DOI:
10.1039/C9SM02247C
(Paper)
Soft Matter, 2020,
16, 3536-3547
Numerical simulations of self-diffusiophoretic colloids at fluid interfaces
Received
12th November 2019
, Accepted 5th March 2020
First published on 9th March 2020
Abstract
The dynamics of active colloids is very sensitive to the presence of boundaries and interfaces which therefore can be used to control their motion. Here we analyze the dynamics of active colloids adsorbed at a fluid–fluid interface. By using a mesoscopic numerical approach which relies on an approximated numerical solution of the Navier–Stokes equation, we show that when adsorbed at a fluid interface, an active colloid experiences a net torque even in the absence of a viscosity contrast between the two adjacent fluids. In particular, we study the dependence of this torque on the contact angle of the colloid with the fluid–fluid interface and on its surface properties. We rationalize our results via an approximate approach which accounts for the appearance of a local friction coefficient. By providing insight into the dynamics of active colloids adsorbed at fluid interfaces, our results are relevant for two-dimensional self assembly and emulsion stabilization by means of active colloids.
I. Introduction
The dynamics of synthetic or biological, self-propelled objects is strongly affected by the presence of boundaries and interfaces.1–5 For example, sperm cells have been observed to accumulate at solid walls6 and bacteria swim in circles when close to substrates7 or fluid interfaces.8 Moreover, synthetic swimmers have been shown to be sensitive to both solid boundaries9–17 and liquid interfaces.18–24 Concerning synthetic swimmers, the presence of boundaries and interfaces is particularly relevant for self-phoretic colloids. Since these active colloids attain their net displacement by generating local gradients of intensive thermodynamic quantities, such as temperature or the (electro)chemical potential,25–28 their active displacement is sensitive to barriers and interfaces, which affect the profile of the local temperature and the (electro)chemical potential gradients.
In this contribution we analyze the dynamics of self-diffusiophoretic colloids (i.e., particles inducing local gradients in the chemical potentials of certain suspended species) which are adsorbed at a fluid–fluid interface. The fluid flow induced by the local stresses is caused by the chemical reaction on the surface of the particle and it will be affected by the presence of two, phase separated fluid phases. The description of such a system via the standard coarse-grained approach, within which the relative velocity between the particle and the fluid is accounted for by the so-called phoretic slip velocity on the surface of the particle,9,25–28 might be insufficient. Indeed, the phoretic slip velocity has been invoked in those cases in which the imbalance in the local chemical potential is confined to a thin shell around the particle where the Stokes equation is solved analytically.25 This approach becomes more complicated if the active colloid is adsorbed at a fluid interface in the presence of a three-phase contact line. In this context, we present a novel approach, based on numerical simulations, in which the motion of the self-diffusiophoretic colloid is obtained by using the lattice Boltzmann method in order to construct approximate solutions of the Navier–Stokes equation directly. In our scheme this hydrodynamics solver is combined with an advection and diffusion equation for the reactants. Such an approach allows us to discuss the reliability of the slip-velocity approach by comparison with previous approximate analytical results.19 In particular, in the present study we focus on the case in which the two fluids have the same viscosity, for which the approximate analytical model19 predicts the absence of any torque on the particle. Interestingly, our results show that, even in this case, self-phoretic colloids trapped at fluid interfaces reorient their symmetry axis. This reorientation occurs whenever the axis of symmetry of the particle is not perpendicular or parallel to the interface. Indeed, for these cases the presence of the interface affects the velocity profile and leads to net torques on the particle.
The presentation of our study is organized as follows. In Section II we present the diffusiophoretic model under study. In Section III we describe our numerical method based on lattice Boltzmann simulations. In Section IV we report our results for the dynamics of self-diffusiophoretic colloids trapped at fluid interfaces, and in Section V we summarize our main findings.
II. Model
We describe the active colloid via the diffusiophoretic model.25–28 We assume that a chemical reaction occurs at the surface of the colloid with a rate which depends on the local chemical properties of the surface. Accordingly, in the regime of small Péclet numbers, the mass density ρrp of the reaction product† obeys the following differential equation:| |  | (1) |
where W is the interaction potential between the reaction product and the colloid, D is the diffusion coefficient of the reaction product, and β−1 = kBT is the thermal energy. Eqn (1) is to be solved with the boundary conditions| | | J·n||r−rc|=R = αRρrξ(|r − rc = R|) | (2) |
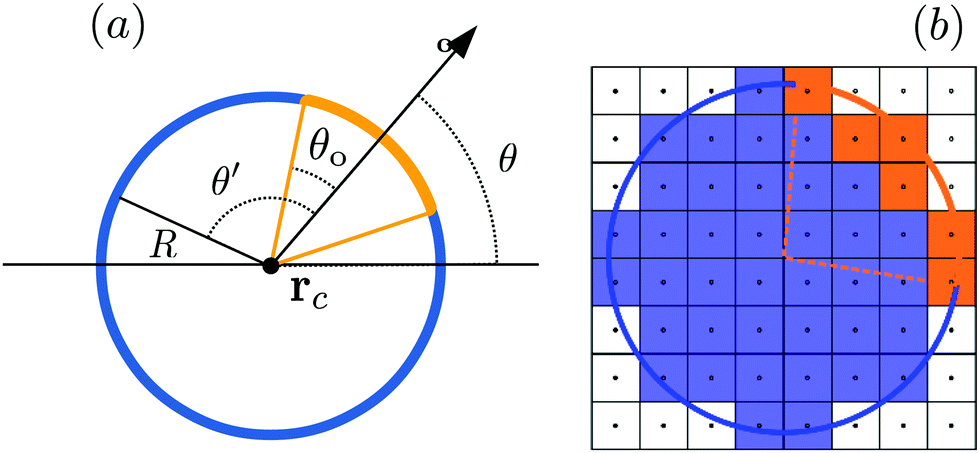
andwhere |r − rc| is the distance of a fluid cell from the center of mass of the colloid, located at rc and characterized by its radius R, α is the reaction rate,‡ξ is dimensionless and identifies the portion of the surface of the active colloid which is catalyzing the chemical reaction, and ρr is the mass density of the reactant (which we assume to be spatially homogeneous).
The spatial variation of the mass density of the reaction product along the surface of the colloid induces a net motion of the fluid which, in the low Reynolds number regime, is governed by the Stokes equation
with
| |  | (5) |
where
Ptot is the local pressure,
mrp is the mass of the reaction product molecule and

