
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-healing, luminescent metallogelation driven by synergistic metallophilic and fluorine–fluorine interactions†
Kalle
Kolari
a,
Evgeny
Bulatov
 a,
Rajendhraprasad
Tatikonda
a,
Kia
Bertula
b,
Elina
Kalenius
a,
Nonappa
*bc and
Matti
Haukka
*a
a,
Rajendhraprasad
Tatikonda
a,
Kia
Bertula
b,
Elina
Kalenius
a,
Nonappa
*bc and
Matti
Haukka
*a
aDepartment of Chemistry, University of Jyväskylä, P. O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: matti.o.haukka@jyu.fi
bDepartment of Applied Physics, Aalto University School of Science, Puumiehenkuja 2, FI-02150 Espoo, Finland. E-mail: nonappa@aalto.fi
cDepartment of Bioproducts and Biosystems, Aalto University School of Chemical Engineering, Kemistintie 1, FI-02150 Espoo, Finland
First published on 20th February 2020
Abstract
Square planar platinum(II) complexes are attractive building blocks for multifunctional soft materials due to their unique optoelectronic properties. However, for soft materials derived from synthetically simple discrete metal complexes, achieving a combination of optical properties, thermoresponsiveness and excellent mechanical properties is a major challenge. Here, we report the rapid self-recovery of luminescent metallogels derived from platinum(II) complexes of perfluoroalkyl and alkyl derivatives of terpyridine ligands. Using single crystal X-ray diffraction studies, we show that the presence of synergistic platinum–platinum (Pt⋯Pt) metallopolymerization and fluorine–fluorine (F⋯F) interactions are the major driving forces in achieving hierarchical superstructures. The resulting bright red gels showed the presence of highly entangled three-dimensional networks and helical nanofibres with both (P and M) handedness. The gels recover up to 87% of their original storage modulus even after several cycles under oscillatory step-strain rheological measurements showing rapid self-healing. The luminescence properties, along with thermo- and mechanoresponsive gelation, provide the potential to utilize synthetically simple discrete complexes in advanced optical materials.
Introduction
Supramolecular chemistry involving metal–ligand (M–L) coordination-induced self-assembly has opened new avenues in the field of stimulus-responsive soft materials.1–3 This is attributed to the possibilities to access a diverse range of metal components (metal ions, clusters, and nanoparticles) and rationally designed organic ligands with well-defined coordination sites.4,5 Among the metal components containing soft materials, self-healing and stimulus-responsive metallogels have gained considerable attention recently.6,7 The presence of metal components alters the gelation process, gel strength, mechanical properties, and morphological features.8–11 More importantly, metal components also impart properties such as conductivity, redox activity, magnetism, photophysical properties, antimicrobial properties, and thixotropic behaviour.12–18 Furthermore, certain metal ions also serve as a source for in situ metal nanoparticle formation.19–22 Therefore, metallogels find potential applications in chiral recognition, light-emitting materials, soft conductive materials, wearable electronics, energy storage, self-healing devices, catalysis, and antimicrobial systems, and can act as artificial enzyme mimics.23–27 Metallogels contain metal components and organic components with appropriate binding sites. Low molecular weight organic ligands, biopolymers and synthetic polymers having functional groups such as carboxylic acid, amines, thiols and alkynes have been used as coordination sites for a diverse range of mono-, di-, and trivalent metal ions (Ag+, Fe2+, Cu2+, Zn2+, Co2+, Ni2+, Pd2+, Pt2+, Fe3+, and Au3+), metal clusters or nanoparticles,28–31 wherein a combination of metal chelation and other non-covalent interactions such as H-bonding, van der Waals interaction, electrostatic interaction, π-stacking and metallophilic interactions has been explored to achieve hierarchical self-assembly.32–34 Metallogels involving low molecular weight organic ligands result in discrete coordination complexes or coordination polymerization upon complexation.19–22 When the gelation consists of the self-assembly of discrete complexes, the supramolecular interactions between the organic ligands act as the primary driving force, whereas, for coordination polymerization-induced gelation, the metal–ligand interaction forms the main driving force.35,36 For supramolecular metallogels, the gelation can be induced by adding metal salt into a solution containing organic ligands with appropriate binding sites or by dissolving a premade metal complex. In the latter case, the main driving forces are metallophilic (metallopolymerization) interactions along with other supramolecular interactions of organic moieties.Gold(III) and platinum(II) complexes are known to undergo metallophilic interaction-induced self-assembly.37,38 Au(III) and square planar Pt(II) complexes also offer unique optoelectronic properties.39,40 The ability to undergo a dramatic colour change upon self-assembly of Pt(II) complexes is attributed to the presence of Pt⋯Pt interaction.41 Therefore, self-assembly of various mononuclear and binuclear Pt(II) polypyridine complexes has been explored. Examples include alkynylplatinum(II) complexes with terpyridine ligands.42–44 Such complexes due to strong σ-donating alkynyl ligands have been shown to possess interesting aggregation behaviour. Depending on the substituents, chiral and helical morphologies, substitution dependent colour change, luminescence, vapochromism, and vapoluminescence have been reported.45,46 Such properties occur due to changes in weak metal–metal interactions, π-stacking or hydrogen bonding upon exposure to vapour (analyte).47 Furthermore, features such as self-assembly-induced luminescence provide promising applications in optoelectronics and in developing vapochromic sensors. Platinum(II) complexes have also been shown to form metallogels showing significant changes in the colour and spectroscopic properties upon sol ⇔ gel transition.48 Though such systems are known to form various superstructures, metallogelation using Pt⋯Pt interactions is limited in the literature.
