
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn air CO2 capture system based on the passive carbonation of large Ca(OH)2 structures
J. Carlos
Abanades
 *,
Yolanda A.
Criado
and
José Ramón
Fernández
*,
Yolanda A.
Criado
and
José Ramón
Fernández
CSIC-INCAR, C/Francisco Pintado Fe, 26, 33011, Oviedo, Spain. E-mail: abanades@incar.csic.es; Fax: +34 985297662; Tel: +34 985118980
First published on 17th February 2020
Abstract
Direct Air Capture (DAC) requires contacting a vast flow of air with a functional surface, which must be accommodated in a large and costly CO2 capture device (i.e. at least 0.2–0.4 m3 of the reactor volume per tCO2 per year). We propose in this paper a low-cost alternative that involves contactor volumes that are one or two orders larger, but require only the passive CO2 carbonation of purpose-built porous structures of Ca(OH)2. Such low-cost materials can be manufactured from natural limestone and/or from recycled carbonated structures by using oxy-calcination technologies, and then simply stacked in such a way as to leave gaps for air to pass through. On the basis of an analysis of the rate controlling factors of the carbonation reaction, we employed as the structural element sintered Ca(OH)2 plates with an area of 2 × 2 m2 and 3 cm thick with a porosity of 0.5, which can be fully carbonated in about 6 months. The cost of CO2 captured from air is estimated to be between 140 and 340 $ per tCO2 depending on assumed cost values for fuel, land use, structural materials manufacture & transport, and process and project contingencies.
1. Introduction
Direct CO2 capture from air (DAC), combined with permanent CO2 storage, is one of the negative emission technologies (NETs) that may be needed in the future to limit global warming to 1.5 °C.1,2 The first conceptual designs of DAC systems were proposed 20 years ago,3 and research into novel concepts and materials is still continuing today.3–22 The direct capture of CO2 from air requires vast contacting surfaces with a great affinity and selectivity towards CO2 (i.e. through a gas–liquid interface, the internal surface of a solid sorbent or through a membrane). Absorption methods generally use a strong alkaline aqueous solvent solution such as Ca(OH)2,3,10,11 NaOH12–14 or KOH15 that reacts with CO2 to yield carbonated salts. Such alkaline solutions are corrosive, require costly gas–liquid contactors and require a large amount of energy for their regeneration.8,16 Adsorbent solids with a high selectivity for CO2, such as amines supported on porous materials, ion-exchange resins, metal–organic frameworks, zeolites or activated carbons, have also been explored.8,9,17,18 Other gas–solid contact options are membranes combined with KOH solutions,19 dispersed amines or ion-exchange resins.20 An entirely different approach is to extract CO2 from large flows of seawater by electrodialysis, after which the water is returned to the ocean to reabsorb more CO2 from the air.21,22Some DAC technologies are being tested on increasingly larger scales to assess their technical and economic viability. Carbon Engineering operates a pilot plant designed to capture around 300 t CO2 per year using aqueous solutions of NaOH or KOH coupled to a Ca(OH)2/CaCO3 loop.15 The estimated cost of the CO2 captured in this process is around 230 $ per ton of CO2 (in the case of “no revenues” for the CO2 captured) depending on the type of fuel used, the cost of electricity or the fate of the CO2 (storage or fuel synthesis).15 Climeworks has built a modular plant able to capture almost 1000 ton CO2 per year using filters made up of porous granulates modified with amines. The process can deliver pure CO2 for subsequent use in greenhouses or for geological storage/use at a cost of around 600 $ per tCO2.23 The largest DAC plant built to date, which is operated by Global Thermostat,24 has been designed to capture around 4000 tCO2 per year.25 This facility is equipped with amine-based ceramic honeycombs able to produce a CO2-rich gas of 98% purity.26
The references to costs noted above were provided by developers and they are subject to a great degree of uncertainty. As pointed out in several studies dedicated to the analysis of the viability and cost of advanced DAC systems,27–34 there are strong arguments for claiming that the cost of CO2 captured from air will be in the range of 500–1000 $ per tCO2, which is about one order of magnitude higher than that of their counterpart technologies for CO2 capture from flue gases. Particularly relevant to the discussion in this work is the cost of the enormous capture device required to achieve the close contact of highly diluted molecules of CO2 in the air with the surface of the material with a high affinity and selectivity towards CO2. Fig. 1 represents the mass balance for a generic air capture device where 1 MtCO2 per year is captured. As can be seen, a huge flow of air (about 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m3 s−1 STP) must flow through the DAC device even if a large capture efficiency (ΔppmvCO2 ≈ 400 ppm between the inlet and the outlet will require deep reactors) is assumed. The reduction of the cross-sectional area of the contactor (Acontact) by increasing the air velocity will be limited by kinetic constraints at the gas/sorbent (or solvent) interface, by the pressure drop along the contactor, and by other phenomena such as water and solvent losses due to evaporation or entrainment.35
000 m3 s−1 STP) must flow through the DAC device even if a large capture efficiency (ΔppmvCO2 ≈ 400 ppm between the inlet and the outlet will require deep reactors) is assumed. The reduction of the cross-sectional area of the contactor (Acontact) by increasing the air velocity will be limited by kinetic constraints at the gas/sorbent (or solvent) interface, by the pressure drop along the contactor, and by other phenomena such as water and solvent losses due to evaporation or entrainment.35
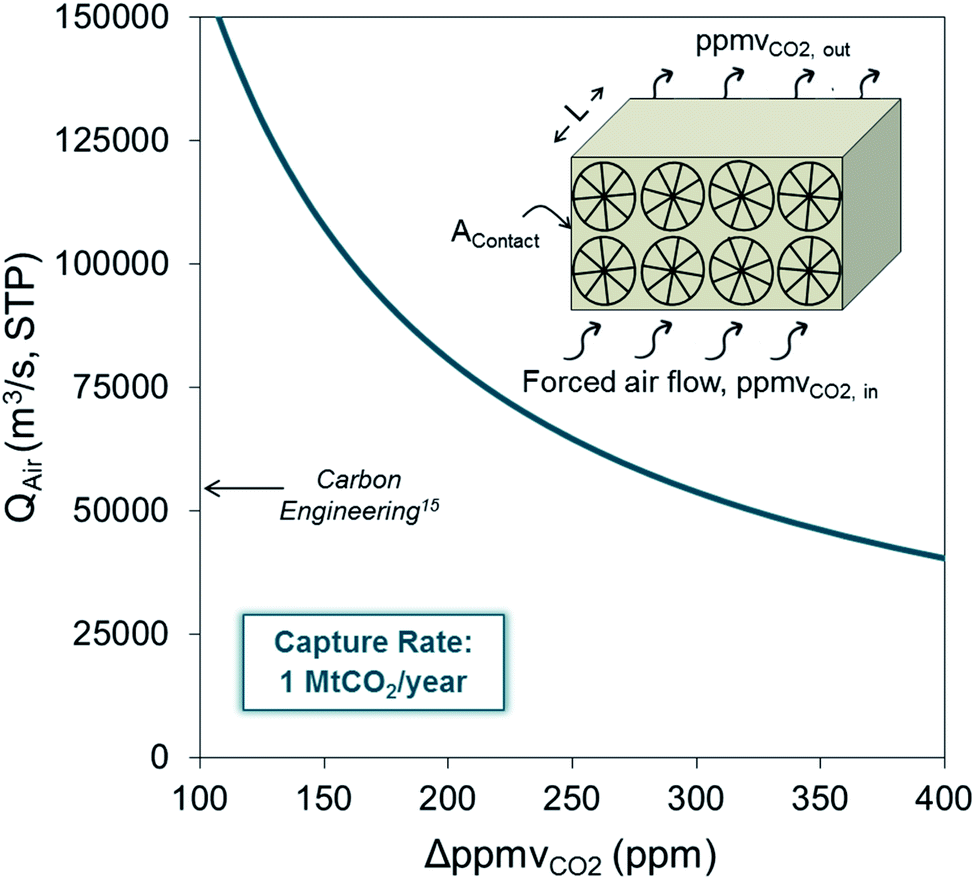 | ||
| Fig. 1 Volumetric flow of air (Qair in m3 s−1 STP) required to achieve a CO2 capture rate of 1 MtCO2 per year for a reduction in the CO2 concentration of ΔppmvCO2 (=ppmvCO2in − ppmvCO2out) in air as it circulates through the CO2 capture system (arrow marks the particular design point of Carbon Engineering,15 in the design of Climeworks36 the air flows range between 67000 and 150000 m3 s−1). | ||
In an attempt to overcome the costly barrier entailed by the capture device in large scale DAC systems, we propose in this work a passive CO2 capture system involving the carbonation of large purpose-built porous structures of low-cost Ca(OH)2. A basic cost analysis, which is mainly based on relatively well-known costs of oxy-combustion and hydration processes for obtaining Ca(OH)2 from CaCO3, demonstrates the economic feasibility of the proposed DAC process for a capture target of 1 MtCO2 per year.
