
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid–liquid equilibrium in polymer–fullerene mixtures; an in situ neutron reflectivity study†
E. L.
Hynes
 a,
P.
Gutfreund
b,
A. J.
Parnell
c and
A. M.
Higgins
*a
a,
P.
Gutfreund
b,
A. J.
Parnell
c and
A. M.
Higgins
*a
aCollege of Engineering, Swansea University, Bay Campus, Fabian Way, Swansea SA1 8EN, Wales, UK. E-mail: a.m.higgins@swansea.ac.uk
bInstitut Laue-Langevin, 71 avenue des Martyrs, 38000 Grenoble, France
cDepartment of Physics and Astronomy, The University of Sheffield, Sheffield S3 7RH, UK
First published on 31st March 2020
Abstract
The composition profiles of a series of model polystyrene/fullerene bilayers are measured, before, during and after thermal annealing, using in situ neutron reflectometry. In combination with grazing-incidence X-ray diffraction measurements, these experiments, which quantify layer compositions as a function of molecular weight using changes in both scattering length density and layer thickness, extend and corroborate recent measurements on ex situ annealed samples and demonstrate that the composition profiles rapidly formed in these systems correspond to two co-existing liquid–liquid phases in thermodynamic equilibrium. The measurements also demonstrate a clear and systematic onset temperature for diffusion of the fullerenes into the PS layer that correlates with the known glass-transition temperatures of both the polymer (as a function of molecular weight) and the fullerene, revealing that the molecular mobility of the fullerenes in these systems is controlled by the intrinsic mobility of the fullerenes themselves and the ability of the polymer to plasticise the fullerenes at the interface. Over the temperature range investigated (up to 145 °C), measurements of equilibrated composition profiles as a function of temperature (during gradual cooling) reveal no significant changes in composition profile, other than those associated with the known thermal expansion/contraction of polystyrene thin-films.
Introduction
The mixed amorphous phases1–5 that are typically found within solution-processed organic photovoltaics (OPVs) can have an important influence on the efficiency and stability of these devices.3,6–8 OPVs made from polymer/small-molecule blends can now achieve power-conversion efficiencies (PCEs) of over 12% in binary9–16 and ternary17 devices, and 17% in tandem solar cells18 (including over 10% PCE for binary OPVs fabricated in air16). This corresponds to a substantial rise in OPV performance over the last twenty years or so, that has mainly resulted from the synthesis of new materials11 and the characterisation/optimisation of OPV devices.19 Considerable effort has been focussed on characterising the morphology and domain composition of the bulk heterojunctions that make up the active layer of working devices,19,20 as these two factors can both have a large impact on charge generation and transport within devices.7,21–23 In recent years, several investigations on model systems have also been performed, in parallel with device optimisation studies, to probe fundamental aspects of polymer/small-molecule mixtures; these have included studies of simplified architectures (most notably bilayers2,4,5,24–27) and model material systems (e.g. polystyrene (PS)/fullerenes28–34). The aim of much of the PS/fullerene work was to systematically investigate both the kinetics and the equilibrium behaviour in relation to mixing/de-mixing, fullerene aggregation/crystallisation and film formation pathways in a range of well-controlled, low polydispersity, model amorphous-polymer/small-molecule systems; the motivation was to produce fundamental knowledge of thin-film and solution behaviour that can provide a framework for the understanding of aspects of OPV device behaviour during and immediately following fabrication, and also under operating conditions.Ye et al.8 recently discovered a strong correlation between fundamental parameters characterising the miscibility in equilibrium polymer/small-molecule systems and OPV performance; Flory–Huggins theory was used to model phase compositions in bilayers and quantitatively link the interaction parameter, χ,35 to the fill-factor, for an amorphous-polymer/fullerene and a range of polymer/small-molecule OPVs. As well as enabling the extraction of χ, the study of equilibrated systems is also important in terms of device stability, as non-equilibrium morphologies and domain compositions, that have been carefully optimised during fabrication, may evolve towards equilibrium under operation, if the components can attain sufficient mobility.3,6,7
Given the importance of understanding equilibrium in mixed amorphous polymer/small-molecule systems, we recently performed an in-depth examination of the hypothesis that polymer/fullerene thin-film systems can form composition profiles consisting of two co-existing phases in liquid–liquid equilibrium.34 We investigated model bilayer systems consisting of (initially pure) PS layers on top of two different fullerene layers; phenyl-C60-butyric acid methyl ester (PCBM) and bis-adduct phenyl-C60-butyric acid methyl ester (bis-PCBM). This investigation was carried out via neutron reflectometry experiments as a function of polymer molecular weight (Mw), annealing temperature, annealing time and layer thickness, using samples that were ex situ annealed and then measured using neutrons. Theoretical predictions of the effect of Mw on layer composition and interface width, based on composition measurement in the high Mw limit, were found to be in very good agreement with experiments for both PCBM/PS and bis-PCBM/PS. In both systems, a significant increase in interfacial width upon decreasing Mw occurred. In the PCBM/PS system little change in layer composition was found with decreasing Mw, whereas in bis-PCBM/PS a much more significant systematic change in layer composition was found on decreasing Mw, indicative of increased mixing. These findings were rationalised (quantitatively) using Flory–Huggins theory.
In this paper we present in situ annealing and neutron reflectivity (NR) measurements on PCBM/PS and bis-PCBM/PS bilayers. These experiments were performed for three reasons; (i) to investigate the effect of polymer Mw and fullerene chemistry on the onset of mass transfer, in relation to the molecular mobilities (glass transition temperatures, Tg) of the component materials, (ii) to probe for any temperature dependent behaviour at elevated temperature (as a function of temperature, during gradual cooling) in the equilibrated systems and (iii) to corroborate (or otherwise) measurements on ex situ annealed samples that we have performed previously (in which samples were annealed and then quenched to room temperature before measuring). As in situ annealing involves the measurement of the composition profile before and after annealing each sample, it not only enables the volume fractions of the components in the co-existing phases to be determined directly from the layer compositions (as for ex situ measurements), but also allows layer volume fractions to be determined from changes in the layer thicknesses on annealing. In situ measurements enable us to examine the time-scales and temperatures required for equilibration, and to also discern whether there is any temperature-dependent behaviour (which cannot be definitively determined using ex situ annealing because of the potential for changes to the samples on cooling). Measurement of the onset temperatures for fullerene diffusion is particularly important in relation to the morphological stability of polymer/small-molecule OPVs in operation.7,36,37 The molecular mobility of the polymer, as a function of Mw,38 is likely to be of key significance here. This is because of the potential for preferential segregation of low-Mw fractions to interfaces39,40 within devices, given the typically broad Mw-distributions of conjugated polymers. Measurements using model PS/fullerene systems with narrow Mw-distribution PS allow us to examine this behaviour in well-controlled polymer/small-molecule thin-film systems. The particular Mws studied herein were chosen for two reasons. Firstly, they encompass the range of PS Mws over which equilibrium thermodynamic theory suggests one is most likely to observe significant changes in the interfacial width as a function of Mw.34,41,42 Secondly, they reflect the range of Mws over which the segmental relaxation times of the PS, as a function of temperature exhibit considerable Mw-dependence.38
