
High performance polyanthraquinone/Co–Ni(OH)2 aqueous batteries based on hydroxyl and potassium insertion/extraction reactions†
Chang
Liu
a,
Ting
Ma
a,
Kexin
Xia
a,
Xuesen
Hou
a,
Qingshun
Nian
a,
Yichao
Cai
a and
Jing
Liang
 *ab
*ab
aKey Laboratory of Advanced Energy Materials Chemistry, Ministry of Education, College of Chemistry, Nankai University, Tianjin 300071, PR China
bRenewable Energy Conversion and Storage Center, Nankai University, Tianjin 300071, PR China. E-mail: jingliang@nankai.edu.cn
First published on 16th October 2019
Abstract
Aqueous potassium ion batteries (APIBs) are safe, robust, and environmentally friendly energy storage equipments. In this work, a high performance K+/OH− hybrid ion aqueous battery, using Co–Ni(OH)2 as the cathode and poly(1,4-anthraquinone) (P14AQ) as the anode, is proposed and characterized. By tuning the polymerization degree of P14AQ, the P14AQ/Co–Ni(OH)2 battery delivers a high energy/power density of 93 W h kg−1 at 1040 W kg−1 with an improved cycle life. Moreover, this P14AQ/Co–Ni(OH)2 battery shows a wide temperature operation window ranging from −30 °C to 25 °C. This proposed system is expected to be a promising battery for large-scale energy storage.
Introduction
Rechargeable aqueous batteries have attracted much attention for electrochemical energy storage because of their low cost, good safety and fast kinetics.1–11 Among them, aqueous potassium ion batteries (APIBs) stand out owing to the high natural abundance and wide availability of potassium.12,13 The relatively higher ionization potential and larger ionic radius of potassium ions, however, limit their further application.14–16 Prussian Blue Analogues (PBAs) are widely investigated as promising cathode materials in APIBs, but their low capacity and conductivity lead to a severe capacity fade or over potential.17,18 Consequently, it is crucial to seek electrode materials possessing a high capacity and an excellent cycling stability.Low capacity is always a crucial problem in APIBs. The specific capacities of inorganic cathodes and anodes reported are lower than 125 mA h g−1.13,17,18 The organic compounds are potential candidates due to their diversity and the multi-electron reaction mechanism.19–24 Most suitable small organic molecules for electrodes are generally soluble in electrolytes.24 Polymerization is an efficient method to solve the dissolution issue by increasing the molecular weight of organic molecules. Recently, polymers of anthraquinones (PAQs) as the anode have been investigated to develop aqueous rechargeable PAQs/Ni(OH)2 batteries in APIBs, such as poly(anthraquinonyl sulfide) (PAQS),25 oligo(anthraquinonyl sulfide) (OAQS) and oligohexane(anthraquinone sulfide) (OHAQS).26 Typically, functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in these analogues that have a similar chemical environment should present a similar capability in batteries. In contrast, their electrochemical performances were different, which was ascribed to different molecular weights.27 So it is important to inhibit the dissolution process through the control of the polymer's polymerization degree. However, it is hard to test the PAQS molecular weight by common measurements such as matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS).28 Therefore, the relationship between the molecular weight and electrochemical performance cannot be investigated. As a result, we chose poly(1,4-anthraquinone) (P14AQ) as the anode material whose molecular weight can be easily tested.29 This polymer has been used in lithium ion batteries and showed excellent electrochemical performance.30 In this work, P14AQ was used in APIBs for the first time. P14AQ has a two-electron transfer reaction with a redox potential at −0.6 V vs. SHE (standard hydrogen electrode) before H2 evolution in alkaline solution. The solubility of P14AQ in electrolyte is dependent on the polymerization degree of the polymer,27 which can be controlled by changing the temperature of the synthesis reaction.
