
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePersistent, highly localized, and tunable [4]helicene radicals†
Aslam C.
Shaikh
 a,
Jules
Moutet
a,
José M.
Veleta
a,
Md Mubarak
Hossain
a,
Jan
Bloch
b,
Andrei V.
Astashkin
a and
Thomas L.
Gianetti
*a
a,
Jules
Moutet
a,
José M.
Veleta
a,
Md Mubarak
Hossain
a,
Jan
Bloch
b,
Andrei V.
Astashkin
a and
Thomas L.
Gianetti
*a
aDepartment of Chemistry and Biochemistry, University of Arizona, Tucson, AZ, USA. E-mail: tgianetti@arizona.edu
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Zürich, Switzerland
First published on 23rd September 2020
Abstract
Persistent organic radicals have gained considerable attention in the fields of catalysis and materials science. In particular, helical molecules are of great interest for the development and application of novel organic radicals in optoelectronic and spintronic materials. Here we report the syntheses of easily tunable and stable neutral quinolinoacridine radicals under anaerobic conditions by chemical reduction of their quinolinoacridinium cation analogs. The structures of these [4]helicene radicals were determined by X-ray crystallography. Density functional theory (DFT) calculations, supported by electron paramagnetic resonance (EPR) measurements, indicate that over 40% of spin density is located at the central carbon of our [4]helicene radicals regardless of their structural modifications. The localization of the charge promotes a reversible oxidation to the cation upon exposure to air. This unusual reactivity toward molecular oxygen was monitored via UV-Vis spectroscopy.
Introduction
Stable organic radicals are of continued interest to the scientific community because of their potential applications in (photo)catalysis,1–3 energy storage, bioimaging, organic light-emitting diodes (OLEDs),4–7 and other materials applications.8–11 In particular, carbon-centered radicals are considered highly promising due to their structural diversity and the tunability of their physicochemical properties. However, organic radicals, especially carbon-centered, are often highly reactive, resulting in hydrogen abstraction, dimerization, recombination, and polymerization,12–15 which makes them short-lived, and difficult to handle, and isolate. As a result, a tradeoff between reactivity and functionality is often required. Incorporation of bulky substituents can kinetically stabilize the radical by isolating the unpaired electron from the molecular surroundings. This helps to prevent/minimize the σ-dimerization, but at a cost of also reducing the access to certain kinds of radical-related reactivity.16 Alternatively, carbon-centered radicals can be thermodynamically stabilized via spin delocalization across a highly conjugated π-system and incorporation of heteroatoms.16 In fact, the majority of persistent carbon-centered radicals are highly delocalized, which promotes π–π interactions and spin–spin communication.17–19 On the other hand, localization of spin density can increase spin polarization20–22 and foster longer excited state lifetime,23 faster spin mixing rate,24,25 and higher reversibility of the cation-radical redox processes under air.26,27 Thus, the syntheses of persistent carbon-centered radicals with localized, yet exposed, spin are highly desirable.In this context, helical organic molecules represent an attractive framework. In addition to their inherent chirality, useful for optoelectronic and spintronic materials, their lack of planarity hampers the radical dimerization without the need for bulky substituents. However, examples of carbon centered [n]helicene neutral radicals remain scarce (n = 4, 5, 6, 7 indicates the number of fused aromatic rings).28–31 The unpaired electron in these systems is stabilized through delocalization over the π-system, and molecules with a higher number of fused aromatic rings show higher thermodynamic stability. Thus, [4]helicenes represent the least stable and least studied radicals of this class. To the best of our knowledge, only five examples of [4]helicene carbon-centered radicals have been reported.32–36 Of these radicals, two showed limited stability and two were formed by bulk electrolysis, and no crystal structures were reported (Scheme 1).
 | ||
| Scheme 1 [4]Helicene radicals reported in the literature (a) and in this work (b). | ||
In 1958, Neunhoffer and Haase reported the first [4]helicene radical, the doubly ortho-bridged radical (I) (see Scheme 1).34 Later, Aulmich and co-workers synthesized a [4]helicene radical (II) with dimethyl-methylene units as bridging groups.32,36 These two radicals showed low stability, readily decomposing when exposed to air, and no electronic structures were reported for them. More recently, Laursen et al. generated a quinolinoacridinium [4]helicene radical (III) by in situ electrochemical reduction of N,N′-dialkyl-1,13-dimethoxyquinacridinium (DMQA+) and studied it by UV-Vis and electron paramagnetic resonance (EPR) spectroscopies.33 However, this species was never isolated and its electronic structure was not reported. In 2012, Morita and co-workers reported the first air-stable chiral phenalenyl [4]helicene radical (IV) stabilized by both an extensive spin delocalization and sterically hindering groups.37 Finally, in 2015, Bucher et al. used bulk electrolysis to produce an air-stable helical phenalenyl radical stabilized without steric hindering group. Electronic structure analysis revealed that the spin density is mostly delocalized across the phenalenyl and pyrilinium moieties.38 Importantly, while the crystal structures of air-stable triaryl porphyrin [4]helicene radicals35 and some heteroatom-centered [4]helicene radicals are known,39,40 the structure of metal-free carbon-centered [4]helicene radical has not been obtained (although the structure of the diamagnetic precursor of radical IV from Scheme 1 is available37).