Another important non-covalent interaction is fluorine–fluorine interaction. Fluorinated analogues of hydrocarbons have been shown to exhibit simultaneous hydro- and lipophobicities, altered aggregation behaviour, steric bulk, stiffness, stability, and lower critical aggregation concentration.49 Therefore, organic ligands containing fluorinated analogues of hydrocarbons offer a unique opportunity to design materials with excellent material properties. Recently, it has been shown that perfluoroalkyl substituted 4-aminophenyl-2,2′,6,2′-terpyridine can act as a metallosupramolecular gelator resulting in rapid self-healing and anion selectivity.50 The F⋯F interactions have been shown to affect the rheological and mechanical properties of the metallogels. In this work, we show that Pt(II) complexes of alkyl and fluoroalkyl containing terpyridine derivatives undergo a synergistic Pt⋯Pt and F⋯F interaction-driven self-assembly leading to rapid gelation. The compounds also exhibit luminescence properties in their solid state, gel state, and solution state. We demonstrate the rapid self-healing of gels containing low solid contents with recovery up to 87% of their storage modulus values even after several cycles. Morphologically, the gels contain a highly entangled 3D network of helical fibres with both handedness.
Results and discussion
Crystal structures and transformations in the solid state
First, we discuss the synthesis and solid state properties of the complexes studied in this work. The synthesis of ligands L1 and L2 and their platinum complexes {[Pt(L1)Cl]Cl} (1) and {[Pt(L2)Cl]Cl} (2) was carried out following a reported literature procedure (see Fig. 1 and ESI† for details and characterization data).51,52 Yellowish plate-like single crystals suitable for X-ray diffraction were obtained upon recrystallization of both the metal complexes from hot ethanol. In their solid state structures, the complexes 1 and 2 show cationic square-planar units self-assembled to form dimers (Fig. 1b and c) via Pt⋯Pt interactions with a distance of 3.4096(5) Å for 1 and 3.3031(2) Å for 2, respectively. These dimer units are further extended to form supramolecular polymeric structures facilitated by π⋯π contacts of terpyridine core. The cationic charges in both the complexes are compensated by the presence of chloride anions. The chloride counter ions are involved in hydrogen bonding with ethanol molecules (i.e. solvate) thus filling voids in crystal packing (Fig. S7 and S8, ESI†). Additionally, terpyridine substituents form lipophilic fluorine and hydrophobic phases in the crystal packing of complexes 1 and 2, respectively. Due to a severe disorder of perfluorinated substituents, the determination of the definite fluorine–fluorine distances remained a challenge. However, the sphere-packing model for the solid state structure of 1 indicates the presence of intermolecular fluorine–fluorine contacts (Fig. 1d), forming lipophilic phases in the crystal structures. In the case of complex 2, a similar arrangement is observed, i.e. hydrophobic phases are formed due to close packing of substituent chains between adjacent molecules (Fig. 1e). | ||
| Fig. 1 Chemical structures and single crystal X-ray structures. (a) Chemical structures of gelator molecules {[Pt(L1)Cl]Cl} (1) and {[Pt(L2)Cl]Cl} (2). (b) X-ray single crystal diffraction-based dimeric structure of gelator 1 showing Pt⋯Pt interaction and (d) showing higher-order packing driven by F⋯F interactions. (c and e) X-ray single crystal diffraction-based dimeric structure of complex 2 indicating Pt⋯Pt interaction and higher-order packing in the solid state. | ||
The yellow crystals of complexes 1 and 2 recrystallized from ethanol underwent a colour change to orange when exposed to air, presumably due to the evaporation of solvent molecules, the crystals of 1 being noticeably less stable (Fig. 2a and Movies S1, S2, ESI†). In both cases, the resulting orange solids are amorphous, as confirmed by powder X-ray diffraction studies (Fig. S9, ESI†). Upon drying under vacuum, a further colour change to dark purple was observed for complex 1, whereas complex 2 remained orange.