O in these analogues that have a similar chemical environment should present a similar capability in batteries. In contrast, their electrochemical performances were different, which was ascribed to different molecular weights.27 So it is important to inhibit the dissolution process through the control of the polymer's polymerization degree. However, it is hard to test the PAQS molecular weight by common measurements such as matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF-MS).28 Therefore, the relationship between the molecular weight and electrochemical performance cannot be investigated. As a result, we chose poly(1,4-anthraquinone) (P14AQ) as the anode material whose molecular weight can be easily tested.29 This polymer has been used in lithium ion batteries and showed excellent electrochemical performance.30 In this work, P14AQ was used in APIBs for the first time. P14AQ has a two-electron transfer reaction with a redox potential at −0.6 V vs. SHE (standard hydrogen electrode) before H2 evolution in alkaline solution. The solubility of P14AQ in electrolyte is dependent on the polymerization degree of the polymer,27 which can be controlled by changing the temperature of the synthesis reaction.
Meanwhile, for the cathode, it is noted that the specific capacity of Ni(OH)2 (289 mA h g−1 for β-Ni(OH)2) is over twice that of potassium intercalation compounds (less than 120 mA h g−1).18 The Ni(OH)2 cathode with OH− insertion/extraction reactions is widely used in aqueous batteries, such as nickel-metal hydride batteries.10 So we used Ni(OH)2 as the cathode in APIBs. A K+/OH− hybrid ion battery is consequently formed.
Herein, we designed a P14AQ/Co–Ni(OH)2 rechargeable battery in aqueous K+ electrolyte by using β-Ni(OH)2 as the cathode and P14AQ as the anode, based on OH− insertion/extraction reactions and reversible CO to C–O–K transformation. A small amount of Co in Ni(OH)2 played the role of the conductor.32 During the charge process, K+ reacted with the P14AQ anode to produce P14AQ-2K, while OH− was simultaneously inserted into the Ni(OH)2 cathode (Fig. 1a). P14AQ was synthesized with different molecular weights by controlling synthesis conditions. With M-9000 (molecular weight is 9000), the P14AQ/Co–Ni(OH)2 battery presented a high energy/power density of 93 W h kg−1 at 1040 W kg−1 (based on the mass of active materials on electrodes and KOH reacted) and a good cycling stability (83.2% capacity retention after 500 cycles). The battery temperature tolerance was also investigated, and it demonstrated a very excellent cycling stability with a wide temperature range from −30 °C to 25 °C.
 | ||
| Fig. 1 (a) The redox mechanism of Co–Ni(OH)2 and P14AQ. (b) CV curve of P14AQ in 13.0 M KOH at 50 mV s−1. (c) CV curve of Co–Ni(OH)2 in 13.0 M KOH at 5 mV s−1. | ||
Results and discussion
By using 1,4-dichloroanthraquinone (1,4-DCAQ) as the monomer, P14AQ was synthesized through a simple one-step condensation polymerization reaction.29,31 The detailed synthesis procedures are provided in the ESI.† P14AQ was characterized by nuclear magnetic resonance spectrometry (NMR) and attenuated total reflectance-fourier transform infrared spectrometry (ATR-FTIR) (see Fig. S2–S4 in the ESI†). The results indicated that P14AQ was pure.Finally, three P14AQ samples with different polymerization degrees were obtained by altering temperatures of the reaction.31 MALDI-TOF-MS confirmed that the largest molecular weights of the as-obtained three polymers were about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 9000 and 8000, respectively, (correspondingly denoted as M-10000, M-9000 and M-8000) (Fig. S5†). In other words, the repeating units of polymers were 48, 43 and 38. Their morphologies and composition were observed by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and EDS-mapping (Fig. S6 and S7†). The prepared P14AQ exhibited a homogeneous spherical morphology with a diameter of about nearly 200 nm. With the increase of molecular weight, the sizes of the three samples remained similar. Thermogravimetric analysis (TG) profiles (Fig. S8†) showed that their thermal stability was superior.
000, 9000 and 8000, respectively, (correspondingly denoted as M-10000, M-9000 and M-8000) (Fig. S5†). In other words, the repeating units of polymers were 48, 43 and 38. Their morphologies and composition were observed by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and EDS-mapping (Fig. S6 and S7†). The prepared P14AQ exhibited a homogeneous spherical morphology with a diameter of about nearly 200 nm. With the increase of molecular weight, the sizes of the three samples remained similar. Thermogravimetric analysis (TG) profiles (Fig. S8†) showed that their thermal stability was superior.