We recently reported the use of [4]helical carbenium ions, DMQA+, as efficient photocatalysts to generate [4]helicene radicals in situ, using a red LED as the energy source.41 The quinacridyl scaffold, first introduced by Laursen,42 contains bridging amine groups that allow facile structural modification, and its heterocyclic core can be easily functionalized.43 The isolation and characterization of a series of [4]helicene radicals would provide a valuable insight for the design of new [4]helical carbenium-ion photocatalysts. Herein, we demonstrate the syntheses and structural tunability of [4]helicene quinacridyl neutral radicals and report their isolation, characterization, and electronic structure determination. The electronic properties of these radicals were systematically studied by 1H NMR, continuous wave (CW) EPR, electron-nuclear double resonance (ENDOR), cyclic voltammetry, UV-Vis absorption spectroscopy, and density functional theory (DFT) calculations. This allowed us to investigate the effect of the substituent group in the amine bridging unit (NR) on the physicochemical properties of these radicals. We also present the first X-ray structures of a metal-free carbon-centered [4]helicene radicals. Additionally, we demonstrate that this unique scaffold results in highly localized radicals that are persistent under inert atmosphere. Finally, the stability of all of these radicals under aerobic conditions was investigated by UV-Vis absorption spectroscopy. While most radicals react irreversibly with molecular oxygen, we observe the reversible oxidation of the presented carboradicals to their carbocation analogs.
Results and discussion
Synthesis
The precursor [4]helicenium cations (2+–5+) were synthesized by adapting literature protocols (see Scheme 2 and the ESI†),42,44 the tris(2,6-dimethoxyphenyl)methylium tetrafluoroborate (1),45 in the presence of a corresponding primary amine, was stirred in acetonitrile at 85 °C for 12 h. This resulted in the formation of the helicenium cations 2-H+, 3+, and the previously reported 4+, with high yields (Scheme 2, condition (i)). The helicenium 2-NO2+ was obtained by nitration of 2-H+ following the procedure reported by Lacour (Scheme 2, condition (ii)). Finally, the helicenium cation 5+ was synthesized in a two-step process (Scheme 2, condition (iii)). First, 1 was reacted with one equivalent of 2-(pyridin-2-yl)ethan-1-amine at room temperature for 2 h, resulting in a color change from purple to red, representative of the formation of the known acridinium intermediate, S1.46S1 was then reacted with excess 3-(dimethylamino)-1-propylamine in acetonitrile at 85 °C for 12 h, affording 5+ with high yield (Scheme 2). | ||
| Scheme 2 (a) Synthesis of cations, 2+–5+: (i) synthetic route for 2-H+, 3+ and 4+: 1.0 equiv. of (1), 25.0 equiv. of amine, CH3CN, 85 °C, 12 h; (ii) synthetic route for 2-NO2+: 1.0 equiv. of (2+), HNO3 (67 w%), 30 min; (iii) synthetic route for 5+: 1.0 equiv. of (1), 1.2 equiv. of 2-(2-dimethylaminoethyl)pyridine, EtOAc, rt, 1.5 h; followed by 15.0 equiv. of 3-(dimethylamino)-1-propylamine, CH3CN, 85 °C, 12 h. (b) Synthetic route for radicals, 2˙–5˙: 1.0 equiv. of (2+–5+), 1.1 equiv. of K or KC8 in THF, rt, overnight. | ||
The formation of [4]heliceniums 2-H+–5+ was confirmed by 1H and 13C NMR (see ESI†) and X-ray crystallography (vide infra). The 1H NMR spectra of the carbocations 2-H+ and 3+–5+ are well resolved and sharp. In contrast, the 1H NMR spectrum of 2-NO2+ shows broad and poorly resolved resonances at room temperature, suggesting a dynamic exchange. Variable temperature (VT) 1H NMR spectroscopy analysis of 2-NO2+ in CD2Cl2 over a temperature range of 193–333 K is consistent with the presence of an equilibrium between two conformers (Scheme 3). At 193 K, six methylenic protons are observed (Fig. S15†), three of which are diastereotopic, according to the low-temperature 1H–1H COSY NMR spectrum (Fig. S17†). These data suggest that 2-NO2+ favors the conformation of the propyl chain neighboring the nitro group that brings the terminal NMe2 group into a close intramolecular contact with the central carbon (Scheme 3).
 | ||
| Scheme 3 2-NO2+–nPr-NMe2 interaction with the central C+ suggested by VT 1H NMR spectroscopy. | ||
Radicals 2˙–5˙ were obtained by reduction of 2+–5+, respectively, with potassium metal in THF at room temperature overnight (Scheme 2 and ESI†). The reaction mixture turned from a dark green suspension to a dark magenta solution. After filtration of the formed insoluble KBF4 salt and removal of THF under vacuum, the resultant solids were extracted with toluene. Crystallization from a toluene/hexane mixture at −35 °C afforded 2˙–5˙ as dark brown crystals in good yields (Scheme 2). The formation of radicals 2˙–5˙ was confirmed by EPR spectroscopy (vide infra). Remarkably, the radicals formed are stable under inert atmosphere at room temperature in both the solid and solution over several months (see ESI†). The molecules 2˙–5˙ were analyzed by paramagnetic 1H NMR spectroscopy to determine if the pendant arms of the molecule have any effect or interaction with the radical system (Fig. S25 and S26†). Due to the paramagnetic nature of these compounds, the aromatic protons of the helicene radical scaffold are not observable. However, the α- and β-methylene protons of the pendant arms consistently produce broad lines at 17 and −7 ppm, respectively, for all radicals (the α and β positions are defined with respect to the bridging nitrogen, see ESI†). The 1H NMR signals from the terminal R groups (CH2NMe2, Py or CH3), while broad, are located in the diamagnetic 10–0 ppm region. This suggests that the unpaired electron is delocalized across the fused heterocyclic rings and that the nature of the pendant arms has little influence on the radical character of 2˙–5˙.