is determined either by imposing fluid incompressibility (∇·
v = 0) or by the equation of state for the fluid (as in our numerical scheme, see below). The boundary conditions for the fluid velocity are
| | | v(r − rc) = U + Ω × (r − rc), for |r − rc| = R | (6) |
and
where
U(
Ω) is the linear (angular) velocity of the colloid, and
v(
zI)·
n is the component of the fluid velocity perpendicular to the interface located at
zI.
§
III. Numerical methods
Our system is composed of two phase separated fluids (e.g., oil and water) acting as solvents, the reactants, and the products of the chemical reaction (such as the decomposition of hydrogen peroxide into water and oxygen), and the colloid (see Fig. 1). In order to determine the dynamics of the system, we put forward diverse numerical approaches for describing each of these components.
 |
| | Fig. 1 Illustration of the system of a self-diffusiophoretic colloid (sphere) at a fluid–fluid interface (red, solid line) between two fluids with dynamic viscosities η1 and η2 and reactant mass densities ρ1 and ρ2. The blue line represents the inert region of the colloid surface, while the orange, solid line represents the catalytic region with opening angle θo. The colloid is partially wetted by both fluids and its position with respect to the interface is captured by the contact angle θc. The orientation of the colloid with respect to the interface is characterized by the angle θ between the axis o of the colloid and a line parallel to the interface (black, solid line). Here the orientation of the particle corresponds to θo > 0. | |
A. The lattice Boltzmann method
In order to solve the dynamics of the fluids we use the lattice Boltzmann method (LBM) as it is implemented in the LB3D package.29,30 Within the LBM the fluid phases are described by their discretized single particle distribution functions fσi(r,t), which give the probability of finding a fluid particle of component σ at position r with velocity ci. Our systems consists of two species, such as σ1 = oil and σ2 = water, forming two fully separated phases. Here, we use a so-called D3Q19 lattice with 19 discrete velocities ci, i = 1,…,19, in three dimensions. We measure times in units of the integration time step Δt and lengths in units of the lattice spacing Δx. These microscopic auxiliary quantities have no physical meaning and their values are chosen to be smaller than any other physically relevant length or time scale. Eventually, it turns out to be convenient to fix the magnitudes of Δt and Δx to unity and to measure times and lengths in units of Δt and Δx, respectively. Accordingly, the actual dimensional values of Δt and Δx in actual units follow from the smallest length and time scale with physical meaning.¶ The particle distribution functions fσi(r,t) evolve in time due to advection from the neighboring lattice sites and due to collisions among particles at the same lattice site. In the following we use the so-called Bhatnagar–Gross–Krook (BGK) collision operator.31 After some algebra involving the discretization of space, time, and velocities32 the time evolution of the distribution functions fσi(r,t) follows as| |  | (9) |
where the rhs of eqn (10) is the BGK collision operator, τσ is the relaxation time of the fluid component σ, and fσ,eqi(r,t) is the local equilibrium distribution function which, in the small Mach number limit, is given by33| | 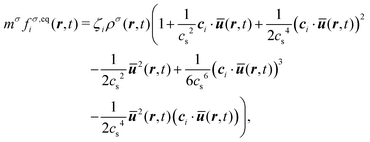 | (10) |
where ζi are the lattice weights29,30 and mσ is the mass of species σ. The relaxation time is related to the kinematic viscosity of the fluid as  , where
, where  is the speed of sound in the phase dominated by species σ. Our numerical approach, in the present form, requires all species to have the same mass mσ = m, and hence the same speed of sound cσs = cs. In the following we choose to fix the lattice time step Δt and the lattice spacing Δx to unity. In these units, it is common to choose
is the speed of sound in the phase dominated by species σ. Our numerical approach, in the present form, requires all species to have the same mass mσ = m, and hence the same speed of sound cσs = cs. In the following we choose to fix the lattice time step Δt and the lattice spacing Δx to unity. In these units, it is common to choose  as the lattice speed of sound in terms of the time step Δt and the lattice spacing Δx.32 Once the distribution functions fσ,eqi(r,t) are known, it is possible to compute the local mass density of the fluid:
as the lattice speed of sound in terms of the time step Δt and the lattice spacing Δx.32 Once the distribution functions fσ,eqi(r,t) are known, it is possible to compute the local mass density of the fluid:| |  | (11) |
where mσ is the mass of a single particle of species σ. The barycentric velocity of the fluid mixture is| |  | (12) |
Finally, the velocities of the individual fluid components are given by| |  | (13) |
In order to account for multiple solvent phases we follow the method introduced by Shan and Chen.33 Within this method the interaction force density acting among distinct species has the form| |  | (14) |
where gσσ′ denotes the interaction strength between the components σ and σ′; ψσ is a dimensionless pseudo-potential, which is a functional of the mass density. Here, the functional form of ψσ is chosen as||| | 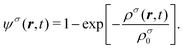 | (15) |
where ρσ0 is a reference density which is related to the bulk properties of the phase dominated by species σ. The force density in eqn (14) is applied to the fluid by adding a shift to ū:| |  | (16) |
Accordingly, in the expression for the equilibrium distribution functions fσ,eqi(r,t), ū is replaced by ū′ (see eqn (16)). Values of the interaction strengths gσσ′ between distinct species, i.e., σ ≠ σ′, which exceed a threshold, eventually lead to their separation, whereas values of gσσ′ for σ ≠ σ′ exceeding the threshold give rise to the separation of the liquid and the vapor phases of a certain species.33 In the following we take gσσ′ = 0.1 and gσσ = 0 which leads to an interface with a thickness of ca. 5 lattice units and to a surface tension of the order of 0.1 in lattice units. We note that this value of the surface tension is sufficient to keep the colloid trapped at the interface. In fact, the typical velocity, in lattice units, which the active colloid attains in the present numerical study is v ≈ 10−3 and hence the drag force Fdrag = 6πηRv is much smaller (taking  ) than the force induced by the surface tension Fsurf ≈ 2πRγ where γ = 0.1 is the magnitude of the surface tension.
) than the force induced by the surface tension Fsurf ≈ 2πRγ where γ = 0.1 is the magnitude of the surface tension.
In order to study the behavior of a colloid suspended at a fluid–fluid interface, a scheme is needed for treating objects, which are large compared with the particles forming the fluids. The separation of length scales between the mesoscopic colloidal size and the molecular size of the fluid particles allows one to keep the coarse-grained description for the fluid (via LBM) while simultaneously treating the colloid as a spherical object characterized completely by its size, position, orientation, and its linear and angular velocities. The interaction between the colloid and the fluid gives rise to forces and torques acting on both the colloid and the fluid. The technical details of the implementation of the coupling between the colloid and the fluid within LBM are discussed in ref. 34–38. In the following we shall outline only the basic features of this method.
The colloid occupies those lattice cells which are inside the spherical colloid (see Fig. 2). As the colloid moves, the configuration of lattice cells occupied by the colloid is updated. Solid impenetrability is accounted for by bouncing back those contributions to the fluid flow which attempt to invade the solid boundaries.37,39 This is implemented by updating the distribution function after the streaming step according to
| | | fσi(r,t + Δt) = fσi(r − ciΔt,t) | (17) |
for all
i, if the lattice site
r −
ciΔ
t is occupied by fluid particles. In contrast, if
r −
ciΔ
t is occupied by the colloid, the fluid particles are bounced back,
i.e., their velocity is flipped:
| |  | (18) |
where
i′ is defined as the index corresponding to
ci′ = −
ci. This procedure leads to a no-slip boundary condition at the surface of the colloid and to a momentum transfer between the fluid species
σ and the colloid, which induces a local force density
| |  | (19) |
and a torque density
| |  | (20) |
where
r(
t) is the vector pointing from the center of the colloid to the site where bounce-back occurs. In order to be consistent the above mentioned bounce-back rule (see
eqn (18)) has to be modified by accounting for the motion of the colloid.
37,39 In order to do so, a correction is added to the fluid distribution functions:
37,39| |  | (21) |
where
usurf is the local velocity at the surface of the colloid,
r is the position of the fluid lattice site. (Note that
eqn (21) holds explicitly in the limit Δ
t → 0 due to
eqn (18).) Consequently, the force density acting on the colloid is modified, too:
| |  | (22) |
Finally, when the colloid moves it occupies pristine cells at its front and it releases cells at its back. In the case of newly occupied cells, the fluid located therein is deleted and its momentum is transferred to the colloid by adding
| |  | (23) |
to the rhs of
eqn (22). In the case of cells being
released by the colloid, fresh fluid is created at the corresponding sites with velocity
usurf(
r,
t) and density
![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) σ
σ, where
σ is obtained by averaging the fluid composition in the direct neighborhood:
37| |  | (24) |
where the sum
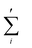
is restricted to those values of
i for which
r +
ciΔ
t is a fluid site;
N is the number of these sites. In addition, in order to account for the unknown exact density profile in the close vicinity of a colloid, we apply a density correction algorithm as explained in
ref. 37 and 38. In order to conserve momentum, a contribution is added to the force density (
eqn (22)):
| | 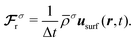 | (25) |
 |
| | Fig. 2 The solid sphere (a) represents the idealized colloid, while the cubic cells represent the lattice Boltzmann cells (b). Fluid cells are white, colloid cells are blue, and catalytically active cells are orange. The center of the cells is indicated by a black dot. | |
In order to avoid artifacts during the computation of the Shan–Chen forces acting on the colloid and to tune the wetting properties of the colloid, the outermost layer of lattice sites occupied by the colloid is filled with a virtual fluid which is only used during the computation of the Shan–Chen forces acting in the direct vicinity of the colloid. Its density is obtained similarly to eqn (24), but can be tuned by adding an offset Δρ to eqn (24), which controls the wettability of the colloid. Finally, as usual the Shan–Chen forces are used to compute the force acting on the colloid:
| |  | (26) |
Accordingly, by tuning the magnitude of Δ
ρ, it is possible to control the contact angle of the colloid.
37,38
B. Dynamics of the reactant and the reaction product
We assume that the two fluids forming the adjacent phases act as reservoirs of the reactant and of the reaction product such that the mass density ρr of the reactant is regarded as to be homogeneous in the bulk of both fluid phases. Close to the interface the mass density of the two fluid phases varies. In the following we assume that the ratios| |  | (27) |
of the number densities frσ of the reactant suspended in the fluid consisting of species σ, and those of the fluid phases,| |  | (28) |
are kept constant throughout both fluid phases. Accordingly, the mass density of reactants at a given position is defined as:| |  | (29) |
where the sum runs over all fluid species σ and mr is the mass of a reactant molecule. Eqn (29) implies that ρr(r) = κmrfr(r) where κ is the number of fluid species. Concerning the interaction between the reactant molecules and the colloid, we assume that the surface of the colloid is covered by a catalyst with axial symmetry (see Fig. 2(a)). The strength of the chemical reaction is controlled by the surface activity ξ, which determines the rate at which reactant molecules are converted into solute molecules.
A simple choice of the surface activity ξ is given by
| |  | (30) |
The prefactor
ξ0 is the base activity,
θ′ is the azimuthal angle (see
Fig. 2),
θo is the opening angle which defines the size of the catalytic cap (see
Fig. 2(a)), and
Δ is an interpolation length. This corresponds to full activity for angles well within
θo, zero activity outside, and a linear interpolation in between. Without this linear interpolation (
i.e., for
Δ = 0), small rotations of the colloid may not change the activity, due to the roughness of the colloid surface.
** Optimally, the value of
Δ should correspond to
ca. one lattice cell, which is accomplished if
Δ ≈ Δ
x/
R, where
R is the radius of the colloid. This is the quantity which we have used in our simulations.
In the following we specialize on the case of a single reaction product (our approach can be easily extended to an arbitrary number of them) which is equally soluble in both fluid phases and which diffuses in both fluid phases with equal diffusion coefficient. Moreover we focus on the small Péclet limit, in which the advection contribution to the time evolution of the density of the reaction product is negligibly small as compared with the diffusion contribution. Accordingly, the dynamics of the mass density of the reaction product is decoupled from that of the two fluid phases. The former appears as an additional scalar field defined on the LBM lattice, such that a cell at lattice position r contains a mass density ρrp(r):
| |  | (31) |
where, as for the reactant (see
eqn (29)), we have introduced the number density ratios

