
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyclic nitronate olefin cycloaddition (ANOC): regio- and stereospecific synthesis of isoxazolines†
Liang
Ma
,
Luyao
Kou
,
Feng
Jin
,
Xionglve
Cheng
,
Suyan
Tao
,
Gangzhong
Jiang
,
Xiaoguang
Bao
 * and
Xiaobing
Wan
*
* and
Xiaobing
Wan
*
Key Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: xgbao@suda.edu.cn; wanxb@suda.edu.cn
First published on 6th November 2020
Abstract
We report the first demonstrations of intra- and intermolecular acyclic nitronate olefin cycloaddition (ANOC) reactions that enable the highly efficient syntheses of isoxazolines bearing various functional groups. This general approach to accessing γ-lactone fused isoxazolines was hitherto unprecedented. The room temperature transformations reported herein exhibit wide substrate scopes, as evidenced by more than 70 examples, including the generation of five tricyclic isoxazolines. The robustness of this methodology was confirmed by a series of trials that afforded highly functionalized isoxazolines. Both experimental results and density functional theory calculations indicate that these transformations proceed via the in situ formation of acyclic nitronates together with concerted [3+2] cycloaddition and tert-butyloxy group elimination processes to give regio- and stereospecificity.
Introduction
Fused bicyclic isoxazolines are present in many natural products and biologically relevant molecules,1 and can also serve as versatile intermediates in synthetic chemistry.2 Intramolecular nitrile oxide olefin cycloaddition (INOC) provides a classical route to the synthesis of these bicyclic moieties,3 especially those found in biologically important molecules.4 Silyl nitronates can serve as synthetic equivalents of nitrile oxides in 1,3-dipolar cycloadditions with olefins. Thus, intramolecular silyl nitronate olefin cycloaddition (ISOC) has emerged as a powerful alternative to INOC for the syntheses of isoxazoline-fused bicycles.5 A representative process is shown in Scheme 1a. Here, a Michael addition provides nitro compound A, having a tethered alkene moiety. The in situ conversion of A to nitrile oxide B with a stoichiometric amount of a dehydrating reagent (PhNCO/Et3N or (Boc)2O/4-dimethylaminopyridine) is followed by 1,3-dipolar cycloaddition to give the desired fused bicyclic isoxazoline compound (representing the INOC process). Alternatively, the reaction of A with TMSCl and Et3N affords silyl nitronate C, which undergoes [3+2] cycloaddition and the subsequent elimination of a silyloxy group in the presence of TBAF or an acid to form a fused bicyclic isoxazoline (the ISOC process). The ISOC process typically provides better regio- and stereo-selectivities than the INOC process as a result of the near-linear geometry of nitrile oxides. However, both mechanisms suffer from various drawbacks, including the need for stoichiometric quantities of toxic reagents, harsh reaction conditions, labor-intensive procedures and undesired side reactions.6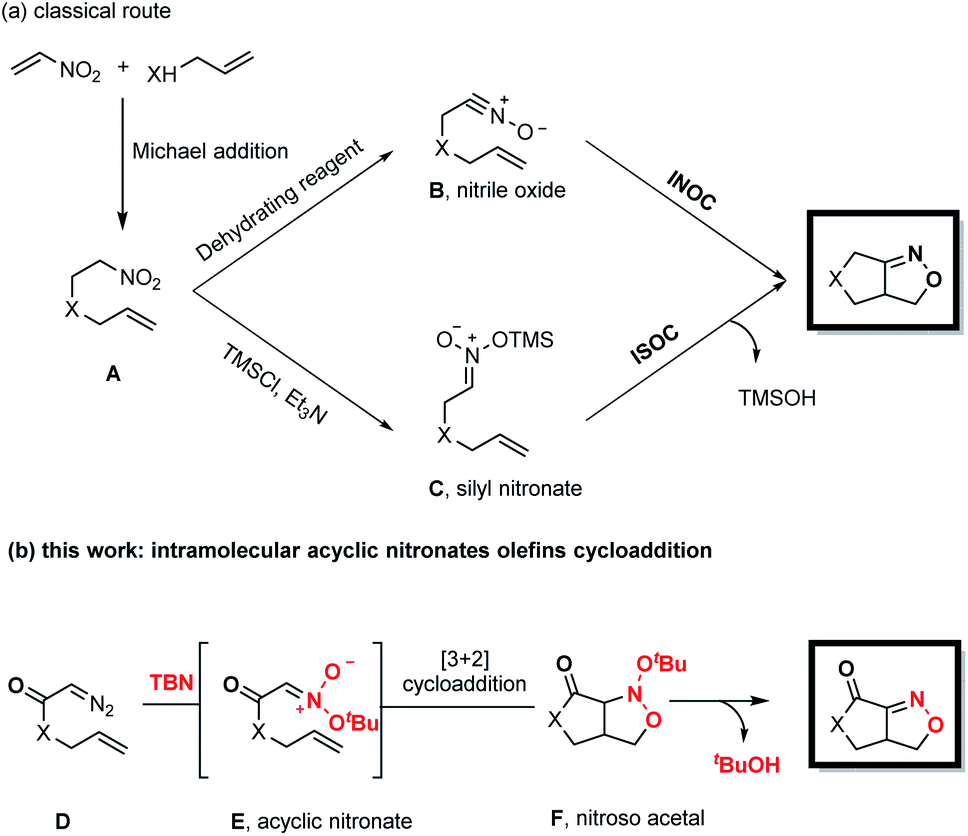 | ||
| Scheme 1 Synthesis of isoxazoline fused bicyclic compounds. | ||
Fused bicyclic isoxazolines are highly important, especially because of their unique functionalities, and so it would be beneficial to devise creative yet practical strategies for the construction of a new library of such compounds. In the present work, we developed a novel methodology for this purpose, as shown in Scheme 1b. This new approach consists of a coupling reaction between diazo compound D bearing a tethered alkene and tert-butyl nitrite to give an acyclic nitronate, E. The subsequent intramolecular [3+2] cycloaddition of E provides nitroso acetal F, after which the elimination of a tert-butyloxy group generates the desired fused bicyclic isoxazoline.
Denmark and coworkers developed an elegant strategy for the rapid construction of polycyclic nitroso acetal systems through the tandem [4+2]/[3+2] cycloaddition of nitroalkenes, employing cyclic nitronates as key intermediates.7 In sharp contrast, the synthetic utility of the analogous reactions of acyclic nitronates has been seriously limited8 by a lack of general methods for the preparation of these compounds and by their low thermal stability. This thermal instability presented several potentially severe challenges with regard to the goal of the present research. Specifically, it was not certain that acyclic nitronates could be readily obtained from the coupling of diazo compounds and tert-butyl nitrite. In addition, it was uncertain if these acyclic nitronates, after being generated in situ, would be successfully intercepted by the olefins rather than undergoing undesired side reactions.9 In addition, it was vital to identify suitable conditions to promote elimination of the tert-butyloxy group of the nitroso acetals so as to obtain the desired fused bicyclic isoxazolines.
Results and discussion
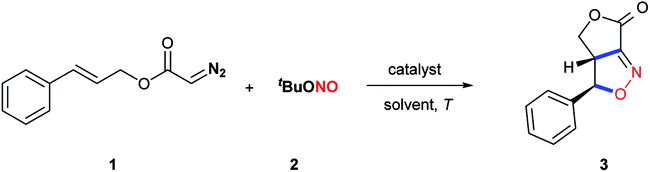
Recently, we reported the Cu(OAc)2·H2O-catalyzed multicomponent reaction of diazo compounds, alkenes and tert-butyl nitrite in the presence of DABCO to construct diverse isoxazoline derivatives.10 However, using this approach, the γ-lactone fused isoxazoline 3 (entry 1, Table 1) was not obtained in any significant amount when diazo compound 1 (derived from cinnamyl alcohol) was used as the substrate. Interestingly, a moderate yield was achieved in MeCN in the absence of Cu(OAc)2·H2O and DABCO (entry 4), indicating that different mechanisms were involved in these two systems. In addition, the data showed that lowering the temperature did not affect the reaction. After extensive screenings, the optimized conditions were determined to comprise the use of DMF as the solvent under catalyst free conditions at room temperature, which gave the trans-cycloadduct 3 in a high 85% yield (entry 7). The use of solvents other than DMF inhibited the formation of the desired product 3 (entries 8–19). Notably, no other diastereomer was observed in this cycloaddition reaction, indicating that this intramolecular acyclic nitronate olefin cycloaddition (ANOC) proceeded through a concerted process.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Solvent | Yieldb (%) |
| a Reaction conditions: diazo compound 1 (0.2 mmol, 1.0 equiv.), tert-butyl nitrite 2 (0.6 mmol, 3.0 equiv.) and solvent (2.0 mL) at 25 °C for 12 h unless otherwise noted. b Isolated yields. c Cu(OAc)2·H2O (0.02 mmol, 10 mol%), and DABCO (0.2 mmol, 1.0 equiv.) were used. d DABCO (0.2 mmol, 1.0 equiv.) was used. | ||||
| 1 | Cu(OAc)2·H2O | 80 | Toluene | <5c |
| 2 | — | 80 | Toluene | <5d |
| 3 | — | 80 | Toluene | <5 |
| 4 | — | 80 | MeCN | 53 |
| 5 | — | 80 | MeNO2 | 37 |
| 6 | — | 80 | DMF | 33 |
| 7 | — | 25 | DMF | 85 |
| 8 | — | 25 | MeNO2 | 64 |
| 9 | — | 25 | MeCN | 58 |
| 10 | — | 25 | Toluene | <5 |
| 11 | — | 25 | DMSO | 63 |
| 12 | — | 25 | Acetone | <5 |
| 13 | — | 25 | Dioxane | <5 |
| 14 | — | 25 | DCE | <5 |
| 15 | — | 25 | THF | <5 |
| 16 | — | 25 | NMP | <5 |
| 17 | — | 25 | EA | <5 |
| 18 | — | 25 | CHCl3 | <5 |
| 19 | — | 25 | H2O | <5 |
Both γ-lactones11 and isoxazolines1,2 are key pharmacophores in many biologically active compounds, both synthetic and naturally occurring, and can serve as versatile synthons for organic chemistry. However, γ-lactone fused isoxazolines remain an elusive subclass of heterobicycles primarily as a result of the inherent difficulty in constructing this framework.12 This has hindered the detailed study of the potential biological activity and synthetic utility of such compounds. Fortunately, the ANOC process reported herein provides a general approach to accessing new γ-lactone fused isoxazolines.
With this goal in mind, we explored the substrate scope of this new process, with the results summarized in Table 2. Diazo compounds obtained from a variety of substituted cinnamyl alcohol derivatives were employed in this transformation and the desired fused bicyclic isoxazolines 4–15 were achieved in moderate to excellent yields. The presence of both electron-withdrawing (CN, CF3, F, Cl, Br, and I) and electron-donating (MeS, alkyl, and ether) groups on the phenyl ring was well tolerated in this intramolecular cycloaddition reaction. Notably, the cycloadduct 16 was generated as the exclusive product among four possible stereoisomers in a high 80% yield when the diazo compound obtained from 4-phenyl-3-buten-2-ol was used as the substrate. The relative stereo configuration of 16 was confirmed by single crystal X-ray diffraction. Diazo compounds derived from alkyl-substituted allyl alcohols were also determined to be suitable substrates for this intramolecular ANOC reaction, and gave the corresponding fused bicyclic isoxazolines 17–25 in satisfactory yields. Notably, when we attempted to use a diazo ketone in the reaction, the fused bicyclic isoxazoline cyclopentanone 26 was obtained with complete regio- and stereoselectivity. The phenyl group could also be replaced with a furan ring in this ANOC process to generate the corresponding cycloadduct 27 in a moderate yield.
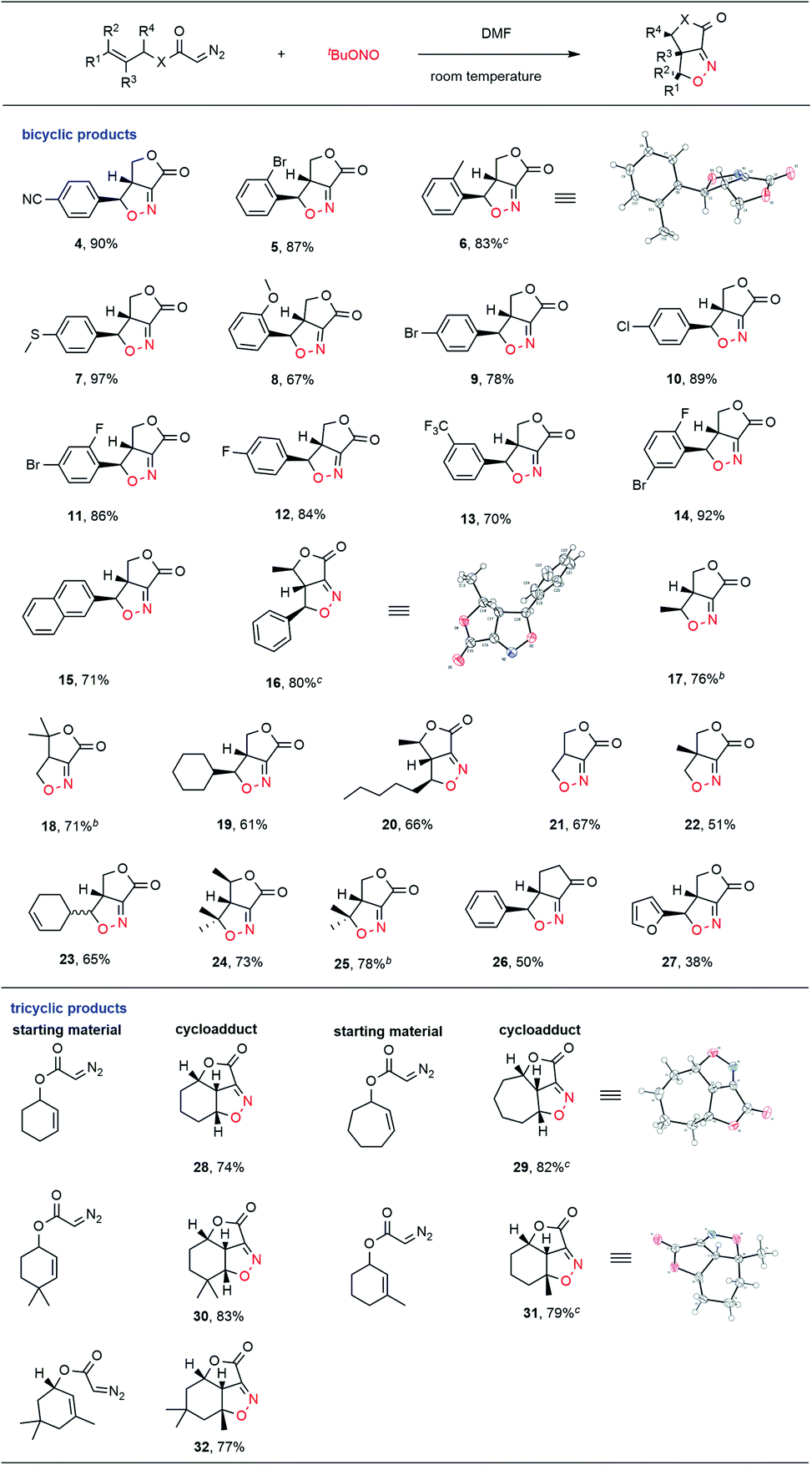
To further demonstrate the utility of this annular chemistry as a means of allowing the one-pot construction of a wide range of isoxazolines, we applied the optimized conditions to generate diazo compounds bearing cyclic olefins. As anticipated, a variety of tricyclic compounds (28–32) were obtained, each as a single stereoisomer, with regio- and stereo-specificity and in good yields. The present methodology was found to allow the rapid construction of tricyclic heterocycles bearing three contiguous stereogenic centers starting from simple substrates. The exact structures of the fused tricyclic products 29 and 31 were further verified by X-ray crystallography, which confirmed the stereochemistry at the bridgehead carbon atoms in each.
The scalability of this novel methodology was demonstrated by performing a gram-scale experiment (using 10 mmol of the substrate), in which the cycloadduct 12 was obtained in a high 79% yield (Scheme 2). This product was subsequently derivatized to illustrate potential synthetic applications. These derivatizations including hydrolysis (33), esterification (34), ammonolysis (35) and ring expansion (36) reactions, all of which readily proceeded to give highly functionalized isoxazolines in high yields.
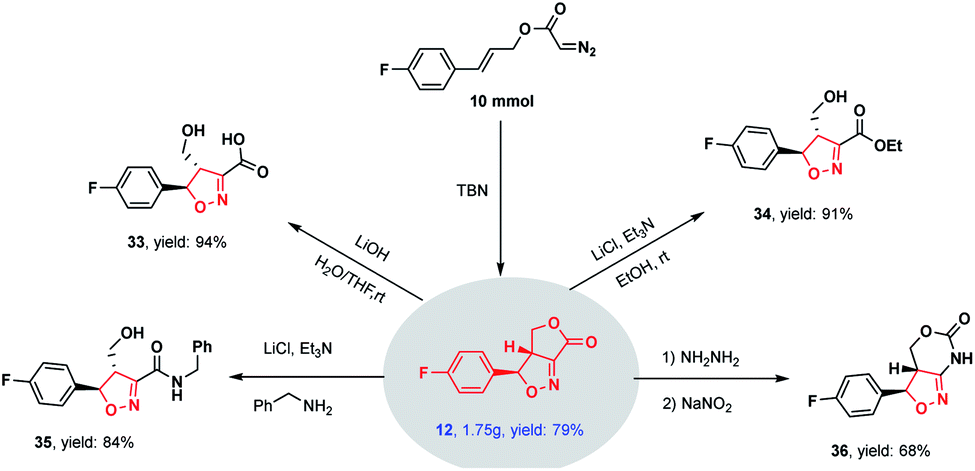 | ||
| Scheme 2 Synthetic utility. | ||
Furoxan, a dimer of a highly reactive nitrile oxide, could not be detected in the products of these reactions, indicating that nitrile oxide did not serve as the reaction intermediate in the formation of these γ-lactone fused isoxazolines.12b Further control experiments were carried out to elucidate the reaction pathway involved in these transformations (Scheme 3). In these trials, butylated hydroxytoluene was added to the reaction as a radical trapping reagent and the desired product 3 was still obtained in a high 72% yield. This result indicated that radical intermediates were not involved in the formation of the fused bicyclic isoxazolines. We next prepared diazo compound 37 from (Z)-2-bromo-3-phenylprop-2-en-1-ol and subjected it to this transformation. As shown in Scheme 3b, a small amount of acyclic nitronate 39 was detected along with the formation of the fused bicycle isoxazole 38 in 13% yield. As expected, the cycloaddition of 39 evidently generated 38, indicating that acyclic nitronates serve as key reaction intermediates in this ANOC process. The most common approach for the synthesis of acyclic nitronates involves the reaction between aliphatic nitro compounds and electrophilic reagents, which typically produces a mixture of stereoisomers. In contrast, the present work successfully demonstrated the stereospecific synthesis of trans-acyclic nitronates based on the room temperature coupling of diazo compounds and tert-butyl nitrite under neutral conditions.
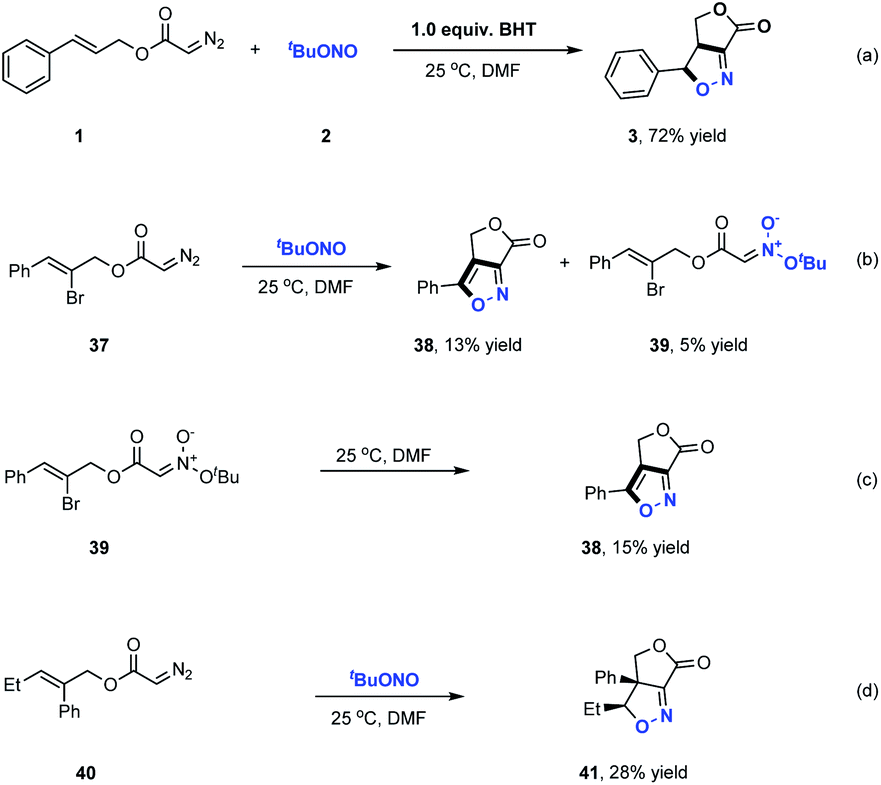 | ||
| Scheme 3 Probe for the possible mechanism. | ||
Computational studies were performed to gain mechanistic insights into the ANOC reaction. First, a transition state (TS) of nucleophilic substitution between the diazo compound (1) and the tert-butyl nitrite (2) is located as TS1, in which the N1⋯C1 distance is shortened to 1.86 Å, while the distance of C1⋯N2 is lengthened to 1.38 Å (Fig. S1 in the ESI†). Subsequently, the acyclic nitronate intermediate with a trans configuration (INT1) is afforded along with the dissociation of N2. In addition, the possible cis configuration of acyclic nitronate (INT1′) was also considered. The calculated activation barrier leading to the cis structure is 1.4 kcal mol−1 higher than that of the trans one and the formed INT1′ is 3.7 kcal mol−1 higher in energy than INT1. Thus, the trans configuration of acyclic nitronate is more feasible to generate. Moreover, a computational study suggests that the process leading to the acyclic nitronate intermediate could be promoted by a trace amount of water, which was further verified by experimental results (for details, see Table S1 in the ESI†). The TS of nucleophilic substitution can be stabilized by water molecule(s) via H-bonding interactions (Fig. S2 in the ESI†). Next, the formed INT1 could undergo intramolecular [3+2] cycloaddition between the nitronate and the alkenyl moieties. The corresponding transition state is located as TS2, in which the C1⋯C2 and O1⋯C3 distances are shortened to 2.13 and 2.56 Å, respectively. Thus, a bicyclic intermediate INT2 is given via a concerted step. It should be noted that the two hydrogen atom attached to the two bridge carbons, respectively, are in a cis configuration in INT2. The corresponding TS leading to the trans configuration, INT2′, is shown as TS2′, which is 2.3 kcal mol−1 higher than TS2. The formation of INT2viaTS2 is favorable because the C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 and the NO moieties are in the same orientation, which is ready to undergo cyclization. For TS2′, however, the aformentioned two moieties are far away due to the trans orientation, which is not good for cyclization. Consequently, the formation of the cis configuration INT2viaTS2 is more favorable to occur, which is consistent with experimental observation. The favored cis configuration of INT2 implies that this [3+2] reaction might be sensitive to the steric hindrance caused by the groups attached to the two bridge carbons. When a Br or Ph group is attached to C2 (37/40), the calculated activation barriers for the intramolecular [3+2] cycloaddition are 24.6 and 24.5 kcal mol−1 (Fig. S3 and S4 in the ESI†), respectively, which are considerably higher than that of the unsubstituted substrate. The repulsive interaction between the bigger atoms/groups present at the bridge carbons is mainly responsible for the less effective reactions in Scheme 3b–d. Finally, the elimination of tBuOH from INT2 leads to the desired product 3. This process can be assisted by a water molecule that serves as a shuttle to transfer a proton viaTS3 (Fig. S1 in the ESI†).
C3 and the NO moieties are in the same orientation, which is ready to undergo cyclization. For TS2′, however, the aformentioned two moieties are far away due to the trans orientation, which is not good for cyclization. Consequently, the formation of the cis configuration INT2viaTS2 is more favorable to occur, which is consistent with experimental observation. The favored cis configuration of INT2 implies that this [3+2] reaction might be sensitive to the steric hindrance caused by the groups attached to the two bridge carbons. When a Br or Ph group is attached to C2 (37/40), the calculated activation barriers for the intramolecular [3+2] cycloaddition are 24.6 and 24.5 kcal mol−1 (Fig. S3 and S4 in the ESI†), respectively, which are considerably higher than that of the unsubstituted substrate. The repulsive interaction between the bigger atoms/groups present at the bridge carbons is mainly responsible for the less effective reactions in Scheme 3b–d. Finally, the elimination of tBuOH from INT2 leads to the desired product 3. This process can be assisted by a water molecule that serves as a shuttle to transfer a proton viaTS3 (Fig. S1 in the ESI†).
Based upon our experimental data and density functional theory (DFT) calculations, a plausible mechanism was developed, as shown in Scheme 4. In this process, a nucleophilic substitution reaction between the diazo compound and the tert-butyl nitrite triggers the formation of acyclic nitronate intermediate G, having a trans configuration. This compound subsequently undergoes a concerted [3+2] cycloaddition to produce the nitroso acetal H. Finally, elimination of a tert-butyloxy group delivers the desired fused bicyclic isoxazoline with regio- and stereo-specificity.
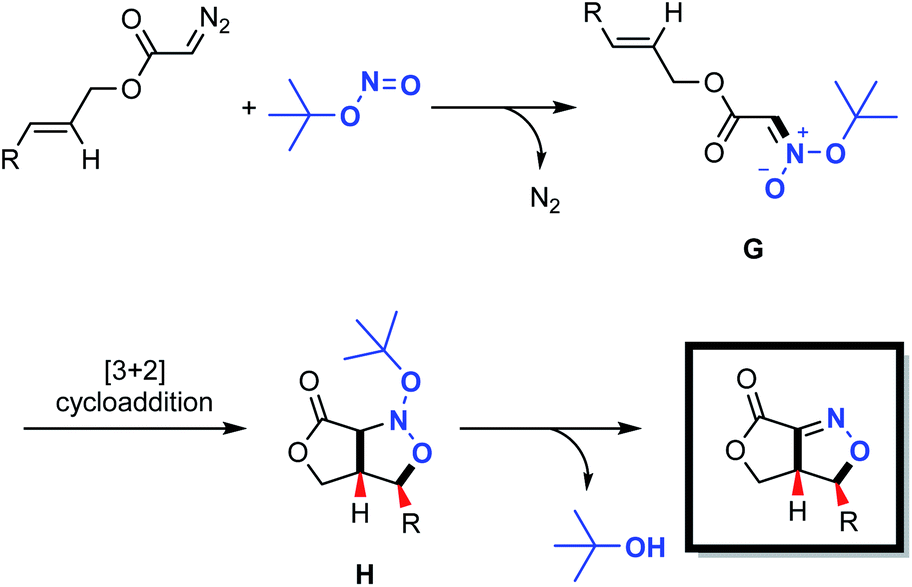 | ||
| Scheme 4 Proposed catalytic cycle. | ||
We next proceeded to evaluate the intermolecular version of this ANOC process (Table 3). A small amount of the cycloadduct 42 was detected following the multicomponent reaction between 4-tert-butylstyrene, ethyl diazoacetate and tBuONO. After examining a variety of reaction parameters, we found that the desired product (42) could be obtained in an 80% yield using BF3·Et2O as a Lewis acid catalyst in MeCN at room temperature (for details, see Table S2 in the ESI†). As expected, computations suggested that the presence of BF3·Et2O promoted the nucleophilic substitution process to provide the acyclic nitronate intermediate (Fig. S5 in the ESI†). Subsequently, the substrate scope for this intermolecular acyclic nitronate olefin cycloaddition was assessed. A set of substituted styrenes bearing electron-donating and – withdrawing groups, such as halide atoms and alkyl, alkoxyl, ester, nitro, cyano and trifluoromethyl groups, were found to be well tolerated, affording the corresponding isoxazolines in moderate to high yields (43–56, 38–75% yields). In addition, 2-vinylnaphthalene could be used to obtain the desired product (57) in a satisfactory yield of 74%. Moreover, regioselectivity could be achieved by employing appropriate substrates. As an example, methyl (E)-3-(4-vinylphenyl)acrylate gave complete regioselectivity based on cycloaddition at the terminal double bond even in the presence of the internal unsaturation (58, 64% yield). A styrene substrate modified with a borate ester moiety was also found to provide product 59, demonstrating that this methodology could be suitable for the late-stage derivatization of complicated molecules. Acrylates and an acrylamide also took part in the reaction to furnish the corresponding products in good yields (61–67). Notably, several vulnerable functional groups, including thioether, hydroxyl and amide moieties, were compatible with this cycloaddition protocol, albeit providing moderate yields (60, 40% yield; 62, 51% yield; 67, 55% yield). Interestingly, vinyl silanes and a vinyl sulfone were also viable substrates and gave the corresponding cycloadducts in synthetically useful yields (68, 61% yield; 69, 55% yield; 70, 28% yield). Pentafluoro styrene also functioned well to give the cycloadduct 71 in a 56% yield. The unsaturated heterocycle thiophene was also observed to deliver the corresponding product 72 in a 64% yield. In addition, aliphatic 1-octene and 1-hexene were found to participate in the transformation to generate the anticipated products in moderate yields (73, 33% yield; 74, 48% yield).
| a Reaction conditions: alkenes (0.5 mmol), ethyl diazoacetate (1.0 mmol, 2.0 equiv.), tert-butyl nitrite (1.0 mmol, 2.0 equiv.), BF3·Et2O (0.05 mmol, 10 mol%) and MeCN (2.0 mL) at 25 °C under air for 12 h. |
|---|
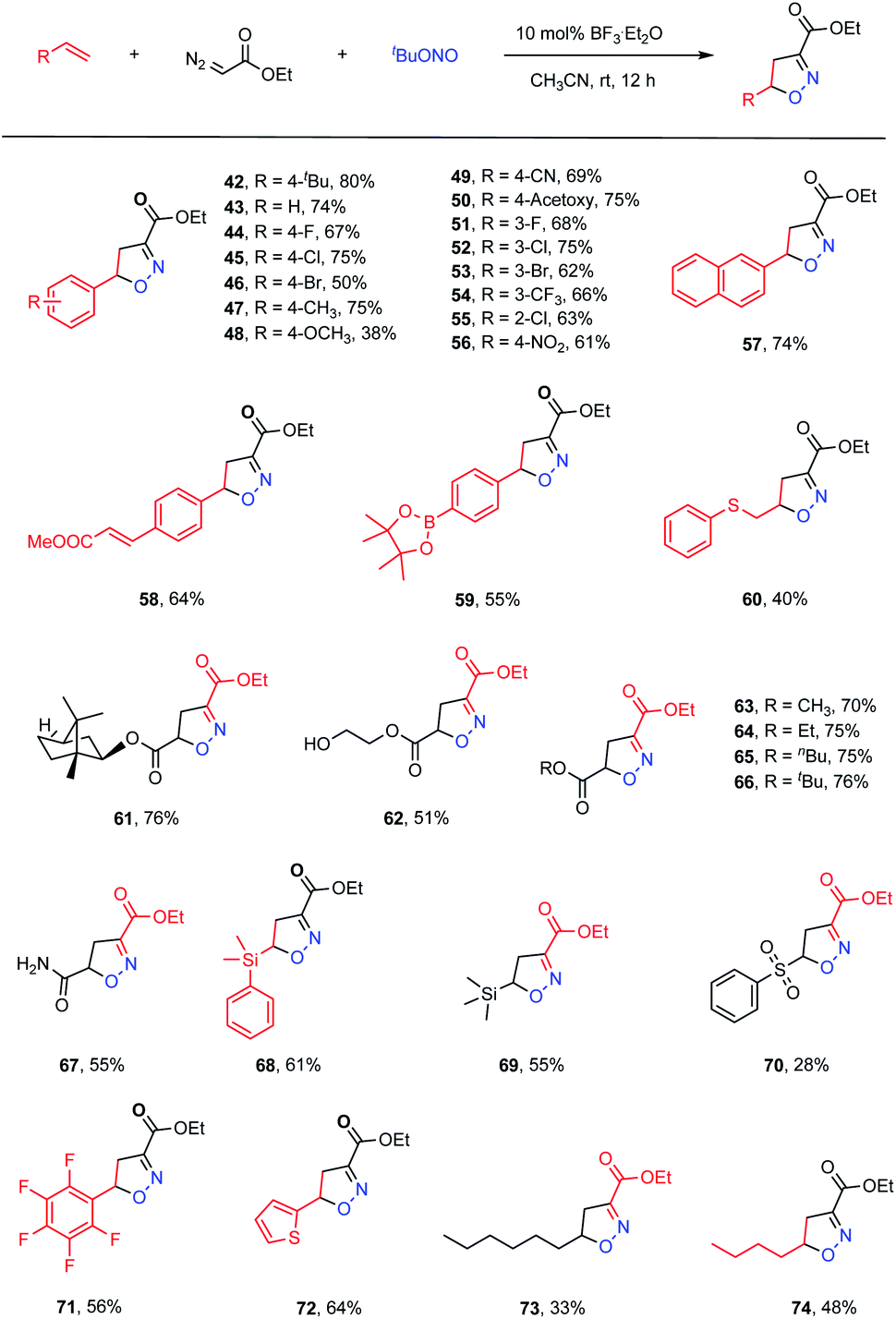
|
To further demonstrate the utility of this transformation, we examined the applicability of diazo compounds derived from various alcohols and acids (Table 4). The diazo ester derivatives of phenol and phenylmethanol provided good yields of the corresponding adducts 75 (67% yield) and 76 (92% yield), respectively. Substrates containing potentially bioactive skeletons, such as furan, trifluoromethyl and thiophene moieties, were also well tolerated (77–79, 45–86% yields). With respect to diazo ketones, phenyl and alkyl analogues reacted smoothly, although the latter showed a lower efficiency (80, 69% yield; 81, 31% yield). In addition, an α-diazo ketone bearing a phenylsulfonyl group reacted with a good yield (82, 72% yield).