X-ray diffraction
Slow DCM/hexane layering afforded crystals of the cationic precursors 2-H+ and 2-NO2+ suitable for X-ray diffraction (XRD) analysis. In both structures (Fig. 1), the steric clash between the MeO groups results in a significant twist deformation characterized by the angles between the o-(MeO)-phenyl moieties of 41.93° and 38.37° in 2-H+ and 2-NO2+, respectively (see Table S5†). The difference in torsion angles between 2-H+ and 2-NO2+ is also evident from the O1–O2 distances of 2.743 Å and 2.659 Å, respectively. The two –nPr-NMe2 arms in 2-H+ extend away from the carbocation scaffold. In 2-NO2+, the –nPr-NMe2 arm adjacent to the NO2 group is significantly out of C4–C1–C2 plane due to a steric interaction and is folded over the carbocation center, shortening the C1–N1 distance to 3.194 Å (Fig. 1). This observation supports the NMe2–C+ interaction deduced from VT 1H NMR spectroscopic results. Such an interaction suggests an increased Lewis acidity conferred on the carbocation center by the presence of the m-(NO2) group.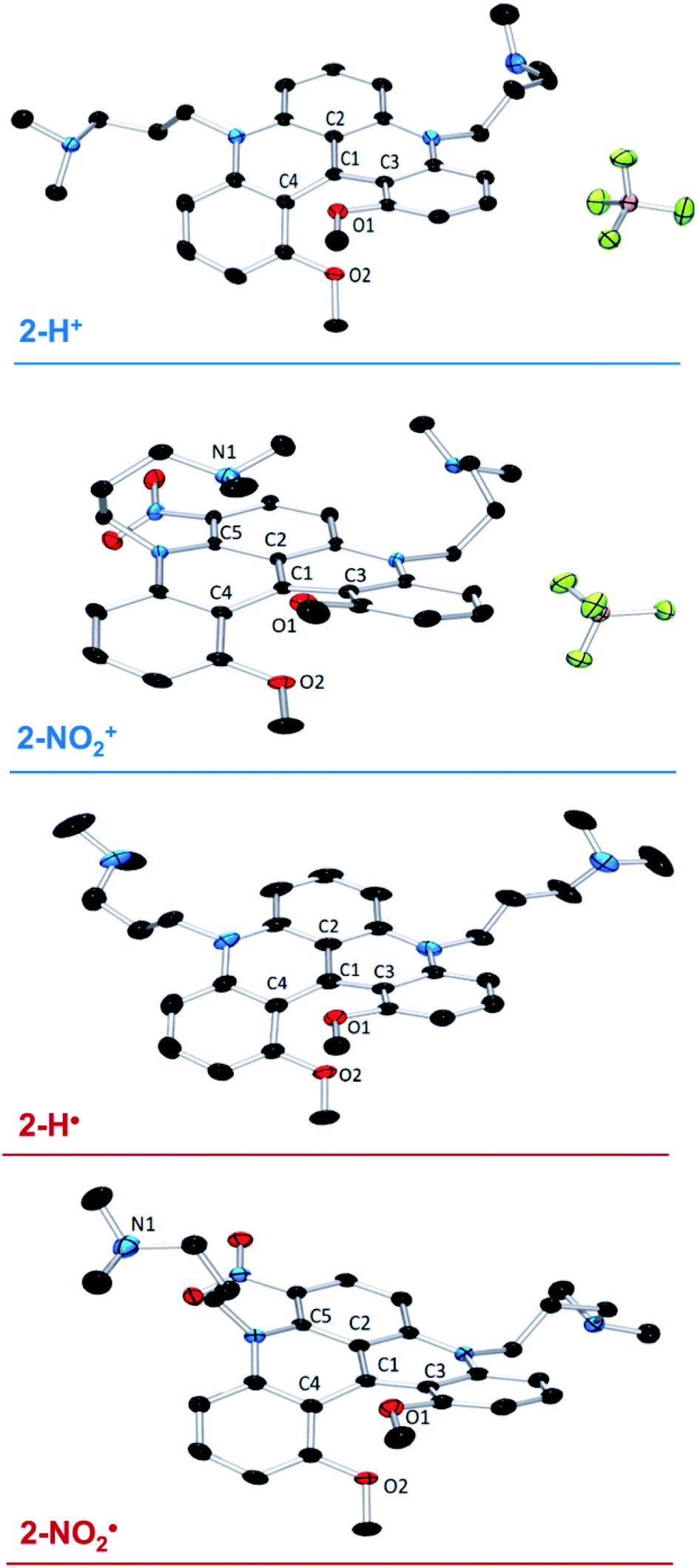 | ||
| Fig. 1 X-ray structures of 2-H+, 2-H˙, 2-NO2+, and 2-NO2˙. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
The crystals of the neutral radicals 2-H˙ and 2-NO2˙ were grown from concentrated toluene solutions by slow layering diffusion of hexane and analyzed by XRD. The absence of BF4− counterion in the crystal structures (Fig. 1) confirms the neutral electric charge of the molecules. It is of note that the packing structure of both radicals revealed the presence of close contact and π-stacking interactions, with distances between 3.2–3.5 Å, producing a 1D spin network (Fig. S27 and 28†). The torsional distortion of the o-(MeO)-phenyl groups is somewhat greater than in the cationic precursors: 45.92° vs. 41.93° in 2-H˙ and 52.05° vs. 38.37° in 2-NO2˙ (Fig. S29†), resulting in similar O1–O2 distances for both molecules: 2.772 Å in 2-H˙ and 2.773 Å in 2-NO2˙ (Table S5†). The increased C1–C2, C1–C3, and C1–C4 interatomic distances in 2-H˙ (1.439(2), 1.444(3), and 1.446(3) Å, respectively) relative to the cation 2-H+ (1.406(2), 1.435(2) and 1.431(2) Å) reflect a change in the electrostatic interactions between C1 and its surrounding atoms. Similarly, the C1–C3 bond in 2-NO2˙ (1.429(3) Å) was elongated when compared to 2-NO2+ (1.413(1) Å). In contrast, other bond distances around the central carbon (C1) in 2-NO2˙ were shortened compared to those in 2-NO2+ (C1–C2 = 1.422(3) vs. 1.431(1) Å) or unaffected (C1–C4 = 1.438(3) vs. 1.440(1) Å), suggesting a structural or electronic effect of the NO2 group. Finally, same as in the corresponding cation, the –nPr-NMe2 arm adjacent to the nitro group in 2-NO2˙ is significantly out of plane due a steric clash with the NO2 moiety (Fig. S30†). However, in contrast to 2-NO2+, the NMe2 is oriented away from the electron-rich π-system of the neutral radical molecule, and no intramolecular interaction is observed.