and we have accounted for their spatial variation. The dynamics of the reaction product is governed by
eqn (1) and (2). However, in numerical simulations implementing boundary conditions at
r → ∞ is challenging. In order to ensure that
ρrp(
r) reaches a steady state within the simulation box with periodic boundary conditions along the three spatial directions, we introduce a homogeneous sink term with constant decay rate
††χ:
| |  | (32) |
with the boundary condition (identical to
eqn (2))
| | | J·n||r−rc|=R = αRρrξ(|r − rc| = R), | (33) |
where we have introduced the sink term
χ and |
r −
rc| is the distance of a fluid cell from the center of mass of the colloid, located at
rc (see
Fig. 2). The interaction potential between the colloid and the reaction product is chosen as
| | 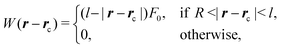 | (34) |
where
F0 and
l are the strength and the range of the potential, respectively. In the following we assume
l = 4 lattice units and
βF0l = 5 × 10
−5(Δ
x)
3 where
β is the inverse thermal energy. This potential has no angular dependence,
i.e., it is the same for the catalytic and the inert side of the colloid surface.
Eqn (32) is solved
via a finite-difference scheme on the same grid as the one used by the LBM. Finally, the potential
W, together with the non-equilibrium mass density profile
ρrp of the reaction product, induces a pressure gradient on the fluid phases as
25| |  | (35) |
Eqn (35) is applied as an external body force acting on the fluid nodes of the LBM fluid species. In the discrete difference scheme, the local flow
J of the reaction product, produced by the chemical reaction at the surface of the colloid, into a fluid cell at
r per time is given by
where
α is the reaction rate,
n is the local normal at the colloid surface pointing towards the fluid phase,
ρr is the reactant mass density, and
Ξ(
r) denotes the activity of the colloid cells neighboring the fluid cell at
r:
| |  | (37) |
where the sum

