
Molecular recognition with soft biomaterials
 a
and
Nicholas A.
Peppas
*abcdef
a
and
Nicholas A.
Peppas
*abcdef
Abstract
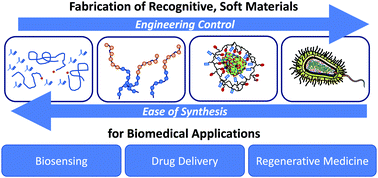
Biomacromolecules and engineered materials can achieve molecular recognition if they engage their ligand with properly oriented and chemically complementary moieties. Recently, there has been significant interest in fabricating recognitive soft materials, which possess specific affinity for biological analytes. We present a summary and evaluation of current recognitive materials for biosensing, drug delivery, and regenerative medicine applications. We highlight the impact of material composition on the extent and specificity of ligand adsorption, citing new theoretical and empirical evidence. We conclude with a guide for synthesizing and characterizing novel recognitive materials, as well as recommendations for ligand selection and experimental design.
- This article is part of the themed collection: Soft Matter 15th Anniversary Perspectives
 Please wait while we load your content...
Please wait while we load your content...