2. Passive direct air capture process concept
Fig. 2 shows the general scheme of the concept investigated in this work. The design target is to estimate the necessary volume and cost of the porous passive carbonation infrastructure (represented as a blue box on the left-hand side of Fig. 2), which is aimed at capturing 1 MtCO2 per year, and the size, cost and energy requirements of the units needed to manufacture such structures in a continuous manner.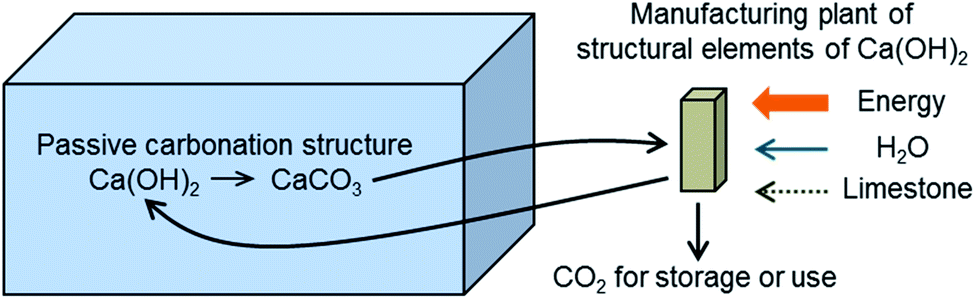 | ||
| Fig. 2 The concept of passive CO2 capture from air by the carbonation of a suitable structure of solid Ca(OH)2 exposed to ambient air. | ||
It should be noted that although in Fig. 2 we use the expression “CO2 for storage or use”, the negative emission character of DAC technologies necessarily requires the permanent storage of CO2, which must be added as a cost to account for avoided cost.37 If CO2 was used for the production of synthetic fuels with renewable energy, this would only lead to a maximum of 50% CO2 captured,38 which might be insufficient in future scenarios demanding negative emission technologies.2
The DAC concept shown in Fig. 2 is based on the known tendency of porous Ca(OH)2 solids to slowly carbonate in contact with ambient air. Anecdotally, the passive carbonation of solid beds composed of Ca(OH)2 is already commercially exploited for removing ambient CO2 in order to preserve fruit in storage halls.39,40 As a climate mitigation tool, the ambient carbonation of concrete and alkaline wastes obtained from steel, cement, lime and other solids containing free CaO and Ca(OH)2 is another passive method for sequestering CO2.2,41–49 However, to our knowledge, a comprehensive design and cost analysis of a purpose-built passive DAC system, such as that represented in Fig. 2, has not yet been reported in the literature. As will be explained below in more detail, the very low specific cost and widespread geographical availability of natural limestone make it possible to manufacture derived calcium-based materials cheaply by means of calcination and hydration. In this study, porous Ca(OH)2 has been chosen as the functional material for CO2 capture due to its suitable mechanical stability when pelletized and favorable carbonation kinetics compared to CaO.47,50,51
The slow kinetics of large porous carbonating structures can be modelled52 by accounting for the advance rate of a neat carbonation front according to Fick's law. In our case, where CO2 diffuses through the porous CaCO3 layer resulting from the carbonation of Ca(OH)2, assuming that there is no expansion of the porous solid during carbonation (i.e. ρCaCO3(1 − εCaCO3) = ρCa(OH)2(1 − εCa(OH)2)), the mass balance can be represented as follows:
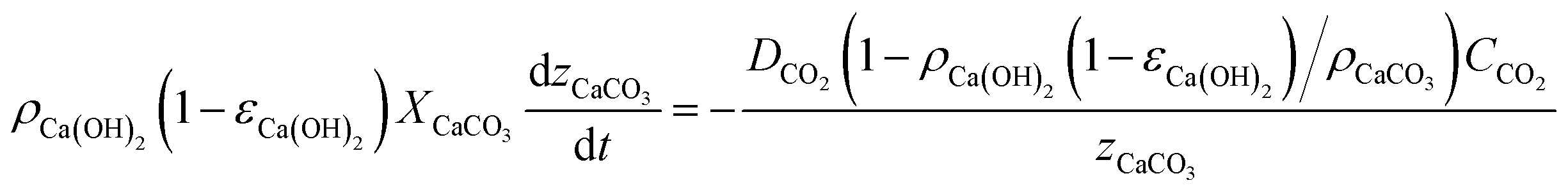 | (1) |
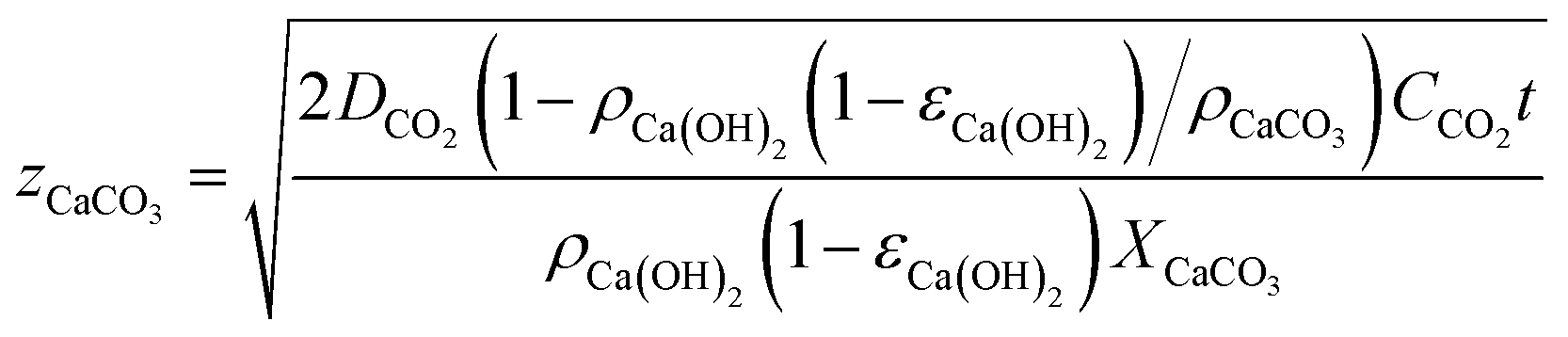 | (2) |
Fig. 3 shows the evolution with time of the carbonated layer in a Ca(OH)2-based solid according to eqn (2) for different values of Ca(OH)2 solid porosity, εCa(OH)2, and assuming a ppmvCO2 gradient of 400 ppm (CCO2 = ppmvCO2/(RT × 106) in mol m−3) between the external surface and the carbonation front. As can be seen, individual Ca(OH)2 solids of thickness 5 mm (assuming εCa(OH)2 = 0.5) would require around 15 days to approach total carbonation. This timescale is consistent with the experimental results of Erans et al.,47 who obtained carbonation conversions of around 70% after 12.5 days in particles of hydrated lime exposed to ambient air in layers of 5 mm thickness. Fig. 3 extends to an arbitrary scale of 1 year the results for thicker and less porous solids.