is performed only over neighboring cells inside the colloid (see
Fig. 2). This recipe prevents fluid cells in contact with more than one catalyst cell to be exposed to a larger flux, induced by the discretization of the shape of the colloid.
Eqn (36) and (37) enforce a constant flux per area even in the discrete representation of the colloid.
C. Dynamics of the colloid
The dynamics of the colloid is governed by the equation of motion| |  | (38) |
where m is the mass of the colloid and  is given by eqn (26). The last term in eqn (38) stems from the interaction of the colloid with the reaction product. We emphasize that the force applied to the colloid (eqn (38)) equals in magnitude the force applied on the fluid phases and on the solute such that the total force on the system is zero. This equation is integrated numerically via a leap-frog scheme.40,41
is given by eqn (26). The last term in eqn (38) stems from the interaction of the colloid with the reaction product. We emphasize that the force applied to the colloid (eqn (38)) equals in magnitude the force applied on the fluid phases and on the solute such that the total force on the system is zero. This equation is integrated numerically via a leap-frog scheme.40,41
IV. Results
After validating our numerical scheme against analytical predictions for the velocity of a self-diffusiophoretic colloid in a homogeneous fluid42 (see Appendix A), we study the dynamics of a self-diffusiophoretic colloid adsorbed at a fluid interface. We perform numerical simulations with a colloid of radius R = 10Δx. This implies that the particle size corresponds to roughly four times the interface width. Obviously, this is expected to be insufficient in order to separate the length scales, and the particles should be bona fide nanoparticles. However, we found in numerous previous studies, using the same algorithm, that the interface width has a surprisingly small influence on the particle dynamics (see e.g., ref. 37 and 40).
All simulations are initialized by equilibrating the interface after the colloid has adsorbed; then the chemical reaction at the surface of the colloid is turned on. In the following, we analyze the dynamics of the colloid in the case that the viscosities of the two fluids are equal, η1 = η2 = η, and for various values of the opening angle θo of the catalytic cap and of the contact angle θc (see Fig. 1), which characterizes the adsorption of the colloid at the interface. Concerning the mass density of the reactant we consider two situations. In the first one, both fluids have the same mass fraction of reactant molecules: i.e., their mass fractions are assumed to be equal
| |  | (39) |
In the second case one has
| |  | (40) |
so that one fluid does not contain any reactant molecules. Finally, we study two different scenarios: one, in which the colloid can move freely along the interface, and another one, in which the lateral colloid position is fixed by an external force.
A. Equal reactant mass fractions
First, we consider the case in which the reactants are suspended with equal mass fraction in both fluid phases (eqn (39)). In this case certain symmetries can be identified, depending on the relative orientation θ of the axis of the colloid with respect to the plane of the interface: (i) the fore-aft symmetry for θ = ±π/2, i.e., if the axis of the colloid is parallel to the normal of the interface. (ii) For c2 = c1 ≠ 0 the velocity and the density profile of the reaction product acquire an additional mirror symmetry about θ = 0, i.e., if the axis of the colloid lies within the plane of the interface.
As a first case, we study the dynamics of a colloid which is partially covered by catalyst, i.e., here θo = π/4, and has a contact angle θc = π/2. Fig. 3 shows the orientation θ(t) of the colloid as a function of time for a free colloid (Fig. 3(a)) as well as for a colloid the center of mass of which is kept at a fixed position by an external force (Fig. 3(b)). Interestingly, the dynamics in the two setups are quite similar, in that the catalytic reaction induces a net torque on the colloid which leads θ to approach a steady state with θ(t = ∞) ≡ θ∞ = 0. We note that, while for the moving colloid θ∞ = 0 is the only steady state, for the fixed colloid θ∞ = π/2 is also a steady state. This difference is due to the fact that if the colloid is not fixed, its center of mass will move also along the direction normal to the interface. Hence, the distinction between the two cases emphasizes that θ∞ = π/2 is not stable with respect to fluctuations of the position of the center of mass of the colloid.
 |
| | Fig. 3 Angle θ of the orientation of the colloid with respect to the interface (see Fig. 1) as a function of time, normalized by the time t0 it takes for a half-covered colloid to move a distance which equals its own radius, t0 = R/v0, for the case of a free colloid (panel (a)) and for the case of a fixed colloid (panel (b)). The velocity v0 is the velocity the very same colloid attains in a homogeneous fluid characterized by the same viscosity (see Appendix A). The lines correspond to various initial orientations θ(t = 0) of the colloid [θ(t = 0) = 0, π/8, π/4, 3π/8, π/2] for θo = π/4. In the case of the free colloid, the trajectories have been smoothed by integrating over a shifting time-window 104Δt wide. The lines are symmetric with respect to θ = 0. | |
Next, we study the dependence of the dynamics of the colloid on the areal size of the catalytic cap, characterized by θo, and on the contact angle θc. Fig. 4 shows the stable steady state orientations θ∞ as a function of the opening angle θo for various contact angles of a free colloid (Fig. 4(a)) and of a colloid the center of mass of which is kept fixed (Fig. 4(b)). Interestingly, both panels of Fig. 4 show that for opening angles θo ≠ π/2 the steady state orientation of the colloid is θ∞ ≠ π/2, i.e., the colloid attains a steady translation along the interface, because the driving force points into the axial direction of the colloid and thus provides a lateral component. The normal component of the velocity is compensated by the force induced by the deformation of the interface. This ensures a steady distance of the center of mass of the colloid from the interface. In particular, θ∞ grows upon increasing θo for both θc = 0.55π (red upward triangles) and θc = 0.6π (green downward triangles). Finally, for θo = π/2, the stable steady state is θ∞ = ±π/2 for all contact angles θc which leads to a vanishing velocity along the interface. We note that the case of contact angle θc = π/2 (blue circles) is peculiar because in this case the steady state orientation is θ∞ = 0 which implies maximum velocity along the interface. This result holds for all opening angles θo < π/2. For θo = π/2 with θc = π/2 we face numerical difficulties due to the symmetry of the problem. In fact, for θc = π/2 the center of mass of the colloid lies at the interface and if the colloid is half-covered (θo = π/2) the only non-motile state requires θ = π/2. However, when θ → π/2, the typical size of the active site being wetted by one of the two fluid phases becomes comparable to the lattice constant and, for θc = π/2 and θo = π/2, our discrete numerical approach would require larger ratios of the colloid size and the lattice constant which, however, we could not explore.
 |
| | Fig. 4 Steady state angle θ(t = ∞) ≡ θ∞ as a function of the opening angle θo. Blue circles, red upward triangles, and green downward triangles correspond to θc = π/2, 0.55π, and 0.6π, respectively. Panels (a) and (b) correspond to a free and a fixed colloid, respectively. The data are symmetric with respect to θ∞ = 0 (not shown). | |
In the case in which the colloid can move freely, at steady state its lateral velocity along the interface depends on both the contact angle and the opening angle. Fig. 5 shows that for θc = π/2 the maximum speed is obtained for θo = π/2 and it equals the one (v0) obtained in a homogeneous fluid. This tells that, under the condition of equal viscosity among the two fluid phases, due to the symmetry of the problem, the interface does not affect the fluid flow and hence the motion of the colloid. In contrast, Fig. 5 shows that, for θc ≠ π/2, v has a non-monotonous dependence on the opening angle. Indeed, v vanishes for θo = 0 (i.e., for a passive colloid) and for θo = π/2, and it attains a maximum for θo ≃ π/4. This non-monotonous dependence of v on θo can be understood by relating the data in Fig. 5 to those shown in Fig. 4(a). Indeed, for θo → 0 at steady state the axis of the colloid lies in the plane of the interface (i.e., θ(t = ∞) → 0), whereas upon increasing θo the steady state orientation, θ∞, also grows and so does the steady state velocity. For even larger values of θo the value of θ∞ increases until, for θo → π/2, one has θ∞ → π/2 which implies v → 0.
 |
| | Fig. 5 Lateral velocity v along the interface, normalized by the velocity v0 = v(θo = π/2) (v0 = 0.00175 in LBM units), of a free Janus colloid (θo = π/2) in a homogeneous fluid as a function of the opening angle θo for various contact angles: blue circles, red upward triangles, and green downward triangles correspond to θc = π/2, 0.55π, and 0.6π, respectively. The blue line represents the velocity of a free Janus colloid in a homogeneous bulk fluid as function of θo. | |
The rotation of the symmetry axis of the colloid we have just described is rather counter-intuitive, in particular for θc = π/2 and η1 = η2. Indeed, the equality of the viscosity of the two fluids and the lack of accumulation of the reaction product at the interface enforce the symmetry of the mass density profile of the reaction product about the symmetry axis of the colloid and therefore of the local pressure gradients acting on the fluid. Hence, at first glance, in this scenario one might expect no net torque. Actually, for this case a simplified analytical model,19 which disregards the fluid flow in the vicinity of the three-phase contact line, predicts that there is no reorientation of the colloid at all. In the following, we show that the observed torque is due to the fact that the mobility is not homogeneous along the surface of the colloid. Indeed, the boundary conditions imposed on the fluid velocity by the presence of the interface lead to an effective local mobility the spatial variation of which generates the torque. In order to highlight this relationship, we determine the net velocity, which a passive colloid attains if it is pushed by an external local force density fr0(r), localized at r0 near the colloid surface and acting solely on the fluid and being directed towards the center of the colloid:
| |  | (41) |
where
δ(
r −
r0) is the Dirac delta function,
F is a force, and
rc is the position of the center of mass of the colloid. In these corresponding simulations, the colloid attains a steady state in which it rotates with constant angular velocity
ω. We are interested only in its component
ωz(
t = ∞) =
ez·
ω(
t = ∞) along the
z-direction.
‡‡ We introduce the dimensionless inverse hydrodynamic mobility coefficient
| |  | (42) |
as the ratio of the angular velocity and the magnitude of the applied force,
§§ where
R is the radius of the colloid,
γ0 = 6π
ηR is the friction coefficient of the colloid;
γ(
r0) depends on
r0via ω(
t = ∞).
In Fig. 6, the dimensionless inverse mobility coefficient is shown as a function of the x and y positions in a plane perpendicular to the interface (black horizontal line in Fig. 6) and passing through the colloid center of mass. This highlights the point symmetry of the dimensionless inverse mobility and that forces closer to the interface give rise to larger torques and hence higher values of the steady angular velocity ωz. In the small Reynolds number regime, which is valid for the motion of the present diffusiophoretic colloid, the Navier–Stokes equation reduces to the (linear) Stokes equation. Hence, the local mobility allows us to determine the torque on the colloid in z-direction for any given force density f(r), as long as the force acts on the colloid along the radial direction. For the case of a self-diffusiophoretic colloid, the force density distribution, due to phoresis, is given by  (see eqn (35)) where here r indicates the distance from the center of mass of the colloid. Accordingly, the local mobility allows us to determine the torque τz = τ·ez exerted on the colloid by the density profile of the reaction product:
(see eqn (35)) where here r indicates the distance from the center of mass of the colloid. Accordingly, the local mobility allows us to determine the torque τz = τ·ez exerted on the colloid by the density profile of the reaction product:
| |  | (43) |
where
B is a dimensional fitting parameter, and
L is the size of the simulation box. Actually,
eqn (43) approximates the total torque in that we are considering solely the contributions to the torque stemming from the plane perpendicular to the interface and passing through the center of mass of the colloid (see
Fig. 6). Contributions stemming from the rest of the surface of the colloid are accounted for by the fitting parameter
B. We emphasize that the same value of the parameter
B,
i.e.,
B = 5.2Δ
x, fits well the data for colloids with various opening angles
θo as shown in
Fig. 7, 10, 11 and 12. The validity of this approach will be discussed
a posteriori,
i.e., by comparing it with the values extracted from the corresponding lattice Boltzmann simulations.
 |
| | Fig. 6 Inverse mobility coefficient (red/blue color) γ (eqn (42)) as a function of the position in the x–y plane through the colloid center (black dot) for a colloid characterized by θc = π/2. The interface lies in the x–z plane located at y = 0 (black horizontal line). The blue color tells that a force at this position leads to a negative angular velocity ωz < 0, whereas a force in cells with red color leads to a positive angular velocity ωz > 0. The inverse mobility coefficient is only shown for positions which are at most l = 4 lattice units away from the colloid (R < r < R + l), where l is the range of the interaction potential (see eqn (34)). The blue circle represents the colloid. | |
 |
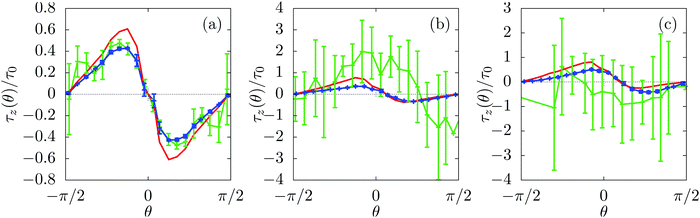
| | Fig. 7 Component τz in the z-direction of the torque on the colloid as a function of the rotation θ. Blue (green) solid lines correspond to the LBM measured torque for a fixed (free) colloid; the error bars indicate the standard error. (Note that for certain values of θ the blue, green, and red lines totally overlap and thus are not visible.) Red solid lines correspond to the torque calculated viaeqn (43) for θo = π/4 with (a) θc = 0.5π, (b) θc ≈ 0.55π, and (c) θc ≈ 0.6π. The torque components are normalized by τ0 ≡ τ(θ = −π/4, θc = π/2) as obtained from eqn (43) with θc = π/2 (τ0 = 0.04 in LBM units). The error bars are due to the discretization of the colloid surface (for both fixed and mobile colloids) and due to the motion normal to the interface (mobile colloid, green lines). | |
Fig. 7 provides a comparison of the torque calculated viaeqn (43) with the torque obtained directly from the lattice Boltzmann simulations for a colloid with opening angle θo = π/4 and for three contact angles. (For a discussion of these kind of results for various opening angles see Appendix B.) Interestingly, Fig. 7(a) shows that for a contact angle θc = π/2 the prediction of eqn (43) agrees very well with the torque obtained from the simulations for both a free and a fixed colloid. As expected, Fig. 7(a) shows that the torque on the colloid is zero for the steady states characterized by 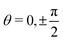 .
.
For contact angles θc ≠ π/2 (see Fig. 7(b) and (c)), the torques on the free and on the fixed colloid are no longer equal. This is expected, because for these contact angles the lateral motion along the interface leads to an additional contribution to the torque.43 This explains the mismatch between the prediction of eqn (43) and the torque of the free colloid obtained from the simulations shown in Fig. 7(b) and (c). However, this additional torque is absent for the fixed colloid. This explains the good agreement between the prediction of eqn (43) and the torque on the fixed colloid calculated from the simulations shown in Fig. 7(b) and (c).
1. Inhomogeneous reactant densities.