DFT calculations and EPR/ENDOR measurements
The electronic structure of radicals 2˙–5˙ was studied using DFT calculations (see ESI†). Natural bond order (NBO) analysis revealed a considerable spin population (about 40%) localized on the central carbon (C1) in all radicals. The calculated spin density distributions were similar, regardless of the nature of the alkyl groups on the amino substituent or the presence of the electron withdrawing group NO2 (Fig. S31†). In contrast, a significant change in the SOMO stabilization energy has resulted from introduction of the nitro group: −5 eV in 2-NO2˙vs. −4.4 eV in 2-H˙ (Fig. S32†).Fig. 2a shows the calculated spin populations on the atoms of the conjugated ring system (the numbers shown in magenta) in radicals 2-H˙ and 3˙–5˙. Note that only one half of the molecule is shown, because the ring system has an approximate C2 symmetry. Fig. 2b shows the spin populations for the radical 2-NO2˙, where a ring hydrogen on one side of the molecule (marked by an asterisk in Fig. 2a) was substituted by the NO2 group. As noted above, the spin population distributions in 2-NO2˙ and 2-H˙, as well as 3˙–5˙, are similar.
 | ||
| Fig. 2 a) Spin populations and 1H hfi constants in radicals 2-H˙ and 3˙–5˙. Only one half of the ring system is shown. The other half is related by C2 symmetry with respect to the C–C bond shown by the thick brown line. The spin populations (in % units) of the ring atoms calculated by DFT are shown in magenta. The calculated 1H hfi constants are shown in green, and those determined by ENDOR are in light blue. The ENDOR line assignments (see Fig. 3b) are in red color and in parentheses. The ring carbon spin populations estimated from the experimental hfi constants using the McConnell equation are shown in dark blue. (b) The same information for the radical 2-NO2˙, where the hydrogen marked by an asterisk in panel a was substituted by the NO2 group on one side of the molecule only. | ||
EPR measurements were conducted in toluene solutions at room temperature to further probe the electronic structure of these radicals. A Gaussian line centered at g ≈ 2.003, with the width of about 0.76 mT was observed, with a poorly resolved hyperfine structure with a splitting of 0.088 mT for 2-H˙ and 3˙–5˙ (trace 1, Fig. 3a) and unresolved structure for 2-NO2˙ (trace 2, Fig. 3a).
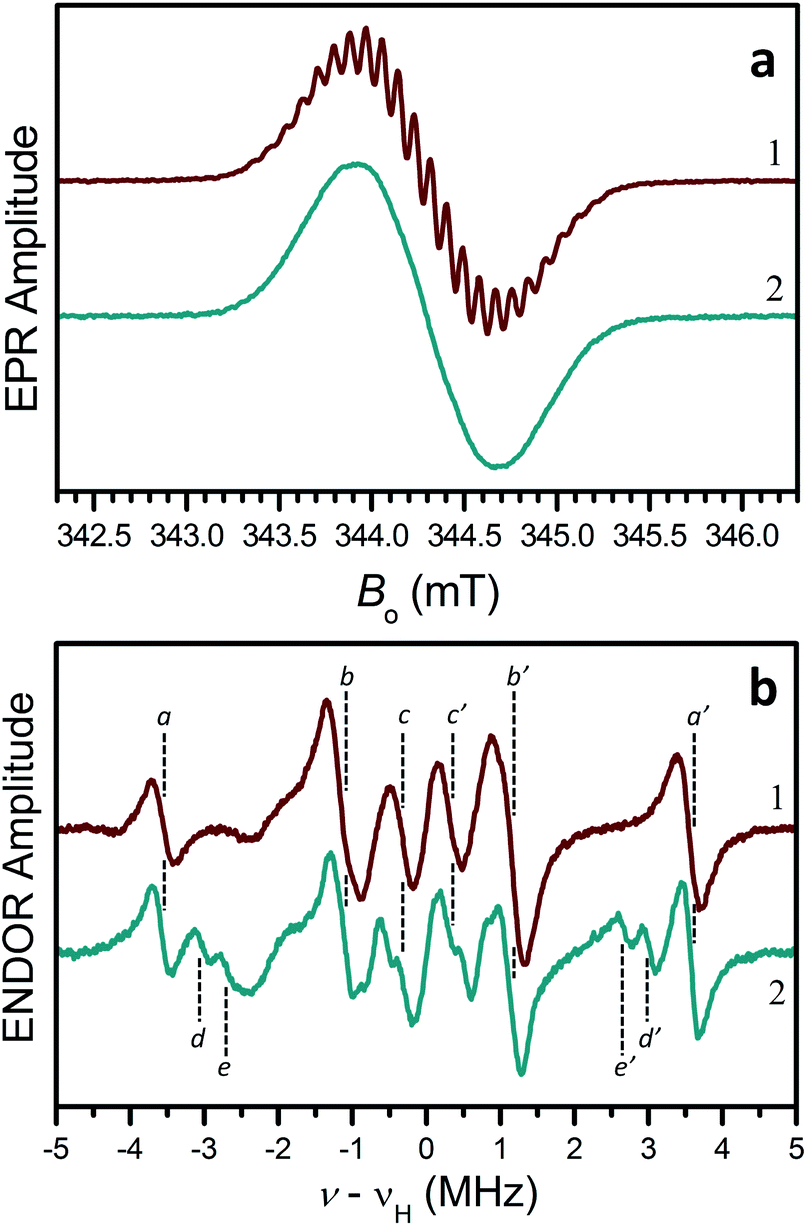 | ||
| Fig. 3 a) CW EPR spectra of radicals 2-H˙ (trace 1) and 2-NO2˙ (trace 2) in liquid toluene solutions at room temperature. Experimental conditions: mw frequency, 9.651 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.01 mT. (b) CW ENDOR spectra of radicals 2-H˙ (trace 1) and 2-NO2˙ (trace 2) in liquid toluene-d8 solutions. Experimental conditions: mw frequency, 9.558 GHz; mw power, 32 mW; magnetic field, 340.9 mT (center of the EPR spectrum); radiofrequency (rf) power, 200 W; rf modulation amplitude, 100 kHz (frequency modulation); temperature, 210 K. Note that both the EPR and ENDOR spectra of 3˙–5˙ were similar to those of 2-H˙. | ||
