DOI:
10.1039/D0SC04826G
(Edge Article)
Chem. Sci., 2020,
11, 11055-11059
Phosphinoborylenes as stable sources of fleeting borylenes†
Received
2nd September 2020
, Accepted 9th September 2020
First published on 10th September 2020
Abstract
Base-stabilised borylenes that mimic the ability of transition metals to bind and activate inert substrates have attracted significant attention in recent years. However, such species are typically highly reactive and fleeting, and often cannot be isolated at ambient temperature. Herein, we describe a readily accessible trimethylphosphine-stabilised borylborylene which was found to possess a labile P–B bond that reversibly cleaves upon gentle heating. Exchange of the labile phosphine with other nucleophiles (CO, isocyanide, 4-dimethylaminopyridine) was investigated, and the binding strength of a range of potential borylene “ligands” has been evaluated computationally. The room-temperature-stable PMe3-bound borylenes were subsequently applied to novel bond activations including [2 + 2] cycloaddition with carbodiimides and the reduction of dichalcogenides, revealing that PMe3-stabilised borylenes can effectively behave as stable sources of the analogous fleeting dicoordinate species under mild conditions.
Introduction
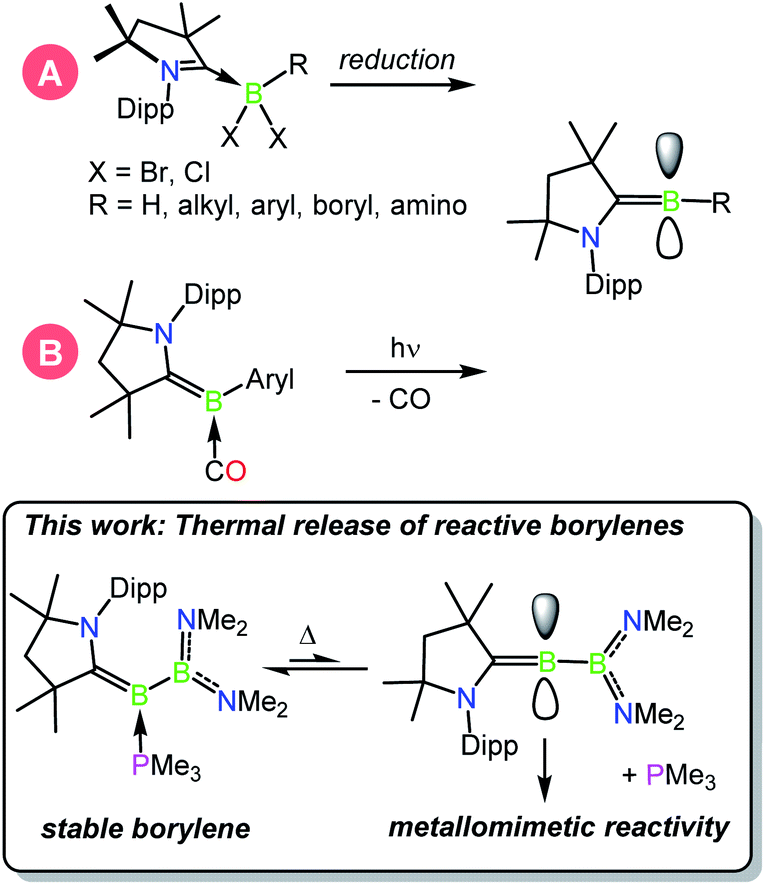
In the continuing effort to replace environmentally deleterious metals in chemical reactions, p block compounds that mimic the unique properties of transition metals in the activation of strong bonds have recently come to the forefront of main-group chemical research.1–3 In particular, low-valent boron compounds in the form of bimolecular frustrated Lewis pairs,4–8 B–B multiply bonded species,9–11 and borylenes12,13 have captured significant attention as their unique bonding patterns and orbital arrangements allow for favorable interactions with a number of substrates often