Co–Ni(OH)2 was reused from a commercially available NiMH battery (PISEN AA/HR 15/51). Fig. S9† displayed the powder X-ray diffraction (XRD) patterns of the Co–Ni(OH)2 sample. We observed that the Co–Ni(OH)2 sample showed multiple peaks of varying intensity, which matched well with Ni(OH)2 (JCPDS no. 1-1047). The SEM image of the Co–Ni(OH)2 sample in Fig. S10a† showed homogeneous sheets. The TEM elemental mappings of the Co–Ni(OH)2 sample in Fig. S10c† indicated that the elements of Ni, O and Co distributed homogeneously in the sample. The molar ratio of Ni:O:Co was 32.96:64.99:2.05 revealed by energy-dispersive spectrometry (EDS) image in Fig. S10b.† The ratio of Ni:O was close to 1:2, which was in accordance with the Ni(OH)2 composition. The XPS spectra in Fig. S11† also showed the existence of Co in the Ni(OH)2 sample.
Electrochemical characterization of P14AQ and Co–Ni(OH)2 was performed by employing cyclic voltammetry (CV) and galvanostatic measurements in three-electrode cells. Then, all potentials were presented versus the Hg/HgO reference electrode. The current–voltage profiles of P14AQ (red) and Co–Ni(OH)2 (blue) are shown in Fig. 1b and c. The CV data indicated that reversible redox reactions of P14AQ underwent at −0.7 V (versus Hg/HgO) in 13.0 M KOH. Under the same conditions, the redox potential of Co–Ni(OH)2 was around 0.3 V. This indicated that the average voltage of a full cell would be above 1.0 V, which is quite competitive in aqueous batteries. The galvanostatic profile of Co–Ni(OH)2 is shown in Fig. S12.† At 260 mA g−1, Co–Ni(OH)2 exhibited a reversible capacity of 200 mA h g−1.
The electrochemical performance of P14AQ was tested based on various distributions of the polymerization degree. It was reported that polymerization is an efficient method to inhibit the dissolution of the active material.32 To discuss this issue, the electrochemical performances of the as-prepared M-8000, M-9000 and M-10000 were explored.
First, M-8000, M-9000 and M-10000 were used as working electrodes in three-electrode cells separately. In the second cycle, the galvanostatic profiles of M-8000, M-9000 and M-10000 showed well-defined discharge (potassiation) plateaus at −0.8 V and charge (depotassiation) plateaus at −0.6 V (Fig. S13†), which fitted well with CV data. Based on the theoretical capacity when two K+ ions bind with one anthraquinone (AQ) molecule, 1C was defined as 260 mA g−1. At 1C, M-8000, M-9000 and M-10000 exhibited reversible capacities of 198, 230 and 166 mA h g−1 in the first cycle. Among the three samples, M-9000 gave the highest discharge capacity.
