
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-phase fluorescent BODIPY–peptide synthesis via in situ dipyrrin construction†
Yue
Wu
a,
Wing-Sze
Tam
a,
Ho-Fai
Chau
a,
Simranjeet
Kaur
a,
Waygen
Thor
a,
Wei Shen
Aik
a,
Wai-Lun
Chan
 *abc,
Markus
Zweckstetter
*bc and
Ka-Leung
Wong
*a
*abc,
Markus
Zweckstetter
*bc and
Ka-Leung
Wong
*a
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong SAR, China. E-mail: kulicechan@hkbu.edu.hk; klwong@hkbu.edu.hk; wai-lun.chan@mpibpc.mpg.de
bDepartment for NMR-based Structural Biology, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany. E-mail: mazw@nmr.mpibpc.mpg.de
cGerman Center for Neurodegenerative Diseases (DZNE), Von-Siebold-Str. 3a, 37075 Göttingen, Germany. E-mail: markus.zweckstetter@dzne.de
First published on 24th September 2020
Abstract
Traditional fluorescent peptide chemical syntheses hinge on the use of limited fluorescent/dye-taggable unnatural amino acids and entail multiple costly purifications. Here we describe a facile and efficient protocol for in situ construction of dipyrrins on the N-terminus with 20 natural and five unnatural amino acids and the lysine's side chain of selected peptides/peptide drugs through Fmoc-based solid-phase peptide synthesis. The new strategy enables the direct formation of boron–dipyrromethene (BODIPY)–peptide conjugates from simple aldehyde and pyrrole derivatives without pre-functionalization, and only requires a single-time chromatographic purification at the final stage. As a model study, synthesized EBNA1-targeting BODIPY1–Pep4 demonstrates intact selectivity in vitro, responsive fluorescence enhancement, and higher light cytotoxicity due to the photo-generation of cytotoxic singlet oxygen. This work offers a novel practical synthetic platform for fluorescent peptides for multifaceted biomedical applications.
Introduction
Fluorescent peptide/peptidomimetic-based targeting and visualization of the subcellular localization,1,2 and probing and modulation of intra/inter-biomolecular interactions,3,4 of the protein target of interest provides a versatile platform, either in vitro/vivo or in assays,5–7 for fundamental biochemistry/chemical biology research,8–10 as well as drug discovery and development.11–14 With the growing significance and recent successes in peptide (and peptide-containing) therapeutics,15–20 by virtue of their (i) scalable modular synthetic accessibility and tunability, (ii) target-specific selectivity, potency and efficacy, (iii) good biocompatibility, low systemic toxicity and easy clearance, there is a great demand for more general and flexible synthetic methodologies and ligation protocols to make peptides fluorescent, upon functional de novo design, for in-depth biomedical investigations.To date, despite the development of automated flow-based approaches,21,22 solid-phase peptide synthesis (SPPS) remains the most common method for peptide preparation at both academic and industrial research settings.23–27 In SPPS, amino acids with the protected N-terminus (as well as the protected side chain if necessary) are used as building blocks: upon the covalent attachment onto insoluble resin beads and the iterative amidation and N-Fmoc deprotection, tailor-designed peptides of known sequence can be prepared in very good to excellent yields. Meanwhile, all the excess reactants and unbound side-products can be removed by simple washing after each single step, with the only purification step being required until the final cleavage of the peptide from the resin. However, when it comes to synthetic fluorescent peptides, i.e. fused with fluorescent proteins (FPs) by molecular biology and site-specific protein labelling protocols,28–30 or labelled/modified with small-molecule fluorophores (e.g. FITC, TRITC, AMCA, 5/6-FAM, Cy3/5, Lucifer Yellow, etc.) at either the N-terminus or at the internal lysines/cysteines/unnatural amino acids' linkable/clickable side chains with straightforward functional group coupling reactions and bioorthogonal ligations,31–38 multiple transformation steps followed by tedious and costly purifications are required. In addition, relatively large fusion FPs and steric fluorophores can alter the physio–chemical properties of short peptides and adversely interfere with their interactions with targets in vitro/vivo.39 Therefore, employing an optimal spacer/linker is technically a key routine solution for this “fluorescent post-labelling” approach (Fig. 1A).
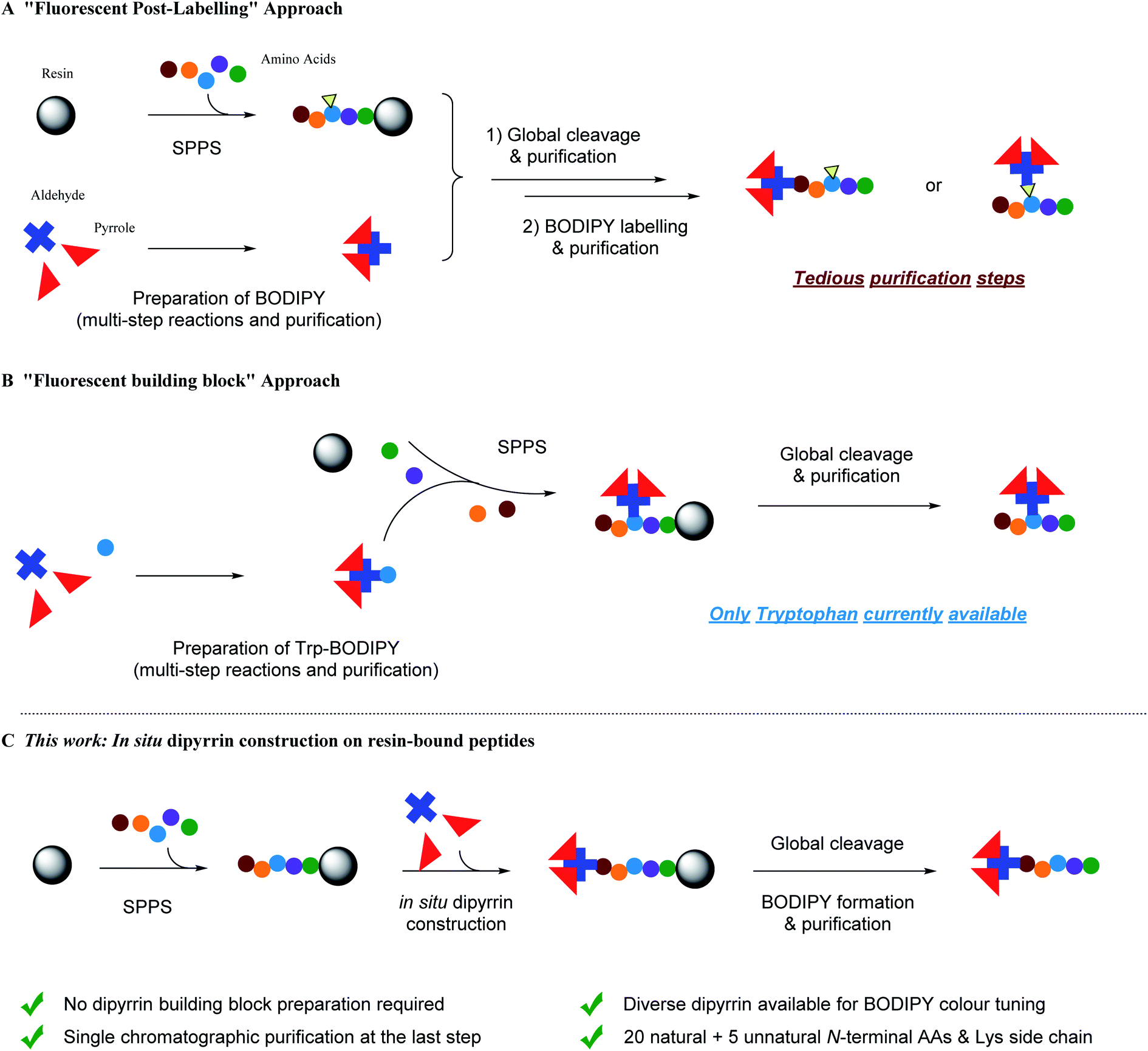 | ||