found in TM-catalysed reactions. While the chemistry of metalloborylenes,14–16 B–B multiple bonds, and particularly that of frustrated Lewis pairs are more mature fields of study, the field of metal-free borylenes has more recently emerged due to early challenges faced in the study of these oft-fleeting species.17–19 The study of isolable metal-free borylenes began in 2011 with the characterisation of a stable borylene supported by two cyclic alkyl(amino)carbenes (CAACs)20,21 by Bertrand and co-workers.22 These and other similar borylenes are nucleophilic at boron, and have been shown to act both as donor ligands and reducing agents toward a variety of main-group and transition metal species.23–30 In 2014, a significant advance in the synthesis of the first isolable, singly base-stabilised borylene was reported by Bertrand and Stephan via the introduction of electronic push and pull substituents at the boron centre.31 With boron-centred filled and vacant orbitals, this species was the first crystalline borylene with an electronic structure similar to that of a transition metal, and was able to effect metallomimetic reactivity in the binding of CO and scission of dihydrogen at ambient temperature.32 In another advance, in 2018 we reported the capture of dinitrogen by a fleeting singly base-stabilised borylene,33,34,42,43 revealing the powerful ability of these emerging species to activate highly inert bonds (Scheme 1).37
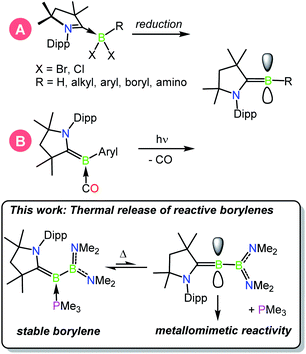 |
| | Scheme 1 Known methods of generating CAAC-stabilised borylenes: (A) reduction, (B) photolytic CO extrusion.18 | |
Ligand exchange is a hallmark of transition metal chemistry, allowing reversible bond activations that are crucial steps in a multitude of catalytic processes, and is an emerging property of low-valent main group species.35,36 It was recently reported that metallomimetic reactivity could be