Then the full cell performances of three polymer anodes were tested. In the first cycle, the charge capacity of M-8000, M-9000 and M-10000 was 250, 228 and 156 mA h g−1 (Fig. 2a). The charge capacity of the first cycle increased when the molecular weight decreased. This was because active sites would be harder to react with when the polymerization degree was higher. The discharge capacity of the first cycle was 198, 217 and 154 mA h g−1. M-8000 gave a much lower discharge capacity than M-9000. This was due to the dissolution problem of active materials in the charge process. In the charge process, P14AQ transformed into P14AQ-2K (the mechanism will be verified later) (Fig. 1a). The cause of capacity loss was that P14AQ-2K dissolved in electrolyte and couldn't be oxidized to P14AQ on the electrode. In the charge process, M-8000–2k dissolved more than M-9000–2k. As a result, the discharge capacity of M-8000 was smaller than that of M-9000. To verify this point, ultraviolet-visible spectroscopy (UV-Vis) tests were performed. After charging to 1.2 V for the first time, the separators were removed, then oxidized in air and immerged in dichloromethane to perform the UV-Vis test. For polymers with a larger polymerization degree, weaker absorption peaks appeared, proving the suppression of the dissolution process (Fig. S14†). We also noticed the growth of M-8000 after the first cycle (Fig. S15†), which occurred through the dissolution-reprecipitation process. The growing crystal may expand the electrochemically inactive region of the electrode, which caused capacity fading.33Fig. 2b and c present the rate capabilities of M-8000, M-9000 and M-10000 electrodes in full cells from 1C to 16C (4.16 A g−1). The potentials of full cells were more than 1.0 V, which were proved by CV at 0.1 mV s−1 (Fig. S16†). Among them, M-9000 showed the best rate performance. It had a remarkably high specific capacity of 228 mA h g−1 at a current density of 1C and maintained specific capacities of 216, 204, 190 and 174 mA h g−1 at current densities of 2, 4, 8 and 16C (4.16 A g−1), respectively (the current densities and capacities are calculated based on the mass of P14AQ in the anode). Meanwhile, M-10000 exhibited relatively poor rate capability and it may be due to the low conductivity.
 | ||
| Fig. 2 (a) Initial discharge–charge profiles of full cells of M-8000, M-9000 and M-10000. (b) Initial discharge–charge profiles of full cells of M-9000 at current rates of 1, 2, 4, 8 and 16C. (c) Rate performance of P14AQ/Co–Ni(OH)2 batteries of M-8000, M-9000 and M-10000. (d) The cycling performance and coulombic efficiencies of P14AQ/Co–Ni(OH)2 batteries of M-8000, M-9000 and M-10000 at 8C (2.08 A g−1). | ||
To fully illustrate the advantages of M-9000, the cycling performances of the three polymers with different molecular weights were investigated at 8C (2.08 A g−1) (Fig. 2d). All three polymers needed the activation process. It can be seen that the M-8000 electrode suffered from fast capacity decay and the capacity was just only 124 mA h g−1 (∼74.0%) after 500 cycles owing to the dissolution during potassiation/depotassiation. After 500 cycles, the capacity retentions of M-9000 and M-10000 were 83.2% and 93.2%, respectively, which were much higher than those of M-8000 (∼74.0%). The different electrochemical performances among three polymers could be attributed to different molecular weights. On the one hand, the low molecular weight of P14AQ (e.g. M-8000) would lead to its dissolution throughout the whole cycles as previously indicated, and the dissolution rate was the largest in the first charge process. This result would inevitably aggravate the capacity fading. On the other hand, long chains of P14AQ (e.g. M-10000) would result in increasing inactive sites and reducing electrical conductivity. Although its cycling durability has been improved, its specific capacity was much lower than that of M-9000. Besides, the long cycling life was due to the stability of Co–Ni(OH)2 and M-9000. After 500 cycles at 8C, H-NMR of M-9000 and XRD of Co–Ni(OH)2 were performed. As shown in Fig. S17 and 18,† the compositions of M-9000 and Co–Ni(OH)2 didn't change.
The M-9000/Co–Ni(OH)2 battery had a specific energy of 93 W h kg−1 and a specific power of 1040 W kg−1 (based on the mass of active materials on electrodes and reacted KOH) (Fig. 3a). The formula for calculating energy density is in eqn S1.† These properties were notable because high specific power could be simultaneously achieved along with high specific energy, thus making these batteries potentially competitive against other APIBs13,17,18,25,26 and lead-acid batteries34 (Table S1†). As Fig. 3b indicates, the “AOB” (Aqueous Organic Batteries)-shaped LED lights could be lit by connecting two full cells in series.