Up to now, we have studied the case in which both fluid phases contain reactant molecules. Here, we consider the system in which one of the two fluids does not contain the reactant (eqn (40)), yet the product of the chemical reaction can diffuse, with equal diffusivity, in both fluid phases. Clearly, in this case the point symmetry of τz(θ) at θ = 0 is broken even for the contact angle θc = π/2, whereas its mirror-symmetry at θc = ±π/2 remains.
Since we have not observed major discrepancies between the case of a moving colloid and that of a colloid fixed in space, in the following we focus on the case of a fixed colloid, because it is computationally easier. If the catalytic cap is fully immersed in that fluid phase which contains the reactant, the dynamics of the colloid is identical to that observed if the reactant is dissolved in both fluid phases. From geometric considerations (see Fig. 1), it follows that the catalytic cap is exposed to the fluid without reactant if θ ≥ θ1 with
| | | θ1 = (θc − π/2) − θo, | (44) |
and it is fully submerged in the fluid without reactants for
θ ≥
θ2 with
| | | θ2 = (θc − π/2) + θo. | (45) |
Accordingly, as long as
θ <
θ1 the catalytic cap does not get into contact with the fluid without reactant and hence the colloid will behave exactly as in the case in which both fluids contain the reactant. If
θ >
θ1, a large gradient of the solute concentration occurs close to the interface due to the lack of reaction in the other fluid phase. This drives the rotation of the colloid further towards the fluid without reactant, until the catalytic cap becomes fully submerged in this fluid,
i.e.,
θ ≥
θ2. At this point the system becomes “passive” because there is no longer production of solute and thus the colloid does not move anymore.
Fig. 8 shows the steady states θ∞ of the colloid as a function of the opening angle. We remark that Fig. 8 does not show all stable steady states with θ > θ2, i.e., when the system becomes passive due to the lack of chemical reactions. Interestingly, the steady states shown in Fig. 8 are in good agreement with our simple geometrical estimate (eqn (45)) except for a constant offset. We speculate that this offset is a numerical artifact because a small concentration of reactant molecules can occur in the interface region. (We recall that in the lattice Boltzmann scheme the fluid–fluid interface has a finite thickness, which in the present simulations is ca. 5 lattice constants.) In particular, within the Shan–Chen model the density of the fluid, which contains the reactant, is not zero everywhere because the two fluids do not completely demix. Therefore, when the colloid protrudes far into the fluid without reactant, there is a torque rotating it back towards the fluid with reactant, in line with the results from Section IV A. Finally, we note that for a Janus colloid, i.e., θo = π/2, and with θc > π/2 the steady state θ∞ = π/2 is independent of the contact angle, because the Janus colloid is always in contact with the fluid with reactant.
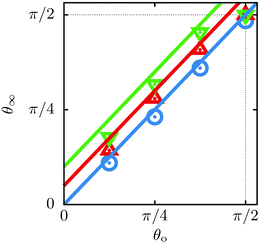 |
| | Fig. 8 Steady state angle θ∞ as a function of the opening angle θo. Symbols are simulation data; blue circles: θc = π/2; red upward triangles: θc ≈ 0.55π; green downward triangles: θc ≈ 0.6π. The blue, red, and green lines show the threshold angle θ2 (eqn (45)) for the corresponding contact angle θc as a function of the opening angle θo. | |
V. Conclusions
We have presented a novel numerical approach which is capable of capturing the dynamics of self-phoretic colloids even in the presence of a fluid interface which affects the boundary condition of the fluid velocity close to the surface of the colloid. In particular, we have characterized the dynamics of a self-diffusiophoretic colloid adsorbed at a fluid–fluid interface for the case in which the colloid is let free to move as well as when the colloid center of mass is kept at a constant position by an external force. We have found that a rotation of the axis of symmetry of the colloid arises even for fluids with equal viscosity. In particular, we have found that the steady state orientation depends on both the opening angle θo of the catalytic cap and the contact angle θc.
In order to understand the origin of such a reorientation we have calculated the local mobility by applying locally external, constant forces on a passive colloid. We have extracted the local value of the mobility by taking the ratio between the applied force and the steady-state angular velocity. This local mobility has been used to determine the effective torque acting on the self-diffusiophoretic colloid. Interestingly, we have found that, for θc = π/2, the mobility matrix predicts quite well the effective torque on a free colloid as well as on a colloid the center of mass of which is kept spatially fixed. If θc ≠ π/2 we have found good agreement for the case of the fixed colloid, whereas an additional torque arises in the case of a mobile colloid. Finally, we have discussed the case in which only one fluid contains the reactant. In this case we have found that the stable steady-state orientation is the one with the cap immersed in the fluid without reactant so that the colloid becomes inactive.
Conflicts of interest
There are no conflicts to declare.
Appendix A: validation of the numerical scheme
In order to validate our numerical scheme we characterize the dynamics of a self-diffusiophoretic colloid in a homogeneous bulk fluid. In this case, after an initial transient, the colloid attains a steady state in which it moves with a constant velocity v = ezv. We compare the results of simulations performed for various values of the opening angle θo with analytic results.42
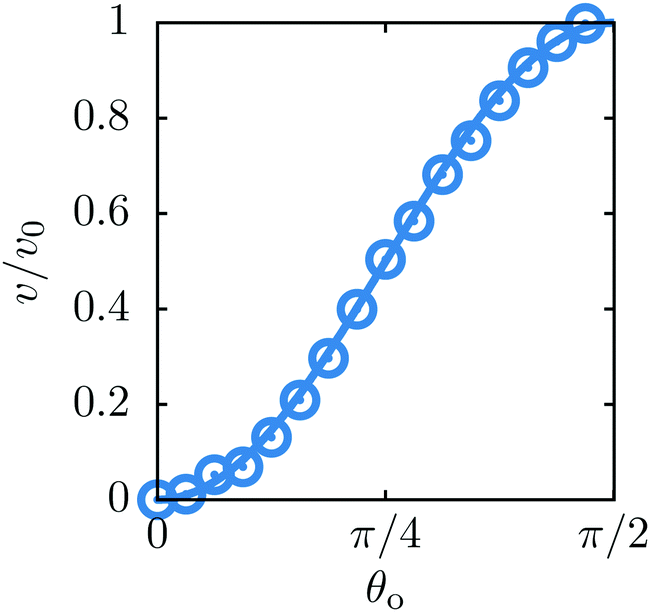
Fig. 9 shows the reduced steady state velocity of the colloid as a function of the opening angle. As shown in the figure, the agreement with the analytic results is very good.
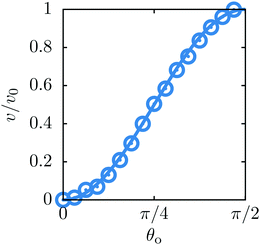 |
| | Fig. 9 The velocity v of a self-diffusiophoretic colloid as a function of the opening angle θo, normalized by v0 = v(θo = π/2) (see Fig. 5). The symbols are simulation data whereas the solid line provides the analytical prediction.42 | |
Appendix B: additional results
In Fig. 7, 10, 11 and 12 the z-component of the torques on self-diffusiophoretic colloids with opening angles θo = π/4, π/2, 3π/8, and π/8, respectively, are shown as function of the colloid rotation for three contact angles θc.
 |
| | Fig. 10 Same as in Fig. 7 for θo = π/2. | |
 |
| | Fig. 11 Same as in Fig. 7 for θo = 3π/8. | |
 |
| | Fig. 12 Same as in Fig. 7 for θo = π/8. | |
In Fig. 10–12 there is still qualitative agreement between the measured torque on the fixed colloid and the torque calculated viaeqn (43), although the quantitative agreement is weaker. As before, the torque on the free colloid and the torque on the fixed colloid agree very well for all opening angles if the contact angle is θc = π/2, because in this case the lateral motion does not induce a torque on the colloid.
Acknowledgements
Open Access funding provided by the Max Planck Society.
References
- S. J. Ebbens and J. R. Howse, In pursuit of propulsion at the nanoscale, Soft Matter, 2010, 6, 726 RSC.
- R. Kapral, Perspective: nanomotors without moving parts that propel themselves in solution, J. Chem. Phys., 2013, 138, 202901 CrossRef PubMed.
- E. Lauga and T. Powers, The hydrodynamics of swimming microorganisms, Rep. Prog. Phys., 2009, 72, 096601 CrossRef.
- J. Elgeti, R. G. Winkler and G. Gompper, Physics of microswimmers, Rep. Prog. Phys., 2015, 78, 056601 CrossRef CAS.
- C. Bechinger, R. Di Leonardo, H. Löwen, C. Reichhardt, G. Volpe and G. Volpe, Active particles in complex and crowded environments, Rev. Mod. Phys., 2016, 88, 045006 CrossRef.
- L. Rotschild, Non-random Distribution of Bull Spermatozoa in a Drop of Sperm Suspension, Nature, 1963, 198, 1221 CrossRef.
- P. D. Frymier, R. M. Ford, H. C. Berg and P. T. Cummings, Three-dimensional tracking of motile bacteria near a solid planar surface, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 6195 CrossRef CAS PubMed.
- R. Di Leonardo, D. Dell'Arciprete, L. Angelani and V. Iebba, Swimming with an Image, Phys. Rev. Lett., 2011, 106, 038101 CrossRef CAS PubMed.
- M. N. Popescu, S. Dietrich and G. Oshanin, Confinement effects on diffusiophoretic self-propellers, J. Chem. Phys., 2009, 130, 194702 CrossRef CAS PubMed.
- S. Das, A. Garg, A. I. Campbell, J. R. Howse, A. Sen and D. Velegol,
et al., Boundaries can steer active Janus spheres, Nat. Commun., 2015, 6, 8999 CrossRef CAS PubMed.
- W. E. Uspal, M. N. Popescu, S. Dietrich and M. Tasinkevych, Self-propulsion of a catalytically active particle near a planar wall: from reflection to sliding and hovering, Soft Matter, 2015, 11, 434 RSC.
- J. Simmchen, J. Katuri, W. E. Uspal, M. N. Popescu, M. Tasinkevych and S. Sánchez, Topographical pathways guide chemical microswimmers, Nat. Commun., 2016, 7, 10598 CrossRef.
- K. Schaar, A. Zöttl and H. Stark, Detention Times of Microswimmers Close to Surfaces: Influence of Hydrodynamic Interactions and Noise, Phys. Rev. Lett., 2015, 115, 038101 CrossRef.
- S. Spagnolie and E. Lauga, Hydrodynamics of self-propulsion near a boundary: predictions and accuracy of far-field approximations, J. Fluid Mech., 2012, 700, 105 CrossRef.
- D. Pimponi, M. Chinappi, P. Gualtieri and C. M. Casciola, Hydrodynamics of flagellated microswimmers near free-slip interfaces, J. Fluid Mech., 2016, 789, 514 CrossRef CAS.
- P. Malgaretti and H. Stark, Model microswimmers in channels with varying cross section, J. Chem. Phys., 2017, 146, 174901 CrossRef PubMed.
- A. Morin, D. Lopes Cardozo, V. Chikkadi and D. Bartolo, Diffusion, subdiffusion, and localization of active colloids in random post lattices, Phys. Rev. E, 2017, 96, 042611 CrossRef PubMed.
- X. Wang, M. In, C. Blanc, M. Nobili and A. Stocco, Enhanced active motion of Janus colloids at the water surface, Soft Matter, 2015, 11, 7376 RSC.
- P. Malgaretti, M. N. Popescu and S. Dietrich, Active colloids at fluid interfaces, Soft Matter, 2016, 12, 4007 RSC.
- A. Dominguez, P. Malgaretti, M. N. Popescu and S. Dietrich, Effective interaction between active colloids and fluid interfaces induced by Marangoni flows, Phys. Rev. Lett., 2016, 116, 078301 CrossRef PubMed.
- A. Dominguez, P. Malgaretti, M. N. Popescu and S. Dietrich, Collective dynamics of chemically active particles trapped at a fluid interface, Soft Matter, 2016, 12, 8398 RSC.
- J. Simmchen and P. Malgaretti, Active Particle Accumulation at Boundaries:
A Strategy to Measure Contact Angles, ChemNanoMat, 2017, 3, 790 CrossRef CAS.
- K. Dietrich, D. Renggli, M. Zanini, G. Volpe, I. Buttinoni and L. Isa, Two-dimensional nature of the active Brownian motion of catalytic microswimmers at solid and liquid interfaces, New J. Phys., 2017, 19, 065008 CrossRef.
- P. Malgaretti, M. N. Popescu and S. Dietrich, Self-diffusiophoresis induced by fluid interfaces, Soft Matter, 2018, 18, 1375 RSC.
- J. L. Anderson, Colloid Transport by Interfacial Forces, Annu. Rev. Fluid Mech., 1989, 21, 61 CrossRef.
- R. Golestanian, T. B. Liverpool and A. Ajdari, Propulsion of a Molecular Machine by Asymmetric Distribution of Reaction Products, Phys. Rev. Lett., 2005, 94, 220801 CrossRef PubMed.
- F. Jülicher and J. Prost, Generic theory of colloidal transport, Eur. Phys. J. E: Soft Matter Biol. Phys., 2009, 29, 27 CrossRef PubMed.
-
W. C. K. Poon, From Clarkia to Escherichia and Janus: The physics of natural and synthetic active colloids, in Proceedings of the International School of Physics “Enrico Fermi” Course CLXXXIV “Physics of Complex Colloid”, ed. C. Bechinger, F. Sciortino and P. Ziherl, SIF, Bologna, 2003, p. 317 Search PubMed.
- R. Benzi, S. Succi and M. Vergassola, The lattice Boltzmann equation: theory and applications, Phys. Rep., 1992, 222, 145 CrossRef.
- J. Harting, J. Chin, M. Venturoli and P. V. Coveney, Large-scale lattice Boltzmann simulations of complex fluids: advances through the advent of computational Grids, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 1895 CrossRef PubMed.
- P. L. Bhatnagar, E. P. Gross and M. Krook, A model for collision processes in gases. I. Small amplitude processes in charged and neutral one-component systems, Phys. Rev., 1954, 94, 511 CrossRef CAS.
-
T. Krüger, H. Kusumaatmaja, A. Kuzmin, O. Shardt, G. Silva and E. M. Viggen, The Lattice Boltzmann Method, Springer, Berlin, 2017 Search PubMed.
- X. Shan and H. Chen, Lattice Boltzmann model for simulating flows with multiple phases and components, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1993, 47, 1815 CrossRef PubMed.
- A. J. C. Ladd, Numerical simulations of particulate suspensions via a discretized Boltzmann equation. Part 1. Theoretical foundation, J. Fluid Mech., 1994, 271, 285 CrossRef CAS.
- A. J. C. Ladd, Numerical simulations of particulate suspensions via a discretized Boltzmann equation. Part 2. Numerical results, J. Fluid Mech., 1994, 271, 311 CrossRef CAS.
- C. K. Aidun, Y. Lu and E. J. Ding, Direct analysis of particulate suspensions with inertia using the discrete Boltzmann equation, J. Fluid Mech., 1998, 373, 287 CrossRef CAS.
- F. Jansen and J. Harting, From bijels to Pickering emulsions: A lattice Boltzmann study, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 046707 CrossRef PubMed.
- S. Frijters, F. Günther and J. Harting, Effects of nanoparticles and surfactant on droplets in shear flow, Soft Matter, 2012, 8, 6542 RSC.
- A. J. C. Ladd and R. Verberg, Lattice–Boltzmann Simulations of Particle-Fluid Suspensions, J. Stat. Phys., 2011, 104, 1194 Search PubMed.
- G. B. Davies, T. Krüger, P. V. Coveney and J. Harting, Detachment energies of spheroidal particles from fluid-fluid interfaces, J. Chem. Phys., 2014, 141, 154902 CrossRef PubMed.
-
W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes: The Art of Scientific Computing, Cambridge University Press, Cambridge, 2007 Search PubMed.
- M. N. Popescu, S. Dietrich, M. Tasinkevych and J. Ralston, Phoretic motion of spheroidal particles due to self-generated solute gradients, Eur. Phys. J. E: Soft Matter Biol. Phys., 2010, 31, 351 CrossRef CAS PubMed.
- C. Pozrikidis, Particle motion near and inside an interface, J. Fluid Mech., 2007, 575, 333 CrossRef.
Footnotes |
| † In this contribution we specialize to the case of a single reaction product. Our approach can be easily extended to an arbitrary number of them. |
‡ In the numerical simulations the values of α, of the range δ of the potential W, and D are taken such that the relevant Damköhler number is 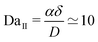 . . |
| § In our numerical scheme, see below, the boundary condition in eqn (8) will be accounted for by the explicit microscopic interactions among the two fluid species. |
| ¶ For example, in the case of a single colloid suspended in a Newtonian fluid the smallest relevant length scale is the size of the colloid and the relevant time scale is its mobility. In the case of a diffusiophoretic colloid the smallest length and time scales are set by the characteristic length and time involved in the diffusion process of the diffusing species, i.e., the range L of the interaction potential W between the solute and the colloid and the diffusion time across this length, i.e., t = L2/D where D is the diffusion coefficient of the diffusing species. |
| || The masses of the reactants, the reaction product, and the colloid enter into the description via their respective equation of motion. |
| ** This “roughness” is due to the discrete nature of the LBM. |
†† This term induces an additional decay length  . The contribution of the sink term to the velocity of the colloid is disregardable provided that Λ ≫ λ where λ is the decay length of the strength of the interaction between the colloid and the reaction product. . The contribution of the sink term to the velocity of the colloid is disregardable provided that Λ ≫ λ where λ is the decay length of the strength of the interaction between the colloid and the reaction product. |
| ‡‡ Note that in general, the mobility coefficient is a tensor because both the applied force and the angular velocity are vectors. However, for the self-phoretic colloid the local forces are always perpendicular to the colloid surface, and we are interested only in the z-component of the angular velocity. For this purpose the description of eqn (42) suffices. |
| §§ Since the force is acting radially, the torque on the colloid is zero. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
N.
Rivas
c,
A.
Scagliarini
*ab,
N.
Rivas
c,
A.
Scagliarini
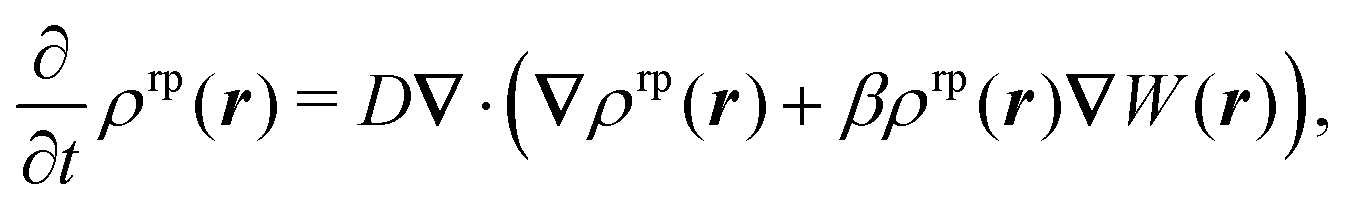