achieved by the photolytic extrusion of CO from carbonyl-borylenes,28,37 providing the first methods to generate fleeting singly base-stabilised borylenes from a doubly base-stabilised borylene precursor. Herein, we report that the facile thermal release of trimethylphosphine from phosphine-stabilised borylborylenes can likewise be used to generate reactive borylenes that effect metallomimetic transformations under mild conditions.
Results and discussion
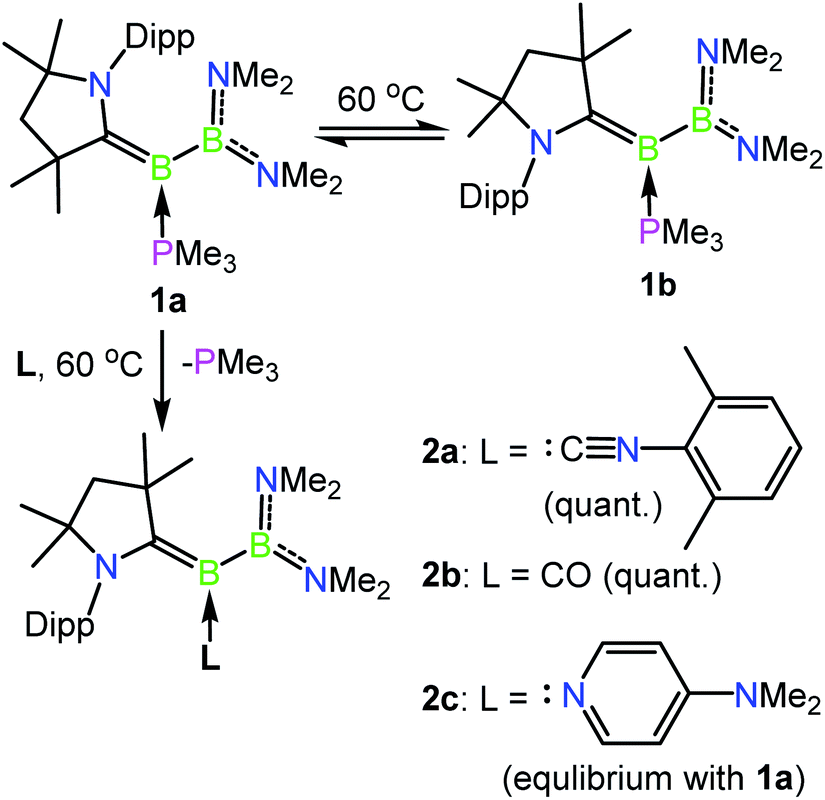
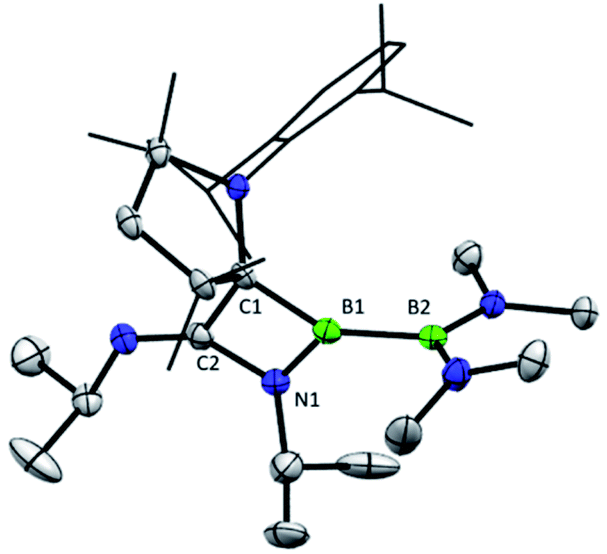
The previously reported preparation of the phosphine-stabilised borylene 1a involves reduction of the CAAC-coordinated aminoboryl(amino)borenium salt [(MeCAAC)B(NMe2)BBr(NMe2)][BArCl] with KC8 at −80 °C, followed by subsequent addition of a slight excess of PMe3.38 After warming to room temperature, filtration and concentration of the solution, the product was crystallised from the reaction mixture, affording a single isomer 1a with 11B NMR spectroscopic signals at 40.1 and 0.7 ppm.38 However, it was noticed that when volatiles were rapidly removed from the reaction mixture such that crystallisation was largely avoided, a second isomer 1b was also present, distinguishable by its 11B NMR shifts at 42.3 and −1.7 ppm. While pure solutions of isomer 1a are stable at ambient temperature, isomer 1b could also be formed in small amounts by heating solutions of 1a, with approximately 23% conversion to 1b observed after heating a C6D6 solution of 1a at 60 °C for 30 minutes. Single crystals of 1b were obtained by kinetic resolution of a mixture of 1a and 1bvia reaction with 2,6-dimethylphenylisocyanide (vide infra), and its molecular structure was determined crystallographically (Fig. 1). Metric parameters of 1a and 1b are similar and indicate significant delocalisation of the borylene lone pair electrons towards the carbene carbon centre.