Experimental
Materials
PCBM and bis-PCBM with a purity of 99.5% were purchased from Solenne, Netherlands. PS batches were purchased from Agilent Technologies, UK. The nominal Mws of these were 2, 3, 5, 20 and 100 kg mol−1. The weight average molecular weights (Mw) of these batches were 1.86, 2.93, 4.73, 18.5 and 111.4 kg mol−1 respectively, with polydispersity indices (Mw/Mn, where Mn is the number average molecular weight) of 1.04, 1.04, 1.04, 1.03 and 1.03 respectively. These batches are referred to as 2k, 3k, etc. throughout this manuscript. Silicon substrates of orientation 100 (with a native oxide layer), with a diameter of 2 inches and a thickness of 1150 microns were purchased from Siltronix, France. Mica sheets of size 65 mm × 65 mm and thickness 0.15 mm were purchased from Goodfellow, UK. Toluene, chlorobenzene, acetone and isopropanol (all of purity 99.9%) were all purchased from Sigma Aldrich, UK.Sample fabrication
Fullerenes were dissolved in chlorobenzene and then spin-coated onto silicon substrates that had been sonicated in acetone and isopropanol (for 15 minutes each) and then rinsed in de-ionised water. The PS layers were spin-coated from toluene solutions onto freshly-cleaved mica substrates, and then floated on top of the silicon/fullerene in a bath of de-ionised water (further details can be found in Môn et al.29 and Hynes43). Table S1 in the ESI† shows the spin-coating speeds and solution concentrations used to fabricate each of the layers. The samples were then placed under vacuum at room temperature (and in the dark) for at least 2 hours.Sample annealing
In situ annealing was performed under vacuum in the neutron beam at the reflectometer D17,44 at the Institut Laue Langevin (ILL), Grenoble, France. There is an offset between the set-point temperature and the sample (surface) temperature of a few °C. Details of the sample temperature calibration during heating and cooling are given in the ESI.†NR
The time-of-flight (TOF) methodology in which a broad range of wavelengths is incident on the sample was used for all NR measurements. Incident angles of 0.8° and 3.2° were used to obtain the full reflectivity measurements before and after annealing, with measurement times of 15 and 80 minutes respectively. In the full reflectivity measurements the percentage instrumental resolution (dqz/qz × 100, where dqz is the instrumental resolution at momentum transfer qz) varied from 1.7% to 5.8% across the qz range of approximately 0.007–0.2 Å−1. Kinetic measurements were performed using 30 s time slices, at an incident angle of 0.87° and an instrumental resolution of between 3% and 7.3%, over the qz range of approximately 0.008–0.09 Å−1. To calculate the specular reflectivity from the raw data obtained from the incident and reflected neutron beams, we used the COSMOS data reduction program.45 In the data analysis a fixed resolution of 4% was used when fitting the full reflectivity curves and 5% when fitting the kinetic data for all samples. These values reflect a combination of the instrumental resolution (set by the slit geometry and chopper settings of the instrument) and any effective resolution effects that may occur due to the samples themselves, such as slight layer thickness variation across the samples which will tend to reduce the resolution of the sharpest minima in the reflectivity curve.Reflectivity curves were fitted using motofit46 with bilayer models that were parameterised by Gaussian roughness at the buried fullerene/PS-rich interface and the sample surface. Bilayer fits of the NR data involved six adjustable parameters; the SLD and thickness of each layer, the interfacial roughness, and the sample surface roughness. The model composition profiles also included a silicon oxide layer, which was fixed for all samples (thickness = 12 Å, SLD = 3 × 10−6 Å−2, roughness = 3 Å). The layer thicknesses, layer SLDs and surface/interface roughnesses were not constrained during fitting of the full reflectivity curves. Since the Gaussian roughness represents the width of an error function SLD profile at the sample surface and buried interface, it was also necessary to check that surface and interface roughness parameters returned by the fits remained small in comparison to the layer thicknesses within the sample. In all measurements (full reflectivity curves and kinetic measurements) the bottom layer thickness was a factor of at least 3 times larger than the Gaussian roughness of the buried interface. The robustness of the full reflectivity measurements and the extracted fit parameters from these bilayers has been investigated previously in a number of ways, including performing repeat measurements, assessing the quality of the samples using real space (imaging) methods and carefully examining the robustness of the fitting procedures.34,43 However, given the restricted qz range, lower resolution and shorter counting times we were careful in the current study to pay particular attention to the robustness of the fits to the kinetic measurements. Of key importance here is the assessment of the robustness of the four main fit parameters that are known to evolve during annealing, given the existence of six fit parameters overall. Details of these assessments, that involve comparison of 30 s and two min time slices, and comparison of fits with six adjustable parameters and fits with only four or five, are given in the ESI.† The outcome of these assessments is that the overall behaviour of the bilayer fits during annealing is robust, but that there is sometimes the potential for what looks like over-parameterisation when using six adjustable parameters. This may be the cause of the slight offsets of some fit parameters (interfacial roughness and top layer SLD) between the full reflectivity curves, and the kinetic fits at neighbouring timesteps (e.g.; as seen in Fig. 3e at the end of annealing). The potential for too many adjustable parameters in the kinetic fits is most evident in the 20k-PS/PCBM sample (see ESI†), in which two local minima in the goodness of fit parameter, χ2, with slightly different layer thicknesses are seen, with consequent non-physical behaviour in the kinetic fits as a function of time. For this reason the kinetic fits for this sample, shown in Fig. 3j–l, have only five adjustable parameters, with the bottom layer SLD fixed at 4.65 × 10−6 Å−2 (unlike the other samples in Fig. 3 which all have six adjustable parameters). To further test robustness, fits were performed using both Genetic and Levenburg–Marquardt algorithms,47 with kinetic fits carried out using the Levenburg–Marquardt algorithm in two ways; (i) by starting the fit at each time point from the fit to the preceding time point, and (ii) starting from a different location in parameter space.