 is determined either by imposing fluid incompressibility (∇·v = 0) or by the equation of state for the fluid (as in our numerical scheme, see below). The boundary conditions for the fluid velocity are
is determined either by imposing fluid incompressibility (∇·v = 0) or by the equation of state for the fluid (as in our numerical scheme, see below). The boundary conditions for the fluid velocity are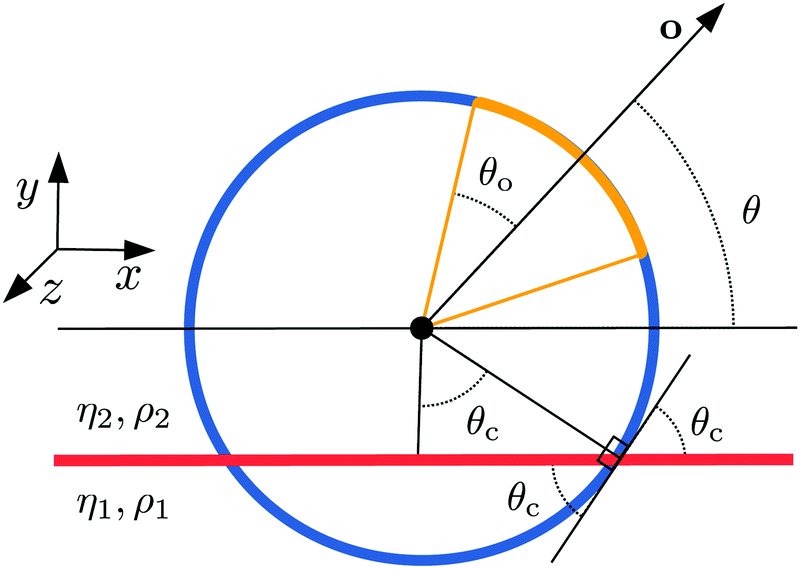