 | ||
| Fig. 3 (a) Energy density comparison of the M-9000/Co–Ni(OH)2 battery with several reported APIBs and other devices. (b) Photograph of small LED lights powered by two full cells connected in series. (c) M-9000/Co–Ni(OH)2 charge/discharge profiles of the second cycle at −30 °C, −15 °C and 25 °C. (d) Discharge capacity versus the cycle number of the M-9000/Co–Ni(OH)2 battery at 1C at −30 °C, −15 °C and 25 °C. | ||
Besides, the temperature effect on the M-9000's cycles in the P14AQ/Co–Ni(OH)2 battery was evaluated from −30 °C to 25 °C at 1C. Fig. 3c presents full cell charge/discharge profiles of the second cycle at different temperatures. It could be noticed that when reducing the temperature, the reversible capacity decreased slightly and the potential difference between charge and discharge plateaus increased. This could be attributed to the decreased conductivities and increased polarization at lower temperature.35Fig. 3d presents the cycling performance of the M-9000/Co–Ni(OH)2 battery at different temperatures at 1C. When temperatures were −15 °C and −30 °C, the discharge capacities were 185.9 mA h g−1 and 148.7 mA h g−1, respectively. The capacity retention was nearly 100% over 50 cycles. At −30 °C, the M-9000/Co–Ni(OH)2 battery was able to cycle for 300 times at 1C with 100% capacity retention (Fig. S19†). The excellent performance at low temperature was because the electrochemical reaction of P14AQ depended less on temperature.25 This reaction would be discussed in the following part. In addition, the electrolyte (13.0 M KOH) didn't freeze even at −40 °C for 5 hours (Fig. S20†), so it would result in good battery performances at low temperature.
To uncover the origin of the M-9000/Co–Ni(OH)2 system's improved performance, a series of ex situ structural and elemental characteristics have been carried out. First of all, by comparing SEM element mapping images of the M-9000 anode at the pristine, fully charged (1.2 V) state and fully discharged (0.1 V) state, a reversible process of K appearance/disappearance on the M-9000 anode could be obviously recognized (Fig. S21†). Furthermore, XPS characterization of the M-9000 anode during a discharge/discharge cycle was performed. Fig. 4c further verifies the appearance of potassium ions on the M-9000 anode during the charge process, and the disappearance after discharged to 0.1 V.
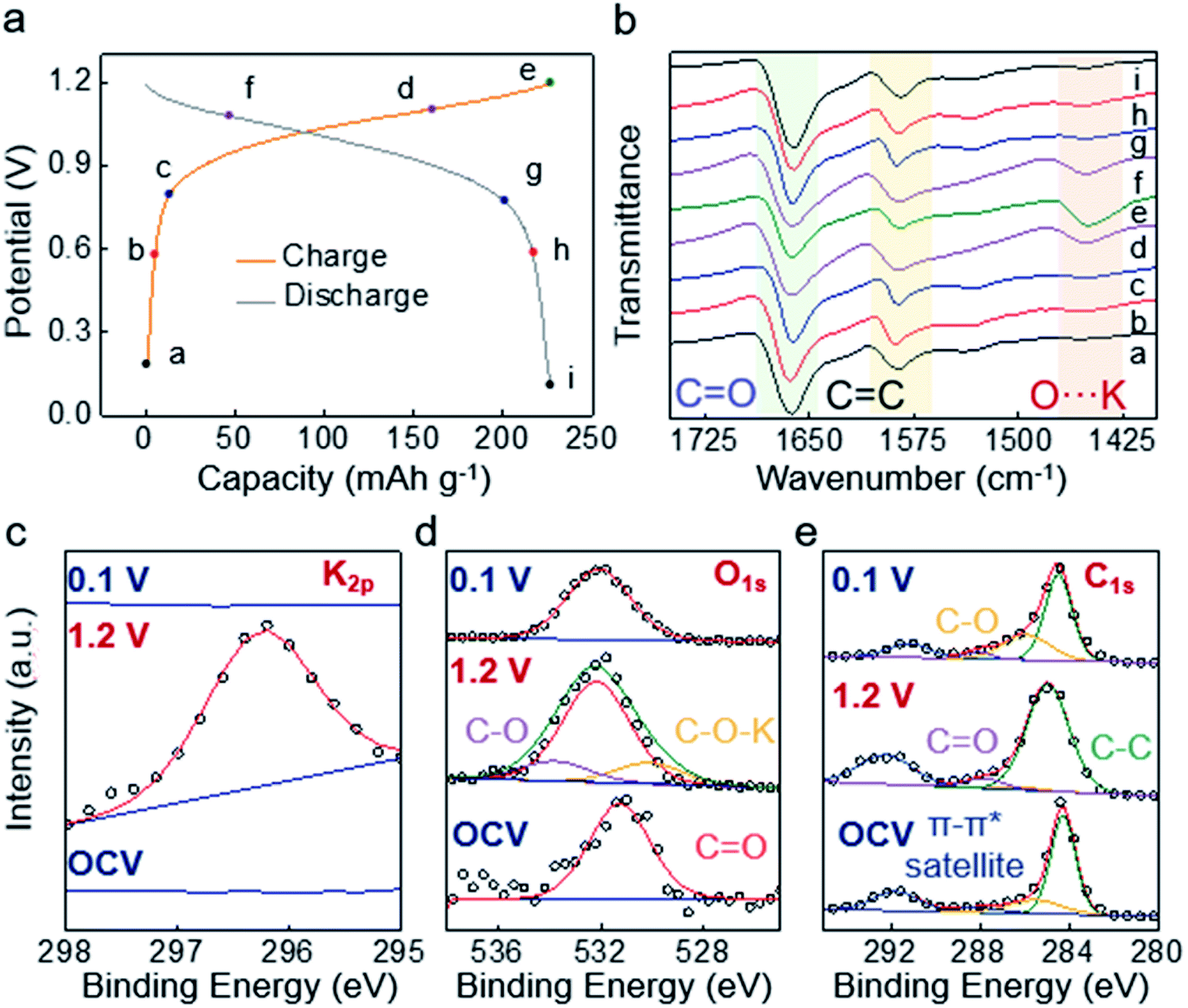 | ||
| Fig. 4 (a) Charge/discharge profiles of the M-9000/Co–Ni(OH)2 battery measured at 1C (260 mA g−1). (b) Ex situ FTIR spectra of the P14AQ electrode at various charged and discharged states in a charge/discharge cycle, corresponding to the marked points on a discharge/charge curve (left). High-resolution (c) K 2p (d) O 1s (e) C 1s XPS spectra (X-ray photoelectron spectroscopy) of the P14AQ electrode at the pristine, fully charged (1.2 V) state and fully discharged (0.1 V) state. | ||