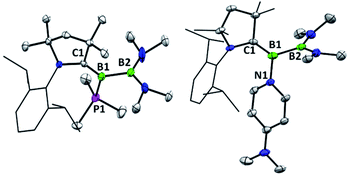 |
| | Fig. 1 Crystallographically-derived molecular structure of the Z-isomer of the phosphinoborylene 1b (left) and the DMAP-stabilised borylene 2c (right). Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms and thermal ellipsoids of the Dipp groups are omitted for clarity. Selected bond lengths (Å) and angles (°) for 1b: B1–B2: 1.732(3), B1–C1: 1.459(3), B1–P1: 1.904(2); C1–B1–P1: 123.5(1), C1–B1–B2: 128.8(2), B2–B1–P1: 106.8(1). For 2c: B1–B2: 1.714(4), B1–C1: 1.463(3), B1–N1: 1.584(3); C1–B1–N1: 122.4(2), C1–B1–B2: 125.7(2), B2–B1–N1: 111.5(2). | |
As such an isomerisation suggested lability of the phosphine, exchange reactions of 1a with more strongly interacting donor–acceptor ligands were attempted. Heating a yellow solution of 1a with 3.0 equiv. of 2,6-dimethylphenylisocyanide at 60 °C resulted in a gradual color change to deep red and quantitative conversion to the isocyanide-stabilised borylene 2a after 6 h (Scheme 2). A similar reaction with 1 atm of CO also resulted in complete conversion to borylene carbonyl 2b. However, upon heating 1a with 2.6 equivalents of the nitrogen base 4-(N,N-dimethylamino)pyridine (DMAP), only partial conversion to the DMAP-stabilised borylene 2c was observed after 6 h, which eventually gave way to decomposition to a mixture of products upon further heating. The formation of 2c was confirmed by its independent synthesis via low-temperature KC8 reduction of [(MeCAAC)B(NMe2)BBr(NMe2)][BArCl] and subsequent addition of DMAP. Compound 2c displays 11B NMR signals at 42.9 and 23.5 ppm, and crystals suitable for X-ray diffraction were grown by slow evaporation of a pentane solution (Fig. 1). A molecular structure confirmed the formulation of 2c and revealed a B–NDMAP bond (1.584(3) Å) and a B–CCAAC bond (1.463(3) Å), indicating population of the B–CCAAC π-bond and the presence of a single dative bond between DMAP and the borylene centre. As with 1a, isomerism about the B–CAAC bond is also observed in solutions of 2c, although only the Z isomer could be crystallographically characterised (see ESI†).
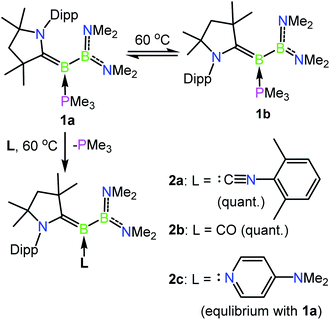 |