GIXD
Measurements were performed at Sheffield University using a Xeuss 2.0 (Xenocs France) SAXS/WAXS laboratory beamline, with a liquid Gallium MetalJet X-ray source (Excillum Sweden) which has an X-ray energy of 9.2 keV and a corresponding wavelength of 1.34 Å. The instrument was calibrated using Silver Behenate (Alfa Aeser). The sample chamber and flight tubes were all evacuated to reduce air scatter. The X-rays were detected using a Pilatus3R 1 M detector.Statistics
The error bars in the NR plots represent the standard deviation (calculated from the neutron counts).Results and discussion
In situ annealing was carried out in discrete temperature steps, an example of which is shown in Fig. 1a. At the end of annealing the heater was turned off and the sample was allowed to cool gradually (under vacuum). High-resolution NR measurements were made before annealing, and again after the samples had cooled to 80 °C (a temperature below which no significant changes in reflectivity were seen). During annealing, lower-resolution ‘kinetic’ measurements were taken, enabling a higher neutron flux. Combined with the collection of data over a restricted, qz range, this allowed us to obtain usable data in much shorter times (on the order of a minute) than is possible when collecting full high-resolution measurements. Our interest in these experiments is to investigate the composition profile in bilayers. However, annealing these systems at elevated temperatures for extended periods opens up the potential for the formation of lateral inhomogeneities in the samples, for example caused by dewetting of one or other of the two liquid layers or by extensive aggregation or crystallisation of the fullerenes.29 Our annealing methodology is therefore a compromise between temperature-resolution during heating (with many smaller temperature steps, allowing us to more accurately pin-down the onset of diffusion between the layers) and the preservation of bilayer-like samples into the later stages of the experiment (for which fewer, larger steps, and therefore a lower overall annealing time, would be preferable); preservation of bilayer structures until the end of the experiments allows us to compare composition profiles on the same samples before and after annealing, and extract layer composition information from measurements of the changes in layer thicknesses on annealing. These can then be compared with layer composition measurements extracted from the layer SLDs. The examination of behaviour during and after gradual cooling also allows us to compare our results with previous measurements on samples that were ex situ annealed and then rapidly quenched.34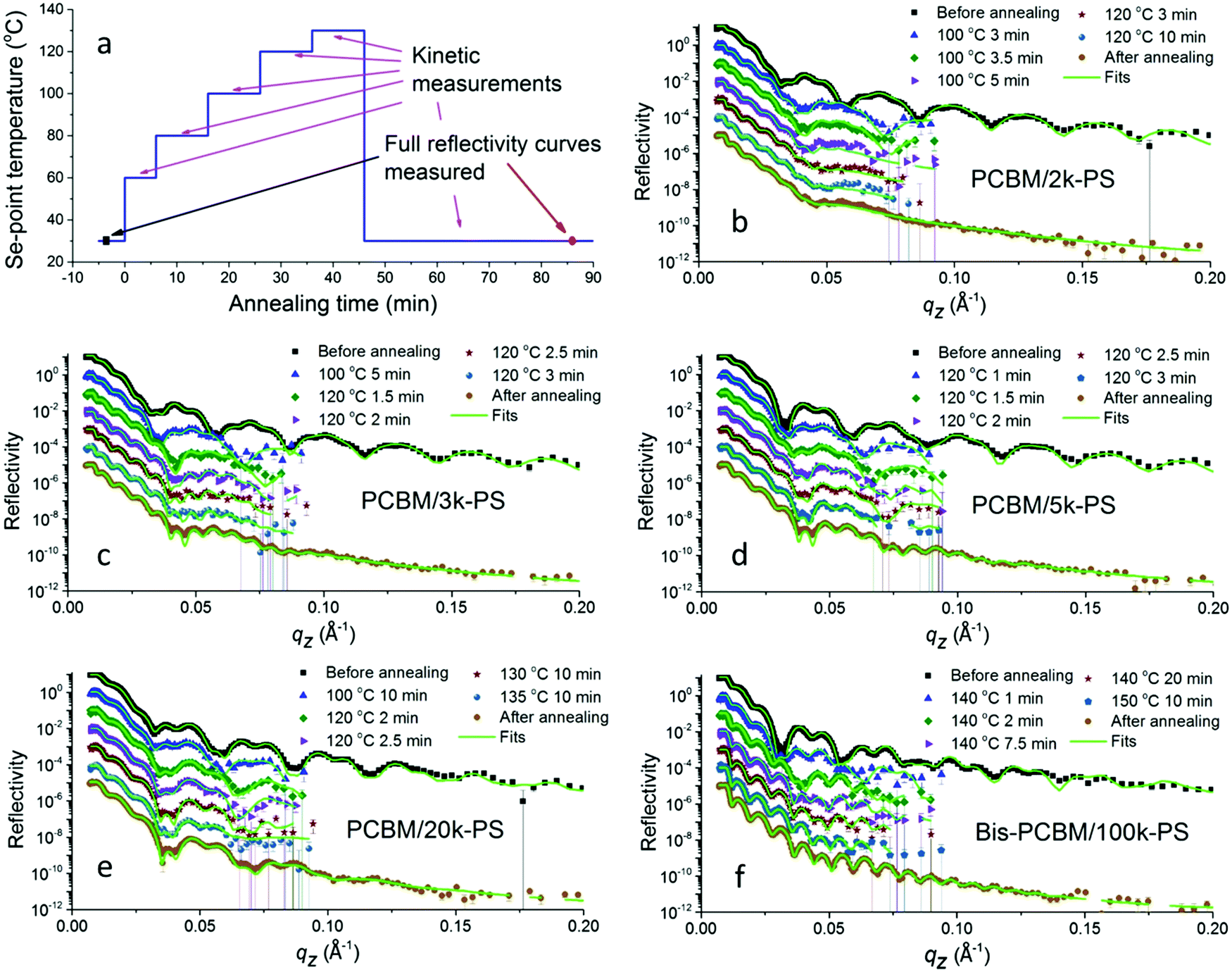 | ||
| Fig. 1 NR measurements and fits, before, during and after in situ thermal annealing; (a) plot showing a typical annealing protocol, (b) PCBM/2k-PS, (c) PCBM/3k-PS, (d) PCBM/5k-PS, (e) PCBM/20k-PS, (f) bis-PCBM/100k-PS. The kinetic data shows measurements and fits for 30 s slices. | ||
A summary of the in situ NR data is shown in Fig. 1b–f. Four PCBM/PS bilayers were investigated, with PS Mws of 1.86, 2.93, 4.73 and 18.5 kg mol−1 (referred to throughout this manuscript as 2k, 3k, 5k and 20k respectively). One bis-PCBM/PS bilayer, with Mw of 111.4 kg mol−1 (referred to as 100k) was also measured. All samples were fabricated by first spin-coating a pure fullerene layer onto a silicon substrate. Pure PS layers were then placed on top of this to make bilayers. Significant changes in the NR curves are seen in all samples following annealing. The kinetic data was collected in 30 s time slices, and fits to the NR from these slices are shown in Fig. 1, alongside fits to the NR curves before and after annealing. Kinetic data was also fitted after combining sets of four consecutive 30 s slices into two minute time slices. The fits shown throughout this paper (before, during and after annealing) correspond to bilayers, where composition profiles are modelled as two layers of uniform scattering length density (SLD) with Gaussian roughness41,48,49 at both the buried interface and the sample surface. In all of the fits shown in Fig. 1, all six parameters that characterise the bilayers (two SLDs, two layer thicknesses and two roughnesses) are adjustable.
Plots of the SLD profiles, corresponding to the fits before and after annealing, are shown in Fig. 2. Significant and consistent changes occur in all five cases. These changes arise due to diffusion of the fullerenes into the PS layers. No significant diffusion of the PS into the fullerene layers is observed in any of these samples. Fullerene diffusion raises the SLD and thickness of the top layer and reduces the thickness of the bottom layer. Given the fully adjustable layer thicknesses and SLDs, there is no constraint on the total sample thickness or the mass per unit area before or after annealing, during fitting. However changes in both of these is at the level of a few percent following annealing; the total sample thickness reduces by between 0.1% and 2.4% following annealing, and the integrated scattering length per unit area changes by between −5% and +4% following annealing. The interfacial roughness also increases significantly in all cases. In previous measurements on ex situ annealed and then measured PCBM/PS samples, we found top-layer (PS-rich) SLDs between approximately 1.5 and 1.7 × 10−6 Å−2 (see Hynes et al.,34Fig. 3b), for an extensive sample set (covering a range of annealing times and temperatures), with no systematic Mw dependence. These findings are confirmed here; the SLDs of the top layers in the PCBM/PS bilayers shown in Fig. 2 are all within this range (2k, 1.64 × 10−6 Å−2; 3k, 1.64 × 10−6 Å−2; 5k, 1.62 × 10−6 Å−2; 20k, 1.60 × 10−6 Å−2). The thickness changes and broadening of the interfaces on annealing also qualitatively confirm the findings from ex situ annealed samples. However, quantitative comparison of the interfacial roughness as a function of PS Mw, reveals some differences with-respect-to the ex situ annealed samples.