| Fig. 1 Schematic representation of strategies for BODIPY-based fluorescent peptide synthesis. (A) Post-SPPS ligation of fluorescent BODIPY dyes on peptides requiring multistep transformations and purifications. (B) Use of BODIPY-derived fluorescent unnatural amino acid/pre-functionalized building blocks during SPPS. (C) This work: in situ dipyrrin construction during SPPS and then boron complexation to form BODIPY dyes with a single chromatographic purification step. | ||
To circumvent the above-mentioned limitations, the “fluorescent building block” approach has emerged as a highly promising alternative for fluorescent peptide production (Fig. 1B).40 For instance, Schultz et al. pioneered the use of genetic encoding to introduce fluorescent unnatural amino acids into proteins at specific sites.41,42 In addition, Imperiali et al. developed an SPPS-based approach for incorporation of fluorescent unnatural amino acids into peptides by design.43–45 Since then, a kaleidoscope of novel organic fluorophore-based fluorescent unnatural amino acids with different photophysical properties and structures have been developed.40 However, interdisciplinary expertise in synthetic biology and chemistry, experience in careful and judicious peptide design, and sufficient research budget are prerequisites for these approaches.
Over the past few decades, fluorinated boron–dipyrromethene (BODIPY) and its derivatives46 have lent themselves to multifarious functional fluorophore scaffolds for diverse biomedical applications,47e.g. fluorescence sensing and imaging (via the Förster resonance energy transfer (FRET) or photoinduced electron transfer (PeT) mechanisms), positron emission tomography imaging, photodynamic therapy, as well as in vitro and in vivo assays, due to their high synthetic availability and cost-effectiveness, tunable photophysical properties and neutral total charge for better cell permeability. BODIPY–peptide conjugates are therefore widely used as dual targeting–imaging tools in life sciences,7,48–54 although a “fluorescent post-labelling” strategy has to be used since BODIPY derivatives are generally vulnerable to strong acid and usually not so adaptable to SPPS conditions.55–57 To this end, Vendrell et al. documented the first “fluorescent building block” approach by developing a fluorogenic Trp–BODIPY amino acid in a spacer-free manner (i.e. direct carbon–carbon bond linkage between a tryptophan's indole moiety at the C2-position and a BODIPY dye by palladium-catalyzed Csp2–Csp2 Heck reaction).58,59 Both linear and cyclic peptides can be prepared.60–62 In addition, Ackermann et al. disclosed a palladium-catalyzed bioorthogonal late-stage Csp3–H activation strategy to append BODIPY dyes to the alanine and phenylalanine residues within a peptide.63
Because tryptophan might not be present in the peptide sequence of interest, while phenylalanine and alanine are often involved in functional interactions and conformational controls of the peptide's secondary/tertiary structures, i.e. should best not be perturbed, complementary and more universal methods are required to create BODIPY–peptide conjugates. For the BODIPY precursor, there are two common ways to synthesize dipyrrin derivatives:64 first, Method A, condensation between one aldehyde and two pyrroles, followed by oxidation, which yields symmetrical dipyrrins; second, Method B, reaction of an α-ketopyrrole/α-formylpyrrole with another pyrrole, giving non-symmetrical dipyrrins. For both methods, a lot of impurities, including excess pyrroles, intra-pyrrole crossing products, and the oxidant and its derivatives, are mixed with desired product, which renders the purification step a laborious and tedious task. Inspired by the advantage of SPPS, we hypothesize that SPPS and dipyrrin synthesis, in this regard, can be perfectly integrated (Fig. 1C).
Herein, we describe a new facile and efficient protocol for in situ dipyrrin construction on the N-terminus and the side chains of peptides through Fmoc-based solid-phase peptide synthesis (SPPS), where simple aldehyde and pyrrole derivatives can be directly utilized as building blocks without pre-functionalization in SPPS, and only one final-stage chromatographic purification step is required. In connection to our on-going research program to develop peptide-based EBNA1 inhibitors against Epstein–Barr virus (EBV)-associated maligenancies,65–67 we constructed dipyrrin moieties on our own library of established EBNA1-targeting peptide Pep4 and synthesized BODIPY–Pep4, which manifests no erosion of biotargeting performance in vitro but enhanced light cytotoxicity over dark cytotoxicity. This can be correlated to the photodynamic therapy (PDT) effect of BODIPY by photoinduced cytotoxic singlet oxygen (1O2) generation. Taken together, this study offers a new pragmatic SPPS methodology for fluorescent peptide production and facilitates a cornucopia of biomedical applications.