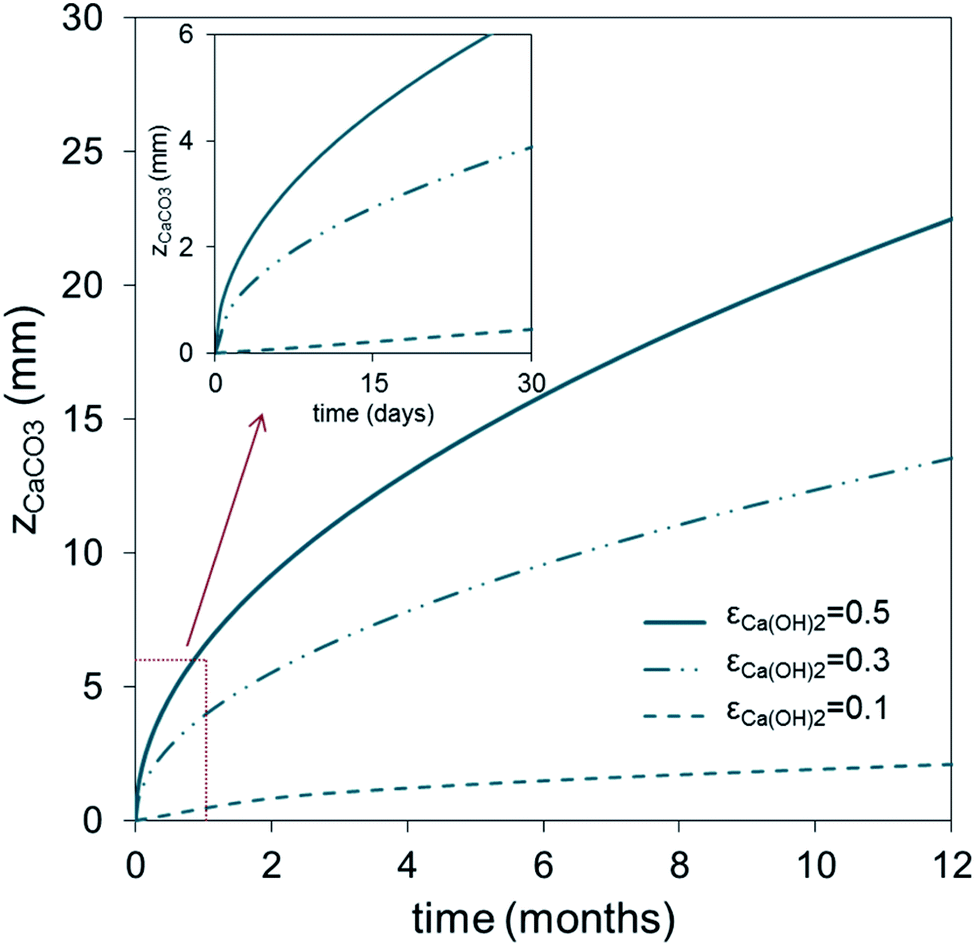 | ||
| Fig. 3 Evolution with time of the carbonated layer (zCaCO3) in carbonated solids according to eqn (2), for different porosities (εCa(OH)2) and assuming a CO2 concentration gradient of 400 ppmv at 20 °C. | ||
To take full advantage of such a slow passive carbonation process in a DAC system targeting 1 MtCO2 per year, amounts of calcium-based sorbent on a megaton scale will be required. Such a carbonating material could take in principle many different forms, ranging from a vast number of pellets of several centimeters thick, simply dispersed on the ground, to purpose-designed porous structures/buildings for passive carbonation and eventual recycling. In the first case, the manufacture of non-recoverable Ca(OH)2 pellets would involve the calcination of natural limestone to generate CaO (and eventually Ca(OH)2) and a pure stream of CO2 for permanent geological storage. Although there is evidence that soil liming provides nutrients for plant growth and contributes to acidity correction,56 further experimental investigation would be required to determine the environmental implications of this option, and the possible constraints related to land availability. For this reason, the present work focuses on large-scale structural alternatives (e.g. stacked plates, beams etc.) that entail the recovery and regeneration of the carbonating solids once they have been completely carbonated. There are already commercial processes for manufacturing solid structures with a high porosity (as in the case of pervious concrete57) and high mechanical stability (provided by the incorporation of cement-type additives58,59). Consequently, details about the structural and mechanical properties of the porous solids have not been discussed in depth in this work. Similarly, the operations designed to produce CaO and a separated stream of pure CO2 from limestone or carbonated solids are sufficiently well known from recent developments in clinker production with oxy-combustion60,61 and CO2 capture by Calcium Looping62,63 and so will only be discussed in the cost section.
3. Estimation of the volume of passive carbonation structures
Of the wide range of possible arrangements for the Ca(OH)2 structures, we have selected, for the purpose of illustration from the results of Fig. 3, flat plates 0.03 m thick with both sides exposed to the atmosphere and with a porosity εCa(OH)2 = 0.5 that can achieve total carbonation in approximately 6 months. An external exposed area of 50.7 × 106 m2 of such solids would be needed to capture 1 MtCO2 per year or, in other words, 761000 m3 of porous plates. With a specific minimum capture contactor volume of almost 0.8 m3 per tCO2 per year, this configuration is already 2–4 times larger than other competing air capture devices noted in the Introduction. However, it is possible to envisage recycling schemes (analogous to those already in operation for glass, metals, plastics etc.) promoting the deployment of disperse, but very large passive carbonation systems. In societies aiming at zero emission objectives, a person could reduce by 1 tCO2 per year his per capita emissions by participating in such a recycling scheme, if he/she were responsible for maintaining exposed to the atmosphere (i.e. on a house, in a garden, terrace or on the roof of a building) 25.4 m2 of plates exposed on both sides, and replaced approximately every 6 months. Much smaller surfaces or volumes of solids could also be considered if the thicknesses of the plates were reduced and the frequency of recycling was increased (see Fig. 3).
For the case of larger and centralized schemes, an important constraint affecting these carbonating structures is that they must be arranged in such a way that a continuous and renovated flow of air is in contact with the structure. To ensure this, further assumptions on the dimensions and the way the plates are stacked, including additional resistances other than those strictly related to the carbonation process inside the porous solids, must be taken into account (see Fig. 4).