To gain insight into the hyperfine structure and the effect of the NO2 group, 1H hyperfine interactions (hfi) were determined by recording the 1H ENDOR spectra (Fig. 3b). The 1H ENDOR spectra of 2-H˙ and 3˙–5˙ were generally similar, showing three pairs of lines denoted by (a,a′), (b,b′), and (c,c′) (Fig. 3b, trace 1). Each pair of lines is located at the frequencies of νH ± αH/2, where νH is the proton Zeeman frequency and aH is the hfi constant (different for different protons). The specific hfi constants estimated for each pair of ENDOR lines are: |aHa| ≈ 7.1 MHz (for a,a′ lines), |aHb| ≈ 2.3 MHz (for b,b′ lines), and |aHc| ≈ 0.65 MHz (for c,c′ lines). The splitting of 0.088 mT (∼2.5 MHz in frequency units) observed in the EPR spectra of 2-H˙ and 3˙–5˙ is obviously determined by aHa and aHb.
The 1H hfi constants were assigned to specific molecular positions by comparing them to the DFT predictions. In Fig. 2, the 1H hfi constants resulting from the calculated spin populations on the hydrogen atoms (aH = 1420ρH [MHz]) are shown in green, and the corresponding experimental hfi constants are shown in light blue. The ENDOR line assignments are indicated in parentheses. The spin populations on the ring carbon atoms estimated from the experimental aH values of the ring protons (α-protons) using the McConnell equation: aH = (−63 MHz)ρC, are shown in dark blue.
For 2-NO2˙, the hydrogen atom marked by an asterisk in Fig. 2a is substituted by the NO2 group (on one side of the molecule only). As discussed above, this leads to a significant conformational distortion and a reorientation of the α-methylene group belonging to the pendant arm in the vicinity of NO2 (Fig. S30†). The spectroscopic result of these modifications is a ∼30% decrease of the relative intensity of (b,b′) lines in the ENDOR spectrum and appearance of (d,d′) and (e,e′) lines corresponding to |aH| = 6.05 and 5.37 MHz, respectively (see trace 2 in Fig. 3b). Such a significant decrease in the (b,b′) lines intensity can be explained by the fact that, according to our hfi assignments shown in Fig. 2, the H to NO2 substitution removes one proton from the |aH| = 2.3 MHz pool ((b,b′) lines). In addition, two more protons (those belonging to the affected α-methylene group) are “relocated” from the (b,b′) lines to the new (d,d′) and (e,e′) ENDOR lines. The three protons removed from the |aH| = 2.3 MHz pool, which originally consisted of 10 protons, thus explain the 30% decrease of the (b,b′) lines intensity. The change of the hfi constants of the α-methylene protons from 2.3 MHz to 6.05 and 5.37 MHz is mostly caused by their repositioning with respect to the spin-carrying electronic orbital.
It is interesting to compare the spin population distributions obtained in this work with the distribution calculated (and supported by EPR and ENDOR results) for the [4]helicene radical IV (see Scheme 1).37 While in our radicals the largest spin population (∼42.5%) is located on the central carbon atom (see Fig. 2), radical IV has the largest spin populations (∼30% each) on the two ring carbons adjacent to the apex of the triangle representing the shape of the molecule (black ring in Scheme 1).37 At the same time, the spin population on the central carbon in radical IV is small and negative.
This dramatic change in the spin population distribution can be traced to the difference in the chemical identity of the ring nitrogens: amino in our radicals vs. imino in radical IV. It is of note that this change occurs in spite of the fact that the nitrogen coordination geometry in our radicals remains close to planar, and thus the change in spin population distribution is likely caused by the difference in the number of electrons donated into the π-system in both cases: two in our radicals and one in radical IV.