Moreover, ex situ ATR-FTIR and XPS (O1s and C1s spectra) were used to investigate the bonding and composition changes of quinone. The peak located at about 1590 cm−1 belonged to CC stretching vibration of the framework. It was nearly unchanged in discharge and charge processes. As observed in Fig. 4a and b, the characteristic peak of carbonyl group stretching vibration at about 1663 cm−1 gradually weakened from the initial state (a) to state (e) during the charge process. At the same time, the O⋯K peak (1455 cm−1) heightened. Both of them indicated the reduction reaction of the quinone group to the enolate group and the coordination of K+ with the carbonyl group.24 In other words, P14AQ transformed into P14AQ-2K in the charge process (Fig. 1a). The high-resolution O1s spectra in Fig. 4d also showed the formation of C–O–K (530.1 eV) during the charge process.36 Similarly, C1s spectra in Fig. 4e supported the conclusion that CO transformed into C–O–K. Since the majority of M-9000 was carbon, the changes of CO and C–O peaks were not easy to observe. The changes of the oxygen bond and carbon bond were based on XPS data of the reported Mg/P14AQ battery.37 Note that enolate would be spontaneously reoxidized to quinone in air (the disassembly of the battery, washing and drying of the charged electrode were carried out in a glove box, but the testing was inevitably carried out in air for a few minutes), so the CO peak was difficult to be completely eliminated like other systems.30 Another reason was that some carbonyls in the polymer didn't react due to steric hindrance. In the discharge process to 0.1 V, the CO peaks fully recovered their original positions and intensities in both FTIR and XPS spectra, demonstrating the reversible K+ coordination/incoordination in Fig. 4 (the full FTIR spectra are given in Fig. S22†). Therefore, the redox mechanism involving reversible CO to C–O–K transformation could be established from our FTIR and XPS experiments and is similar to the proposed redox mechanism of reported quinone-based Li or Na–organic battery systems.38,39
In addition, ex situ1H NMR spectra in Fig. S23† were also used to investigate the reaction process. Compared with the 1H NMR spectra of the M-9000 anode at the pristine, charged state and discharged state, reappearance of the peaks of M-9000 could be obviously recognized. The reduced P14AQ-2K was difficult to dissolve in dichloromethane, so no obvious peaks existed. After oxidation in the discharge process, the peaks reappeared, which means that the discharge/charge reaction is reversible (the full NMR spectra are given in Fig. S24†).