| | Scheme 2 Isomerisation and exchange reactions of phosphine-borylene 1a. Dipp = 2,6-diisopropylphenyl, quant. = quantitative. | |
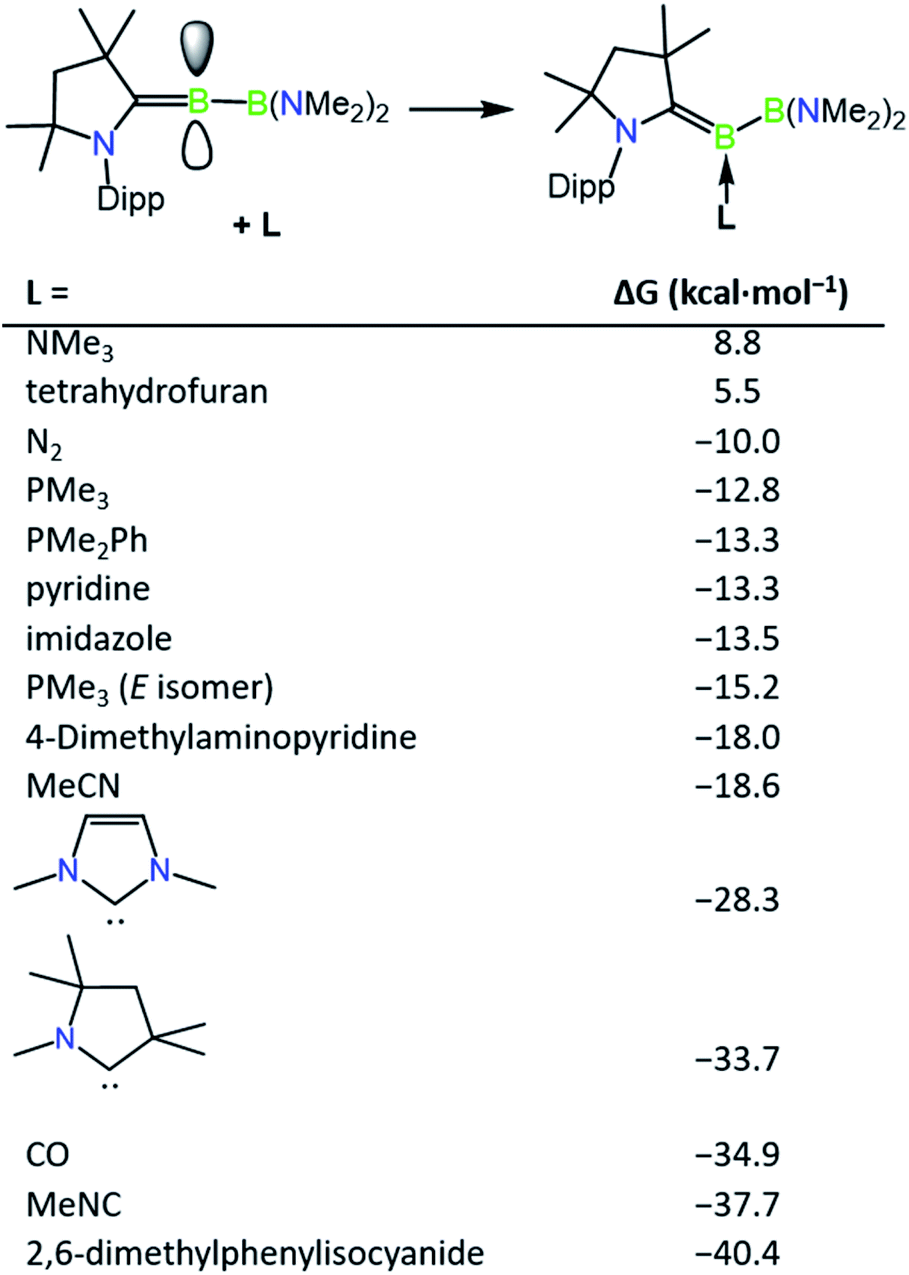
The incomplete conversion of 1a to 2c despite the presence of an excess of DMAP suggested that an equilibrium is formed between these two species, and therefore we turned to computations to evaluate the relative thermodynamic stabilities of a range of base-stabilised borylenes (Fig. 2). It was found that both phosphorus and nitrogen bases feature weak interactions with the borylene centre (ca. −10 to −18 kcal mol−1) and might be anticipated to form thermally labile complexes. Despite the calculated bonding strength of −10.0 kcal mol−1 for molecular nitrogen, we were unable to observe formation of an N2 product by exchange with 1a, likely due to a low kinetic barrier to its decomposition at ambient temperature. Conversely, more strongly donating NHCs and more strongly π-acidic bases such as CO and isocyanides feature stronger bonds with the borylene centre, which would be anticipated to form robust complexes. It was found that the energy of formation of 1a (−15.2 kcal mol−1) and 2c (−18.0 kcal mol−1) are similar with respect to the “free” unsaturated dicoordinate borylene, accounting for the partial conversion to 2c observed upon heating solutions of 1a. The implied lability of the DMAP ligand in 2c was further confirmed by an exchange reaction with 4.0 equivalents of 2,6-dimethylphenylisocyanide which quantitatively converted to 2a after 3 h at ambient temperature. The partial isomerisation of 1a into 1b upon heating can also be accounted for by their similar thermodynamic stability, which differ by only 2.4 kcal mol−1. Despite this, the barrier for PMe3 loss from 1b seems to be greater than that of 1a, as heated reactions of 1a with 2,6-dimethylphenylisocyanide become enriched in 1b as the reaction progresses, from which single crystals of 1b can be obtained as a mixture with 2a. The borylenes 2a and 2b have significantly more exergonic formation energies (−40.4 and −34.9 kcal mol−1) and were found to be inert to thermal exchange or decomposition even at temperatures as high as 100 °C.