UV-Vis spectroscopy and electrochemistry
The UV-Vis spectra of the cations and radicals (2–5) were studied to further probe the electronic structures of these molecules. All studied cations and radicals possess three distinct absorption features in the visible region. Compounds 2-H+ and 3+–5+ show similar spectra, with a sharp peak around 620 nm, a shoulder at 570 nm, and a broad absorption around 430 nm (Fig. 4a). However, the low energy transition of the helicenium 2-NO2+ is blue-shifted compared to 2-H+. Similar observations were made for the [4]helicene radicals (Fig. 4b). Compounds 2-H˙ and 3˙–5˙ have comparable spectra, whereas the low energy transition of 2-NO2˙ is blue-shifted. These observations suggest that the nature of the nitrogen bridge substituents has a negligible effect on the transition energies. However, the introduction of the electron-withdrawing NO2 group induces a moderate hypsochromic shift of the lower energy transition and an increase of molar extinction coefficient for the high energy transition in both 2-NO2+ and 2-NO2˙ compared to 2-H+ and 2-H˙ respectively (Fig. 4a and b). | ||
| Fig. 4 (a) UV-Vis spectra of heliceniums 2+–5+ in PhCF3, and (b) UV-Vis spectra of helicene radicals 2˙–5˙ in PhCF3. | ||
Interestingly, while all radicals exhibit a weak broad transition in the long-wavelength region (between 600 and 750 nm), their three main absorptions are being significantly blue-shifted from their cation analogs (Fig. S34 and Table S8†). Laursen previously reported that such blue shift indicates a reduced effect of heteroatoms of the conjugated framework and a reduced conjugation across the π-system,33 consistent with the increased torsional distortion observed in solid state and a predominant localization of the spin density on the central carbon.
The stability of these radicals to air was investigated using UV-Vis spectroscopy. Solutions of 2˙–5˙ in CF3-toluene prepared in an N2-filled glove box were exposed to air and monitored over time. Upon exposure to air, the absorption spectra of the radicals 2-H˙ (Fig. 5) and 3˙–5˙ (Fig. S40–S45†) slowly (within hours) transformed to those of the cationic analogs. Unlike most organic radicals, these DMQA radicals do not undergo oxygen insertion or dimer formation. Instead, a clean reversible oxidation to DMQA+ is observed.47 This is explained by the fact that the reactivity of the unpaired electron (predominantly localized at the central carbon) is kept in check by the helical molecular structure maintained by the steric interaction between the two ortho-methoxy groups, which hampers the dimerization and formation of the oxo-analog via oxidative degradation.32,48 The first-order decay of 2-H˙ is characterized by the half-life time (t1/2) of 26 min (Fig. S35†). The t1/2 values for 3˙–5˙ were significantly larger: 51 min, 54 min, and 44 min, respectively (Fig. S35†). The 2-NO2˙ radical shows a dramatic increase in t1/2 to about 210 min (Fig. S35†), but a less selective oxidation (Fig. S39†). The reduced reactivity of 2-NO2˙ is attributed to the inductive effect of the electron-withdrawing NO2 groups (vide supra, cyclic voltammetry and SOMO energy).49–52
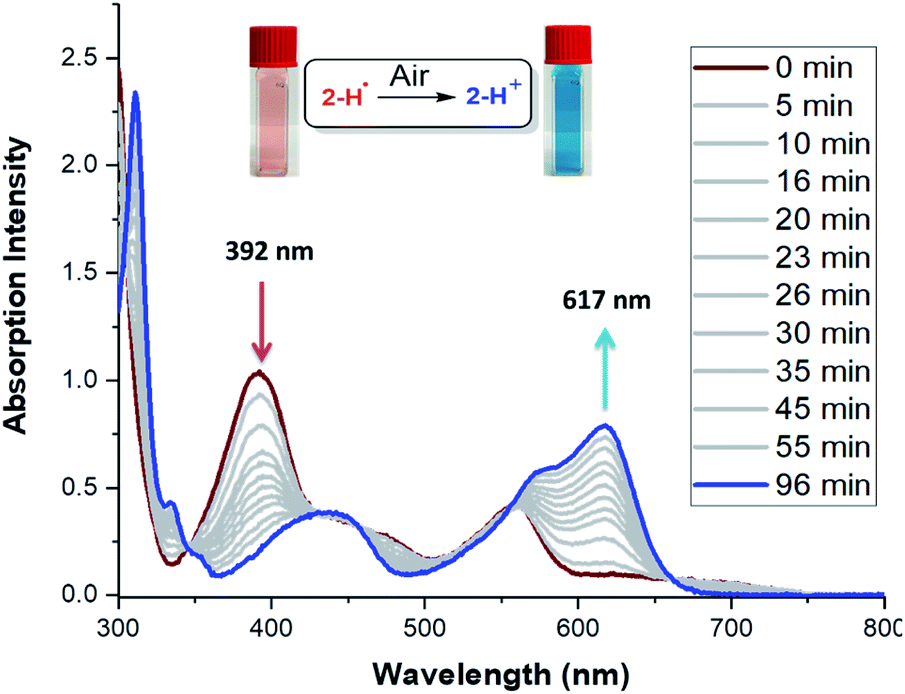 | ||
| Fig. 5 Change of UV-Vis absorption spectra of 2-H˙ in PhCF3 upon exposure to air. | ||
To further understand the reactivity differences between these [4]helicene radicals, the electrochemical behavior of 2+–5+ was investigated by cyclic voltammetry (see Fig. 6 and ESI† for details). Under reductive conditions, a reversible event is observed around E1/2 = −1.25 V vs. Fc/Fc+ in 2+–5+. This event, which correspond to the reduction of DMQA+ to DMQA˙,33 is observed at a higher potential for 2-NO2+ (−1.05 V vs. Fc/Fc+), consistent with a more electron-deficient scaffold and a reduced reactivity toward oxygen due to the presence of the NO2 group. A second redox event corresponding to the reduction of DMQA˙ to DMQA− was also present.33 This was observed as a reversible process at E1/2 = −1.85 V for 2-NO2+ and as an irreversible process for 2-H+ and 3+–5+ at E1/2 = −2.3 V vs. Fc/Fc+.33