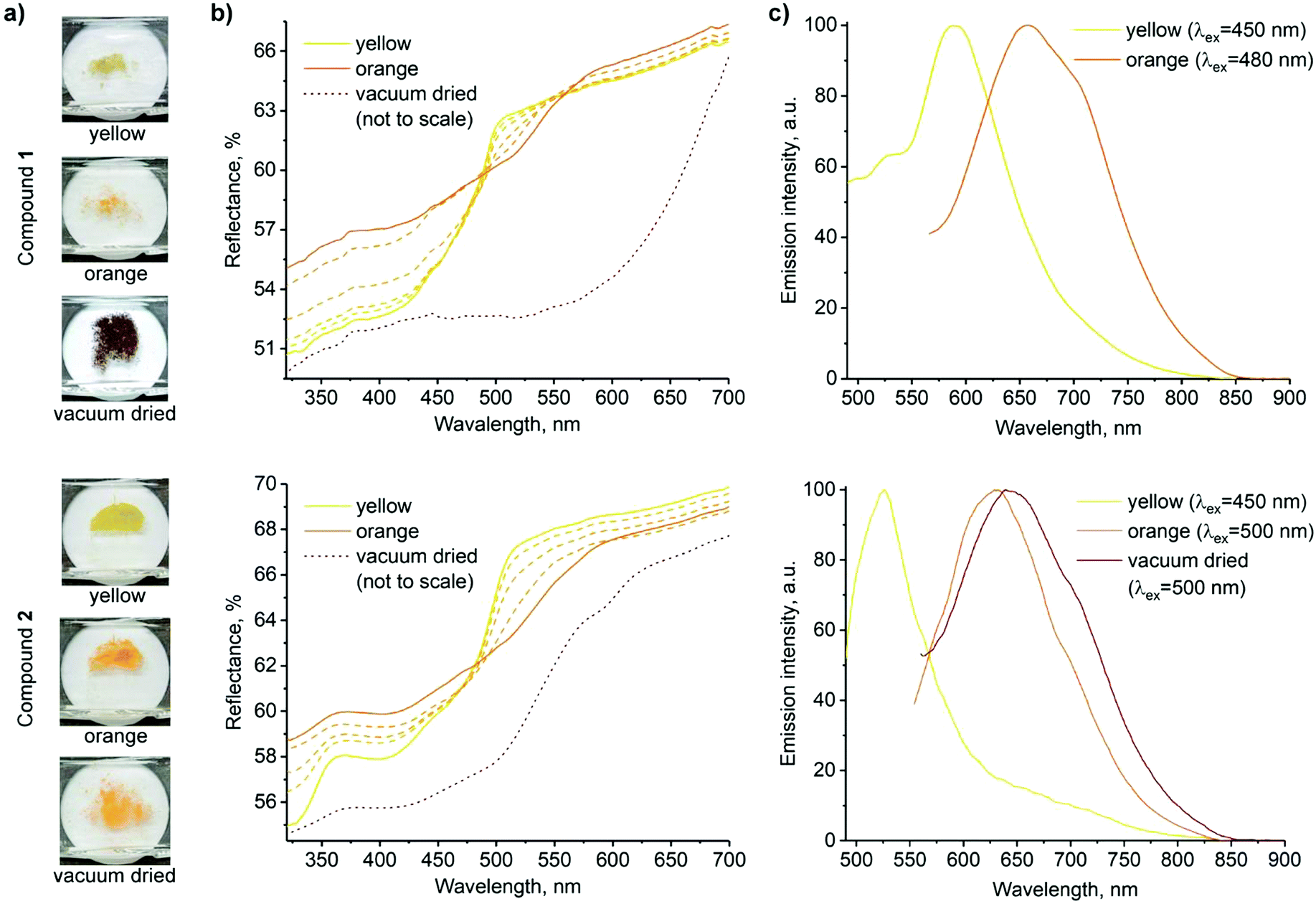 | ||
| Fig. 2 Solid state forms of 1 (top) and 2 (bottom). (a) Photographs of samples of the three forms used for spectroscopic measurements. (b) Reflectance spectra (transition from yellow to orange forms is shown in dashed lines; spectra of the vacuum dried powders are adjusted to match the scale, and the original spectra are presented in the ESI,† Fig. S10 and S11). (c) Normalized emission spectra (the decrease in emission intensity along with the red shift is demonstrated in ESI,† Fig. S12). | ||
Fig. 2 shows the photographs and corresponding reflectance and normalized emission spectra of solids of 1 and 2 in different forms. The visual colour changes of the solids are manifested in their reflectance spectra by the appearance of shoulders at λ ≈ 515 nm and 530 nm upon transformations from the yellow to air-dried orange forms of 1 and 2, respectively. At the same time, luminescence emission bands of 1 and 2 decrease in intensity and undergo a redshift (λem = 590 → 650 and 525 → 630 nm, respectively). Vacuum drying results in a further redshift of reflectance, with a broad reflectance band being formed between λ ≈ 500 and 650 nm in the case of dark purple 1. However, only a subtle band at λ ≈ 600 nm was observed in the case of orange 2. The effect of vacuum drying on the luminescence is also much stronger for 1, which displays no detectable emission, whereas the emission of 2 is only slightly red-shifted compared to the air-dried form (Fig. 2b and c). Intermolecular π⋯π and Pt⋯Pt interactions in stacked terpyridine complexes of platinum(II) (Fig. 1) are known to alter the photophysical properties of the complexes. Particularly, metal to ligand charge transfer (MLCT) absorption and emission bands of single molecules change to more red-shifted metal–metal to ligand charge transfer (MMLCT) bands within the stacks. Therefore, the observed redshifts in reflectance and emission spectra of 1 and 2 in the solid state upon drying are attributed to the MMLCT bands. While the amorphous nature of the orange forms prevents direct determination of their molecular arrangements by X-ray diffraction methods, the MMLCT bands indicate the presence of continuous Pt⋯Pt interactions, in contrast with the dimeric units in the yellow crystalline forms. Similar transformations between yellow and red forms have previously been observed for other terpyridine complexes.47,53
Aggregation in solution and gelation
After solid state characterization of complexes 1 and 2, we studied the self-assembly behaviour in polar organic solvents. A bright yellow solution of complex 1 was obtained in dimethyl sulfoxide (DMSO) at lower concentrations (C ≤ 1 mM). However, heating was required to dissolve the complex at higher concentrations. Importantly, allowing a supersaturated DMSO solution of complex 1 to attain room temperature resulted in a change of colour to bright red, accompanied by the gelation, which showed resistance to flow upon inversion (Fig. 3a and Movie S3, ESI†). The gelation was observed for a concentration as low as 0.6 w/v% (note: from hereafter, all gelator/solvent w/v% ratios are denoted as %). Similar results were also obtained in N,N-dimethylformamide (DMF). Complex 1 remained insoluble in water and partially soluble when a mixture of DMSO/water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was used for gelation. Importantly, no such colour change was observed in ethanol, and the attempts to form gels produced an unstable gelatinous precipitate. On the other hand, complex 2 only formed an unstable gel in ethanol at concentrations above 1.5% and no gelation or colour change was observed either in DMSO or DMF. Such a difference in the behaviours of 1 and 2 is attributed to their different aggregation capabilities. Apart from the intermolecular π⋯π interactions between the terpyridine units and Pt⋯Pt interactions, the aliphatic chains in 1 and 2 further promote aggregation via lipophilic stacking, as observed in the crystal structures (Fig. 1d and e). The perfluoroalkyl chain in 1 is expected to cause stronger aggregation due to F⋯F interactions, as previously reported in the literature.51 Therefore, the change of colour for complex 1 from yellow to red in solution at higher concentrations and upon gelation is attributed to the appearance of the MMLCT band, associated with the stacking of the terpyridine units assisted by F⋯F interactions. On the other hand, complex 2 demonstrates much less capability for aggregation.54–56
