J. Appl. Polym. Sci., 2016, 133, 1–8 CrossRef.
- P. Raffa, P. Brandenburg, D. A. Z. Wever, A. A. Broekhuis and F. Picchioni, Macromolecules, 2013, 46, 7106–7111 CrossRef CAS.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- K. Letchford and H. Burt, Eur. J. Pharm. Biopharm., 2007, 65, 259–269 CrossRef CAS PubMed.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS.
- J. Xu, Y. Sun, J. Chen and S. Zhong, New J. Chem., 2018, 42, 3028–3034 RSC.
- S. Nehache, C. C. Yeh, M. Semsarilar, A. Deratani, Y. Chang and D. Quemener, Macromol. Biosci., 2016, 16, 57–62 CrossRef CAS.
- H. Zhao, B. Wang, Q. Li, L. Wang, J. Sun, Y. Zhang, L. Ji and Z. Cao, RSC Adv., 2017, 7, 10124–10131 RSC.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2019, 52, 3697–3715 CrossRef CAS.
- T. Elkin, S. M. Copp, R. L. Hamblin, J. S. Martinez, G. A. Montaño and R. C. Rocha, Materials, 2019, 12, 601 CrossRef CAS PubMed.
- P. Alexandridis, Curr. Opin. Colloid Interface Sci., 1997, 2, 478–489 CrossRef CAS.
- P. Kaewsaiha, K. Matsumoto and H. Matsuoka, Langmuir, 2005, 21, 9938–9945 CrossRef CAS PubMed.
- C. Ramireddy, S. E. Webber, P. Munk, Z. Tuzar and K. Procházka, Macromolecules, 1992, 25, 2541–2545 CrossRef CAS.
- O. Colombani, M. Ruppel, F. Schubert, H. Zettl, D. V. Pergushov and A. H. E. Müller, Macromolecules, 2007, 40, 4338–4350 CrossRef CAS.
- M. Meijerink, F. van Mastrigt, L. E. Franken, M. C. A. Stuart, F. Picchioni and P. Raffa, Pure Appl. Chem., 2017, 89, 1641–1658 CAS.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- S. B. Lee, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2003, 4, 1386–1393 CrossRef CAS PubMed.
- D. D. Bendejacq, V. Ponsinet and M. Joanicot, Langmuir, 2005, 21, 1712–1718 CrossRef CAS PubMed.
- T. J. Neal, D. L. Beattie, S. J. Byard, G. N. Smith, M. W. Murray, N. S. J. Williams, S. N. Emmett, S. P. Armes, S. G. Spain and O. O. Mykhaylyk, Macromolecules, 2018, 51, 1474–1487 CrossRef CAS.
- A. A. A. Smith, A. Hall, V. Wu and T. Xu, ACS Macro Lett., 2019, 8, 36–40 CrossRef CAS.
- Y. Miura, N. Nakamura, I. Taniguchi and A. Ichikawa, Polymer, 2003, 44, 3461–3467 CrossRef CAS.
- B. Lessard, S. C. Schmidt and M. Marić, Macromolecules, 2008, 41, 3446–3454 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483–491 CAS.
- X. Zhang, J. Xia and K. Matyjaszewski, Macromolecules, 2000, 33, 2340–2345 CrossRef CAS.
- G. Martínez, M. Sánchez-Chaves, C. M. Rocha and G. Ellis, Polym. Degrad. Stab., 2002, 76, 205–210 CrossRef.
- R. J. Schaefgek and I. M. Sarasohn, J. Polym. Sci., 1962, 58, 1049–1061 CrossRef.
- J. Kötz, S. Kosmella and T. Beitz, Prog. Polym. Sci., 2001, 26, 1199–1232 CrossRef.
- D. T. N. Chen, Q. Wen, P. A. Janmey, J. C. Crocker and A. G. Yodh, Annu. Rev. Condens. Matter Phys., 2010, 1, 301–322 CrossRef CAS.
- W. K. Ng, K. C. Tam and R. D. Jenkins, Polymer, 2001, 42, 249–259 CrossRef CAS.
- V. G. Babak, J. Desbrières and V. E. Tikhonov, Colloids Surf., A, 2005, 255, 119–130 CrossRef CAS.
- C. Tsitsilianis, I. Iliopoulos and G. Ducouret, Macromolecules, 2000, 33, 2936–2943 CrossRef CAS.
- M. C. A. Stuart, J. C. Van De Pas and J. B. F. N. Engberts, J. Phys. Org. Chem., 2005, 18, 929–934 CrossRef CAS.
- M. Aoudia and M. A. J. Rodgers, Langmuir, 2005, 118, 10355–10361 CrossRef PubMed.
- R. M. F. Fernandes, E. F. Marques, B. F. B. Silva and Y. Wang, J. Mol. Liq., 2010, 157, 113–118 CrossRef CAS.
- F. M. Menger and L. Shi, J. Am. Chem. Soc., 2009, 131, 6672–6673 CrossRef CAS PubMed.
- Z. Ge, Y. Zhou, Z. Tong and S. Liu, Langmuir, 2011, 27, 1143–1151 CrossRef CAS PubMed.
- B. A. Deyerle and Y. Zhang, Langmuir, 2011, 27, 9203–9210 CrossRef CAS PubMed.
- E. Leontidis, Curr. Opin. Colloid Interface Sci., 2002, 7, 81–91 CrossRef CAS.
- Y. Yuan, W. Zhan, H. Yi, Y. Zhao and S. Song, Colloids Surf., A, 2018, 539, 80–84 CrossRef CAS.
- H. Zhou, Y. Liang, P. Huang, T. Liang, H. Wu, P. Lian, C. Jia, Y. Zhu and H. Jia, J. Mol. Liq., 2018, 249, 33–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm00153h |
| This journal is © The Royal Society of Chemistry 2020 |