 | ||
| Fig. 2 GIXD image after in situ annealing and SLD profiles before and after in situ annealing; (a) GIXD image from the in situ annealed PCBM/2k-PS sample at an incident angle of 0.16°, (b) PCBM/2k-PS SLD profiles, (c) PCBM/3k-PS SLD profiles, (d) PCBM/5k-PS SLD profiles, (e) PCBM/20k-PS SLD profiles (f) bis-PCBM/100k-PS SLD profiles. Qxy and Qz in (a) are the in-plane (horizontal) and out-of-plane (vertical) components of the momentum transfer respectively.52 | ||
 | ||
| Fig. 3 Fit parameters versus time for in situ annealed PCBM/PS bilayers; (a–c) layer thicknesses, top layer SLD and interfacial roughness fit parameters respectively for the 2k-PS sample; (d–f) fit parameters for the 3k-PS sample, (g–i) fit parameters for the 5k-PS sample; (j–l) fit parameters for the 20k-PS sample. The kinetic data parameters are for fits to 2 min slices. | ||
The most significant difference with-respect-to samples that have been ex situ annealed (for 1–10 min at various temperatures34) is seen in the PCBM/2k-PS sample. Fig. 1b shows that the fit for this sample after annealing, does not reproduce some of the fringes seen in the data well. This is particularly evident for the high frequency fringes that sit on top of the lower frequency fringe between qz of approximately 0.04 and 0.08 Å−1. Similar damping of fringes has been seen in some 2k-PS samples that were ex situ annealed, with those annealed at higher temperatures or longer times particularly susceptible.43 Optical microscopy on these samples revealed extensive development of lateral inhomogeneities, that roughen the sample surface,43 and this also occurs here on the PCBM/2k-PS sample (see Fig. S16c, ESI†). Atomic force microscopy (AFM) reveals that these inhomogeneities consist of protrusions through the bilayer sample surface (up to a few hundred nanometers in height in the PCBM/2k-PS sample; i.e. significantly larger than the bilayer thickness itself) that sometimes display clear anisotropic features, indicative of small needle-like PCBM crystals.29,34,43 Grazing-incident X-ray diffraction (GIXD) measurements were performed on this sample after annealing, to assess whether any significant fullerene crystallisation had occurred within the sample. PS molecular mobility in these systems has been previously shown to strongly influence fullerene crystal growth rates,29 and therefore (for a given annealing protocol) the higher-mobility 2k-PS sample38 represents the sample likely to have the highest crystal content. PCBM crystals in thin films (blends, bilayers and single layers) produce a very specific GIXD pattern.29,50,51 However, the GIXD detector map shown in Fig. 2a shows only an amorphous ring, with no evidence of any significant fullerene crystallisation. This leads us to conclude that (i) the fullerenes in these samples remain largely amorphous and (ii) the lateral inhomogeneities shown in Fig. S16c (ESI†) are either fullerene ‘aggregates’ that do not give rise to crystalline diffraction, or they contain a very small volume fraction of crystals, diffraction from which is dominated by the amorphous PCBM in the sample. The key impact that the lateral inhomogeneities appear to have on the fit to this sample after annealing, concerns the interfacial roughness parameter. The (Gaussian) roughness of this sample after annealing is 38 Å which is above the range of 28–35 Å found for ex situ annealed samples (across a range of annealing temperatures and times).34 Smaller-sized lateral inhomogeneities also occur on the 3k, 5k and 20k-PS/PCBM bilayers, and on the 100k-PS/bis-PCBM bilayer (see Fig. S16d–f and S17, ESI†). In these samples, however, the interfacial roughness parameters (25 Å, 22 Å and 19 Å for 3k, 5k and 20k-PS/PCBM bilayers and 27 Å for the 100k-PS/bis-PCBM bilayer respectively) are all within the ranges found for ex situ annealed samples (that are annealed for much shorter times);34 NB – for the 100k-PS/bis-PCBM in situ annealed bilayer this comparison is with 20k-PS/bis-PCBM ex situ bilayers, with the justification that these samples are of sufficiently high Mw that the composition profiles are essentially independent of Mw. For comparison, Fig. S16a and b (ESI†) show two ex situ annealed 5k PS samples with very different levels of lateral inhomogeneity, that have very similar reflectivity curves, fits and SLD profiles.34,43 Given the agreement in SLD profiles (in particular the interfacial roughness) between the two ex situ samples shown in Fig. S16a and b (ESI†), and between the 3k-100k PS in situ samples (shown in Fig. S16d–f, ESI†) and the corresponding ex situ 3–100k PS samples in Hynes et al.,34 it is clear that the level of lateral inhomogeneities on these samples is sufficiently low that the NR is dominated by the reflections from the bilayer regions of the sample that are in-between the inhomogeneities. Only for the 2k-PS/PCBM in situ annealed bilayer (in Fig. S16c, ESI†) is the level of lateral inhomogeneity high enough to significantly influence the NR and the fitted interfacial roughness parameter. It also seems likely that the increased fullerene layer SLD after annealing in this sample shown in Fig. 2b (a value of 4.93, which is at the extreme end of all of the values measured in ex situ34 and in situ annealed samples) is an artefact of the relatively poor fit for this NR curve, as a result of the lateral inhomogeneities. However, even for this sample the layer thickness parameters and the top layer SLD are reliably determined, enabling investigation of the fullerene diffusion process.
Although the bilayer models contain six adjustable parameters, two of these are of low significance; (i) there is no significant reduction in the bottom layer SLD on annealing (Fig. 2b–f), and this layer therefore remains pure fullerene after annealing, and (ii) the fits are much less sensitive to the sample surface roughness than to the interfacial roughness (see Fig. S4, ESI†). We therefore now focus on the four key parameters of top layer thickness, bottom layer thickness, top layer SLD and interfacial roughness, before, during and after annealing. These parameters are shown in Fig. 3 for the four PCBM/PS bilayers, with the parameters during annealing representing two-minute time slices. Some plots of parameters versus time using 30 s slices (corresponding to the kinetic fits shown in Fig. 1) are also shown in the ESI,† Fig. S8 and S9. The behaviour of the parameters with time for the 30 s and two-minute slices is very similar in all cases. All samples shown in Fig. 3 show the same characteristic behaviour, in which there is a rapid change in the layer thicknesses, top layer SLD and interfacial roughness following a step in the set-point temperature. The mean sample temperature (the temperature measured at the surface of the silicon/fullerene/polymer sample) is of order 5 °C lower than the set-point, and there is a typical overshoot of the set-point by around 2 °C for a 20 °C temperature step (see ESI,† Fig. S5 and S6 and Table S2). Depending on the Mw, the next temperature step may also produce significant changes in the four key parameters. Fig. S5 (ESI†), which shows the bottom layer thickness as a function of time overlaid with the sample temperature plot for this annealing protocol for one of the samples, more closely examines the kinetics at play here. In this (3k-PS/PCBM) sample it is clear that the nature of the temperature overshoot on increasing the set-point to 100 °C, combined with the relatively low molecular mobility in the system (by definition) near to the temperatures where the components first becomes mobile at all, means that the diffusion process is not able to conclude before the temperature drops down to its steady state value after the temperature step. Depending on the temperature at which the (fullerene in the) system becomes mobile in comparison to the temperatures of the steps and the overshoot, fullerene diffusion may begin, then slow down and only resume following a further temperature step. As is shown in Fig. S5 (ESI†), mass transfer is then complete with relatively few changes occurring after any further temperature steps.