:1 v/v) was used for gelation. Importantly, no such colour change was observed in ethanol, and the attempts to form gels produced an unstable gelatinous precipitate. On the other hand, complex 2 only formed an unstable gel in ethanol at concentrations above 1.5% and no gelation or colour change was observed either in DMSO or DMF. Such a difference in the behaviours of 1 and 2 is attributed to their different aggregation capabilities. Apart from the intermolecular π⋯π interactions between the terpyridine units and Pt⋯Pt interactions, the aliphatic chains in 1 and 2 further promote aggregation via lipophilic stacking, as observed in the crystal structures (Fig. 1d and e). The perfluoroalkyl chain in 1 is expected to cause stronger aggregation due to F⋯F interactions, as previously reported in the literature.51 Therefore, the change of colour for complex 1 from yellow to red in solution at higher concentrations and upon gelation is attributed to the appearance of the MMLCT band, associated with the stacking of the terpyridine units assisted by F⋯F interactions. On the other hand, complex 2 demonstrates much less capability for aggregation.54–56
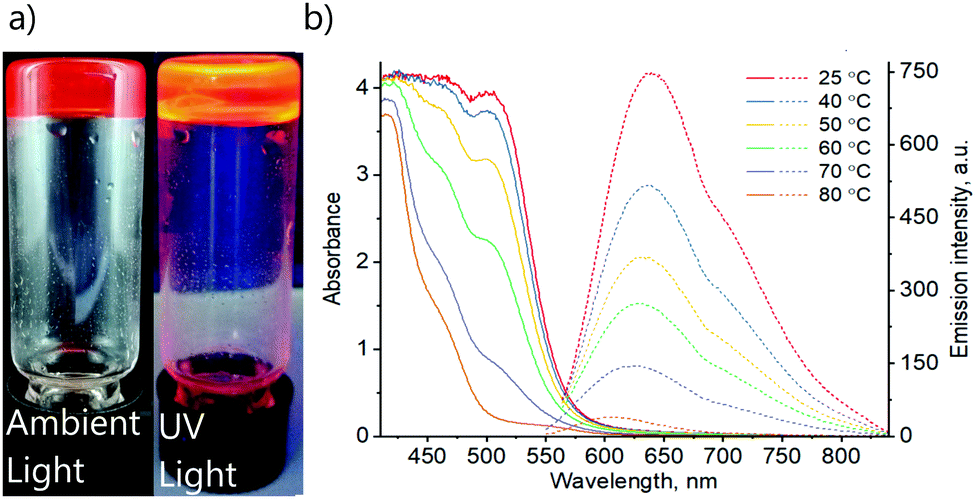 | ||
| Fig. 3 (a) The photographs of the DMSO gel of complex 1 under ambient light and under UV-radiation. (b) Absorption (solid lines) and emission (dashed lines) spectra of DMSO gel of 1 (0.6%) at various temperatures. Excitation wavelengths λex = 550 and 530 nm were used for temperatures 25–60 °C and 70–80 °C accordingly. | ||
Nuclear magnetic resonance (NMR) spectroscopy has been utilized extensively to study gels.57–59 In the literature, it has often been shown that the 1H NMR resonance peaks of gels at room temperature show similar chemical shift values to that of the solution state (after gel melting).60–62 It has been hypothesized that for gels at room temperature, the observed 1H resonance peaks predominantly originate from the free molecules that are not bound to the gel network, whereas the molecules in the aggregated state remain NMR silent.60,61 We have performed variable temperature (VT) 1H and 19F NMR spectroscopy measurements of the DMSO-d6 gel of 1 to investigate the sol ⇔ gel transition and interactions involved in the gelation. The 1H NMR resonance signals remained invisible and featureless in temperature range of 30–50 °C, a property that is typical for low molecular weight gels below their melting temperature. This is attributed to increased viscosity and reduced molecular tumbling in the gel state. However, above 60 °C, the signal to noise ratio improved and the aliphatic and aromatic region displayed a clear splitting pattern at 90 °C (see ESI,† Fig. S13a and b). The signals at the same time underwent a slight downfield shift (Δδ between 0.01 to 0.09 ppm) (see ESI,† Table S2). The gel ⇔ sol transition observed in VT 1H NMR experiments is in agreement with the gel melting temperature (Tgel = 69 °C) of the 0.6% DMSO gel of 1 determined using an inverted test tube method. Variable temperature 19F NMR spectroscopy measurements displayed broad peaks at 30 °C. A downfield shift in the 19F resonance peaks was revealed upon heating the gel from 30 °C to 90 °C. Furthermore, upon heating, the 19F signals also became sharp and showed an apparent splitting, indicating changes in F⋯F interactions in the transformation from gel to solution state (ESI,† Fig. S13b). Unlike the VT 1H NMR spectra, the VT 19F NMR spectra showed a clear change in chemical shift values with Δδ between 0.15 and 1.61 ppm (see ESI,† Table S3). Fourier transformed infrared (FT-IR) spectroscopy of complex 1 in its synthetic solid, gel (1%) and solution states (0.4%) was performed to further probe the fluorine–fluorine interactions by monitoring the νs(CF2) stretching frequencies. The FT-IR spectra of solid powder form revealed the C–F stretching frequency at 1144 cm−1 (ESI,† Fig. S14). The gel of complex 1 showed an increased stretching frequency at 1148 cm−1. Finally, further shifting was detected in solution (1153 cm−1). Thus, it can be concluded that fluorine–fluorine interactions are present in solid, gel and solution with decreasing strength, affecting the self-assembly of monomeric units and gelation.