Results and discussion
To construct dipyrrin on the N-terminus of resin–bound Pep1 (H2N–YFMVF–COOH), which was previously used as an EBNA1-targeting peptide,65–67 the benzaldehyde moiety was installed on the N-terminus through routine SPPS, followed by condensation with 2,4-dimethylpyrrole under the catalysis of BF3·OEt2 on the resin. Subsequently, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was added to oxidize the newly formed dipyrrolomethane into dipyrrin in situ: the resin turned red after this step indicating the formation of dipyrrin. After global cleavage, the dipyrrin–peptide conjugate DP1–Pep1 was obtained with 51% yield, which is comparable to the yield of the unconjugated peptide (Pep1, 60%). The reaction was monitored by HLPC, ESI-MS and 1H-NMR (Fig. 2).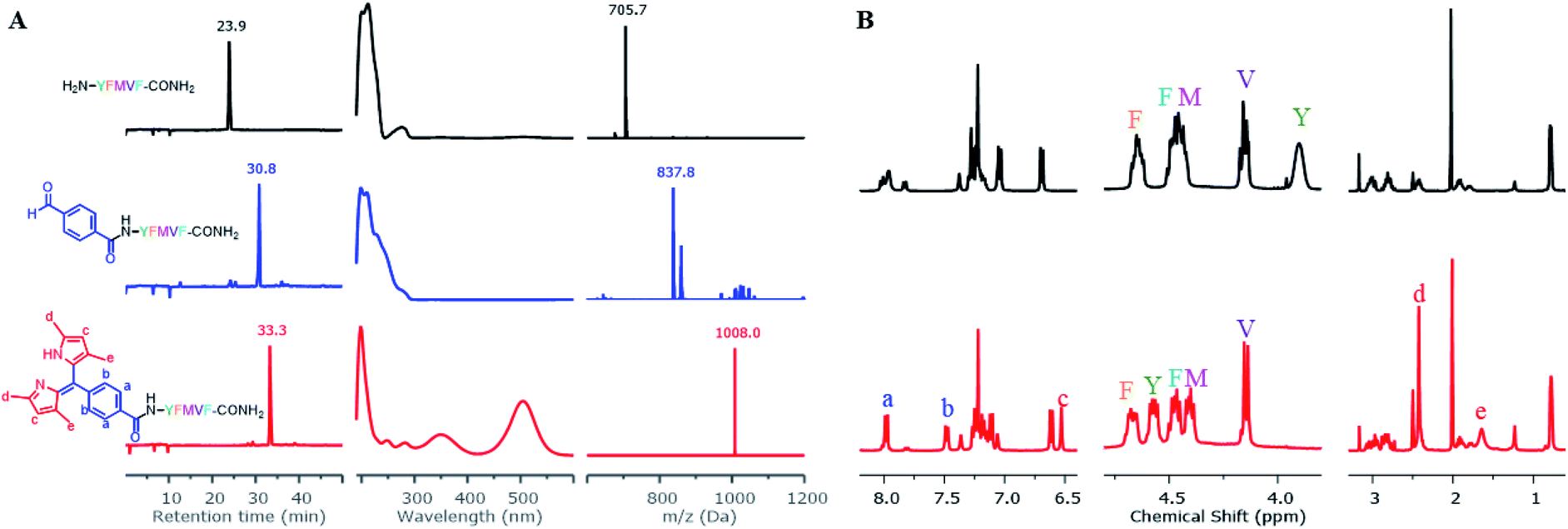 | ||
| Fig. 2 Direct construction of dipyrrin on resin–bound Pep1. (A) The crude samples (resin) were taken and cleaved, with the resulting solutions being monitored by HPLC and ESI-MS. From top to bottom: unconjugated peptide (black), aldehyde–peptide conjugate (blue), dipyrrin–peptide conjugate DP1–Pep1 (red); from left to right: their HPLC chromatographs, UV-Vis spectra corresponding to the peak on HPLC, and ESI-MS spectra. (B) Comparisons of 1H-NMR spectra of Pep1 and DP1–Pep1: four protons from the aldehyde building block (a and b) and two protons from the pyrrole (c) were found in the aromatic region; the α-H of tyrosine (Y, near the N-terminal) shifts largely after conjugation; 12 protons from the four methyl groups of dipyrrin (d and e) were found at 2.4 and 1.7 ppm. | ||
We next explored the substrate diversity of various aldehydes and pyrroles under the above-mentioned procedure to synthesize a series of dipyrrin conjugates of Pep1 (Fig. 3). Various simple pyrrole derivatives and indole derivatives were tried, and most of them yielded corresponding symmetrical dipyrrin conjugates in good both absolute and relative yields. The failures of DP7 and DP11 suggest that the reaction can be hindered by the steric effect, while the trace amount of DP8/17 hints towards the sensitivity of the conjugated vinyl group towards the conditions. Since functionalized α-ketopyrroles/α-formylpyrroles (e.g. –COOH group containing) have very few commercial sources and are hard to synthesize as building blocks, 5-formyl-2,4-dimethyl-3-pyrrolecarboxylic acid (FDMPA) attracted our attention for its commercial availability and low cost (below 10 USD per g). FDMPA was able to act as an aldehyde analogue to prepare DP14. For the unsymmetrical cases, we adopted the Method B. Treated by POCl3 with other pyrrole derivatives, DP15 and DP16 were obtained in good yields.