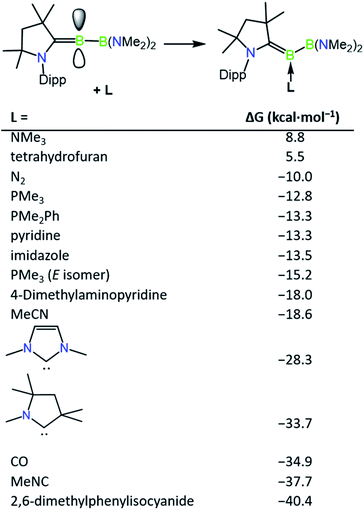 |
| | Fig. 2 Relative thermodynamic stabilities of ligand-stabilised borylenes (M06 (D3)/Def2-SVP). Computed isomer is Z unless otherwise stated. | |
A kinetic study on the conversion of 1a to 2a was also undertaken (see ESI†), which indicated that this reaction is first-order in 1a, but zeroth-order in 2,6-dimethylisocyanide, indicating that dissociative substitution is likely operating. Monitoring reaction rate at variable temperatures has revealed an activation barrier of 29.3 kcal mol−1 for this conversion, which is in line with its steady progress at 60 °C. This thermal release of reactive borylenes complements earlier work where similar species were generated photolytically from carbonyl-stabilised borylenes,28,37 and now permits exchange reactions of both strongly and weakly bound borylene ligands.
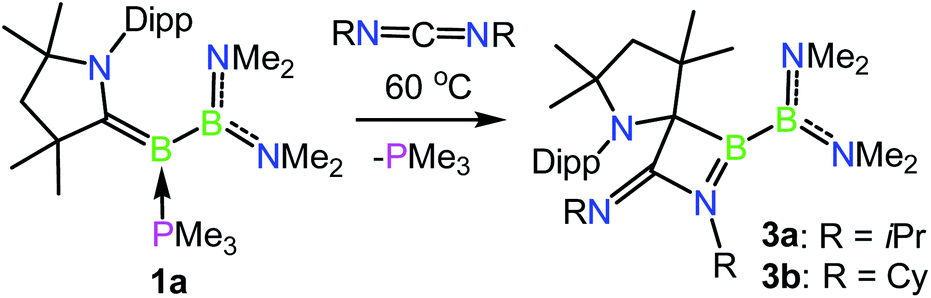
Given the ability of 1a to participate in exchange reactions with nucleophiles, we became interested in its ability to effect further ‘free borylene’ type reactivity in the capture of unsaturated molecules. To this end, heating a solution of 1a with 3.0 equiv. of N,N′-diisopropylcarbodiimide resulted in clean conversion to the [2 + 2] cycloaddition product 3a, whereby a C–C bond has been formed between the formerly carbene carbon center and the central carbon of the carbodiimide (Scheme 3 and Fig. 3). This transformation is unique in that it is not exclusively borylene-centred but involves a 1,2 addition across the B–C double bond, somewhat reminiscent of the formation of the intermediate metallocyclobutane in transition metal-based olefin metathesis.3911B NMR spectroscopy revealed chemical shifts of 61.3 and 34.2 ppm for 3a, which can be assigned to the CAAC–B and B(NMe2)2 fragments, respectively. The reaction of 1a with N,N-dicyclohexylcarbodiimide likewise provided clean conversion to the analogous product 3b, which exhibits similar structural and spectroscopic characteristics to 3a. However, the bulkier N,N′-bis(2,6-diisopropylphenyl)carbodiimide did not react with 1a upon extended heating at 60 °C, likely due to its steric bulk.