To gain more insights, variable temperature absorption and emission spectroscopic analyses of solution and gel states of complex 1 were carried out (Fig. 3). The absorption spectrum of the DMSO gel (0.6%) of complex 1 shows a peak at λabs = 500 nm at 25 °C. Importantly, the intensity decreases upon increasing temperature. Similarly, the corresponding emission also decreases in intensity with increasing temperature and shows a blue shift upon heating from λem = 640 nm at 25 °C to λem = 600 nm at 80 °C. These observations match well with the changes in photophysical properties of solid 1 upon the transformation from yellow to orange form (Fig. 2b and c). Thus, the absorption and emission bands at λabs = 500 nm and λem = 640 nm are accordingly assigned to the MMLCT transitions, which are expected to depend on aggregation and, consequently, temperature. The emission lifetime at room temperature τ = 51 ns suggests the fluorescence nature of the complex. Though such a short excited-state lifetime is not very common for transitions involving Pt(II) centres, previously, it has been reported for a limited number of terpyridine platinum(II) complexes.55
Preliminary studies of absorption and emission spectra of the solution of complex 1 in DMSO at various concentrations and various temperatures indicated the formation of more than one type of aggregate. Therefore, an unambiguous interpretation of the spectra remained a challenge. However, the correlation of photophysical properties of 1 in solid and gel states indicates similarities in intermolecular arrangements, namely the formation of continuous chains with stacked terpyridine units and close Pt⋯Pt contacts within the gel structure. The F⋯F interactions and packing of the perfluoroalkyl chains also appear to take place, based on the NMR and FT-IR spectra, as well as the non-gelling behaviour of complex 2. A detailed study of the aggregation behaviour of complex 1 in solution is beyond the scope of this work and will be the subject of a separate study.
Gelation and morphology
The morphological features of self-assembled superstructures in the gel were studied using electron microscopy imaging. First, the scanning electron microscopy (SEM) imaging of DMSO and DMF gels of complex 1 was performed. For SEM imaging, aerogels were prepared using a liquid propane freeze-drying method.63 Freeze-drying allows minimum drying artefacts during specimen preparation. The representative SEM micrographs of aerogels from DMSO and DMF gels of 1 are shown in Fig. 4. | ||
| Fig. 4 Scanning electron microscopy. (a and b) SEM micrographs of aerogels from the DMSO gel of 1 (1.0%) showing helical screw-like structures. (c) Schematic illustration of helical screw-like fibres in the DMSO gel of 1. (d and e) SEM micrographs of aerogels from the DMF gels of 1 (1.0%) showing helical fibre bundles. (f) Schematic illustration of helical fibres in the DMF gel of 1. For additional SEM images, see ESI† (Fig. S15 and S16). | ||
Interestingly, the SEM micrographs show the presence of helical screw-like fibres with both handedness (P and M) in DMSO (Fig. 4b and c). Similarly, in DMF, helical fibres with both handedness are observed (Fig. 4e and f; see ESI† for additional SEM images, Fig. S15 and S16). It is well documented in the literature that achiral gelators tend to form helical structures with both handedness.64,65 Importantly, triazine-containing alkynylplatinum(II) terpyridine complexes with appropriate substituents on terpyridine units have been shown to assemble into a mixture of right- and left-handed helical fibres in DMSO–water mixtures.66 It has been hypothesized that in such complexes, the terpyridine units are bent with respect to the alkynyl ligand, inducing directional Pt⋯Pt interactions and π-stacking. Furthermore, such assemblies are also due to dominant hydrophobic–hydrophobic interactions in polar solvents. Based on the X-ray crystal structure of the solids, VT NMR spectroscopy and FT-IR studies of complex 1, it is evident that there exists a strong F⋯F interaction in the gel and solid states. Therefore, the presence of Pt⋯Pt, π⋯π and F⋯F interactions results in minimized unfavourable interactions in polar DMSO and drives the formation of helical nanostructures.
From SEM imaging, it is evidenced that the helical fibres with lateral dimensions varying from 100 to 500 nm are composed of smaller fibrils. To further evaluate the nature of the fibrillar structures, a TEM specimen was prepared by drop-casting the freshly prepared hot sol onto holey-carbon film (Fig. 5). TEM micrographs of DMSO gels of 1 clearly show highly entangled fibrillar structures. The fibril diameters varied from 15 to 30 nm with indefinite length. It is important to note that the sample preparation methods for SEM and TEM are different. For SEM, premade gels are freeze-dried to obtain aerogels, whereas TEM studied a drop casted thin film of hot sol. TEM, therefore, allowed the smaller fibrillar structures to be observed. Similarly, the DMF gel of 1 also showed structural features that undergo higher-order assembly into fibres as indicated in the SEM micrographs.
 | ||
| Fig. 5 Transmission electron microscopy. (a and b) TEM micrographs of the dried DMSO gel of 1 (0.6%). (c and d) TEM micrographs of the dried DMF gel of 1 (1.0%). For additional TEM micrographs, see ESI,† Fig. S17 and S18. | ||
Fibrillar structures were also observed when non-gelling complex 2 was dispersed in DMSO or DMF with fibre diameters ranging from 15 to 50 nm.
Rheological properties
Metallogels exhibit unique mechanical properties in rheological experiments such as self-healing and thixotropy.6,50 The mechanical properties of DMSO and DMF gels of complex 1 were studied using dynamic oscillatory rheological measurements on the 1.0% gels. In all experiments, premade gels were used to study the rheological properties. First, the time sweep experiments were performed to evaluate the stability of the gels and in both cases, the storage modulus (G′) is close to an order of magnitude higher than the loss modulus (G′′), suggesting that the systems under investigation are indeed gels and remained stable under experimental conditions (see ESI,† Fig. S19). It is also evident that DMF gels of 1 displayed higher stiffness than that of DMSO gels. The average storage modulus (G′) values were found to be 90 Pa and 2.0 kPa for DMSO and DMF gels of 1, respectively.The rheological properties of soft materials display non-linear behaviour with a rapid decline in their storage modulus above certain strain levels, known as critical strain. Critical strain allows understanding the linearity of a material under investigation. Below critical strain, the structure of the gels remains intact and applying strain levels above critical strain will disrupt the gel structure. This information is useful in understanding the linear viscoelastic regime of the materials. In our experiments, the DMSO and DMF gels of complex 1 show that the structure remains intact until 32% and 40% strain levels, respectively, above which the materials behaved non-linearly and a cross-over between G′ and G′′ was observed.
The frequency sweep measurements provide insights about the junction networks and temporary bonds that hold the networks together. This information can be extracted using frequency-dependent storage and loss moduli (Fig. 6b). The rheological and SEM studies suggest the presence of junction networks. Another interesting property of metallogels is self-healing. Rheologically, self-healing can be studied by using step-strain experiments. In the case of step-strain tests, recovery of the gel is observed after shearing. The step-strain rheological measurements were performed to investigate the reversible gel ⇔ sol transition and self-recovery, upon several cycles. For the step strain experiments, controlled strains of 0.1% and 150% were applied for 60 s, respectively (Fig. 6c). The gels showed an immediate response to increased strain by turning into viscoelastic liquids as indicated by the rapid decrease in G′ well below that of G′′. The application of increased strain also appears to break the structure further during the 60 s experiment, as shown by the slight decreasing elastic modulus values on subsequent cycles. The gels recovered almost instantaneously upon switching to lower strain (0.1%), i.e. rapid self-recovery. Importantly, the process can be repeated for several cycles. However, slightly lower elastic moduli after the first high–low strain cycle and gradual build-up are observed, therefore indicating that the structure build-up to the equilibrium state would require longer periods of “rest” (low strain). Importantly, the DMSO gels recovered up to 87% of their original storage modulus values even after four cycles. On the other hand, DMF gels recovered up to 61% of the initial storage modulus upon repeated step-strain cycles. Finally, we studied the thermal properties using temperature sweep experiments. First, the premade gel was heated, and the moduli were followed. Temperature ramps from 20 °C to 90 °C and from 90 °C to 20 °C were measured with 0.1% strain amplitude and 5 °C min−1 heating rate. Temperature ramps and step-strain experiments present the average of two measurements. Above 70 °C, a rapid decline in the storage modulus was observed when the 1.0% DMSO and DMF gels begin to melt, in agreement with the visual and variable temperature NMR tests described above. However, the thermoreversibility under rheological measurement is prominent for DMSO gels as they are rather robust. Further, unlike DMSO gels, DMF gels were not robust to transfer and scoop processes, and the solvent was often expelled, which might also explain the observed high modulus.
 | ||
| Fig. 6 Rheological measurements: (a) frequency sweep experiments of DMSO and DMF gels (1.0%) of 1. (b) Strain sweep experiments (arrows indicate the strain% above which the gels break). (c) Step-strain experiments showing the self-recovery by alternately applying 0.1% and 150% strain. (d) The thermoreversible nature of the gels is revealed using temperature sweep cycles. See ESI† for additional rheological data with error bars. | ||
Conclusions
Self-assembly of discrete metal complexes offers a range of opportunities to construct structurally and functionally unique soft materials using simple synthetic design. More importantly, simple chemical modification of functional units allows a remarkable change in their structure, function, and interactions. These changes also affect the self-assembly behaviour, morphology, and rheological properties at extremely low solid contents. In this work, we have demonstrated that replacement of the alkyl derivative with a fluoroalkyl derivative in terpyridine ligand allows control over the solid state assembly, luminescence properties, and gelation ability of complexes 1 and 2. Our results show that the presence of synergistic Pt⋯Pt and F⋯F interactions is responsible for the rapid gelation of complex 1. This combination of interactions is also responsible for self-assembly-induced luminescence, rapid gelation, and self-healing. Therefore, this study demonstrates how the influence of metallophilic interactions on luminescence properties can provide insight on intermolecular arrangements and thus potentially allow investigation of supramolecular structures of the gels. The SEM of the cryo-frozen gel of complex 1 displayed an enantiomeric mixture of fibres with screw rotation to left and right. Fibres were up to 200 nm in diameter. The topology of fibres is stabilized with non-covalent interactions including intermolecular platinum–platinum interactions. Additionally, changing the solvent from ethanol to DMSO affects the formation of helical polymeric structures and enables the formation of fibres with screw rotation by affecting the packing of monomeric units.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KK and RT acknowledge the kind financial support from the Magnus Ehrnrooth Foundation, and Academy of Finland (M. H. Proj. no. 295581). Academy of Finland's Centre of Excellence in Molecular Engineering of Biosynthetic Hybrid Materials (HYBER, 2014–2019) and Aalto University Nanomicroscopy Centre (Aalto-NMC) are acknowledged for the use of their facilities.References
- Functional Metallosupramolecular Materials, ed. J. G. Hardy and F. H Schacher, Royal Society of Chemistry, 1st edn, 2015, pp. 1–446 Search PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- D. G. Kurth, Sci. Technol. Adv. Mater., 2008, 9, 014103 CrossRef PubMed.