In a word, SEM-EDS mapping, FTIR, XPS and NMR were used to clearly verify the electrochemical redox reaction of M-9000 via the reversible transformation of conjugated carbonyls and enolates. According to the reported mechanism of the Ni(OH)2 cathode40 (Fig. 1a), the Ni(OH)2 cathode was oxidized to NiOOH on charging and reduced back to Ni(OH)2 during the discharge process. The mechanism of this K+/OH− hybrid ion battery agreed with the redox mechanism proposed in Fig. 1a.
Conclusions
In summary, we have constructed an aqueous full cell with the Co–Ni(OH)2 cathode, P14AQ anode and KOH aqueous electrolyte. Because of the good reversibility and stability, the M-9000/Co–Ni(OH)2 battery demonstrated a superior electrochemical performance, including high energy/power densities (93 W h kg−1 at 1040 W kg−1), a long-term cycling stability (83.2% capacity retention after 500 cycles at 2.08 A g−1) and a high rate capability (87 W h kg−1 at 2080 W kg−1 and 79 W h kg−1 at 4160 W kg−1) (based on the mass of active materials on electrodes and reacted KOH). The excellent electrochemical performance of M-9000 was ascribed to the optimal molecular weight, which contributed to good electric conductivity and insolubility during the cycling process. This K+/OH− hybrid ion battery would enrich the existing rechargeable APIB system and be a promising battery technology for large-scale energy storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program (2017YFA0206700, 2016YFA0202500 and 2016YFB0901502), the National NSFC (21835004, 21673243, 51771094 and 21805141), MOE 111 project (B12015) and Tianjin High-Tech (18JCZDJC31500).Notes and references
- C. Yang, J. Chen, X. Ji, T. P. Pollard, X. Lü, C.-J. Sun, S. Hou, Q. Liu, C. Liu, T. Qing, Y. Wang, O. Borodin, Y. Ren, K. Xu and C. Wang, Nature, 2019, 569, 245–250 CrossRef CAS PubMed.
- F. Wan, Y. Zhang, L. Zhang, D. Liu, C. Wang, L. Song, Z. Niu and J. Chen, Angew. Chem., Int. Ed. Engl., 2019, 58, 7062–7067 CrossRef CAS PubMed.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- Y. Feng, Q. Zhang, S. Liu, J. Liu, Z. Tao and J. Chen, J. Mater. Chem. A, 2019, 7, 8122–8128 RSC.
- A. Eftekhari, Adv. Energy Mater., 2018, 8, 1801156 CrossRef.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed. Engl., 2018, 57, 11737–11741 CrossRef CAS PubMed.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 RSC.
- S. Zhao, B. Han, D. Zhang, Q. Huang, L. Xiao, L. Chen, D. G. Ivey, Y. Deng and W. Wei, J. Mater. Chem. A, 2018, 6, 5733–5739 RSC.
- Q. Li, Q. Zhang, C. Liu, Z. Zhou, C. Li, B. He, P. Man, X. Wang and Y. Yao, J. Mater. Chem. A, 2019, 7, 12997–13006 RSC.
- W. Chen, Y. Jin, J. Zhao, N. Liu and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2018, 428, 223–237 Search PubMed.