| a Reaction conditions: 4-tert-butylstyrene (0.5 mmol), diazo compounds (1.0 mmol, 2.0 equiv.), tert-butyl nitrite (1.0 mmol, 2.0 equiv.), BF3·Et2O (0.05 mmol, 10 mol%) and MeCN (2.0 mL) at 25 °C under air for 12 h. |
|---|
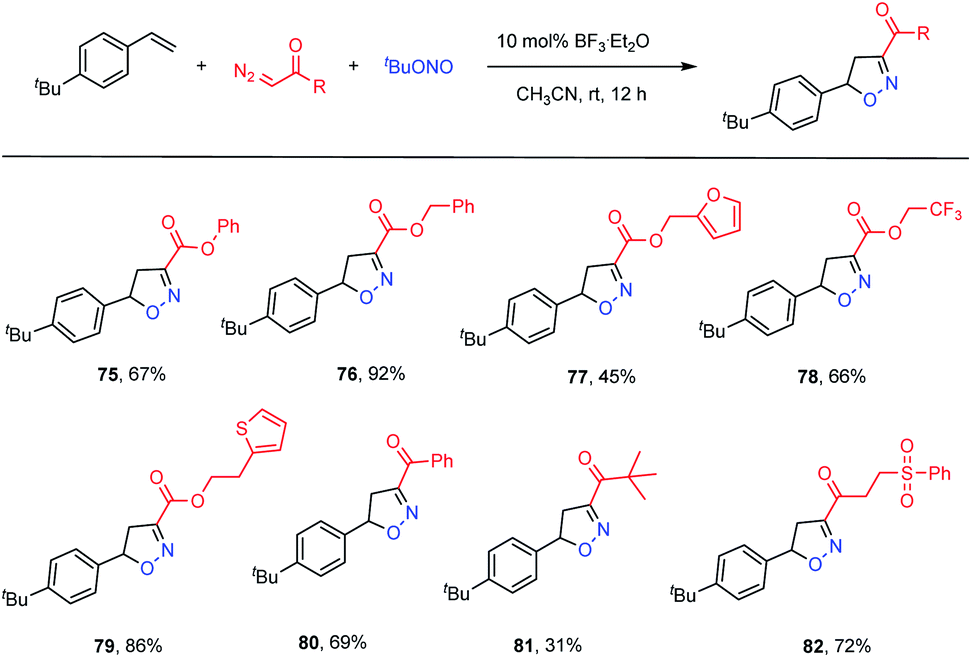
|
Conclusions
In conclusion, this work demonstrated an entirely novel series of intra- and intermolecular ANOC reactions. This methodology provides both regio- and stereo-specificity via a distinct mechanism and offers a powerful means of obtaining a wide range of isoxazolines with excellent functional group tolerance at room temperature. Notably, a new library of γ-lactone fused isoxazolines with important biological and synthetic potential was synthesized, including five tricyclic fused isoxazolines. These compounds are readily transformed into potentially useful, highly functionalized isoxazolines, thus confirming the synthetic utility of this approach. Both mechanistic experiments and DFT calculations indicated that the smooth generation of acyclic nitronates and subsequent interception by carbon–carbon double bonds were responsible for the rapid construction of isoxazolines. The application of this methodology to the total synthesis of complex natural products is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), NSFC (grant numbers 21973068, 21971175, and 21572148). We thank Michael Judge, MSc, from Liwen Bianji, Edanz Editing China (http://www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.Notes and references
- For selected examples on this topic, see: (a) J. Zhang, L. Ren, Y. Wang and X. Fang, RSC Adv., 2019, 9, 39338 RSC; (b) S. S. Prasad and S. Baskaran, J. Org. Chem., 2018, 83, 1558 CrossRef CAS; (c) N. Puttaswamy, G. S. Pavan Kumar, M. Al-Ghorbani, V. Vigneshwaran, B. T. Prabhakar and S. A. Khanum, Eur. J. Med. Chem., 2016, 114, 153 CrossRef CAS.
- For reviews on this topic, see: (a) G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505 CrossRef CAS; (b) G. Desimoni, G. Faita and K. A. Jorgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS; (c) H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151 CrossRef CAS; (d) A. P. Kozikowski, Acc. Chem. Res., 2002, 17, 410 CrossRef.