- R. Dobrawa and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981–4995 CrossRef CAS.
- H. Wu, J. Zheng, A.-L. Kjøniksen, W. Wang, Y. Zhang and J. Ma, Adv. Mater., 2019, 31, 1806204 CrossRef PubMed.
- Y.-Y. Tama and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- M. Häring and D. D. Díaz, Chem. Commun., 2016, 52, 13068–13081 RSC.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, Angew. Chem., Int. Ed., 2014, 53, 12026–12044 CrossRef CAS PubMed.
- H. Svobodová, M. Lahtinen, Z. Wimmer and E. Kolehmainen, Soft Matter, 2012, 8, 7840 RSC.
- J. Ge, E. Neofytou, T. J. Cahill III, R. E. Beygui and R. N. Zare, ACS Nano, 2012, 6, 227 CrossRef CAS PubMed.
- K. Mitsumoto, J. M. Cameron, R.-J. Wei, H. Nishikawa, T. Shiga, M. Nihei, G. N. Newton and H. Oshio, Chem. – Eur. J., 2017, 23, 1502–1506 CrossRef CAS.
- J. Kima and D. Lee, Chem. Sci., 2019, 10, 3864–3872 RSC.
- H. T. P. Anh, C.-M. Huang and C.-J. Huang, Sci. Rep., 2019, 9, 11562 CrossRef.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- H. Liang, Z. Zhang, Q. Yuana and J. Liu, Chem. Commun., 2015, 51, 15196–15199 RSC.
- V. A. Mallia and R. G. Weiss, Soft Matter, 2016, 12, 3665–3676 RSC.
- R. Tatikonda, K. Bertula, Nonappa, S. Hietala, K. Rissanen and M. Haukka, Dalton Trans., 2017, 46, 2793–2802 RSC.
- R. Tatikonda, E. Bulatov, Z. Özdemir, Nonappa and M. Haukka, Soft Matter, 2019, 15, 442–451 RSC.
- M. Tautz, C. Saldías, A. D. Lozano-Gorrín and D. Díaz Díaz, New J. Chem., 2019, 43, 13850–13856 RSC.
- H. Bunzen, Nonappa, E. Kalenius, S. Hietala and E. Kolehmainen, Chem. – Eur. J., 2013, 19, 12978 CrossRef CAS PubMed.
- B. H. Schroeder, A. Guha, A. Lamoureux, G. V. Renterghem, D. Sept, M. Shtein, J. Yang and M. Mayer, Nature, 2017, 214–218 CrossRef PubMed.
- C. Keplinger, J.-Y. Sun, C. C. Foo, P. Rothemund, G. M. Whitesides and Z. Suo, Science, 2013, 341, 984–987 CrossRef CAS PubMed.
- Z. Sun, Z. Li, Y. He, R. Shen, L. Deng, M. Yang, Y. Liang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 13379–13386 CrossRef CAS PubMed.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- C. Wei, J. Su, B. He, G.-B. Wen, Y.-W. Lin and Y. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3504 CrossRef PubMed.
- B. N. Ghosh, S. Bhowmik, P. Mal and K. Rissanen, Chem. Commun., 2014, 50, 734–736 RSC.
- S. Bhowmik, B. N. Ghosh and K. Rissanen, Org. Biomol. Chem., 2014, 12, 8836–8839 RSC.
- J. L. Zhong, X. J. Jia, H. J. Liu, X. Z. Luo, S. G. Hong, N. Zhang and J. Bin Huang, Soft Matter, 2015, 12, 191–199 RSC.
- K. Jie, Y. Zhou, B. Shi and Y. Yao, Chem. Commun., 2015, 51, 8461–8464 RSC.
- S. Datta, M. L. Saha and P. J. Stang, Acc. Chem. Res., 2018, 51, 2047–2063 CrossRef CAS PubMed.
- L. J. Chen and H. B. Yang, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- Y. Zhang, Q. F. Zhou, G. F. Huo, G. Q. Yin, X. L. Zhao, B. Jiang, H. Tan, X. Li and H. B. Yang, Inorg. Chem., 2018, 57, 3516–3520 CrossRef CAS PubMed.
- J. Zhang and C. Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- P. Sutar and T. K. Maji, Chem. Commun., 2016, 52, 8055–8074 RSC.
- E. R. T. Tiekink, Coord. Chem. Rev., 2014, 275, 130–153 CrossRef CAS.
- V. Phillips, F. G. Baddour, T. Lasanta, J. M. López-De-Luzuriaga, J. W. Bacon, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Inorganica Chimica Acta, Elsevier B.V., 2010, vol. 364, pp. 195–204 Search PubMed.
- A. Sathyanarayana, S. Nakamura, K. Hisano, O. Tsutsumi, K. Srinivas and G. Prabusankar, Sci. China: Chem., 2018, 61, 957–965 CrossRef CAS.
- M. H. Y. Chan, M. Ng, S. Y. L. Leung, W. H. Lam and V. W. W. Yam, J. Am. Chem. Soc., 2017, 139, 8639–8645 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed.
- C. Yu, K. M. C. Wong, K. H. Y. Chan and V. W. W. Yam, Angew. Chem., Int. Ed., 2005, 44, 791–794 CrossRef CAS PubMed.
- A. Y. Y. Tam, K. M. C. Wong, N. Zhu, G. Wang and V. W. W. Yam, Langmuir, 2009, 25, 8685–8695 CrossRef CAS PubMed.
- C. Po, Z. Ke, A. Y. Y. Tam, H. F. Chow and V. W. W. Yam, Chem. – Eur. J., 2013, 19, 15735–15744 CrossRef CAS PubMed.
- T. J. Wadas, Q.-M. Wang, Y. Kim, C. Flaschenreim, T. N. Blanton and R. Eisenberg, J. Am. Chem. Soc., 2004, 126, 16841–16849 CrossRef CAS PubMed.
- M. J. Bryant, J. M. Skelton, L. E. Hatcher, C. Stubbs, E. Madrid, A. R. Pallipurath, L. H. Thomas, C. H. Woodall, J. Christensen, S. Fuertes, T. P. Robinson, C. M. Beavers, S. J. Teat, M. R. Warren, F. Pradaux-Caggiano, A. Walsh, F. Marken, D. R. Carbery, S. C. Parker, N. B. McKeown, R. Malpass-Evans, M. Carta and P. R. Raithby, Nat. Commun., 2017, 8, 1800 CrossRef CAS PubMed.
- R. Zhang, Z. Liang, A. Han, H. Wu, P. Du, W. Lai and R. Cao, CrystEngComm, 2014, 16, 5531–5542 RSC.
- L. Ao, T.-F. Fu, Z.-C. Gao, X.-L. Zhang and F. Wang, Chin. Chem. Lett., 2016, 27, 1147–1154 CrossRef.
- R. J. Baker, P. E. Colavita, D. M. Murphy, J. A. Platts and J. D. Wallis, J. Phys. Chem. A, 2012, 116, 1435–1444 CrossRef CAS PubMed.
- L. Arnedo-Sánchez, Nonappa, S. Bhowmik, S. Hietala, R. Puttreddy, M. Lahtinen, L. De Cola and K. Rissanen, Dalton Trans., 2017, 46, 7309–7316 RSC.
- P. Du, Inorg. Chim. Acta, 2010, 363, 1355–1358 CrossRef CAS.
- R. Tatikonda, S. Bhowmik, K. Rissanen, M. Haukka and M. Cametti, Dalton Trans., 2016, 45, 12756–12762 RSC.
- V. C. H. Wong, C. Po, S. Y. L. Leung, A. K. W. Chan, S. Yang, B. Zhu, X. Cui and V. W. W. Yam, J. Am. Chem. Soc., 2018, 140, 657–666 CrossRef CAS PubMed.
- S. W. Lai, M. C. W. Chan, K. K. Cheung and C. M. Che, Inorg. Chem., 1999, 38, 4262–4267 CrossRef CAS.
- S. E. Hobert, J. T. Carney and S. D. Cummings, Inorg. Chim. Acta, 2001, 318, 89–96 CrossRef CAS.
- J. A. Bailey, M. G. Hill, R. E. Marsh, V. M. Miskowski, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1995, 34, 4591–4599 CrossRef CAS.
- B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed.
- Nonappa, M. Lahtinen, B. Behera, E. Kolehmainen and U. Maitra., Soft Matter, 2010, 6, 1748–1757 RSC.
- Nonappa and E. Kolehmainen, Soft Matter, 2016, 12, 6015–6026 RSC.
- Nonappa, D. Šaman and E. Kolehmainen, Magn. Reson. Chem., 2015, 53, 256–260 CrossRef CAS PubMed.
- V. Noponen, Nonappa, M. Lahtinen, A. Valkonen, H. Salo, A. E. Kolehmainen and E. Sievänen, Soft Matter, 2010, 6, 3789–3796 RSC.
- S. Ikonen, Nonappa, A. Valkonen, R. Juvonen, H. Salo and E. Kolehmainen, Org. Biomol. Chem., 2010, 8, 2784–2794 RSC.
- K. Bertula, L. Martikainen, P. Munne, S. Hietala, J. Klefström, O. Ikkala and Nonappa, ACS Macro Lett., 2019, 8, 670–675 CrossRef CAS.
- S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS PubMed.
- H. L. K. Fu, C. Po, S. Y. L. Leung and V. W. W. Yam, ACS Appl. Mater. Interfaces, 2017, 9, 2786–2795 CrossRef CAS PubMed.
- H. L. K. Fu, S. Y. L. Leung and V. W. W. Yam, Chem. Commun., 2017, 53, 11349–11352 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis of ligands and their platinum complexes, NMR spectra, single crystal X-ray data, additional SEM, TEM micrographs and rheological studies. CCDC 1949029 and 1949030. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sm02186h |
| This journal is © The Royal Society of Chemistry 2020 |