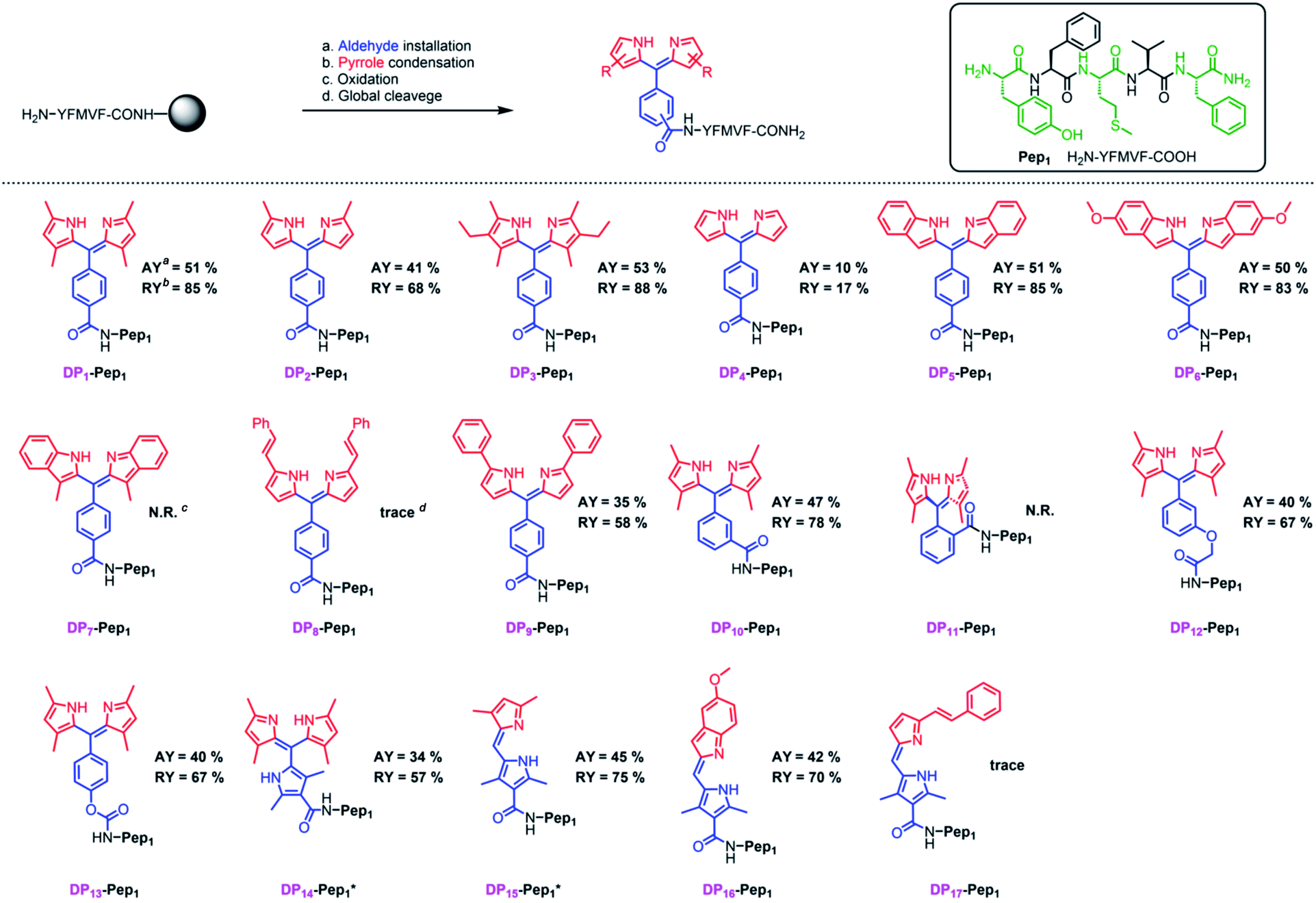 | ||
| Fig. 3 In situ construction of dipyrrin derivatives on the peptide YFMVF. Condition: (a) aldehyde-containing carboxylic acid, PyBOP, DIPEA, DMF, 3 h. (b) Pyrrole derivatives, BF3·OEt2, r.t., DMF, overnight; (c) DDQ, 1 h, DCM; (d) TFA/TIPS/H2O, v/v/v, 95/2.5/2.5, r.t., 2 h. aAbsolute Isolated yield compared with the substitution value of the resin–bound peptide. bRelative yield compared with the isolated yield of the unconjugated peptide.c,d No/Very few desired products were detected by LC-MS. | ||
We then verified the scope of this methodology for peptides with different amino acid compositions (Fig. 4). 4-Formylbenzoic acid and 2,4-dimethylpyrrole were used to construct 1,3,7,9-tetramethyldipyrrin (DP1) for a series of peptides that cover all 20 natural amino acids as well as five commonly used unnatural amino acids. All were obtained with comparable yields as the corresponding unconjugated peptides. The synthesized peptides can be used in a variety of studies, such as EBNA1-targeting peptides (Pep1–Pep6), STAT3-targeting peptides (Pep7 and Pep8), as well as some FDA-approved peptide drugs including cetrorelix (Pep9, as GnRH antagonists for treating prostate cancer, endometriosis, uterine fibroids), angiotensin II (Pep10, used in treatment of sepsis, septic shock, diabetes mellitus, and acute renal failure) and prezatide (Pep11, a copper chelator with potential applications in wound healing and other different functions). In addition to N-terminal conjugation, the dipyrrin moiety can also be installed on the side chain of the lysine residue of Pep11 (the lysine with a removable protecting group on the amine group on the side chain), which substantiates that our procedure can be applied “generally and flexibly” on side chain modifications as well.