- For representative examples on this topic, see: (a) X. T. Li, Q. S. Gu, X. Y. Dong, X. Meng and X. Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 7668 CrossRef CAS; (b) S. L. Bartlett, Y. Sohtome, D. Hashizume, P. S. White, M. Sawamura, J. S. Johnson and M. Sodeoka, J. Am. Chem. Soc., 2017, 139, 8661 CrossRef CAS; (c) C. Kesornpun, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Angew. Chem., Int. Ed., 2016, 55, 3997 CrossRef CAS; (d) M. Gao, Y. Li, Y. Gan and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 8795 CrossRef CAS; (e) B. Han, X. L. Yang, R. Fang, W. Yu, C. Wang, X. Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816 CrossRef CAS.
- For recent examples on this topic, see: (a) Z. Zhou, A. X. Gao and S. A. Snyder, J. Am. Chem. Soc., 2019, 141, 7715 CrossRef CAS; (b) Y. Ji, Z. Xin, H. He and S. Gao, J. Am. Chem. Soc., 2019, 141, 16208 CrossRef CAS; (c) T. Maehara, K. Motoyama, T. Toma, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2017, 56, 1549 CrossRef CAS; (d) S. Fischer, N. Huwyler, S. Wolfrum and E. M. Carreira, Angew. Chem., Int. Ed., 2016, 55, 2555 CrossRef CAS; (e) S. Diethelm and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 8500 CrossRef CAS; (f) S. Yokoshima, T. Ueda, S. Kobayashi, A. Sato, T. Kuboyama, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2002, 124, 2137 CrossRef CAS.
- For representative examples on this topic, see: (a) M. Jiang, L. Feng, J. Feng and P. Jiao, Org. Lett., 2017, 19, 2210 CrossRef CAS; (b) L. Dong, C. Geng and P. Jiao, J. Org. Chem., 2015, 80, 10992 CrossRef CAS; (c) D. Bonne, L. Salat, J.-P. Dulcere and J. Rodriguez, Org. Lett., 2008, 10, 5409 CrossRef CAS; (d) T. Kudoh, T. Ishikawa, Y. Shimizu and S. Saito, Org. Lett., 2003, 5, 3875 CrossRef CAS; (e) W. Dehaen and A. Hassner, Tetrahedron Lett., 1990, 31, 743 CrossRef CAS.
- (a) H. Feuer, Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis, Wiley, Hoboken, 2008 Search PubMed; (b) I. N. N. Namboothiri and A. Hassner, Stereoselective Intramolecular 1,3-Dipolar Cycloadditions, in Stereoselective Heterocyclic Synthesis III, ed. P. Metz, Springer-Verlag Berlin Heidelberg, 2001, pp. 1–49 Search PubMed.
- (a) S. E. Denmark and J. J. Cottell, J. Org. Chem., 2001, 66, 4276 CrossRef CAS; (b) S. E. Denmark and E. A. Martinborough, J. Am. Chem. Soc., 1999, 121, 3046 CrossRef CAS; (c) S. E. Denmark and A. Thorarensen, J. Am. Chem. Soc., 1997, 119, 125 CrossRef CAS; (d) S. E. Denmark and A. Thorarensen, Chem. Rev., 1996, 96, 137 CrossRef CAS.
- (a) S. Kanemasa, S. Kaga and E. Wada, Tetrahedron Lett., 1998, 39, 8865 CrossRef CAS; (b) P. A. Wade, N. V. Amin, H. K. Yen, D. T. Price and G. F. Huhn, J. Org. Chem., 1984, 49, 4595 CrossRef CAS; (c) R. Gree, F. Tonnard and R. Carrie, Tetrahedron, 1976, 32, 675 CrossRef CAS; (d) V. A. Tartakovskii, O. A. Luk'yanov, N. I. Shlykova and S. S. Novikov, Zh. Org. Khim., 1967, 3, 980 CAS.
- For common decomposition reactions of acyclic nitronates, see ref. 6a.
- R. Chen, Y. Zhao, S. Fang, W. Long, H. Sun and X. Wan, Org. Lett., 2017, 19, 5896 CrossRef CAS.
- For selected examples on this topic, see: (a) M. Cianfanelli, G. Olivo, M. Milan, R. J. M. Klein Gebbink, X. Ribas, M. Bietti and M. Costas, J. Am. Chem. Soc., 2020, 142, 1584 CrossRef CAS; (b) A. Hu, Y. Chen, J. J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580 CrossRef CAS; (c) X. Wu, Z. Chen, Y.-B. Bai and V. M. Dong, J. Am. Chem. Soc., 2016, 138, 12013 CrossRef CAS; (d) H. Shigehisa, M. Hayashi, H. Ohkawa, T. Suzuki, H. Okayasu, M. Mukai, A. Yamazaki, R. Kawai, H. Kikuchi, Y. Satoh, A. Fukuyama and K. Hiroya, J. Am. Chem. Soc., 2016, 138, 10597 CrossRef CAS; (e) B. N. Hemric, K. Shen and Q. Wang, J. Am. Chem. Soc., 2016, 138, 5813 CrossRef CAS; (f) X. Xie and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 3767 CrossRef CAS; (g) E. L. McInturff, J. Mowat, A. R. Waldeck and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 17230 CrossRef CAS.
- (a) B. B. Snider and Q. Che, Tetrahedron, 2002, 58, 7821 CrossRef CAS; (b) A. Hassner, K. S. K. Murthy, A. Padwa, U. Chiacchio, D. C. Dean and A. M. Schoffstall, J. Org. Chem., 1989, 54, 5277 CrossRef CAS; (c) L. Garanti and G. Zecchi, J. Heterocycl. Chem., 1980, 17, 609 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and NMR spectra. CCDC 2011278, 2011280, 2011281 and 2021609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05607c |
| This journal is © The Royal Society of Chemistry 2021 |