In general the diffusivity of nanoparticles within polymer melts decreases with Mw, as a result of the increasing melt viscosity.53,54 The exact nature of the behaviour (and in particular its correspondence to the predictions of the Stokes–Einstein relation) depends on a number of factors, including the relative sizes of the nanoparticles and the polymer melt tube diameter (in entangled melts),54 and the occurrence of polymer adsorption (to larger nanoparticles).53 However, the nature of our measurements does not allow us to resolve isothermal fullerene diffusion processes themselves, and our focus is instead on the onset temperatures for diffusion and the equilibration of the bilayer composition profiles. The four PS/PCBM samples show a systematic dependence on the PS Mw for the onset of fullerene diffusion. Fig. 3a and j show that fullerene diffusion begins once the set-point of the 2k-PS/PCBM is raised to 100 °C, but only once the set-point of the 20k-PS/PCBM sample is raised to 120 °C. The 3k and 5k samples show intermediate behaviour. The thickness changes represent of order 10–15% of the thickness of the PS layer and 20–40% of the thickness of the PCBM layer (NB: over this temperature range the thickness change due to thermal expansion of PS is of order 1%, and there is no systematic expansion of PCBM – see ESI,† Fig. S1–S3). These changes are quantified in Fig. 4. For each layer, this shows the ratio of the magnitude of the change in thickness between 80 °C and 100 °C, to the magnitude of the change in thickness between 80 °C and 120 °C. The monotonic dependence on Mw is clear, and this behaviour clearly correlates with the known reduction in the Tg of PS as the Mw is lowered.38
 | ||
| Fig. 4 The ratio of the magnitude of the change in the thickness between 80 °C and 100 °C, Δt80–100°C, to the magnitude of the change in thickness between 80 °C and 120 °C, Δt80–120°C, versus weight-average molecular weight (Mw), for the top and bottom layers in each bilayer. | ||
Significant differences in composition profile with-respect-to PCBM/PS are found when the fullerene chemistry is modified by the addition of a second side-chain to the C60 cage, to make bis-PCBM. The additional side-chain inhibits crystallisation of this fullerene55 and also changes the miscibility with solvents and polymers.26,56,57 Qualitatively similar fullerene diffusion behaviour is seen for bis-PCBM/PS bilayers on annealing, but the layer compositions and interfacial roughness are significantly different, with higher fullerene content in the PS-rich (top) layer and broader interfaces, compared to PCBM/PS bilayers.34 The diffusion onset temperature is also found to be dependent on fullerene chemistry. Fig. 5 shows the changes in the four key fit parameters for a 100k-PS/bis-PCBM bilayer, before, during and after annealing. This shows a very small change in layer thicknesses as the set-point is raised to 120 °C, with the main thickness changes, and changes in the interfacial roughness and top layer SLD occurring at a set-point of 140 °C. The Tg of 100k-PS (100 °C)38 is only a few degrees higher than for 20k-PS, and is well below the sample temperature that occurs at a set-point of 120 °C. It therefore appears that the delayed diffusion onset in bis-PCBM/PS bilayers, in comparison to PCBM/PS, is a consequence of the lower mobility of the fullerene; PCBM has a reported Tg of between 117 and 131 °C,26,58,59 while bis-PCBM has a reported Tg of 145 °C.26 Overall it is therefore clear from these results that the mobility of the fullerene at the polymer/fullerene interface is dependent on both the innate mobility of the fullerene itself, and also the mobility of the PS; i.e. the lower Tg polymer on the other side of the initially sharp interface, effectively plasticises the higher Tg fullerene. This is consistent with the behaviour of polymer/fullerene blends, in which a single Tg, intermediate between that of the pure components, is reported.59,60 It is also consistent with differential scanning calorimetry measurements and ellipsometry experiments on PCBM/polymer and bis-PCBM/polymer bilayers,26 that show fullerene diffusion beginning below the reported bulk Tg of the fullerenes. The results reported herein are the second example of this behaviour that we have found in these amorphous polymer/fullerene systems. In the current study the increase in the mobility of the fullerene with decreasing PS Mw occurs as a result of the Mw-dependent mobility of the PS, while in previous work we discovered that the film-thickness dependent mobility of PS has a strong influence on fullerene crystallisation kinetics within PCBM/PS bilayers.29 Both of these factors have important implications for morphological stability in polymer/small-molecule OPVs under operation, due to the thin-film nature of these devices (and also the nanoscale dimensions of the polymer-rich domains within the active layer61) and the broad Mw distributions in conjugated polymer systems; the Mw-dependent plasticisation of the fullerene reported herein, is of particular significance owing to the potential for preferential segregation of low-Mw polymer fractions to interfaces,39,40 and the potential evolution of domain composition and morphology3,6,7,15 as a result. The implications for OPV performance are clear. The evolution of morphology or domain composition within carefully optimised devices3,7 under operating conditions, can result in efficiency reductions. In addition, component migration in polymer/fullerene blends following annealing below the bulk Tg of the fullerene62 or the bulk Tg of both the fullerene and polymer,36 can also be accompanied by significant decreases in OPV efficiency. In the former case Sachs-Quintana et al.62 see preferential interfacial segregation of the polymer at one of the device electrodes, while Li et al.36 observe fullerene diffusion and crystallite growth in amorphous polymer/PCBM blends. In contrast, in other (amorphous-polymer/fullerene) systems, annealing below the bulk Tg of both components can lead to device efficiency gains, that are attributed, at least in part, to local fullerene diffusion.63 In addition to the knowledge that the Tg of conjugated polymers can deviate from bulk values,64 the Mw-dependent plasticisation of fullerenes by amorphous polymers reported herein, illustrates the importance of understanding how component Mw, Mw distributions and confinement to nanoscale dimensions may combine to influence component mobility within polymer-based OPVs under operation.
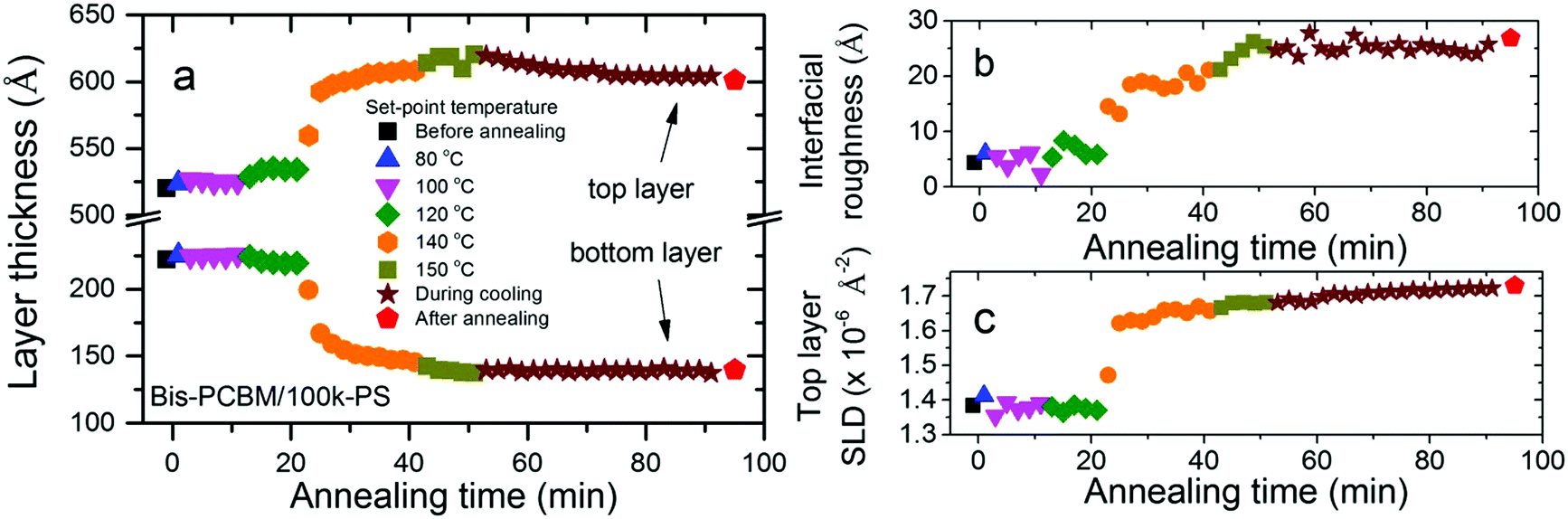 | ||
| Fig. 5 Fit parameters versus time for an in situ annealed bis-PCBM/100k-PS bilayer; (a–c) layer thicknesses, interfacial roughness and top layer SLD fit parameters respectively. | ||
We now examine the layer compositions quantitatively. Ex situ and in situ measurements are complementary here; ex situ measurements allow more controlled heating over shorter periods of time and rapid cooling, while in situ measurements allow us to directly probe structure at elevated temperature. Ex situ measurements enable us to calculate the polymer volume fraction in the (top) PS-rich layer, ϕPS, using layer composition information, while in situ measurements allow such calculations, but also enable the determination of volume fractions from changes in the thicknesses of the top and bottom layers on annealing. The details of the methodology for calculating fullerene/polymer volume fractions from layer compositions (SLDs) are given in Hynes et al. (supplementary information),34 while the second method simply measures ϕPS as the ratio of the change in the bottom (fullerene) layer thickness on annealing, to the final thickness of the top (PS-rich) layer. The former method relies on estimates of the SLDs of pure PS, for which we take two different values, 1.31 × 10−6 Å−2 (our mean measured value for the SLD of pure PS in unannealed samples) and 1.4 × 10−6 Å−2 (a value from the literature48), thus enabling an assessment of the uncertainty in the volume fraction calculations (as similarly performed in our previous ex situ study34). Fig. 6 compares ϕPS as a function of Mw, calculated from top layer compositions and also from changes in layer thickness for the four in situ annealed PCBM/PS bilayers, to estimates from previous ex situ measurements and theoretical predictions (using Flory–Huggins theory in which the interaction parameter, χ, is extracted using measurements at high Mw34). As can be seen in Fig. 6 reasonable agreement for ϕPS is found between the values determined from the in situ measurements (from both layer composition measurements and changes in layer thicknesses) and both the ex situ measurements and the theoretical predictions. In addition, for the 100k-PS/bis-PCBM in situ sample the calculated values for ϕPS using, (i) the measured top layer SLD combined with setting the SLD of pure PS equal to 1.31 × 10−6 Å−2, (ii) the measured top layer SLD combined with setting the SLD of pure PS equal to 1.4 × 10−6 Å−2 and (iii) the changes in layer thickness on annealing, are 0.83, 0.86 and 0.86 respectively, while the 20k-PS/bis-PCBM ex situ samples34 have ϕPS equal to 0.81 and 0.84 for pure PS SLDs equal to 1.31 × 10−6 Å−2 and 1.4 × 10−6 Å−2 respectively, again showing reasonable agreement. This close agreement between the in situ and ex situ annealed samples demonstrates that the measurements are robust, in that the heating and cooling protocols used in these experiments (rapid quenching or slow cooling) do not significantly affect the measured composition profiles or the calculated values of ϕPS. Our findings from the in situ measurements therefore corroborate the conclusions from the ex situ results that these polymer/fullerene bilayers rapidly form a liquid–liquid equilibrium.
 | ||
| Fig. 6 Comparison of composition measurements between in situ and ex situ annealed PCBM/PS samples. Experimental measurements of the top layer volume fraction, ϕPS, from the current study are plotted along with previous ex situ measurements and theoretical plots of the free energy of mixing, for various Mws. The ex situ measurements and free energy plots are the same data that is shown in Fig. 5d in Hynes et al.34 The vertical solid lines show the locations of the predicted co-existing compositions (from the common tangent construction), and the vertical dashed lines show the uncertainties in these locations. The value of χ (1.82) was extracted from ϕPS measurements on 20–300k-PS/PCBM bilayers, and is referenced to the volume of a PCBM molecule.34 | ||
The measurements in Fig. 3 and 5 also allow us to investigate any potential changes in the composition profiles during cooling. The clearest systematic trend seen in all five samples on cooling is a gradual reduction in the thickness of the top layer. This behaviour (and also the minimal changes in PCBM thickness on cooling) is entirely commensurate with the thermal contraction seen in pure PS during in situ measurements that we performed on single layer samples (see ESI,† Fig. S1 and S2). These single layer measurements are entirely consistent with reported thermal expansion measurements on PS thin-films in the literature65 (see ESI,† Fig. S3). Given our cooling time/sample temperature calibration (see ESI,† Fig. S6c), we are able to convert the time axes in Fig. 3 and 5 into sample temperatures and plot the top layer thicknesses changes (as a percentage of the layer thickness at a reference temperature of 125 °C) in the five bilayers as a function of temperature in Fig. 7. Overlaid on the data is a (solid black) line representing the mean thermal expansivity of PS films in the melt, and lines representing the reported uncertainty in the expansivity65 (the two dashed lines). It is clear that the thickness changes in the bilayers, over the temperature range investigated, are well described by simple thermal contraction of the PS melt. In pure PS, this gradient would persist down to a temperature close to Tg, at which point a gradual transition to a lower thermal expansivity would be expected. Pure PS layers (of sufficient thickness to exhibit bulk behaviour, typically greater than around 40 nm65) would have Tgs of 48, 66, 78, 95 and 100 °C respectively for the 2k, 3k, 5k, 20k, and 100k samples used in our study (using eqn (2) in Santangelo and Roland38). Although the data in Fig. 7 is somewhat noisy, there is some indication of a change in gradient in the 20k-PS/PCBM and 100k-PS/bis-PCBM samples (in which the top layers contain around 90% and 80–85% PS respectively). With this in mind we have also plotted the reported expansivity of a 40 nm PS film in the glassy state65 in Fig. 7 (green solid line) with a Tg of 95 °C (occurring at the intersection of the mean glass and melt expansivity lines); this is equal to the PS Tg of the 20k sample (PS Mw = 18.5 kg mol−1).38 Given that there are no adjustable parameters in this plot it is clear that it represents the behaviour of the top layer thickness of the 20k-PS/PCBM sample remarkably well. For the 100k-PS/bis-PCBM sample, the top (PS-rich) layer thickness is also well described by the thermal expansion/contraction of PS; although here the gradient of the slope above 100 °C in Fig. 7 is closer to the lower estimate of the melt expansivity (5.2 × 10−4 K−1), rather than the mean (7.2 × 10−4 K−1), with a change in slope in Fig. 7 apparent at around 100 °C. Overall, the picture is clear; over the temperature range investigated, the changes in composition profile are accounted for well by the known thermal expansivity of PS and PCBM, with no evidence for any other changes to the equilibrium composition profile of the bilayers.
 | ||
Fig. 7 Percentage change in the thickness of the top (PS-rich) layer in PS/fullerene bilayers, as a function of sample temperature, during cooling. The changes are calculated with-respect-to the thickness of the top layer at a reference temperature of 125 °C. A line representing the mean expansivity, αmelt, of a PS melt film (7.2 ± 2 × 10−4 K−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 65) is plotted along with upper and lower estimates of αmelt. The expansivity of a 40 nm glassy PS film (extracted from the fit shown in Fig. 4 in Keddie et al.65), αglass, is plotted such that it intersects the melt expansivity line at 95 °C (corresponding to the Tg of 20k-PS38). 65) is plotted along with upper and lower estimates of αmelt. The expansivity of a 40 nm glassy PS film (extracted from the fit shown in Fig. 4 in Keddie et al.65), αglass, is plotted such that it intersects the melt expansivity line at 95 °C (corresponding to the Tg of 20k-PS38). | ||
Conclusions
In this investigation, we have carried out a series of in situ annealing experiments on PCBM/PS and bis-PCBM/PS bilayers, in which composition profiles before, during and after annealing have been determined. Measurements of the compositions of the (top) PS-rich layers as a function of Mw, based on both SLD measurements and changes in layer thicknesses on annealing, were found to be in agreement with measurements of bilayer samples that were ex situ annealed for a range of different times at a range of temperatures. This amounts to further corroboration of the evidence that the composition profiles rapidly formed in these systems following thermal annealing, correspond to two co-existing liquid–liquid phases in thermodynamic equilibrium. These measurements also demonstrate a clear and systematic onset temperature for diffusion of the fullerenes into the PS layer that correlates with both the Mw-dependent molecular mobility (Tg) of the polymer and also the Tg of the fullerene, revealing that the molecular mobility of the fullerenes in these systems is controlled by the intrinsic mobility of the fullerenes themselves and the ability of the polymer to plasticise the fullerenes at the interface. This has important implications for OPV stability, especially in relation to the control of Mw and Mw-distribution, given the potential for segregation of shorter polymer chains to interfaces in polydisperse systems. Measuring composition profiles at elevated temperatures has allowed us to examine evidence for any changes as a function of temperature; over the temperature ranges examined, we do not see any significant changes in composition profile, other than those associated with the known thermal expansivity of PS films.While our focus here has been on measurements of equilibrated systems, in principle, it would be possible to probe the kinetics of the fullerene diffusion process using NR measurements, by performing isothermal experiments closer to the onset temperature for fullerene mobility within the samples, and thereby tuning the diffusion rates to NR measurement timescales. However, this would require very careful temperature control and probably also careful control over the (thermal) history of the individual layers used to fabricate the bilayers, to ensure the repeatability of nominally duplicate samples that are annealed closer to the point where the system vitrifies.
Finally, we note that the in situ neutron reflectometry protocol used herein may complement measurements of the slow dynamics (such as Tg as a function of cooling/heating rate) in thin polymer films, which are typically measured by ellipsometry66 or flash differential scanning calorimetry,67 and enable novel investigations using the strength of isotope labelling to access buried layers68 in a range of different materials systems.
Author contributions
The overall structure of the project was designed by AH, with help from EH. AH and EH were responsible for the development of sample preparation methodologies and the design of experiments. EH performed the sample preparation, with support from AH. The neutron scattering experiments were performed by EH, AH and PG. GIXD experiments were performed by AP and EH. Data analysis was performed by EH, with support from AH. All authors contributed to the writing of the manuscript.Data availability
Data is available from the corresponding author upon request. The raw neutron reflectivity data is stored on a database at ILL; Doi: http://10.5291/ILL-DATA.9-12-456.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the ILL for the award of beam time (experiment number 9-12-456), and the staff of D17 for help during the experiments. EH acknowledges Swansea University for funding her PhD studentship. AJP thanks Xenocs for their help and ongoing support in the GIXD user program in Sheffield, and we thank the UK Engineering and Physical Science Research Council (EPSRC) for funding the purchase of this instrument. The open access fee was covered by FILL2030, a European Union project within the European Commission's Horizon 2020 Research and Innovation programme under grant agreement No. 731096.References
- W. Yin and M. Dadmun, ACS Nano, 2011, 5, 4756–4768 CrossRef CAS PubMed.
- N. D. Treat, M. a. Brady, G. Smith, M. F. Toney, E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Adv. Energy Mater., 2011, 1, 82–89 CrossRef CAS.
- L. Ye, B. A. Collins, X. Jiao, J. Zhao, H. Yan and H. Ade, Adv. Energy Mater., 2018, 8, 1703058 CrossRef.
- D. Chen, A. Nakahara, D. Wei, D. Nordlund and T. P. Russell, Nano Lett., 2011, 11, 561–567 CrossRef CAS PubMed.
- B. A. Collins, E. Gann, L. Guignard, X. He, C. R. McNeill and H. Ade, J. Phys. Chem. Lett., 2010, 1, 3160–3166 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- N. Li, J. D. Perea, T. Kassar, M. Richter, T. Heumueller, G. J. Matt, Y. Hou, N. S. Guldal, H. Chen, S. Chen, S. Langner, M. Berlinghof, T. Unruh and C. J. Brabec, Nat. Commun., 2017, 8, 14541 CrossRef CAS PubMed.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J. H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J. L. Bredas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- Q. Fan, Q. Zhu, Z. Xu, W. Su, J. Chen, J. Wu, X. Guo, W. Ma, M. Zhang and Y. Li, Nano Energy, 2018, 48, 413–420 CrossRef CAS.
- D. He, F. Zhao, J. Xin, J. J. Rech, Z. Wei, W. Ma, W. You, B. Li, L. Jiang, Y. Li and C. Wang, Adv. Energy Mater., 2018, 8, 1802050 CrossRef.
- Y. Xiong, L. Ye, A. Gadisa, Q. Zhang, J. J. Rech, W. You and H. Ade, Adv. Funct. Mater., 2019, 29, 1806262 CrossRef.
- L. Ye, S. Li, X. Liu, S. Zhang, M. Ghasemi, Y. Xiong, J. Hou and H. Ade, Joule, 2019, 3, 443–458 CrossRef CAS.
- L. Ye, Y. Xiong, Q. Zhang, S. Li, C. Wang, Z. Jiang, J. Hou, W. You and H. Ade, Adv. Mater., 2018, 30, 1705485 CrossRef PubMed.
- Z. Xiao, X. Jia and L. Ding, Sci. Bull., 2017, 62, 1562–1564 CrossRef CAS.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–27 CrossRef CAS PubMed.
- K. Vandewal, S. Himmelberger and A. Salleo, Macromolecules, 2013, 46, 6379–6387 CrossRef CAS.
- J. W. Kiel, A. P. Eberle and M. E. Mackay, Phys. Rev. Lett., 2010, 105, 168701 CrossRef PubMed.
- J. A. Bartelt, Z. M. Beiley, E. T. Hoke, W. R. Mateker, J. D. Douglas, B. A. Collins, J. R. Tumbleston, K. R. Graham, A. Amassian, H. Ade, J. M. J. Fréchet, M. F. Toney and M. D. McGehee, Adv. Energy Mater., 2013, 3, 364–374 CrossRef CAS.
- H. Chen, R. Hegde, J. Browning and M. D. Dadmun, Phys. Chem. Chem. Phys., 2012, 14, 5635–5641 RSC.
- K. H. Lee, P. E. Schwenn, A. R. G. Smith, H. Cavaye, P. E. Shaw, M. James, K. B. Krueger, I. R. Gentle, P. Meredith and P. L. Burn, Adv. Mater., 2011, 23, 766–770 CrossRef CAS PubMed.
- D. Leman, M. A. Kelly, S. Ness, S. Engmann, A. Herzing, C. Snyder, H. W. Ro, R. J. Kline, D. M. DeLongchamp and L. J. Richter, Macromolecules, 2015, 48, 383–392 CrossRef CAS.
- H. W. Ro, B. Akgun, B. T. O’Connor, M. Hammond, R. J. Kline, C. R. Snyder, S. K. Satija, A. L. Ayzner, M. F. Toney, C. L. Soles and D. M. DeLongchamp, Macromolecules, 2012, 45, 6587–6599 CrossRef CAS.
- R. Dattani and J. T. Cabral, Soft Matter, 2015, 11, 3125–3131 RSC.
- D. Môn, A. M. Higgins, D. James, M. Hampton, J. E. Macdonald, M. B. Ward, P. Gutfreund, S. Lilliu and J. Rawle, Phys. Chem. Chem. Phys., 2015, 17, 2216–2227 RSC.
- H. C. Wong and J. T. Cabral, Macromolecules, 2011, 44, 4530–4537 CrossRef CAS.
- H. C. Wong and J. T. Cabral, Phys. Rev. Lett., 2010, 105, 038301 CrossRef PubMed.
- H. C. Wong, A. M. Higgins, A. R. Wildes, J. F. Douglas and J. T. Cabral, Adv. Mater., 2013, 25, 985–991 CrossRef CAS PubMed.
- S. Pont, F. Foglia, A. M. Higgins, J. R. Durrant and J. T. Cabral, Adv. Funct. Mater., 2018, 28, 1802520 CrossRef.
- E. L. Hynes, J. T. Cabral, A. J. Parnell, P. Gutfreund, R. J. L. Welbourn, A. D. F. Dunbar, D. Môn and A. M. Higgins, Commun. Phys., 2019, 2, 112 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, Oxford, 2003 Search PubMed.
- Z. Li, K. Ho Chiu, R. Shahid Ashraf, S. Fearn, R. Dattani, H. Cheng Wong, C. H. Tan, J. Wu, J. T. Cabral and J. R. Durrant, Sci. Rep., 2015, 5, 15149 CrossRef CAS PubMed.
- H. C. Wong, Z. Li, C. H. Tan, H. Zhong, Z. Huang, H. Bronstein, I. McCulloch, J. T. Cabral and J. R. Durrant, ACS Nano, 2014, 8, 1297–1308 CrossRef CAS PubMed.
- P. G. Santangelo and C. M. Roland, Macromolecules, 1998, 31, 4581–4585 CrossRef CAS.
- D. Broseta, G. H. Fredrickson, E. Helfand and L. Leibler, Macromolecules, 1990, 23, 132–139 CrossRef CAS.
- P. Mahmoudi, W. S. R. Forrest, T. M. Beardsley and M. W. Matsen, Macromolecules, 2018, 51, 1242–1247 CrossRef CAS.
- R. A. L. Jones and R. W. Richards, Polymers at Surfaces and Interfaces, Cambridge University Press, Cambridge, 1999 Search PubMed.
- H. Tang and K. F. Freed, J. Chem. Phys., 1991, 94, 6307–6322 CrossRef CAS.
- E. L. Hynes, PhD thesis, Swansea University, 2018.
- T. Saerbeck, R. Cubitt, A. Wildes, G. Manzin, K. H. Andersen and P. Gutfreund, J. Appl. Crystallogr., 2018, 51, 249–256 CrossRef CAS.
- P. Gutfreund, T. Saerbeck, M. A. Gonzalez, E. Pellegrini, M. Laver, C. Dewhurst and R. Cubitt, J. Appl. Crystallogr., 2018, 51, 606–615 CrossRef CAS.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- I. G. Hughes and T. P. A. Hase, Measurements and their Uncertainties, Oxford University Press, Oxford, 2010 Search PubMed.
- J. S. Higgins and H. C. Benoit, Polymers and Neutron Scattering, Oxford University Press, Oxford, 1996 Search PubMed.
- M. Sferrazza, C. Xiao, R. A. L. Jones, D. G. Bucknall, J. Webster and J. Penfold, Phys. Rev. Lett., 1997, 78, 3693–3696 CrossRef CAS.
- P. E. Hopkinson, P. A. Staniec, A. J. Pearson, A. D. F. Dunbar, T. Wang, A. J. Ryan, R. A. L. Jones, D. G. Lidzey and A. M. Donald, Macromolecules, 2011, 44, 2908–2917 CrossRef CAS.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- S. C. Mannsfeld, M. L. Tang and Z. Bao, Adv. Mater., 2011, 23, 127–131 CrossRef CAS PubMed.
- P. J. Griffin, V. Bocharova, L. R. Middleton, R. J. Composto, N. Clarke, K. S. Schweizer and K. I. Winey, ACS Macro Lett., 2016, 5, 1141–1145 CrossRef CAS.
- J. T. Kalathi, U. Yamamoto, K. S. Schweizer, G. S. Grest and S. K. Kumar, Phys. Rev. Lett., 2014, 112, 108301 CrossRef PubMed.
- R. K. M. Bouwer, G.-J. A. H. Wetzelaer, P. W. M. Blom and J. C. Hummelen, J. Mater. Chem., 2012, 22, 15412 RSC.
- G. Bernardo, N. Deb, S. M. King and D. G. Bucknall, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 994–1001 CrossRef CAS.
- J. D. Perea, S. Langner, M. Salvador, J. Kontos, G. Jarvas, F. Winkler, F. Machui, A. Gorling, A. Dallos, T. Ameri and C. J. Brabec, J. Phys. Chem. B, 2016, 120, 4431–4438 CrossRef CAS PubMed.
- T. T. Ngo, D. N. Nguyen and V. T. Nguyen, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2012, 3, 045001 Search PubMed.
- J. Zhao, A. Swinnen, G. Van Assche, J. Manca, D. Vanderzande and B. Van Mele, J. Phys. Chem. B, 2009, 113, 1587–1591 CrossRef CAS PubMed.
- C. Müller, Chem. Mater., 2015, 27, 2740–2754 CrossRef.
- W. Ma, C. Yang and A. J. Heeger, Adv. Mater., 2007, 19, 1387–1390 CrossRef CAS.
- I. T. Sachs-Quintana, T. Heumüller, W. R. Mateker, D. E. Orozco, R. Cheacharoen, S. Sweetnam, C. J. Brabec and M. D. McGehee, Adv. Funct. Mater., 2014, 24, 3978–3985 CrossRef CAS.
- J. Bergqvist, C. Lindqvist, O. Bäcke, Z. Ma, Z. Tang, W. Tress, S. Gustafsson, E. Wang, E. Olsson, M. R. Andersson, O. Inganäs and C. Müller, J. Mater. Chem. A, 2014, 2, 6146–6152 RSC.
- T. Wang, A. J. Pearson, A. D. Dunbar, P. A. Staniec, D. C. Watters, D. Coles, H. Yi, A. Iraqi, D. G. Lidzey and R. A. Jones, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 129 CrossRef PubMed.
- J. L. Keddie, R. A. L. Jones and R. A. Cory, Europhys. Lett., 1994, 27, 59–64 CrossRef CAS.
- Z. Fakhraai and J. A. Forrest, Phys. Rev. Lett., 2005, 95, 025701 CrossRef PubMed.
- N. Shamim, Y. P. Koh, S. L. Simon and G. B. McKenna, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1462–1468 CrossRef.
- R. Inoue, K. Kawashima, K. Matsui, T. Kanaya, K. Nishida, G. Matsuba and M. Hino, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 021801 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm02337b |
| This journal is © The Royal Society of Chemistry 2020 |