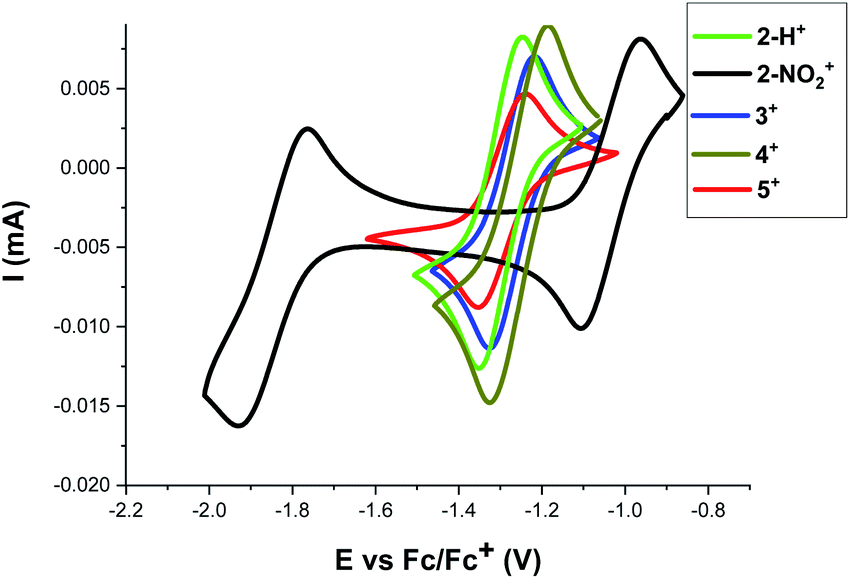 | ||
| Fig. 6 Cyclic voltammograms of 2+–5+ (2 mM) in DCM ([TBA][PF6] 0.1 M) solutions recorded at a platinum working electrode (v = 0.1 V s−1) and Ag/Ag+ as internal reference electrode, Fc/Fc+ was used as secondary reference by setting its E1/2 = 0. The arrows indicate the direction of the scan. The reduction half-wave potentials Ered11/2vs. the Fc/Fc+ redox couple are: −1.31, −1.05, −1.26, −1.27, and −1.29 V for 2-H+, 2-NO2+, 3+, 4+, and 5+, respectively. In addition, the second reduction event with Ered21/2 = −1.85 was observed for 2-NO2+. | ||
Conclusions
In summary, we synthesized and isolated a series of [4]helicene quinacridyl radicals 2˙–5˙. The spectroscopic, computational, and chemical characterization suggests that the nature of the bridging group (NR) has little to no impact on the electronic structure, reactivity, and physical properties of these radicals because the unpaired spin resides predominantly in the heterocyclic π-system. Introduction of an electron withdrawing group, 2-NO2˙, resulted in an enhanced stability demonstrated by the more negative SOMO energy, a less reducing E1/2 potential, and a longer lifetime when exposed to air. However, EPR spectroscopy and DFT calculations show that the electronic structure is virtually unaffected, with the unpaired electron in 2˙–5˙ being predominantly localized on the central carbon. Importantly, despite the strongly localized spin density, no dimerization or decomposition under inert atmosphere were observed. This is a consequence of the helicity of the conjugated scaffold, which can be modified to achieve a desired/optimal kinetic stabilization of carbon-centered radicals. Furthermore, spin localization at the carbon center promotes a clean and reversible oxidation upon exposure to air, as supported by the UV-Vis spectroscopy. The same reversibility of the radical ↔ cation transformation was observed electrochemically. Cyclic voltammograms of 2+–5+ indicated a highly reversible one-electron redox event for this process. Finally, X-ray crystallography analysis of 2-H˙ and 2-NO2˙ revealed that these [4]helicene radicals are efficiently protected from σ-dimerization while being sufficiently exposed to allow intermolecular interactions in the solid state.The DMQA radicals are catalytic intermediates in the light-induced transformations catalysed by DMQA+.41 The structure/properties relationship of these radicals presented in this work will help develop more durable and efficient photocatalysts. Furthermore, due to their high persistence, wide-range optical absorption, reversible redox properties, and spin localization with minimal steric protection, these helical radicals have great potential for photocatalysis, optical, and materials applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the University of Arizona for this work. We thank Professor Rebecca Page and Dr Michael Clarkson from the University of Arizona for Single crystal X-ray diffractions data collection. We thank Professors Elisa Tomat and Jon Njardarson from the University of Arizona, and Prof. Robert G. Bergman from UC Berkeley for helpful discussions. J. M. V. would like to acknowledge the effort of individuals, groups, and institutions working towards a more diverse and inclusive environment in academia.Notes and references
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 Search PubMed
.
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 Search PubMed
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 Search PubMed
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 Search PubMed
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 Search PubMed
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 Search PubMed
- D. A. Wilcox, V. Agarkar, S. Mukherjee and B. W. Boudouris, Annu. Rev. Chem. Biomol. Eng., 2018, 9, 83–103 Search PubMed
- S. Kumar, Y. Kumar, S. Keshri and P. Mukhopadhyay, Magnetochemistry, 2016, 2, 42 Search PubMed
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 Search PubMed
- T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105–3118 Search PubMed
- J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560–2575 Search PubMed
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 Search PubMed
- Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463–1535 Search PubMed
- A. Rajca, Chem. Rev., 1994, 94, 871–893 Search PubMed
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 Search PubMed
- B. Tang, J. Zhao, J. F. Xu and X. Zhang, Chem. Sci., 2020, 11, 1192–1204 Search PubMed
- K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 Search PubMed
- R. S. Ghadwal, Synlett, 2019, 30, 1765–1775 Search PubMed
- T. Kubo, Molecules, 2019, 24, 665 Search PubMed
- T. F. Hunter and M. C. R. Symons, Nature, 1967, 213, 1121–1122 Search PubMed
- J. Melin and P. Fuentealba, Int. J. Quantum Chem., 2003, 92, 381–390 Search PubMed
- S. Mitov, A. Panchenko and E. Roduner, J. Phys. Chem. A, 2007, 111, 5294–5299 Search PubMed
- N. E. Horwitz, B. T. Phelan, J. N. Nelson, C. M. Mauck, M. D. Krzyaniak and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 4455–4463 Search PubMed
- M. Fujita, A. Ishida, T. Majima and S. Takamuku, J. Phys. Chem., 1996, 100, 5382–5387 Search PubMed
- K. L. Ivanov, A. Wagenpfahl, C. Deibel and J. Matysik, Beilstein J. Nanotechnol., 2017, 8, 1427–1445 Search PubMed
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691–1695 Search PubMed
- Z. Sun and J. Wu, J. Mater. Chem., 2012, 22, 4151–4160 Search PubMed
- Y. Wang, H. Zhang, M. Pink, A. Olankitwanit, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2016, 138, 7298–7304 Search PubMed
- P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 Search PubMed
- J. K. Zak, M. Miyasaka, S. Rajca, M. Lapkowski and A. Rajca, J. Am. Chem. Soc., 2010, 132, 3246–3247 Search PubMed
- K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem.–A Eur. J., 2018, 24, 572–575 Search PubMed
- F. A. Neugebauer, D. Hellwinkel and G. Aulmich, Tetrahedron Lett., 1978, 19, 4871–4874 Search PubMed
- T. J. Sørensen, M. F. Nielsen and B. W. Laursen, ChemPlusChem, 2014, 79, 1030–1035 Search PubMed
- O. Neunhoeffer and H. Haase, Chem. Ber., 1958, 91, 1801–1805 Search PubMed
- K. Kato, W. Cha, J. Oh, K. Furukawa, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 8711–8714 Search PubMed
- D. Hellwinkel, M. Melan and G. Aulmich, Tetrahedron Lett., 1976, 17, 4137–4138 Search PubMed
- A. Ueda, H. Wasa, S. Suzuki, K. Okada, K. Sato, T. Takui and Y. Morita, Angew. Chem., Int. Ed., 2012, 51, 6691–6695 Search PubMed
- O. Anamimoghadam, M. D. Symes, D. L. Long, S. Sproules, L. Cronin and G. Bucher, J. Am. Chem. Soc., 2015, 137, 14944–14951 Search PubMed
- B. D. Gliemann, A. G. Petrovic, E. M. Zolnhofer, P. O. Dral, F. Hampel, G. Breitenbruch, P. Schulze, V. Raghavan, K. Meyer, P. L. Polavarapu, N. Berova and M. Kivala, Chem. Asian J., 2017, 12, 31–35 Search PubMed
- M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Inorg. Chem., 2007, 46, 10153–10157 Search PubMed
- L. Mei, J. M. Veleta, T. L. Gianetti, L. Mei, J. M. Veleta and T. L. Gianetti, J. Am. Chem. Soc., 2020, 142, 12056–12061 Search PubMed
- B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 Search PubMed
- I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 Search PubMed
- C. Herse, D. Bas, F. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 Search PubMed
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 Search PubMed
- L. Mei, J. M. Veleta, J. Bloch, H. J. Goodman, D. Pierce-Navarro, A. Villalobos and T. L. Gianetti, Dalton Trans. 10.1039/d0dt00419g
- During the oxidation of the DMQA˙ to DMQA+ upon exposure to air, the nature of the counter ions was not indentified. We hypothesized that the superoxide ion (O2−˙) is formed, which will likely further react with its environment, based on our previous report (ref. 41) and the recent report: M. Karimi, R. Borthakur, C. L. Dorsey, C.-H. Chen, S. Lajeune and F. P. Gabbaï, J. Am. Chem. Soc., 2020, 142, 13651–13656 Search PubMed
- M. J. Sabacky, C. S. Johnson, R. G. Smith, H. S. Gutowsky and J. C. Martin, J. Am. Chem. Soc., 1967, 89, 2054–2058 Search PubMed
- E. Font-Sanchis, C. Aliaga, K. S. Focsaneanu and J. C. Scaiano, Chem. Commun., 2002, 2, 1576–1577 Search PubMed
- E. V. Bejan, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2001, 3, 4059–4062 Search PubMed
- H. D. Hartzler, J. Org. Chem., 1966, 31, 2654–2658 Search PubMed
- J. C. Scaiano, A. Martin, G. P. A. Yap and K. U. Ingold, Org. Lett., 2000, 2, 899–901 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2004974–2004977. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04850j |
| This journal is © The Royal Society of Chemistry 2020 |