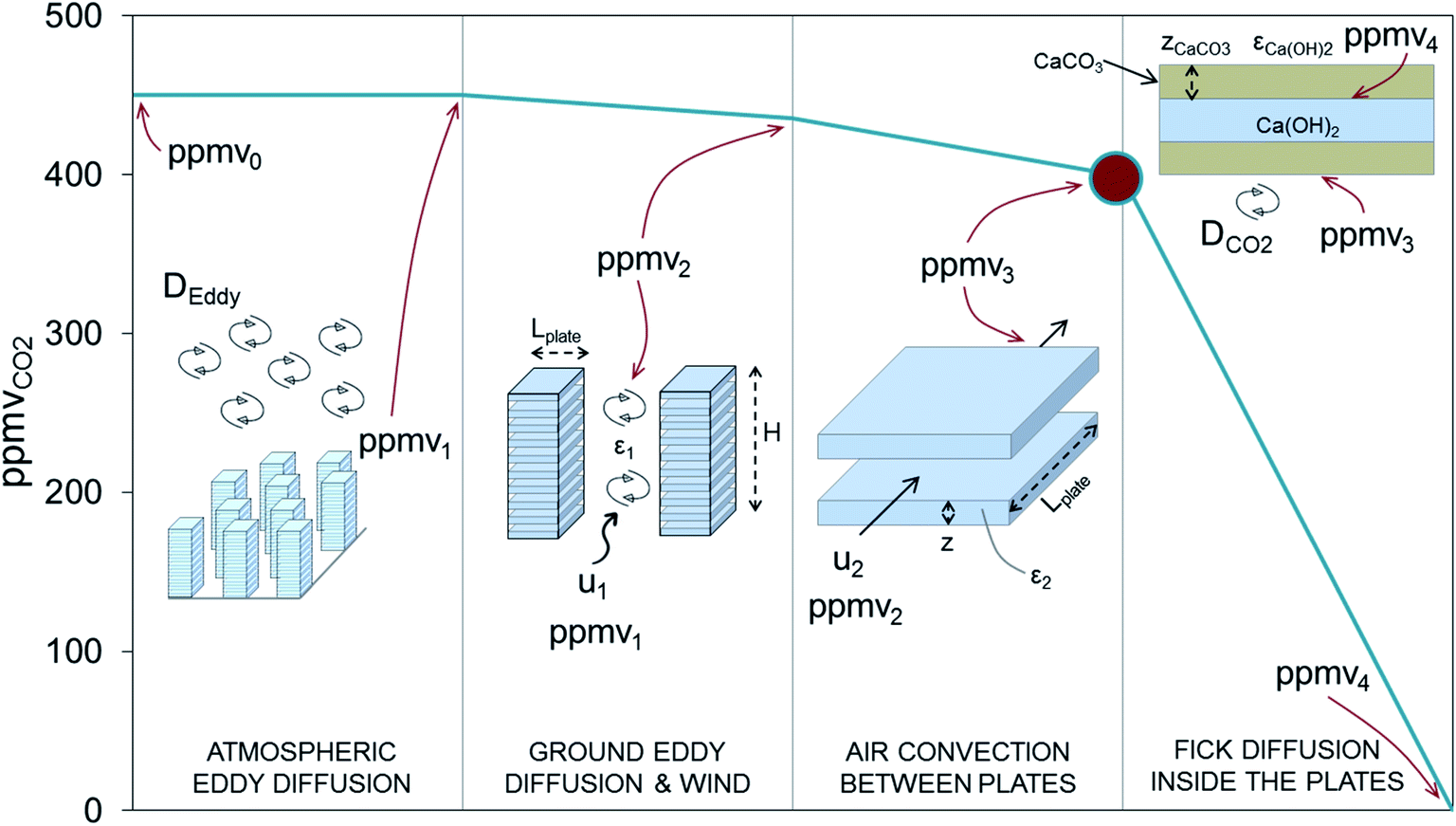 | ||
| Fig. 4 Evolution of the CO2 concentration in air through the different CO2 transport/diffusion stages, taking place at increasing scales from right to left: interior of a plate, individual plates, stacked plates and numerous stacked plates arranged in a field. | ||
The volume of the overall structure increases as additional free space is required to facilitate the access of CO2 from air to the external surface of each plate. Although the design decisions may differ considerably depending on local conditions (i.e. availability of wind and other atmospheric convective phenomena), the target is to design structures that are sufficiently expanded to ensure a high concentration of CO2 on the external surface of each individual plate (large dot in Fig. 4). The volume occupied by the stacks of plates will be Vstack = Vcarb/(1 − ε2), where ε2 is the volume fraction of the plates. In an oversimplification, it is assumed that CO2 is transported horizontally by wind velocity (u1) to the carbonating stacks. The actual velocity of the air circulating between the plates (u2) is given by the net dynamic pressure exerted by the wind (at a velocity u1) on the surface of the structure. This can be calculated by means of Bernoulli's equation and taking into account the friction factor and the geometry of the channel.64 With the aim of achieving a value of u2 > 0.5u1, we have chosen squares that measure 2 × 2 m2, arranged horizontally one on top of the other in the form of open structures 10 m high, with a space in between each plate that is twice their thickness (i.e. ε2 = 0.67). With these dimensions, and a front open stack cross-section of 0.8 Mm2 (=LplateHε2Nstacks), wind velocities u1 below 0.8 m s−1 (i.e. 3 km h−1) are high enough to sustain an overall carbonation process controlled by Fick's diffusion as in Fig. 4 (since ppmv3 − ppmv0 will exceed 50 ppmvCO2 for u1 > 0.8 m s−1). In many open field regions, average wind velocities of between 3 and 7 m s−1 are common.65,66 This will ensure a sufficient supply of air to the carbonating surfaces (except at the very beginning of the carbonation period, where zCaCO3 is close to zero and the rate of carbonation may be limited by the value of u1). Therefore, assuming a utilization factor for the carbonation structures of 1, a mass of 0.84 Mton of Ca(OH)2 will need to be replaced every 6 months to remove from air 1 Mton of CO2 per year. This amount of material will occupy a volume of about 761000 m3 of porous plates, which is equivalent to around 2.3 Mm3 of open stacks. Finally, when the stacked plates are arranged in a specially assigned open field (left hand side of Fig. 4), an extra-large volume of the overall structure (Vstructure = Vcarb/((1 − ε2)(1 − ε1))) will be needed to take into account the resistance linked to Eddy diffusion, which is perceptible in large CO2 sink areas, such as forests, where variations in the CO2 concentration of between 25 and 35 ppm occur.67,68 A value for ε1 of 0.95 may be required to ensure that there is sufficient separation between the stacks to allow isolated roughness flow.69 This will ensure the continuous renovation of CO2 in the vicinity of the stacked plates. The DAC system will therefore need to occupy a total volume of 45.7 Mm3 which means 4.6 Mm2 of land use when considering the height of stacked plates (i.e. H = 10 m).
To sum up, the specific contactor volumes necessary for the proposed passive air capture (which will range from 2.3 to 45.7 m3 per tCO2 per year) for highly dispersed stacked structures and for a purpose-designed DAC field, respectively, will be between one and two orders of magnitude higher than the values reported for the competing DAC systems referred to in the Introduction. However, as it will be claimed in the next section, this DAC scheme could be sufficiently competitive in the context of a carbon-constrained world.