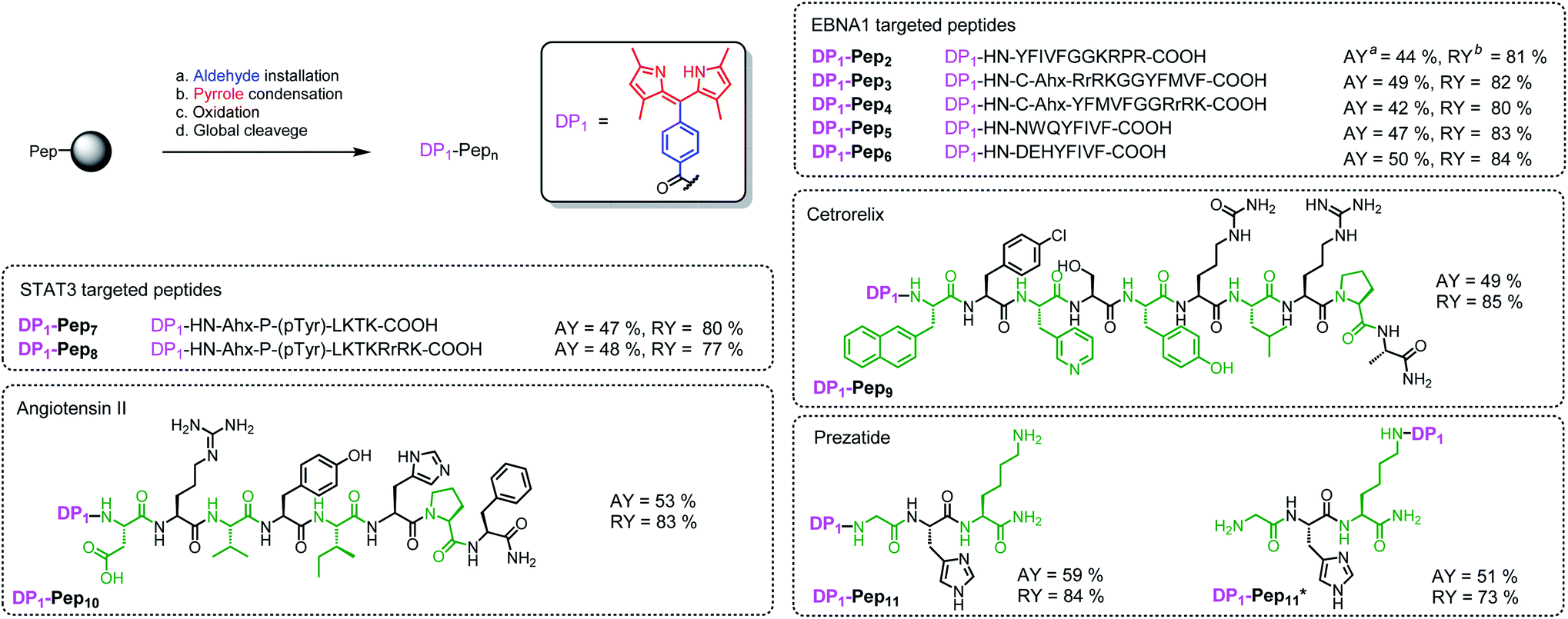 | ||
| Fig. 4 In situ construction of DP1 on different peptides. aAbsolute isolated yield compared with substitution value of the resin–bound peptide. bRelative yield compared with the isolated yield of the unconjugated peptide. | ||
Next, we investigated SPPS boron complexation, which is a long-standing challenge for dipyrrin–peptide conjugates to deliver BODIPY–peptide conjugates (Fig. 5A). Traditionally, this complexation step is conducted in less-polar solvents. However, peptides often suffer from poor solubility in these solvents, while polar solvents resulted in no complexation. Upon systematic solvent screening, we found acetonitrile (ACN), which displays moderate solubility for peptides, to be optimal: both the purified dipyrrin–peptide conjugates and the crude product from SPPS were converted in ACN into the corresponding BODIPY conjugates within 10 minutes with acceptable yields. Notably, bulky base DIPEA should be used and the products should be separated from the reacting mixture rapidly, in order to avoid racemization. 1H-NMR spectra of DP1–Pep1 and BODIPY1–Pep1 are shown in Fig. 5C and S8–S11.† In addition, BODIPY-conjugates with different colours of fluorescence were prepared (Fig. 5B), further illustrating the possibility to achieve fast fluorescence labelling of de novo peptides with diverse BODIPY dyes for bioimaging and biosensing. It is axiomatic that the photophysical properties of BODIPY dyes are largely influenced by the extent of electron delocalization around the boron centre (e.g. most BODIPY dyes emitting around 510 nm in green colour, like BODIPY1–Pep1) by peripherical substitutions on the dipyrrin to (i) extend the π-conjugation system with vinyl/ynyl/aromatic groups (e.g. highly red-shifted BODIPY9–Pep1 to red colour) and (ii) subtly fine-tune it with either electron-donating (e.g. aliphatic groups in BODIPY3–Pep1 for slightly shifting to yellow colour) or electron-withdrawing groups (e.g. halogens).46,68
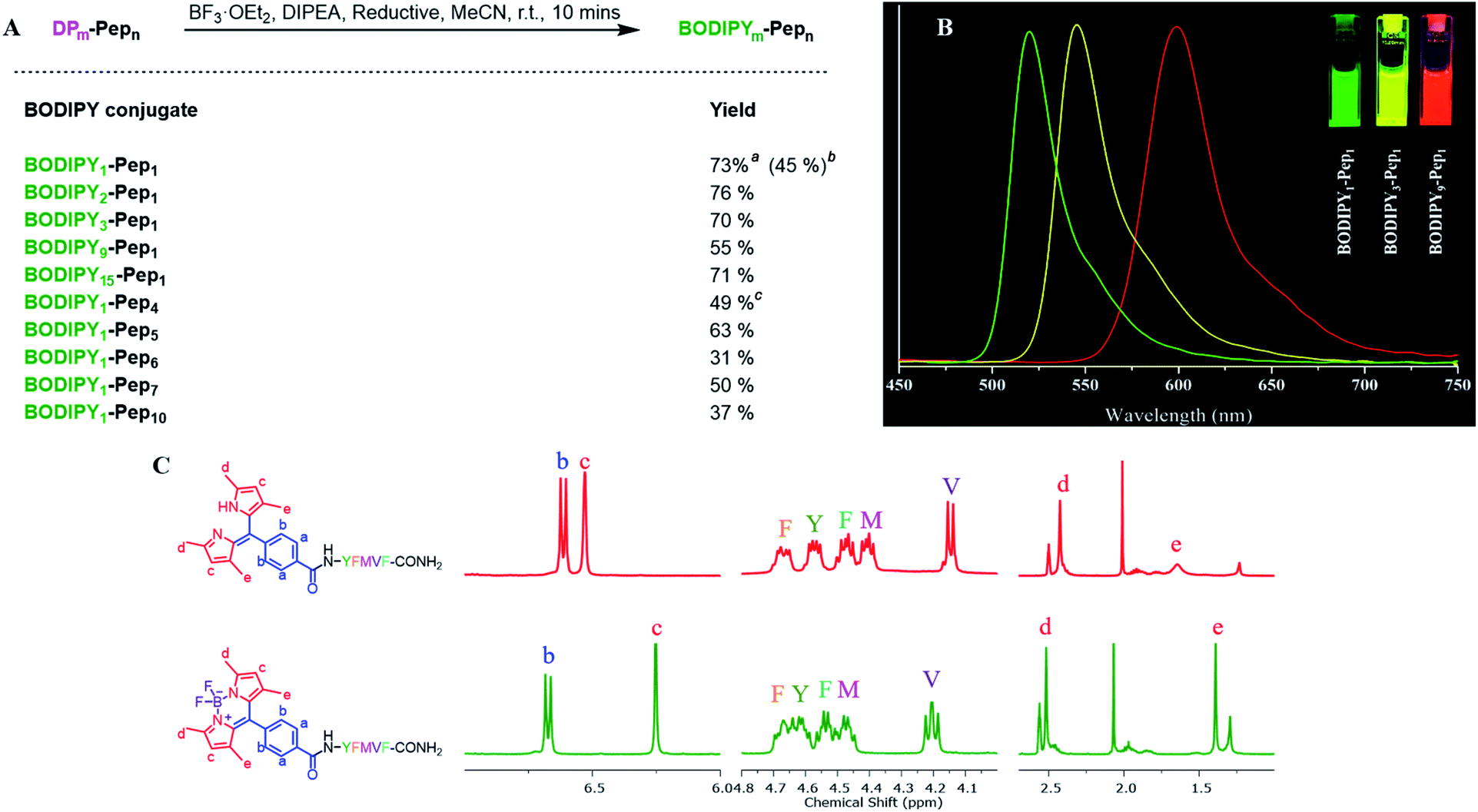 | ||
| Fig. 5 (A) Boron complexation for dipyrrin–peptide conjugates. aPurified dipyrrin conjugate was used; isolated yield compared with the dipyrrin conjugate. bCrude dipyrrin conjugate was used; isolated yield compared with the substitution value of the resin–bound peptide. cEDT was used as the reductant. (B) The fluorescence spectra of 10 μM BODIPY–peptide conjugates (derived from DP1, DP3 and DP9 respectively) in DMSO under 365 nm UV light excitation. (C) Comparison of 1H-NMR spectra of DP1–Pep1 and BODIPY1–Pep1. The chemical shifts of both pyrrole–H (c) and methyl–H (e) are shifted towards the high field. | ||
To support the practical use of the BODIPY–peptide conjugates synthesized by our protocol, we performed confocal imaging for BODIPY1–Pep4 (where Pep4 had been designed as a nucleus-penetrating EBNA1-specific peptide in our previous research)65–67 with both a HeLa cell line (EBNA1−) and the C666 cell line (EBNA1+) (Fig. 6A and S6†). As expected, the signal of BODIPY accumulated relatively fast in the nucleus of C666 cells where the EBNA1 proteins are located, while the uptake of BODIPY into the nucleus of HeLa cells was slow. The confocal imaging experiments confirmed selective EBNA1-targeting performance of BODIPY1–Pep4in vitro despite the use of another chromophore. In addition, BODIPY1–Pep4 demonstrated an almost 5-fold binding-responsive fluorescence enhancement (Fig. 6B, S1 and S2†), as well as a dark/light cytotoxicity difference due to the photo-generation of cytotoxic singlet oxygen (1O2) from the BODIPY moiety (Fig. 6C and D).