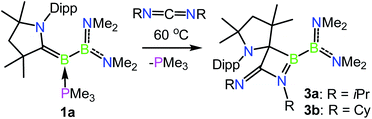 |
| | Scheme 3 Capture of carbodiimides with phosphine-borylene 1a. | |
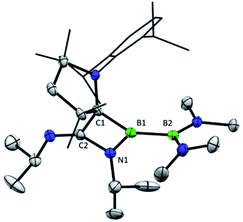 |
| | Fig. 3 Crystallographically-derived molecular structure of the N,N′-diisopropylcarbodiimide trapping product 3a. Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms and ellipsoids of the Dipp groups are omitted for clarity. Selected bond lengths (Å): B1–B2: 1.724(3), B1–N1: 1.428(2), C1–C2: 1.643(2). | |
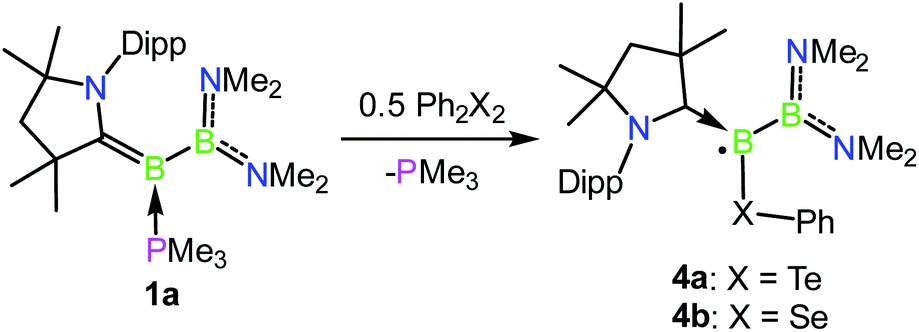
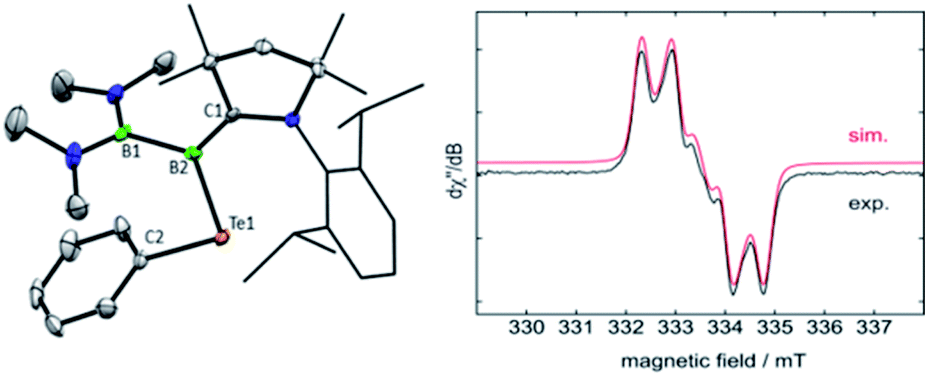
Oxidation reactions were also explored with the phosphine-borylene 1a. While the reaction of 1a with S8 was difficult to control and not particularly selective in any stoichiometry, combination of one equivalent of 1a with 0.5 equivalents of Ph2Te2 resulted in the immediate release of PMe3 coupled with the loss of all other signals in the 1H NMR spectrum, suggesting the formation of a paramagnetic product. Evaporation of the volatiles yielded red single crystals, from which an X-ray crystallographic study identified the product as the CAAC-stabilised telluroborane radical 4a, which was isolated in a 70% yield upon recrystallisation (Scheme 4 and Fig. 4). A molecular structure of 4a confirmed removal of one electron from the borylene lone pair and revealed a partial double bond between the B and C centres, with a C–B bond length of 1.504(5) Å. EPR studies confirmed the radical nature of 4a, and computational investigations indicated that the spin density is located primarily on the boron, carbon and nitrogen atoms (see ESI†), as has been previously observed for related CAAC-stabilised boron-centred radicals.31,32,40,41 The reaction of 1a with Ph2Se2 also yielded the analogous selenoborane radical 4b, which has similar metric parameters to 4a. Given the very short timescale of these oxidation reactions (<1 min) at ambient temperature, it seems probable that phosphine loss is preceded by oxidation in these cases.