4. The cost of the full DAC system
To estimate the cost of capturing and removing from the atmosphere a tonne of CO2 using the full DAC system shown in Fig. 2 we consider the following three main cost elements:(i) The specific cost of capturing and storing CO2 from the calcination of CaCO3, which includes the total capital requirements (TCRs) for the purchase of the oxy-combustion equipment for calcination, energy recovery and compression and purification, all amortized at a certain capital charge factor (CCF). Also, following the standards in major CO2 capture and storage (CCS) cost studies37,70 the CCS cost includes terms for the fixed and variable operating cost (FOM and VOM), the fuel cost (FC), and the cost of transport and permanent geological storage of CO2 (CCO2 T&S).
(ii) The specific cost linked to the handling and transport of structural elements (Chandling) as well as their manufacturing cost (Cmanufacture) from CaO generated in (i) and water.
(iii) The specific cost of the land occupied by the infrastructure (Vstructure/H)Cland, which is amortized as other capital requirements in (i).
Therefore:
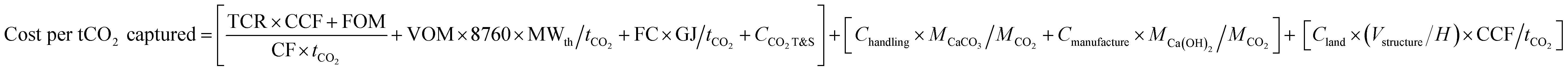 | (3) |
To gain transparency in the cost analysis carried out in this work, a set of minimum and maximum values of the specific cost of each cost component is adopted and justified below using references available in the literature, together with contingencies and other assumptions.
The specific cost of producing pure CO2 and CaO from CaCO3, and then transporting and storing geologically the captured CO2, can be estimated from a basic simulation of the required oxy-combustion process (see Fig. 5). Oxy-fired Circulating Fluidized Bed (CFB) technology has been chosen for the calcination operation, due to the substantial amount of information available in the literature on the cost of these systems for power generation,63,71 Calcium Looping processes62 or even cement plants.61,72 However, other types of calciners (e.g. rotary kilns, entrained beds etc.) with CO2 capture capability and different energy sources (i.e. gas, biomass, H2 or solar energy) may be available for consideration in the future.
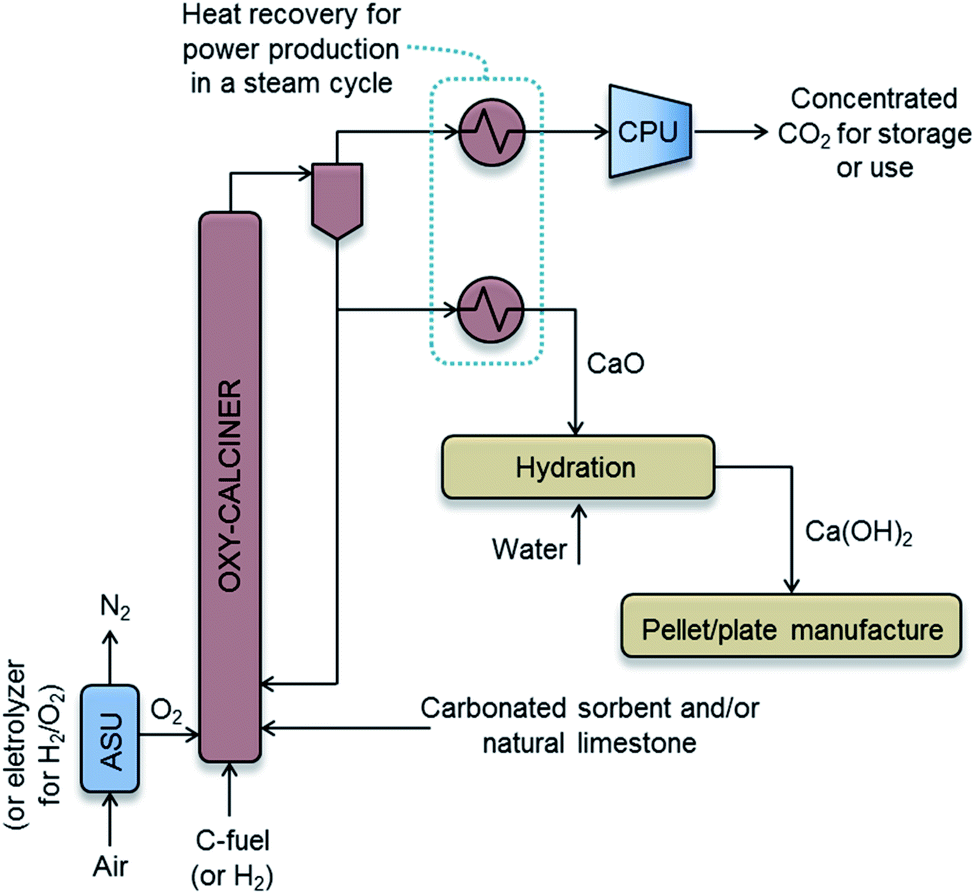 | ||
| Fig. 5 General process scheme of the oxy-fired CFB calcination system and Ca(OH)2 manufacture. | ||
Fig. 5 represents a basic scheme of the proposed process. A flow of 72 kg s−1 of CaCO3 needs to be calcined to achieve the DAC capture target of 1 MtCO2 per year. Following the cost estimation method reported by Guandalini et al.,63 mass and energy balances have been solved, indicating energy requirements for the oxy-calcination operation of 225 MWth (equivalent to 7.1 GJ per tCO2 captured from air), assuming calcination at 900 °C, no-preheating of the inlet streams, biomass as a fuel (assuming a LHV value of 20 MJ kg−1 and 41.4%C for these calculations) burnt with 10 vol% of excess O2, including the use of the Air Separation Unit, ASU, and Compression and Purification Unit, CPU, of 210 kW he per tO2 and 90 kW he per tCO2, respectively, and including also the consumption of about 2.3% fuel input in the auxiliary units.63 The waste heat extracted from the outlet CaO and CO2-rich gas streams (assumed to be at 900 °C) drives a stream cycle (not shown in Fig. 5 for the sake of simplicity) that generates 34 MWe, which is a sufficient power to cover the electricity consumed in the ASU, CPU and other auxiliary units. In other words, the system shown in Fig. 5 is designed as a small oxy-fired combustion boiler where parasitic power losses (mainly ASU and CPU electricity consumption) equal the gross power output, so that no electric power is exported or imported to run the system.