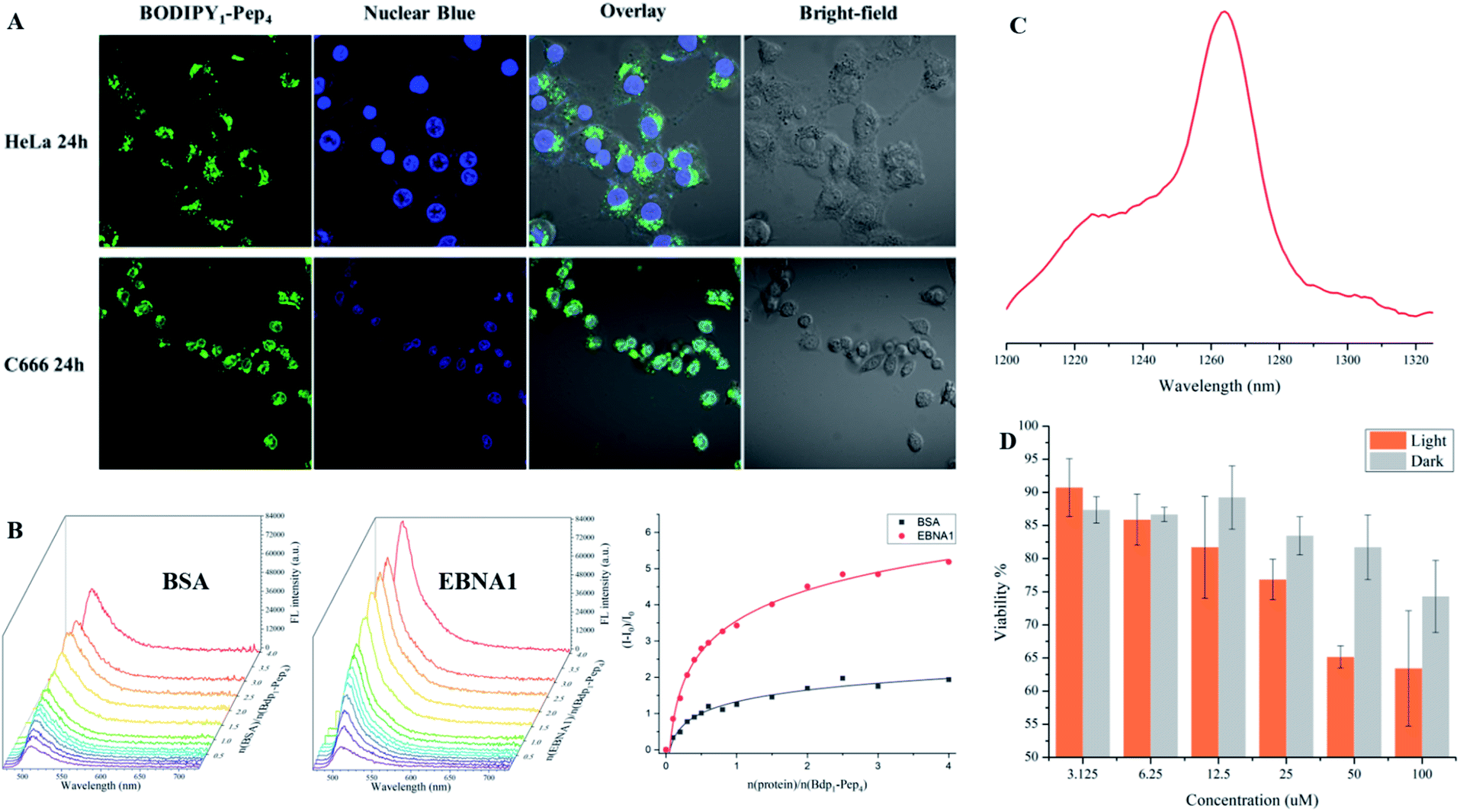 | ||
| Fig. 6 Imaging of fluorescent peptides. (A) Confocal imaging of BODIPY1–Pep4 in HeLa and C666 cell lines. The green fluorescence signal was detected in the nucleus of C666 (EBNA+) but not in the nucleus of HeLa (EBNA−) cells. (B) Fluorescence titration of BSA (left) and EBNA1 (middle) proteins, both at 1.2 μM, with BODIPY1–Pep4. A nearly 5-times enhancement over the course of the titration with EBNA1 was observed (right). (C) Singlet oxygen signal illustrating the potential of BODIPY1–Pep4 as a PDT agent. (D). Comparison of dark/light cytotoxicity of BODIPY1–Pep4 towards C666 cell line. Light toxicity was significantly higher than the dark toxicity. Error bars represent the standard derivation of the mean. | ||
Conclusion
We have developed an efficient and convenient methodology to conjugate peptides with dipyrrin moieties during SPPS that can be further derived into highly emissive bioactive BODIPY–peptide conjugates for multicolour imaging. Various dipyrrin derivatives can be constructed on either the N-terminus or the side chain of peptides in both symmetrical and unsymmetrical ways and the substrate scope comprises all 20 natural and five unnatural amino acids with good yield. The workload and cost for synthesizing dipyrrin/BODIPY–peptide conjugates is greatly reduced by this protocol, which therefore holds tremendous promise for expediting the screening of peptide-based fluorescent probes, as well as for the development of high-throughput (HTS) fluorescence screening platforms and the creation of novel metal–peptide nano-frameworks. The BODIPY-labelled EBNA1-specific peptide (BODIPY1–Pep4) obtained from this methodology exhibited excellent in vitro performance and can serve as a potential photosensitizer for PDT. This work furnishes a new pragmatic alternative SPPS methodology for fluorescent peptide production that can leverage and impact multifaceted biomedical applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (HKBU12300319) and Hong Kong Health and Medical Research Fund (18170022). M.Z. was supported by the advanced grant ‘787679 – LLPS-NMR’ of the European Research Council.Notes and references
- M. Staderini, A. Megia-Fernandez, K. Dhaliwal and M. Bradley, Bioorg. Med. Chem., 2018, 26, 2816–2826 Search PubMed.
- S. Lee, J. Xie and X. Chen, Biochemistry, 2010, 49, 1364–1376 Search PubMed.
- A. D. Cunningham, N. Qvit and D. Mochly-Rosen, Curr. Opin. Struct. Biol., 2017, 44, 59–66 Search PubMed.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 Search PubMed.
- N. N. Patwardhan, Z. Cai, C. N. Newson and A. E. Hargrove, Org. Biomol. Chem., 2019, 17, 1778–1786 Search PubMed.
- J. Zhang, S. J. Yang, F. Gonzalez, J. Yang, Y. Zhang, M. He, N. Shastri and N. Murthy, Chem. Commun., 2018, 54, 7215–7218 Search PubMed.
- E. Pazos, O. Vázquez, J. L. Mascareñas and M. E. Vázquez, Chem. Soc. Rev., 2009, 38, 3348–3359 Search PubMed.