- S. Liu, L. Wang, J. Liu, M. Zhou, Q. Nian, Y. Feng, Z. Tao and L. Shao, J. Mater. Chem. A, 2019, 7, 248–256 RSC.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- Q. Xue, D. Li, Y. Huang, X. Zhang, Y. Ye, E. Fan, L. Li, F. Wu and R. Chen, J. Mater. Chem. A, 2018, 6, 12559–12564 RSC.
- H. Li, Z. Cheng, Q. Zhang, A. Natan, Y. Yang, D. Cao and H. Zhu, Nano Lett., 2018, 18, 7407–7413 CrossRef CAS PubMed.
- A. Eftekhari, J. Power Sources, 2004, 126, 221–228 CrossRef CAS.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2012, 3, 1149 CrossRef PubMed.
- D. Su, A. McDonagh, S. Z. Qiao and G. Wang, Adv. Mater., 2017, 29, 1604007 CrossRef PubMed.
- Y. Luo, L. Liu, K. Lei, J. Shi, G. Xu, F. Li and J. Chen, Chem. Sci., 2019, 10, 2048–2052 RSC.
- Z. Jian, Y. Liang, I. A. Rodríguez-Pérez, Y. Yao and X. Ji, Electrochem. Commun., 2016, 71, 5–8 CrossRef CAS.
- A. Eftekhari, Z. Jian and X. Ji, ACS Appl. Mater. Interfaces, 2017, 9, 4404–4419 CrossRef CAS PubMed.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10, 552–557 RSC.
- G. Dawut, Y. Lu, L. Miao and J. Chen, Inorg. Chem. Front., 2018, 5, 1391–1396 RSC.
- Y. Liang, Y. Jing, S. Gheytani, K. Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS PubMed.
- E. Dražević, A. S. Andersen, K. Wedege, M. L. Henriksen, M. Hinge and A. Bentien, J. Power Sources, 2018, 381, 94–100 CrossRef.
- B. S. Kong, Y. S. Kwon and D. Kim, Polym. J., 1997, 29, 722–732 CrossRef CAS.
- V. S. Nithianandam, F. Chertok and S. Eehan, J. Appl. Polym. Sci., 1991, 42, 2899–2901 CrossRef CAS.
- T. Yamamoto and H. Etori, Macromolecules, 1995, 28, 3371–3379 CrossRef CAS.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed. Engl., 2015, 54, 13947–13951 CrossRef CAS PubMed.
- T. Yamamoto, S. Wakabayashi and K. Osakada, J. Organomet. Chem., 1992, 428, 223–237 CrossRef CAS.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H. D. Lim and K. Kang, Adv. Mater., 2018, 30, e1704682 CrossRef PubMed.
- T. Tomai, H. Hyodo, D. Komatsu and I. Honma, J. Phys. Chem. C, 2018, 122, 2461–2466 CrossRef CAS.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- N. Yesibolati, N. Umirov, A. Koishybay, M. Omarova, I. Kurmanbayeva, Y. Zhang, Y. Zhao and Z. Bakenov, Electrochim. Acta, 2015, 152, 505–511 CrossRef CAS.
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, Angew. Chem., Int. Ed. Engl., 2017, 56, 12561–12565 CrossRef CAS PubMed.
- B. Pan, J. Huang, Z. Feng, L. Zeng, M. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 CrossRef.
- K.-C. Lau, I. A. Shkrob, N. L. Dietz Rago, J. G. Connell, D. Phelan, B. Hu, L. Zhang, Z. Zhang and C. Liao, J. Mater. Chem. A, 2017, 5, 24103–24109 RSC.
- Y. Zhou, B. Wang, C. Liu, N. Han, X. Xu, F. Zhao, J. Fan and Y. Li, Nano Energy, 2015, 15, 654–661 CrossRef CAS.
- A. K. Shukla, S. Venugopalan and B. Hariprakash, J. Power Sources, 2001, 100, 125–148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional results. See DOI: 10.1039/c9se00598f |
| This journal is © The Royal Society of Chemistry 2020 |