Reference costs for a typical oxy-combustion plant have been taken from Guandalini et al.63 and have been updated assuming an inflation rate of 1.7%/annually.73 The specific cost of a reference oxy-combustion plant of 550 MWe (or 1697 MWth) has been estimated to be about 4349 $ per kWe (or 1626 $ per kWth), which results in a total capital requirement (TCR) in 2011 USD of 2392 M$ (ref. 63) including process and project contingencies. Thus, when a scale factor of 0.7 is applied and the cost is updated to 2020 USD, the TCR for the proposed oxy-combustion system of 225 MWth amounts to 671 M$. Assuming a capital charge factor (CCF) of 10.88% per year (ref. 63) and a capacity factor (CF) of 0.9, the CAPEX of the oxy-combustion plant amounts to 81 $ per tCO2. Similarly and by comparison with the cost reported by Guandalini et al.,63 a reference cost of 12 M$ per year has been calculated for fixed operation costs (FOM) by considering a scale factor of 0.65 and 1 for the labor and property taxed and insurance terms, respectively, resulting in a OPEX fixed of 14 $ per tCO2. The variable costs (VOM) reported, without including the limestone cost, are about 9 $ per MW he,63 resulting in about 3 $ per MW hth when the electric efficiency and the inflation rate are applied. Considering that for a capture target of 1 MtCO2 per year are required 225 MWth, a variable OPEX of 7 $ per tCO2 has been calculated. Contingency factors as high as 40–60% are used to account for cost uncertainties in other DAC systems.29,35 We consider for this purpose, a maximum contingency factor of 40%, as the CAPEX and OPEX cost data used from Guandalini et al.63 already include standard process and project contingencies and there is direct industrial experience in oxy-calcination, solids handling and all the energy recovery operations represented in Fig. 5. Note that the CAPEX range (min value of 81 and max 113 $ per tCO2 in Table 1) can be considered conservative cost estimations compared to the CAPEX of the oxy-combustion calciner reported by Keith et al.15 claiming a cost at around 36 $ per tCO2.
| Cost component | Reference values | Cost CO2 captured ($ per tCO2) | %Total min–max | ||||
|---|---|---|---|---|---|---|---|
| Min | Max | Units | Min | Max | |||
| Oxy-combustion plant CAPEX | TCR | 671 | 805 | M$ | 81 | 113 | 59–33 |
| Oxy-combustion plant OPEX fixed | FOM | 12 | 15 | M$ per year | 14 | 20 | 10–6 |
| Oxy-combustion plant OPEX variable | VOM | 3 | 4 | $ per MW hth | 7 | 10 | 5–3 |
| Fuel | FC | 2 | 10 | $ per GJ | 14 | 71 | 10–21 |
| CO2 transport & storage | C CO2 T&S | 5 | 15 | $ per tCO2 | 5 | 15 | 4–4 |
| Handling & transport of structural elements | C handling | 4 | 25 | $ per tCaCO3 | 9 | 57 | 6–17 |
| Pellets/plates manufacture | C manufacture | 5 | 30 | $ per tCa(OH)2 | 8 | 50 | 6–15 |
| Land use | C land | 0 | 10 | $ per m2 | 0 | 5 | 0–1 |
| Total cost of CO 2 captured in $ per tCO 2 | 138 | 341 | |||||
On the other hand, different fuels can be considered to cover the thermal requirements in the oxy-calciner and their cost will vary depending on their type and resource availability. Thus a fuel cost (FC) of between 2 and 10 $ per GJ has been taken as reference numbers, resulting in 14 to 71 $ per tCO2 when considering that 7.1 GJ per ton of CO2 captured from air are required in the subsystem of Fig. 5. Finally for the cost of the CO2 transport and storage (CCO2 T&S), 5 and 15 $ per tCO2 have been taken to be consistent with published values.70
Regarding those cost elements in Table 1 and eqn (3) linked to land use and handling and manufacture of structural elements, the similarity with other existing unit operations facilitates cost estimations. The US average market value of crushed limestone is about 8.4 $ per t.74 Thus the minimum cost has been assumed as half the market value, accounting for a favourable scenario where CaCO3 is provided directly from the quarry with a very short distance transport. Meanwhile, the maximum value for the sensitivity analysis is arbitrarily taken by multiplying by three this figure to 25 $ per t of carbonated material (or 57 $ per tCO2 captured from air).
Regarding the manufacture of the Ca(OH)2 structural elements or pellets, the cost is subjected to larger uncertainties (e.g. small pellets with modest mechanical stability or large porous structures with improved mechanical properties or other design characteristics). A wide cost range from 5 to 30 $ per ton of Ca(OH)2 has been adopted as the manufacture cost (Cmanufacture), which results in 8 to 50 $ per tCO2.
Finally, since there are no special demands on the land to be used for the infrastructures, other than that of facilitating the exposure of a large volume of solids to the atmosphere, the minimum capital cost linked to land use can be assumed as zero. However, for more conservative scenarios, a royalty of Cland = 10 $ per m2 has been taken as the upper bound, consistent with the land cost of extensive energy infrastructures in the US.75 This results only in an increase of 5 $ per tCO2 when the same CCF as for the oxy-combustion plant is applied.
As can be seen in Table 1, the cost of the passive CO2 capture process from air will be between 140 and 340 $ per tCO2. As expected, the main cost driver of the DAC system is the capture and storage of CO2 generated from the oxy-combustion of CaCO3, with a contribution to the total cost of between 67 and 88%. Thus, since there is substantial industrial experience in the operations involved in such operation, the system investigated in this work will be relatively insensitive to additional cost contingencies when compared to other proposed DAC systems involving large contactors for the CO2 capture as discussed in Fig. 1.
5. Conclusions
The cost of Direct Air Capture (DAC) technologies is strongly influenced by the capital cost of a large device (i.e. about 0.2–0.4 m3 of the reactor volume per tCO2 per year) that is needed to bring a vast flow of air into contact with a functional material with an affinity for CO2. In an attempt to overcome this cost barrier, we have proposed design rules for a large-scale, purpose-built, passive carbonation system made up of porous structures of Ca(OH)2 (i.e. plates or pellets with a thickness of 0.03 m and a porosity of 0.5). A DAC system of this type could adopt different forms of viability: from widely dispersed approaches involving the irreversible dissemination of Ca(OH)2 pellets in the soil, or the public recycling of pellets/plates, to centralized schemes where vast structures of elements are stacked up in open fields. In the latter case, the volume of the structures must be able to increase (to up to 46 m3 per tCO2 per year) to ensure that there are enough wind and natural atmospheric convection currents to sustain a given average rate of carbonation. However, the key advantage of the proposed scheme is that the required materials are available at an extremely low cost (i.e. limestone and/or recycled carbonated structures from the DAC scheme) and that the technology required to manufacture them, while generating a pure stream of CO2, is already available (oxy-fuel combustion technologies for CaCO3 calcination). The overall cost estimation of this DAC scheme is between 140 and 340 $ per tCO2 captured, which is considerably less (subject to contingencies) than that of other competing DAC technologies.Nomenclature