- O. T. Johnson, T. Kaur and A. L. Garner, ChemBioChem, 2019, 20, 40–45 Search PubMed.
- J. A. González-Vera, D. Bouzada, C. Bouclier, M. Eugenio Vázquez and M. C. Morris, Chem. Commun., 2017, 53, 6109–6112 Search PubMed.
- D. Baechle, A. Cansier, R. Fischer, J. Brandenburg, T. Burster, C. Driessen and H. Kalbacher, J. Pept. Sci., 2005, 11, 166–174 Search PubMed.
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Methods. Appl. Fluores., 2016, 4, 022001 Search PubMed.
- T. Kim, H. M. Jeon, H. T. Le, T. W. Kim, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 7690–7693 Search PubMed.
- M. Pelay-Gimeno, A. Glas, O. Koch and T. N. Grossmann, Angew. Chem., Int. Ed., 2015, 54, 8896–8927 Search PubMed.
- W. A. Lea and A. Simeonov, Expet Opin. Drug Discov., 2011, 6, 17–32 Search PubMed.
- D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 Search PubMed.
- A. P. Davenport, C. C. G. Scully, C. de Graaf, A. J. H. Brown and J. J. Maguire, Nat. Rev. Drug Discovery, 2020, 19, 389–413 Search PubMed.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2020, 13, 40 Search PubMed.
- A. C. L. Lee, J. L. Harris, K. K. Khanna and J. H. Hong, Int. J. Mol. Sci., 2019, 20, 2383 Search PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 Search PubMed.
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 Search PubMed.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 Search PubMed.
- A. J. Mijalis, D. A. Thomas, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, Nat. Chem. Biol., 2017, 13, 464–466 Search PubMed.
- S. H. M. Gorissen, J. J. R. Crombag, J. M. G. Senden, W. A. H. Waterval, J. Bierau, L. B. Verdijk and L. J. C. van Loon, Amino Acids, 2018, 50, 1685–1695 Search PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 Search PubMed.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 Search PubMed.
- V. Mäde, S. Els-Heindl and A. G. Beck-Sickinger, Beilstein J. Org. Chem., 2014, 10, 1197–1212 Search PubMed.
- M. Amblard, J. A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 Search PubMed.
- X. Chen, J. L. Zaro and W. C. Shen, Adv. Drug Delivery Rev., 2013, 65, 1357–1369 Search PubMed.
- X. Charest-Morin, J. P. Fortin, M. T. Bawolak, R. Lodge and F. Marceau, Pharmacol. Res. Perspect., 2013, 1, e00004 Search PubMed.
- N. A. Bindman and W. A. Van der Donk, J. Am. Chem. Soc., 2013, 135, 10362–10371 Search PubMed.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354 Search PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 Search PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 Search PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 Search PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 Search PubMed.
- R. F. Wissner, S. Batjargal, C. M. Fadzen and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 6529–6540 Search PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 Search PubMed.
- P. Vidal, L. Chaloin, J. Méry, N. Lamb, N. Lautredou, R. Bennes and F. Heitz, J. Pept. Sci., 1996, 2, 125–133 Search PubMed.
- S. F. Hedegaard, M. S. Derbas, T. K. Lind, M. R. Kasimova, M. V. Christensen, M. H. Michaelsen, R. A. Campbell, L. Jorgensen, H. Franzyk, M. Cárdenas and H. M. Nielsen, Sci. Rep., 2018, 8, 6327 Search PubMed.
- Z. Cheng, E. Kuru, A. Sachdeva and M. Vendrell, Nat. Rev. Chem., 2020, 4, 275–290 Search PubMed.
- J. Wang, J. Xie and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 8738–8739 Search PubMed.
- D. Summerer, S. Chen, N. Wu, A. Deiters, J. W. Chin and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9785–9789 Search PubMed.
- P. Venkatraman, T. T. Nguyen, M. Sainlos, O. Bilsel, S. Chitta, B. Imperiali and L. J. Stern, Nat. Chem. Biol., 2007, 3, 222–228 Search PubMed.
- B. Imperiali and M. Sainlos, Nat. Protoc., 2007, 2, 3210–3218 Search PubMed.
- M. E. Vázquez, D. M. Rothman and B. Imperiali, Org. Biomol. Chem., 2004, 2, 1965–1966 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 Search PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 Search PubMed.
- A. Sajid, N. Raju, S. Lusvarghi, S. Vahedi, R. E. Swenson and S. V. Ambudkar, Drug Metab. Dispos., 2019, 47, 1013–1023 Search PubMed.
- N. E. M. Kaufman, Q. Meng, K. E. Griffin, S. S. Singh, A. Dahal, Z. Zhou, F. R. Fronczek, J. M. Mathis, S. D. Jois and M. G. H. Vicente, J. Med. Chem., 2019, 62, 3323–3335 Search PubMed.
- A. C. Schulz-Fincke, M. Blaut, A. Braune and M. Gütschow, ACS Med. Chem. Lett., 2018, 9, 345–350 Search PubMed.
- N. Zhao, T. M. Williams, Z. Zhou, F. R. Fronczek, M. Sibrian-Vazquez, S. D. Jois and M. G. H. Vicente, Bioconjugate Chem., 2017, 28, 1566–1579 Search PubMed.
- A. Paulus, P. Desai, B. Carney, G. Carlucci, T. Reiner, C. Brand and W. A. Weber, EJNMMI Res., 2015, 5, 43 Search PubMed.
- S. Banappagari, A. McCall, K. Fontenot, M. G. H. Vicente, A. Gujar and S. Satyanarayanajois, Eur. J. Med. Chem., 2013, 65, 60–69 Search PubMed.
- J. Yang, H. Chen, I. R. Vlahov, J. X. Cheng and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13872–13877 Search PubMed.
- S. Krajcovicova, J. Stankova, P. Dzubak, M. Hajduch, M. Soural and M. Urban, Chem. -Eur. J., 2018, 24, 4957–4966 Search PubMed.
- A. M. Hansen, A. L. Sewell, R. H. Pedersen, D. L. Long, N. Gadegaard and R. Marquez, Tetrahedron, 2013, 69, 8527–8533 Search PubMed.
- M. Vendrell, G. G. Krishna, K. K. Ghosh, D. Zhai, J. S. Lee, Q. Zhu, Y. H. Yau, S. G. Shochat, H. Kim, J. Chung and Y. T. Chang, Chem. Commun., 2011, 47, 8424–8426 Search PubMed.
- L. Mendive-Tapia, R. Subiros-Funosas, C. Zhao, F. Albericio, N. D. Read, R. Lavilla and M. Vendrell, Nat. Protoc., 2017, 12, 1588–1619 Search PubMed.
- L. Mendive-Tapia, C. Zhao, A. R. Akram, S. Preciado, F. Albericio, M. Lee, A. Serrels, N. Kielland, N. D. Read, R. Lavilla and M. Vendrell, Nat. Commun., 2016, 7, 10940 Search PubMed.
- R. Subiros-Funosas, V. C. L. Ho, N. D. Barth, L. Mendive-Tapia, M. Pappalardo, X. Barril, R. Ma, C. B. Zhang, B. Z. Qian, M. Sintes, O. Ghashghaei, R. Lavilla and M. Vendrell, Chem. Sci., 2020, 11, 1368–1374 Search PubMed.
- R. Subiros-Funosas, L. Mendive-Tapia, J. Sot, J. D. Pound, N. Barth, Y. Varela, F. M. Goñi, M. Paterson, C. D. Gregory, F. Albericio, I. Dransfield, R. Lavilla and M. Vendrell, Chem. Commun., 2017, 53, 945–948 Search PubMed.
- R. Subiros-Funosas, V. C. L. Ho, N. D. Barth, L. Mendive-Tapia, M. Pappalardo, X. Barril, R. Ma, C.-B. Zhang, M. Sintes, O. Ghashghaei, R. Lavilla and M. Vendrell, Chem. Sci., 2020, 11, 1368–1374 Search PubMed.
- W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554–10558 Search PubMed.
- N. A. M. Pereira and T. M. V. D. Pinho E Melo, Org. Prep. Proced. Int., 2014, 46, 183–213 Search PubMed.
- L. Jiang, R. Lan, T. Huang, C.-F. Chan, H. Li, S. Lear, J. Zong, W.-Y. Wong, M. M.-L. Lee, B. D. Chan, W.-L. Chan, W.-S. Lo, N.-K. Mak, M. L. Lung, H. L. Lung, S. W. Tsao, G. S. Taylor, Z.-X. Bian, W. C.-S. Tai, G.-L. Law, W.-T. Wong, S. L. Cobb and K.-L. Wong, Nat. Biomed. Eng., 2017, 1, 1–10 Search PubMed.
- L. Jiang, Y.-L. Lui, H. Li, C.-F. Chan, R. Lan, W.-L. Chan, T. C.-K. Lau, G. S.-W. Tsao, N.-K. Mak and K.-L. Wong, Chem. Commun., 2014, 50, 6517–6519 Search PubMed.
- L. Jiang, H. L. Lung, T. Huang, R. Lan, S. Zha, L. S. Chan, W. Thor, T.-H. Tsoi, H.-F. Chau, C. Boreström, S. L. Cobb, S. W. Tsao, Z.-X. Bian, G.-L. Law, W.-T. Wong, W. C.-S. Tai, W. Y. Chau, Y. Du, L. H. X. Tang, A. K. S. Chiang, J. M. Middeldorp, K.-W. Lo, N. K. Mak, N. J. Long and K.-L. Wong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 26614–26624 Search PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04849f |
| This journal is © The Royal Society of Chemistry 2020 |