 , where
, where 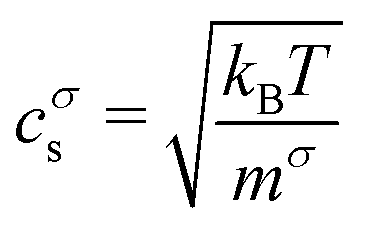 is the speed of sound in the phase dominated by species σ. Our numerical approach, in the present form, requires all species to have the same mass mσ = m, and hence the same speed of sound cσs = cs. In the following we choose to fix the lattice time step Δt and the lattice spacing Δx to unity. In these units, it is common to choose
is the speed of sound in the phase dominated by species σ. Our numerical approach, in the present form, requires all species to have the same mass mσ = m, and hence the same speed of sound cσs = cs. In the following we choose to fix the lattice time step Δt and the lattice spacing Δx to unity. In these units, it is common to choose  as the lattice speed of sound in terms of the time step Δt and the lattice spacing Δx.32 Once the distribution functions fσ,eqi(r,t) are known, it is possible to compute the local mass density of the fluid:
as the lattice speed of sound in terms of the time step Δt and the lattice spacing Δx.32 Once the distribution functions fσ,eqi(r,t) are known, it is possible to compute the local mass density of the fluid:

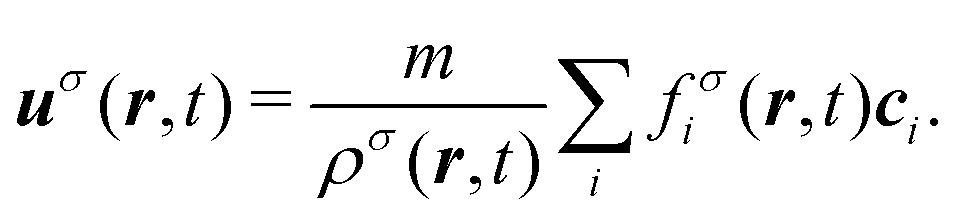

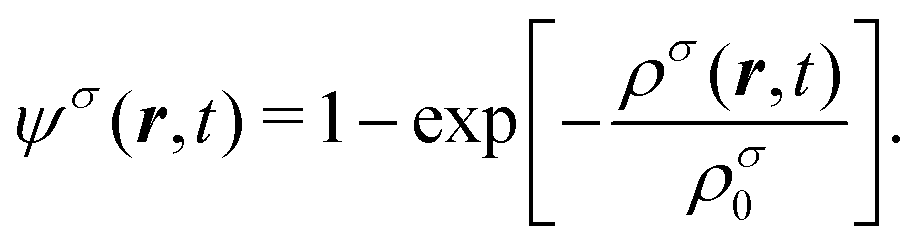

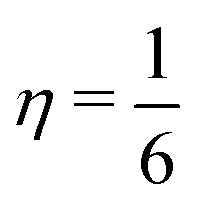 ) than the force induced by the surface tension Fsurf ≈ 2πRγ where γ = 0.1 is the magnitude of the surface tension.
) than the force induced by the surface tension Fsurf ≈ 2πRγ where γ = 0.1 is the magnitude of the surface tension.



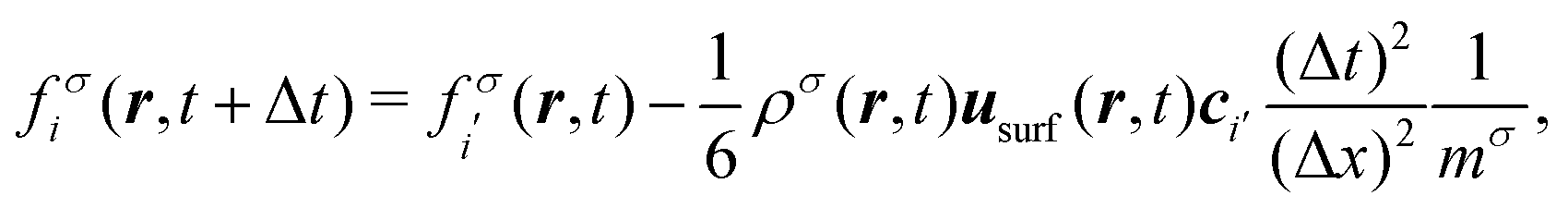



 is restricted to those values of i for which r + ciΔt is a fluid site; N is the number of these sites. In addition, in order to account for the unknown exact density profile in the close vicinity of a colloid, we apply a density correction algorithm as explained in ref. 37 and 38. In order to conserve momentum, a contribution is added to the force density (eqn (22)):
is restricted to those values of i for which r + ciΔt is a fluid site; N is the number of these sites. In addition, in order to account for the unknown exact density profile in the close vicinity of a colloid, we apply a density correction algorithm as explained in ref. 37 and 38. In order to conserve momentum, a contribution is added to the force density (eqn (22)):






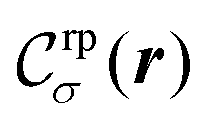 and we have accounted for their spatial variation. The dynamics of the reaction product is governed by eqn (1) and (2). However, in numerical simulations implementing boundary conditions at r → ∞ is challenging. In order to ensure that ρrp(r) reaches a steady state within the simulation box with periodic boundary conditions along the three spatial directions, we introduce a homogeneous sink term with constant decay rate††χ:
and we have accounted for their spatial variation. The dynamics of the reaction product is governed by eqn (1) and (2). However, in numerical simulations implementing boundary conditions at r → ∞ is challenging. In order to ensure that ρrp(r) reaches a steady state within the simulation box with periodic boundary conditions along the three spatial directions, we introduce a homogeneous sink term with constant decay rate††χ: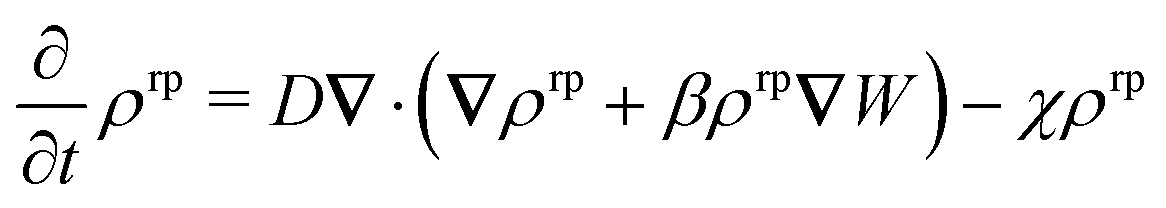



 is performed only over neighboring cells inside the colloid (see Fig. 2). This recipe prevents fluid cells in contact with more than one catalyst cell to be exposed to a larger flux, induced by the discretization of the shape of the colloid. Eqn (36) and (37) enforce a constant flux per area even in the discrete representation of the colloid.
is performed only over neighboring cells inside the colloid (see Fig. 2). This recipe prevents fluid cells in contact with more than one catalyst cell to be exposed to a larger flux, induced by the discretization of the shape of the colloid. Eqn (36) and (37) enforce a constant flux per area even in the discrete representation of the colloid.
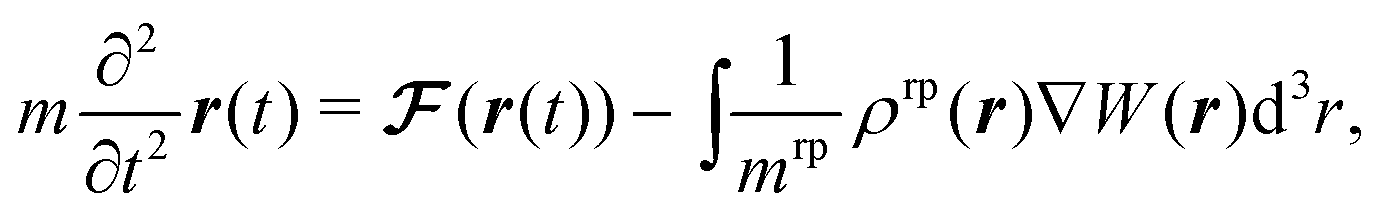
 is given by eqn (26). The last term in eqn (38) stems from the interaction of the colloid with the reaction product. We emphasize that the force applied to the colloid (eqn (38)) equals in magnitude the force applied on the fluid phases and on the solute such that the total force on the system is zero. This equation is integrated numerically via a leap-frog scheme.40,41
is given by eqn (26). The last term in eqn (38) stems from the interaction of the colloid with the reaction product. We emphasize that the force applied to the colloid (eqn (38)) equals in magnitude the force applied on the fluid phases and on the solute such that the total force on the system is zero. This equation is integrated numerically via a leap-frog scheme.40,41







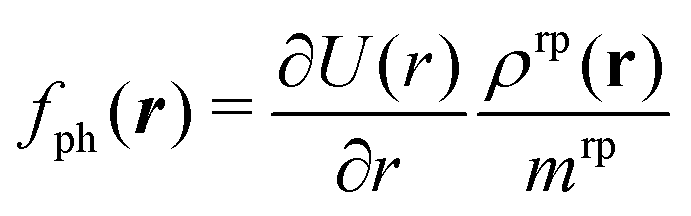 (see eqn (35)) where here r indicates the distance from the center of mass of the colloid. Accordingly, the local mobility allows us to determine the torque τz = τ·ez exerted on the colloid by the density profile of the reaction product:
(see eqn (35)) where here r indicates the distance from the center of mass of the colloid. Accordingly, the local mobility allows us to determine the torque τz = τ·ez exerted on the colloid by the density profile of the reaction product: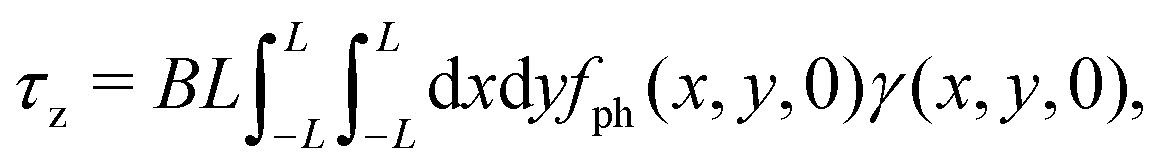


 .
.



 .
. . The contribution of the sink term to the velocity of the colloid is disregardable provided that Λ ≫ λ where λ is the decay length of the strength of the interaction between the colloid and the reaction product.
. The contribution of the sink term to the velocity of the colloid is disregardable provided that Λ ≫ λ where λ is the decay length of the strength of the interaction between the colloid and the reaction product.