| A contact | Air contactor cross-sectional area perpendicular to the air flow, m2 |
| CCF | Capital charge factor, per year |
| C CO2 | CO2 concentration at the external surface of the porous solid, mol m−3 |
| C CO2 T&S | Cost of transport and permanent geological storage of CO2, $ per tCO2 |
| CF | Capacity factor |
| C handling | Specific cost linked to the handling and transport of carbonated structural elements, $ per tCaCO3 |
| C land | Specific cost linked to the land use occupied by the DAC system, $ m−2 |
| C manufacture | Specific cost linked to manufacturing pellets/plates, $ per tCa(OH)2 |
| D CO2 | Diffusion coefficient of CO2, m2 s−1 |
| D Eddy | Eddy diffusion coefficient, m2 s−1 |
| FC | Fuel cost, $ per GJ |
| FOM | Fixed operation costs of the oxy-combustion plant, M$ per year |
| H | Height of the stacked plates, m |
| LHV | Lower heating value of the fuel used in oxy-combustion, MJ kg−1 |
| L plate | Length of the plates, m |
| M i | Molecular weight of the compound i, kg kmol−1 |
| MWth | Required thermal input to the oxy-combustion plant, MWth |
| N stacks | Number of stacks in the DAC system |
| ppmv0 | Atmospheric CO2 concentration, ppm |
| ppmv1 | CO2 concentration in the vicinity of the overall structure, ppm |
| ppmv2 | CO2 concentration outside the stacked plates, ppm |
| ppmv3 | CO2 concentration at the surface of the plates, ppm |
| ppmv4 | CO2 concentration in the carbonation reaction front in the interior of the porous plates or pellets, ppm |
| Q air | Volumetric air flow, m3 s−1 STP |
| R | Universal gas constant, atm L mol−1 K−1 |
| T | Temperature, K |
| t CO2 | CO2 capture rate, MtonCO2 per year |
| TCR | Total capital requirements of the oxy-combustion plant, M$ |
| u 1 | Wind velocity outside the stacked plates, m s−1 |
| u 2 | Velocity of air moving in the space between the plates, m s−1 |
| VOM | Variable operation costs, $ per MW hth |
| V stack | Volume occupied by the stacks of plates, m3 |
| V structure | Volume of the overall DAC structures |
| X CaCO3 | Maximum carbonation conversion of the Ca(OH)2 material |
| z | Flat plate thickness, m |
| z CaCO3 | Thickness of the carbonated product layer, m |
| ε 1 | Volume fraction between stacks |
| ε 2 | Volume fraction between plates |
| ε Ca(OH)2 | Porosity of the Ca(OH)2 layer |
| ε CaCO3 | Porosity of the CaCO3 layer |
| ΔppmvCO2 | Reduction in the CO2 concentration, ppm |
| ρ Ca(OH)2 | Ca(OH)2 molar density, mol m−3 |
| ρ CaCO3 | CaCO3 molar density, mol m−3 |
Conflict of interest
There are no conflicts of interest to declare.References
- IPCC, Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, Intergovernmental Panel on Climate Change, 2018 Search PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- K. S. Lackner, P. Grimes and H. J. Ziock, Presented in part at the 24th international technical conference on coal utilization and fuel systems, Clearwater, Florida (USA), 1999 Search PubMed.
- J. C. Minx, W. F. Lamb, M. W. Callaghan, L. Bornmann and S. Fuss, Environ. Res. Lett., 2017, 12, 035007 CrossRef.
- National Academies of Sciences Engineering and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, The National Academies Press, Washington, DC, 2019 Search PubMed.
- EASAC, Negative emission technologies: What role in meeting Paris Agreement targets? – EASAC policy report 35, European Academies' Science Advisory Council, Germany, 2018 Search PubMed.
- ICEF, Direct air capture of carbon dioxide, Innovation for Coal Earth Forum, 2018 Search PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- S. Elliott, K. S. Lackner, H. J. Ziock, M. K. Dubey, H. P. Hanson, S. Barr, N. A. Ciszkowski and D. R. Blake, Geophys. Res. Lett., 2001, 28, 1235–1238 CrossRef CAS.
- D. W. Keith and M. Ha-Duong, in Greenhouse Gas Control Technologies – 6th International Conference, ed. J. Gale and Y. Kaya, Pergamon, Oxford, 2003, pp. 187–192, DOI:10.1016/B978-008044276-1/50030-1.
- F. Zeman, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS PubMed.
- D. W. Keith, M. Ha-Duong and J. K. Stolaroff, Clim. Change, 2006, 74, 17–45 CrossRef CAS.
- R. Baciocchi, G. Storti and M. Mazzotti, Chem. Eng. Process., 2006, 45, 1047–1058 CrossRef CAS.
- D. W. Keith, G. Holmes, D. St. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- F. M. Brethomé, N. J. Williams, C. A. Seipp, M. K. Kidder and R. Custelcean, Nat. Energy, 2018, 3, 553–559 CrossRef.
- B. Barkakaty, B. G. Sumpter, I. N. Ivanov, M. E. Potter, C. W. Jones and B. S. Lokitz, Environ. Tech. Innovations, 2017, 7, 30–43 CrossRef.
- G. Santori, C. Charalambous, M. C. Ferrari and S. Brandani, Energy, 2018, 162, 1158–1168 CrossRef CAS.
- S. Stucki, A. Schuler and M. Constantinescu, Int. J. Hydrogen Energy, 1995, 20, 653–663 CrossRef CAS.
- K. S. Lackner, Eur. Phys. J.: Spec. Top., 2009, 176, 93–106 Search PubMed.
- C. F. de Lannoy, M. D. Eisaman, A. Jose, S. D. Karnitz, R. W. DeVaul, K. Hannun and J. L. B. Rivest, Int. J. Greenhouse Gas Control, 2018, 70, 243–253 CrossRef CAS.
- M. D. Eisaman, J. L. B. Rivest, S. D. Karnitz, C. F. de Lannoy, A. Jose, R. W. DeVaul and K. Hannun, Int. J. Greenhouse Gas Control, 2018, 70, 254–261 CrossRef CAS.
- Climeworks, Capturing CO2 from airhttp://www.climeworks.com/ Search PubMed.
- Global Thermostat, Global Thermostat – A carbon negative solution, http://www.globalthermostat.com Search PubMed.
- P. Eisenberger, US 9227153 B2, 2016.
- B. Soltoff, Inside ExxonMobil's hookup with carbon removal venture Global Thermostat, https://www.greenbiz.com/article/inside-exxonmobils-hookup-carbon-removal-venture-global-thermostat Search PubMed.
- M. Ranjan and H. J. Herzog, Energy Procedia, 2011, 4, 2869–2876 CrossRef CAS.
- K. Z. House, A. C. Baclig, M. Ranjan, E. A. van Nierop, J. Wilcox and H. J. Herzog, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20428–20433 CrossRef CAS PubMed.
- M. Mazzotti, R. Baciocchi, M. J. Desmond and R. H. Socolow, Clim. Change, 2013, 118, 119–135 CrossRef CAS.
- P. Smith, S. J. Davis, F. Creutzig, S. Fuss, J. Minx, B. Gabrielle, E. Kato, R. B. Jackson, A. Cowie, E. Kriegler, D. P. van Vuuren, J. Rogelj, P. Ciais, J. Milne, J. G. Canadell, D. McCollum, G. Peters, R. Andrew, V. Krey, G. Shrestha, P. Friedlingstein, T. Gasser, A. Grübler, W. K. Heidug, M. Jonas, C. D. Jones, F. Kraxner, E. Littleton, J. Lowe, J. R. Moreira, N. Nakicenovic, M. Obersteiner, A. Patwardhan, M. Rogner, E. Rubin, A. Sharifi, A. Torvanger, Y. Yamagata, J. Edmonds and C. Yongsung, Nat. Clim. Change, 2016, 6, 42 CrossRef CAS.
- S. Fuss, W. F. Lamb, M. W. Callaghan, J. Hilaire, F. Creutzig, T. Amann, T. Beringer, W. de Oliveira Garcia, J. Hartmann, T. Khanna, G. Luderer, G. F. Nemet, J. Rogelj, P. Smith, J. L. V. Vicente, J. Wilcox, M. M. Zamora Dominguez and J. C. Minx, Environ. Res. Lett., 2018, 13, 063002 CrossRef.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- S. Brandani, Energy Environ., 2012, 23, 319–328 CrossRef.