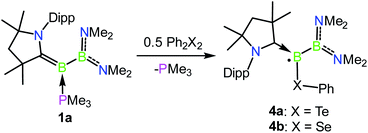 |
| | Scheme 4 Oxidation reactions of 1a with dichalcogenides. | |
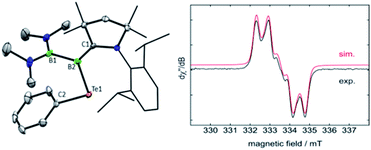 |
| | Fig. 4 Crystallographically-derived molecular structure of the telluroborane radical 5a (left) and its EPR spectrum (right). Thermal ellipsoids are drawn at 50% probability. Hydrogen atoms and ellipsoids of the Dipp groups are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–B2: 1.504(5)/1.514(5), B2–Te1: 2.199(4)/2.196(4), B1–B2: 1.718(5)/1.717(5); C1–B2–B1: 126.4(3)/125.0(3), C1–B2–Te1: 122.9(2)/123.6(2), B1–B2–Te1: 110.7(2)/111.2(2). | |
Conclusions
In summary, we have demonstrated the thermal lability of phosphine- and DMAP-stabilised borylenes, and that such species can effectively behave as convenient and stable sources of reactive two-coordinate borylenes in exchange reactions with other nucleophiles and unsaturated substrates. This method complements existing methods of borylene generation via reduction and photolytic CO extrusion, and provides a facile method to generate these reactive species under mild conditions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. P. would like to acknowledge NSERC and Alexander von Humboldt Postdoctoral Fellowships. C. P., H. B. and A. K. P. would like to acknowledge the DST-DAAD Personnel Exchange Programme. H. B. thanks the Deutsche Foschungsgemeinschaft for funding. A. K. P. thanks the Department of Science and Technology (DST-SERB), New Delhi, for providing financial assistance in the form of a research project (project no. EMR/2016/005294).
Notes and references
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 Search PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 Search PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 Search PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 Search PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 Search PubMed.
- D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 Search PubMed.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 Search PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420–1422 Search PubMed.
- M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 Search PubMed.
- M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 Search PubMed.
- M. Soleilhavoup and G. Bertrand, Chem, 2020, 6, 1275–1282 Search PubMed.
- J. T. Goettel and H. Braunschweig, Coord. Chem. Rev., 2019, 380, 184–200 Search PubMed.
- H. Braunschweig and R. Shang, Inorg. Chem., 2015, 54, 3099–3106 Search PubMed.
- H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 Search PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 Search PubMed.
- P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 Search PubMed.
- A. Hermann, M. Arrowsmith, D. E. Trujillo-Gonzalez, J. O. C. Jiménez-Halla, A. Vargas and H. Braunschweig, J. Am. Chem. Soc., 2020, 142, 5562–5567 Search PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 Search PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 Search PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 Search PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 Search PubMed.
- L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 Search PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 Search PubMed.
- L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 Search PubMed.
- H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436–440 Search PubMed.
- H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 Search PubMed.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098–4102 Search PubMed.
- M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäfer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem.–Eur. J., 2018, 24, 6843–6847 Search PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–13163 Search PubMed.
- C. Pranckevicius, J. O. C. Jimenéz-Halla, M. Kirsch, I. Krummenacher and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10524–10529 Search PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 Search PubMed.
- M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 Search PubMed.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 Search PubMed.
- M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Härterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019, 10, 5095–5103 Search PubMed.
- H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 Search PubMed.
- C. Pranckevicius, C. Herok, F. Fantuzzi, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 12893–12897 Search PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 Search PubMed.
- W. Yang, K. E. Krantz, L. A. Freeman, D. A. Dickie, A. Molino, G. Frenking, S. Pan, D. J. D. Wilson and R. J. Gilliard Jr, Angew. Chem., Int. Ed., 2020, 59, 3850–3854 Search PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360–7363 Search PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem. DOI:10.1038/s41557-020-0520-6.
- R. D. Dewhurst, M.-A. Légaré and H. Braunschweig, Commun. Chem., 2020, 3, 131 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures. CCDC 2018683–2018688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04826g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c and
Holger
Braunschweig
c and
Holger
Braunschweig