- C. Pritchard, A. Yang, P. Holmes and M. Wilkinson, Process Saf. Environ. Prot., 2015, 94, 188–195 CrossRef CAS.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Direct air capture of CO2 with chemicals: A technology assessment for the APS Panel on Public Affairs, American Physical Society, Washington, DC, 2011 Search PubMed.
- C. Gebald, W. Meier, N. Repond, T. Ruesch and A. Wurzbacher, US2017/0106330A1, 2017.
- IPCC, Special report on carbon dioxide capture and storage, Intergovernmental Panel on Climate Change, New York, USA, 2005 Search PubMed.
- J. C. Abanades, E. S. Rubin, M. Mazzotti and H. J. Herzog, Energy Environ. Sci., 2017, 10, 2491–2499 RSC.
- Carmeuse, http://www.carmeuse-agriculture.com/our-products/fruitcal, accessed November 2019.
- J. A. Bartsch, New York Fruit Quart., 2004, 12, 11–12 Search PubMed.
- V. Nikulshina, C. Gebald and A. Steinfeld, Chem. Eng. J., 2009, 146, 244–248 CrossRef CAS.
- A. Kirchofer, A. Becker, A. Brandt and J. Wilcox, Environ. Sci. Technol., 2013, 47, 7548–7554 CrossRef CAS PubMed.
- S. Talukdar and N. Banthia, Constr. Build. Mater., 2013, 40, 775–782 CrossRef.
- F. Xi, S. J. Davis, P. Ciais, D. Crawford-Brown, D. Guan, C. Pade, T. Shi, M. Syddall, J. Lv, L. Ji, L. Bing, J. Wang, W. Wei, K.-H. Yang, B. Lagerblad, I. Galan, C. Andrade, Y. Zhang and Z. Liu, Nat. Geosci., 2016, 9, 880 CrossRef CAS.
- D. P. Hanak and V. Manovic, Energy Convers. Manage., 2018, 160, 455–466 CrossRef CAS.
- D. P. Hanak, B. G. Jenkins, T. Kruger and V. Manovic, Appl. Energy, 2017, 205, 1189–1201 CrossRef CAS.
- M. Erans, S. A. Nabavi and V. Manović, J. Cleaner Prod., 2020, 242, 118330 CrossRef.
- M. Samari, F. Ridha, V. Manovic, A. Macchi and E. J. Anthony, Mitig. Adapt. Strat. Gl., 2019 DOI:10.1007/s11027-019-9845-0.
- P. Renforth, Nat. Commun., 2019, 10, 1401 CrossRef PubMed.
- R. M. Dheilly, J. Tudo and M. Quéneudec, J. Mater. Eng. Perform., 1998, 7, 789–795 CrossRef CAS.
- V. Morales-Flórez, A. Santos, I. Romero-Hermida and L. Esquivias, Chem. Eng. J., 2015, 265, 194–200 CrossRef.
- H. Klopfer, Bautenschutz Bausanier, 1978, 1, 86–97 Search PubMed.
- H. Hilsdorf and J. Kropp, Performance criteria for concrete durability, CRC Press, London, United Kingdom, 1995 Search PubMed.
- B. Lagerblad, Carbon dioxide uptake during concrete life cycle – State of the art, NORDEN – Nordic Innovation Centre project, Stockholm, 2005 Search PubMed.
- W. Ashraf, Constr. Build. Mater., 2016, 120, 558–570 CrossRef CAS.
- D. J. Beerling, J. R. Leake, S. P. Long, J. D. Scholes, J. Ton, P. N. Nelson, M. Bird, E. Kantzas, L. L. Taylor, B. Sarkar, M. Kelland, E. DeLucia, I. Kantola, C. Müller, G. Rau and J. Hansen, Nat. Plants, 2018, 4, 138–147 CrossRef.
- J. Mullaney and T. Lucke, CLEAN – Soil, Air, Water, 2014, 42, 111–124 CrossRef CAS.
- Y. A. Criado, M. Alonso and J. C. Abanades, Ind. Eng. Chem. Res., 2015, 54, 9314–9327 CrossRef.
- K. G. Sakellariou, N. I. Tsongidis, G. Karagiannakis and A. G. Konstandopoulos, Energy Fuels, 2017, 31, 6548–6559 CrossRef CAS.
- M. Voldsund, S. O. Gardarsdottir, E. De Lena, J.-F. Pérez-Calvo, A. Jamali, D. Berstad, C. Fu, M. Romano, S. Roussanaly, R. Anantharaman, H. Hoppe, D. Sutter, M. Mazzotti, M. Gazzani, G. Cinti and K. Jordal, Energies, 2019, 12, 559 CrossRef.
- S. O. Gardarsdottir, E. De Lena, M. Romano, S. Roussanaly, M. Voldsund, J.-F. Pérez-Calvo, D. Berstad, C. Fu, R. Anantharaman, D. Sutter, M. Gazzani, M. Mazzotti and G. Cinti, Energies, 2019, 12, 542 CrossRef CAS.
- J. C. Abanades, B. Arias, A. Lyngfelt, T. Mattisson, D. E. Wiley, H. Li, M. T. Ho, E. Mangano and S. Brandani, Int. J. Greenhouse Gas Control., 2015, 40, 126–166 CrossRef CAS.
- G. Guandalini, M. C. Romano, M. Ho, D. Wiley, E. S. Rubin and J. C. Abanades, Int. J. Greenhouse Gas Control, 2019, 84, 219–231 CrossRef CAS.
- R. B. Perry and D. W. Green, Perry's Chemical Engineers' Handbook, McGraw-Hill, 1999 Search PubMed.
- Global Wind Atlas, https://globalwindatlas.info/ Search PubMed.
- Windy, http://www.windy.com Search PubMed.
- B. Chen, J. M. Chen, J. Liu, D. Chan, K. Higuchi and A. Shashkov, J. Geophys. Res.: Atmos., 2004, 109, D04306 Search PubMed.
- M. K. Dubey, H. Ziock, G. Rueff, S. Elliott, W. S. Smith, K. S. Lackner and N. A. Johnston, Fuel Chemistry Division Preprints, 2002, 47, 81–84 CAS.
- T. R. Oke, Energy Build., 1988, 11, 103–113 CrossRef.
- E. S. Rubin, J. E. Davison and H. J. Herzog, Int. J. Greenhouse Gas Control, 2015, 40, 378–400 CrossRef CAS.
- DOE/NETL, Cost and performance for low rank pulverized coal oxycombustion energy plants, Report DOE/NETL-401/093010 US Department of Energy, National Energy Technology Laboratory, 2010 Search PubMed.
- IEAGHG, CO2 capture in the cement industry, Report 2008/3, International Energy Agency Greenhouse Gas R&D Programme, 2008 Search PubMed.
- Trading Economics, United States Inflation Rate, https://tradingeconomics.com/united-states/inflation-cpi Search PubMed.
- USGS, Crushed stone statistics and information, https://www.usgs.gov/centers/nmic/crushed-stone-statistics-and-information, accessed November 2019 Search PubMed.
- DOE/NETL, Cost and performance baseline for fossil energy plants volume 1a: Bituminous coal (PC) and natural gas to electricity. Revision 3, Report DOE/NETL-2015/1723, US Department of Energy, National Energy Technology Laboratory, 2015 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |