
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic Umpolung of N- and O-substituted alkenes for the synthesis of 1,2-amino alcohols and diols†‡
Stephanie G. E.
Amos
 ,
Stefano
Nicolai
and
Jerome
Waser
*
,
Stefano
Nicolai
and
Jerome
Waser
*
Laboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, Ch-1015, Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
First published on 22nd September 2020
Abstract
We report an organophotocatalytic 1,2-oxyalkynylation of ene-carbamates and enol ethers using Ethynyl BenziodoXolones (EBXs). 1-Alkynyl-1,2-amino alcohols and diols were obtained in up to 89% yield. Photocatalytic formation of radical cations led to Umpolung of the innate reactivity of the alkenes, enabling addition of a nucleophilic benzoate followed by radical alkynylation.
1. Introduction
Accessing 1,2-amino-alcohols and 1,2-diols has been a longstanding target in synthetic methodology. These scaffolds have found multiple applications in pharmaceutical, material and agrochemical sciences.1,2 Alkynes are highly useful building blocks for synthesis, as starting points for product diversification.3 Combining both functionalities, 1-alkynyl-1,2-amino alcohols can be found as intermediates in the synthesis of insecticidal 4-alkynyloxazolines4 and β-erythroidine,5,6 as well as essential structural elements in bioactive antitumoral enediynes.7,8 1-Alkynyl-1,2-diols can be found, for example, in the Petrosiol family of neurotrophic diyne tetraols.9,10Enamides and ene-carbamates are versatile starting materials for the generation of complex aminated building blocks.11–16 In particular, they have been used extensively in atom transfer radical addition (ATRA) reactions.17 Due to their innate nucleophilicity, they are excellent traps for electrophilic radicals, leading to the formation of a nucleophilic α-amino radical I (Scheme 1A, a). The latter can then react with a radical trap,18 undergo oxidation to the α-amino cation,19–21 reduction to the α-amino anion,22 or addition to an organometallic species followed by reductive elimination.23 Despite the efficiency associated to such transformations, all enamide difunctionalizations reported so far are based on the initial addition of a highly reactive electrophilic radical, limiting functional group tolerance and the structural diversity of the obtained products.
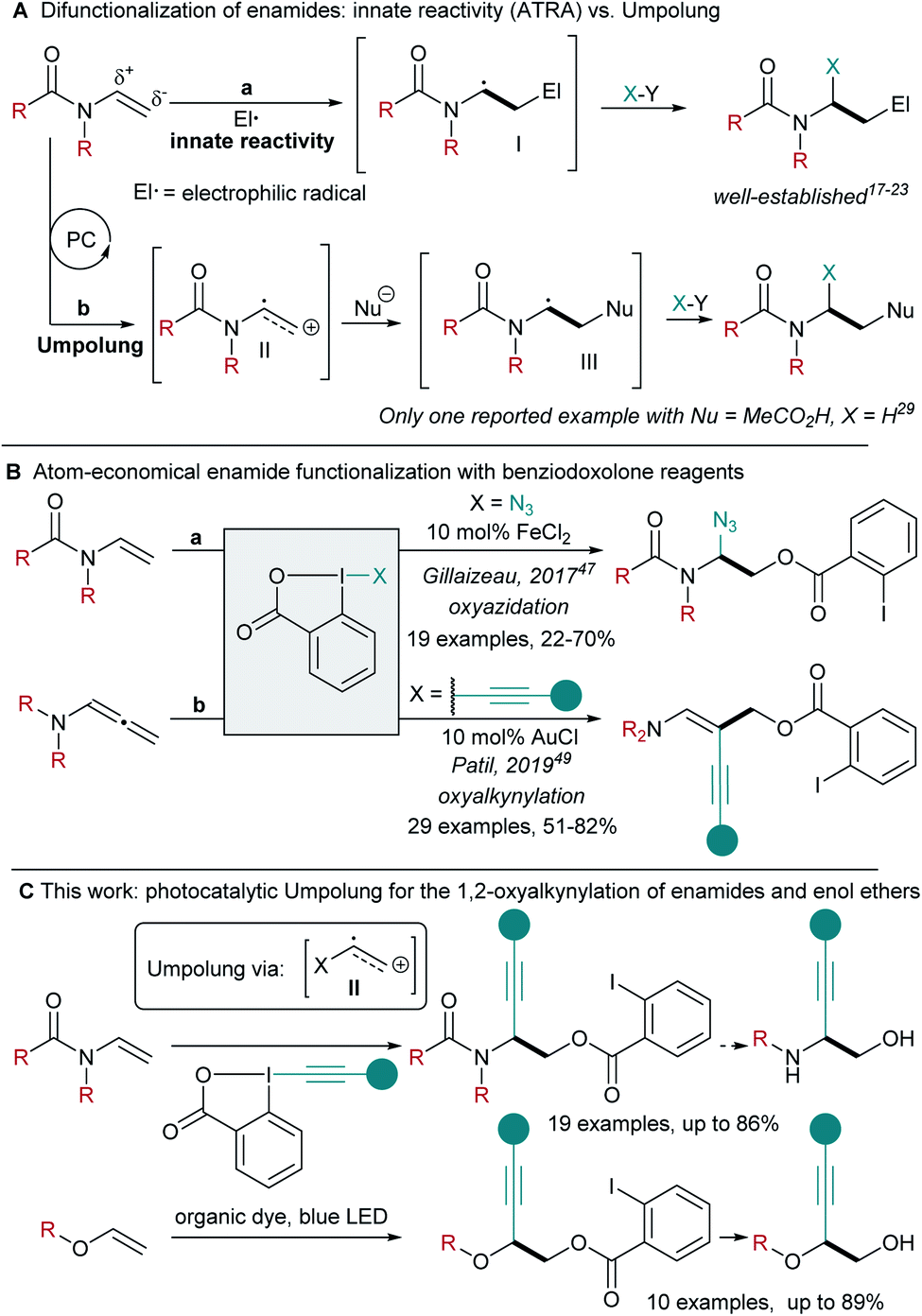 | ||
| Scheme 1 (A) Difunctionalization of enamides: innate reactivity vs. Umpolung. (B) Atom-economical enamide difunctionalization with benziodoxolone reagents. (C) This work: photocatalytic Umpolung enabling the synthesis of 1-alkynyl-1,2-amino alcohols and diols. | ||
Nicewicz and co-workers developed a different approach towards alkene difunctionalization based on oxidation under photoredox conditions for the generation of radical cations.24–26 This highly electrophilic species can then react with various nucleophiles, enabling new types of hydrofunctionalizations.27–32
In the case of enamides or ene-carbamates, such a strategy would result in a neat Umpolung of the reactivity (Scheme 1A, b). It is important to stress that such an approach would completely change the type of transformations accessible, as the first step would involve reaction with a nucleophile, in opposition to the electrophilic radical already intensively investigated.17–23 Although this strategy appears highly attractive to answer current limitations in enamide functionalization, only one example of ene-carbamate hydroacetoxylation has been reported by Nicewicz and co-workers.29 When considering the importance of nitrogen-containing compounds, a difunctionalization of enamides via photocatalytic Umpolung would be highly desirable.
In order to develop such a process, we turned to Ethynyl BenziodoXolone (EBX) hypervalent iodine reagents, which have been identified as efficient traps for radicals.33–37 Their application in radical-mediated olefin alkynylation has also been explored.38–42 Recently, our group has exploited the nucleophilicity of the carboxylate group of EBX reagents in atom-economical reactions such as the 1,1-oxyalkynylation of diazo compounds and the ring-opening/oxyalkynylation of thiiranes.43–46 Therefore, EBX reagents appear ideally suited for the functionalization of radical cations due to their dual nucleophilic/somophilic nature. For what concerns atom economical enamide 1,2-difunctionalization with benziodoxole reagents, the Gillaizeau group has reported an iron-catalyzed enamide oxyazidation (Scheme 1B, a: X = N3) with Zhdankin's reagent.47,48 This reaction was proposed to occur via a classical ATRA mechanism. The Patil group reported a gold catalysed 1,2-oxyalkynylation of allenenamides with EBXs (Scheme 1B, b, X = alkynyl), involving both redox and π-activation by the gold catalyst.49 Consequently, 1,2-oxyalkynylation remains limited to allenenamides as substrates and photocatalytically generated radical cations have never been intercepted with EBX reagents.50
Herein, we show that ene-carbamate radical cations can be generated under oxidative photoredox conditions using 4-CzIPN-derived organic dyes.51–53 The formed intermediates react with Umpolung of the reactivity in an atom-economical fashion with EBX reagents acting as both O-nucleophile and alkynylating radical trap sources (Scheme 1C). This methodology could then be extended to commercially available enamides and enol ethers. The mild oxidative conditions allowed selective reaction of electron-rich alkenes in presence of non-activated ones. This procedure provides easy access to orthogonally protected 1-alkynyl-1,2-amino alcohols and diols, setting the foundations for the development of further difunctionalizations of electron-rich olefins via radical cation intermediates.
2. Results and discussions
Based on previous reports for enamide difunctionalization and α-amino radical alkynylation,18 we started our investigations with N-vinyloxazolidinone (1a)54 and Ph-EBX (2)55 (Table 1). The oxidation potential of 1a was determined to be +1.30 V vs. SCE by cyclic voltammetry. Based on this result, we selected three organic photocatalysts (PC) for their oxidative properties in the excited state: 4-CzIPN (4a*/4a˙−: +1.35 V vs. SCE), 4-ClCzIPN (4b*/4b˙−: +1.58 V) and Mes-Acr+ (5+*/5˙: +2.06 V).27,29 Using DCE as a solvent and 1.5 equivalents of alkene both 4a and 4b enabled product formation (entries 1 and 2). 4b gave a promising 42% yield of the desired compound 3a (entry 2). Highly oxidizing 5 resulted in a 5% yield (entry 3). The yield obtained was dependent on the batch of benziodoxolone 2 when using photocatalyst 4b (entry 4). With a recrystallized batch (as opposed to one purified by trituration only)55,56 the yields were reproducible yet low (entry 5). We speculated that an impurity from the triturated batch was affecting the reactivity. As the most probable impurities were iodine(III) precursors, hydroxy and acetoxy benziodoxolones were examined as additives (BIOH, 6 and BIOAc, 7):57 adding 6 (1.5 equiv., entry 6) improved slightly the yield. With 7 (1.5 equiv., entry 7), the yield increased to 70%. With 0.5 equivalent of 7, the yield remained in the same range (75%, entry 8).58 Both DMSO and DCM could also be used as solvents (entries 9 and 10).59|
|
||||
|---|---|---|---|---|
| Entry | PC | Additive (x equiv.) | Solvent | Yieldb (%) |
| a Reactions conditions: 0.05 mmol 2 (1 equiv.), 1a (1.5 equiv.), additive (x equiv.) and PC (5 mol%) in solvent (0.1 M) unless specified otherwise. Blue led irradiation for 18 h at rt. b 1H NMR yield determined by addition of 0.05 mmol of CH2Br2 as an internal standard after the reaction. c Recrystallized 2. d 2 mol%. e Concentration based on 2: 0.25 M, at 0.2 mmol scale. | ||||
| 1 | 4a | None | DCE | 30 |
| 2 | 4b | None | DCE | 42 |
| 3 | 5 | None | DCE | 5 |
| 4 | 4b | None | DCE | 36–65 |
| 5c | 4b | None | DCE | 34 |
| 6c | 4b | BIOH (6, 1.5) | DCE | 46 |
| 7c | 4b | BIOAc (7, 1.5) | DCE | 70 |
| 8c | 4b | BIOAc (7, 0.5) | DCE | 75 |
| 9c | 4b | BIOAc (7, 0.5) | DMSO | 75 |
| 10c | 4b | BIOAc (7, 0.5) | DCM | 80 |
| 11c | 4b | BIOAc (7, 0.5) | DCMe | 80 |
| 12c | 8 | BIOAc (7, 0.5) | DCMe | 21 |
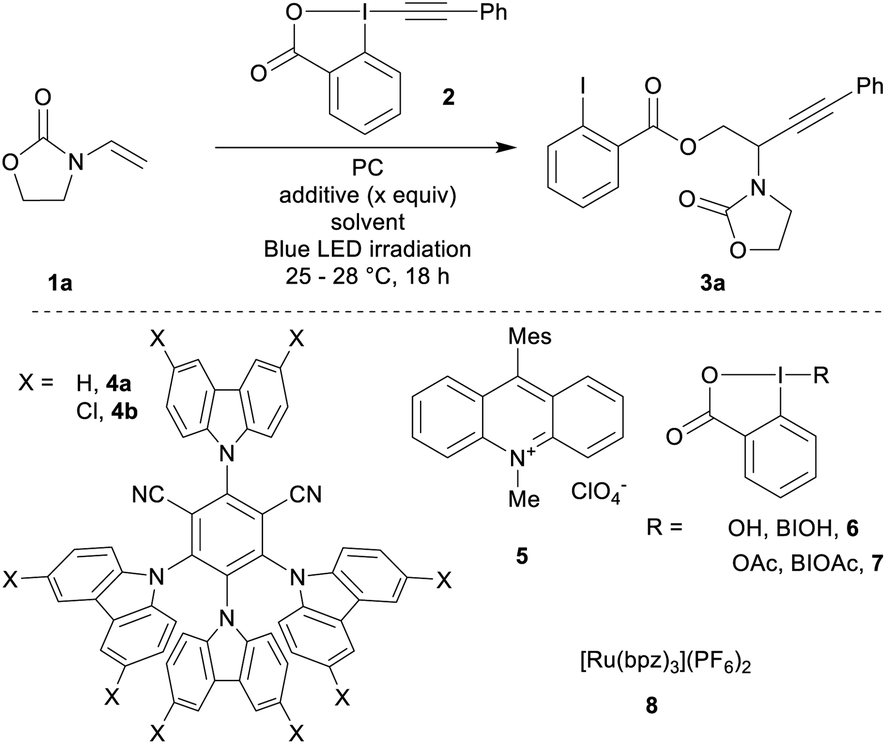
Final adjustments were made on scope scale (0.2 mmol): DCM was used as a solvent with a lower catalyst loading of 2 mol% and an increase of the concentration to 0.25 M. This gave product 3a in 80% yield (entry 11). Finally, we tested ruthenium based photocatalyst 8, which has a comparable oxidation potential (Ru2+*/Ru+: +1.40 V): 3a was only obtained in 21% yield (entry 12). This result may have its origin from the weaker reduction potential of 8 (Ru2+/Ru+: −0.80 V), compared to 4b (4b/4b˙−: −1.10 V).
With the optimized reaction conditions in hand, we explored the scope of the reaction (Scheme 2). Acyclic ene-carbamates were tolerated affording Boc and Cbz protected amines 3b and 3c in 63% and 86% yield. Although N–H vinyl carbamates degraded under the reaction conditions, the orthogonally diprotected ene-carbamate 1d was converted to 3d in 73% yield. A methyl amine worked well under our reaction conditions (3e, 83% yield). An allyl amine was also tolerated affording 3f in 54% yield,60 demonstrating that selective functionalization of ene-carbamates over alkenes was possible. Substrates bearing a silylated alcohol and an ethyl ester yielded the desired compounds 3g and 3h in 52% and 69% yield. The procedure also worked with secondary amines (3i, 65% yield). Commercial N-vinylpyrrolidinone gave compound 3j in 85% yield. α-Substitution of the alkene was not tolerated (1k no conversion), but β-substituted (E)-ene-carbamate afforded 3l as a mixture of diastereoisomers in 80% yield (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr.).
:1 dr.).
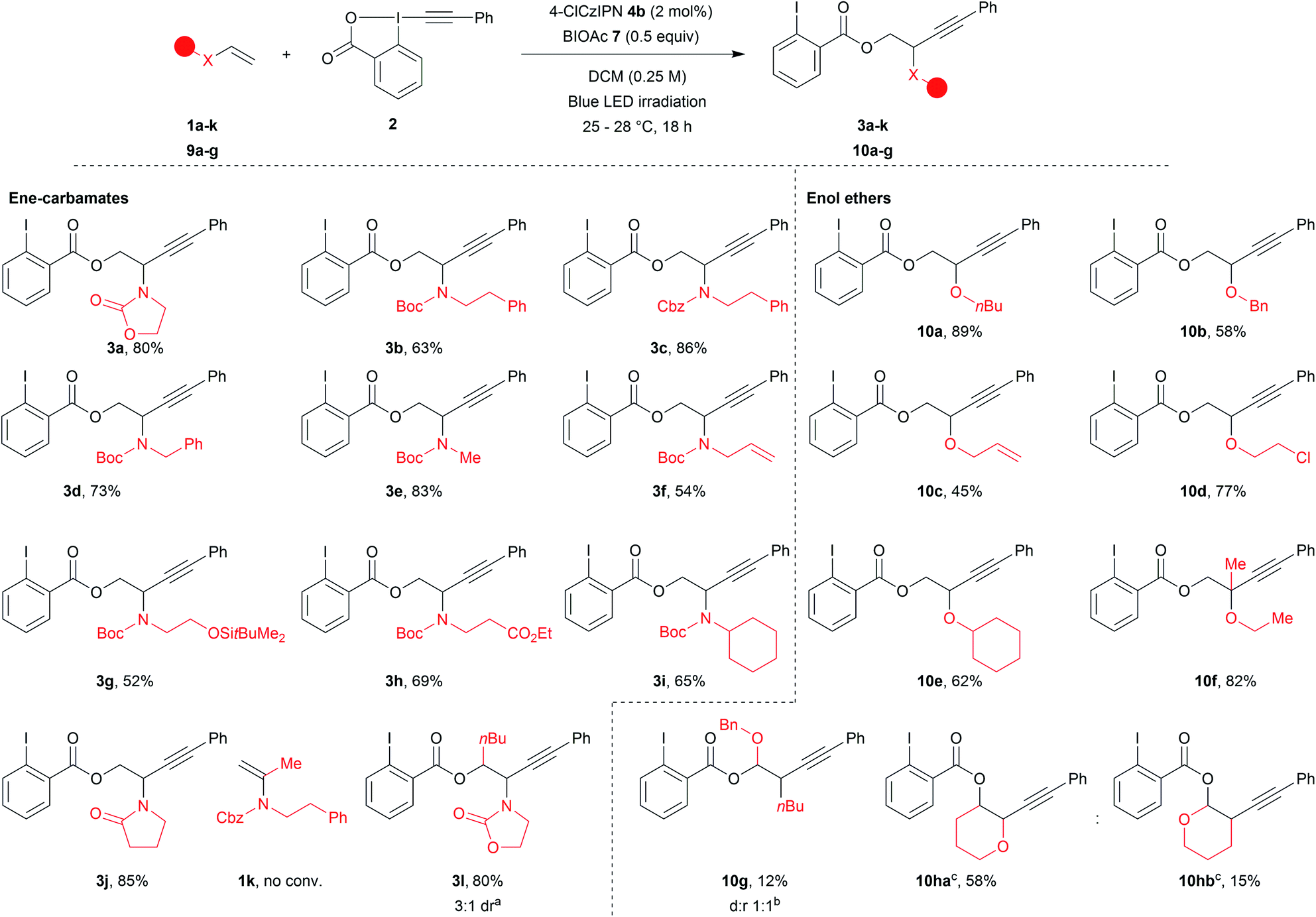 | ||
| Scheme 2 Scope of ene-carbamates and enol ethers. Reactions conditions: 0.20–0.25 mmol 2 (1 equiv.), alkene (1.2–1.5 equiv.), BIOAc (7, 0.5 equiv.) and 4b (2 mol%) in DCM 0.25 M. Blue led irradiation for 18 h at rt. a Isolated ratio. b 1H NMR ratio. c10ha and 10hb were obtained as an inseparable mixture of regioisomers from 9i. | ||
Finally, we examined enolethers, which have comparable oxidation potentials (e.g. dihydropyran (DHP, 9i), 1.51 V vs. SCE). Aliphatic (10a), benzylic (10b) and allylic (10c) ethers were obtained in 89%, 58% and 45% yield. Chlorinated product 10d was obtained in 77% yield. A secondary enol ether afforded 10e in 62% yield. Although α-substituted ene-carbamates were not tolerated (1k, no conv.), tertiary ether 10f was obtained from propen-2-yl enol ether in 82% yield. β-Substitution afforded a mixture of compounds, from which acetal 10g corresponding to Markovnikov addition could be isolated in 13% yield.61 Finally, DHP 9h62 afforded two regioisomers: anti-Markovnikov product 10ha (58% yield) and Markovnikov product 10hb (15% yield).63
Diverse EBX reagents were then examined (Scheme 3). Both electron-poor and electron-rich arenes on the alkyne provided the desired compounds 12a–12e in up to 76% yield. The transfer of a silyl protected alkyne was less efficient and product 12f was obtained in 9% yield only.64 EBXs bearing sensitive functionalities such as an alkyl bromide or a terminal alkene gave the corresponding products 12g and 12h in 51% and 37% yield. Functionalized EBXs could also be used with enol ether 9a affording 12i and 12j in 62% and 52% yield.
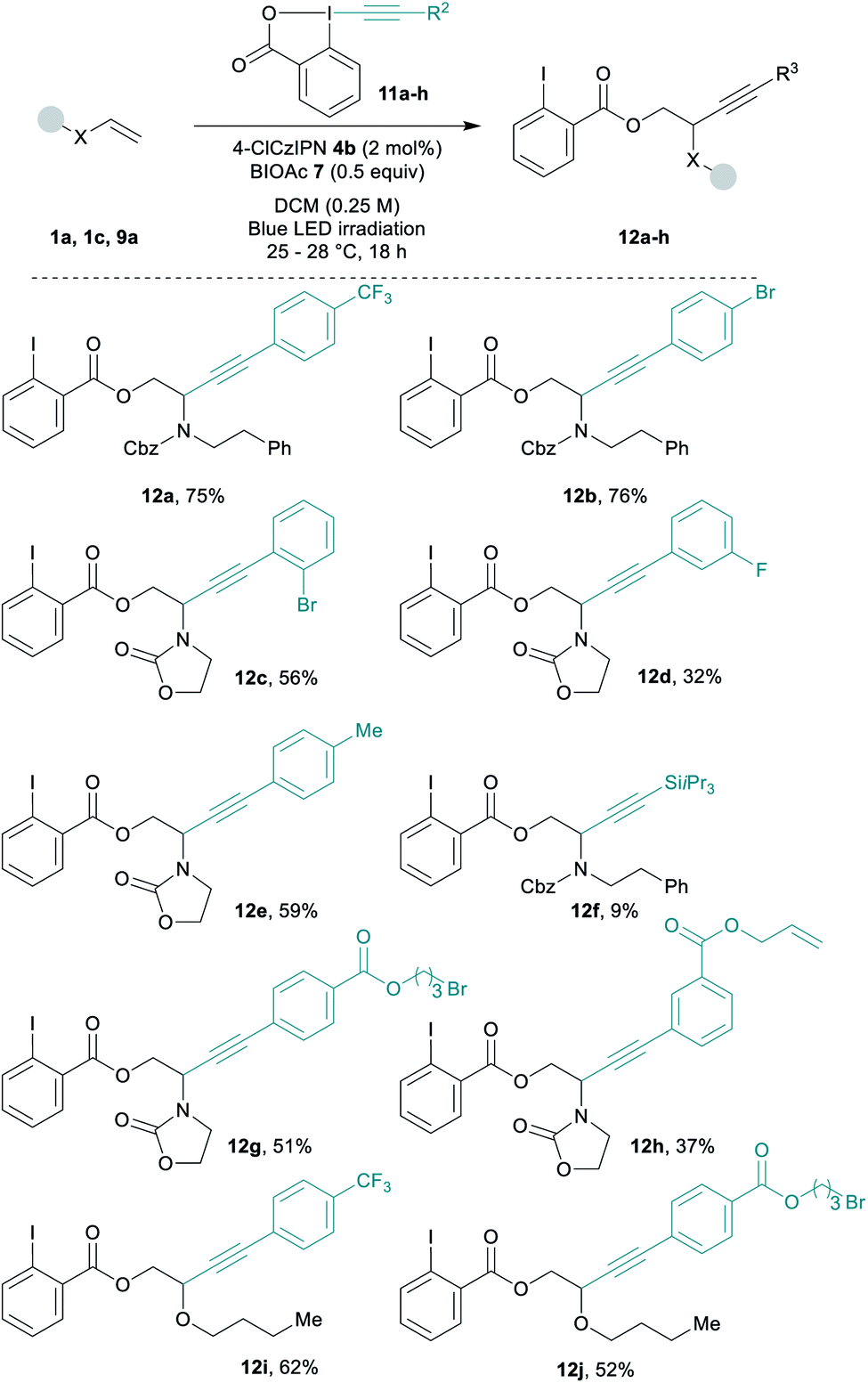 | ||
| Scheme 3 Scope of EBX reagents. Reactions conditions: 0.25 mmol 11a–h (1 equiv.), 1a or 3a (1.5 equiv.), BIOAc (7, 0.5 equiv.) and 4b (2 mol%) in DCM 0.25 M. Blue led irradiation for 18 h at rt. | ||
We then investigated the mechanism of the reaction. Without the photocatalyst and/or a light source, no product was detected (Scheme 4A, eqn (a)). We then considered the possibility of acyloxyl radical Ia (Scheme 4B) adding to the electron-rich olefin 1a. Previously, Ia (a resonance structure of the iodanyl radical Ib) has been reported predominantly as a H-atom abstractor.65–67 To the best of our knowledge, the only proposed report of Ia adding to an alkene is that of the Gillaizeau group.47
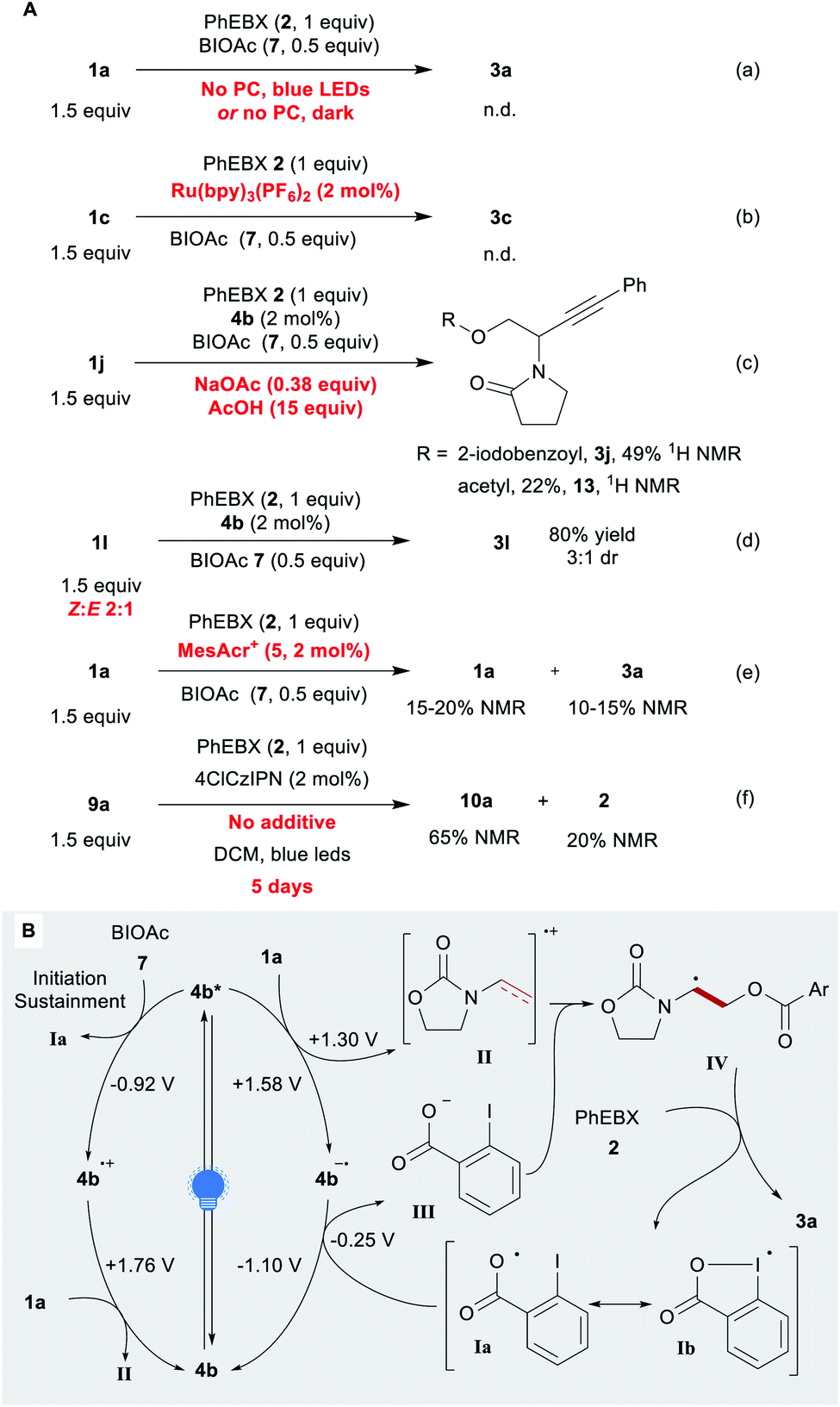 | ||
| Scheme 4 Mechanistic studies. (A) Control experiments Reaction conditions: 0.05–0.25 mmol 2 (1 equiv.), 1 (1.5 equiv.), additive (x equiv.), PC (2 mol%), DCE or DCM (0.25 M). (B) Proposed mechanism. | ||
Chen and co-workers reported the generation of Ia through the reduction of BIOAc (7) by excited Ru(bpy)32+.57 With this photocatalyst, we observed no conversion or product formation (eqn (b)). This suggests that the generation of Ia alone does not lead to product formation. In addition, no conversion was observed with substrate 1k, which is tougher to oxidize (1k˙+/1k ≈ 1.86 V) (Scheme 2). Some conversion would have been expected if the reaction proceeded through the addition of oxygen-centred radical Ia. β-Substituted alkene 1l gave product 3l in 80% yield with the same efficiency as for terminal enamide 1a. An ATRA process would have been more significantly impaired by the substituent. The observed anti-Markovnikov selectivity is in agreement with the reactivity reported for radical cations.26 In order to test our hypothesis, we performed the reaction under the standard conditions in presence of sodium acetate and acetic acid (eqn (c)). The acetate could indeed be introduced (compound 13), but EBX-addition product 3j remains the main product (2:1 ratio). When the reaction was performed with a 2:1 ratio of Z and E isomers of 1l instead of pure E compound, no change in yield and diastereoselectivity was observed, supporting the presence of a radical intermediate (eqn (d)). With the strongly oxidizing catalyst 5, low yields were observed even in presence of BIOAc (7), despite almost full conversion of 1a (eqn (e)). Based on these results, we propose a tentative mechanism for the oxyalkynylation (Scheme 4B). First, the excited photocatalyst 4b* oxidizes 1a generating radical cation II and reduced catalyst 4b˙−. As support for this step, quenching of fluorescence of catalyst 4b by 1a was observed in a Stern–Volmer experiment (see ESI‡ for details).68 Then II is trapped by carboxylate III. This results in the formation of radical IV, which can add to 2 affording the product and iodanyl radical Ib. The latter can close the catalytic cycle by oxidizing 4b˙− to regenerate catalyst 4b.69 We suspect that BIOAc (7) serves as initiator for the reaction by generating Iavia reduction of BIOAc (7) with 4b*. The resulting oxidized catalyst 4b˙+ would be also competent to oxidize 1a. This pathway would also help sustaining the catalytic cycle by ensuring a sufficient concentration of Ia. A final control experiment corroborates this hypothesis: the reaction was performed with no additive (eqn (f)). The desired compound was obtained in 65% 1H NMR yield after 5 days of reaction time with 20% residual PhEBX (2).
We then performed the transformation on gram scale (Scheme 5, eqn (a)), affording 3j in 76% yield (0.998 g). Selective hydrolysis of the ester group from 3j gave 14 in 96% yield (eqn (b)). Hydrolysis of 10a provided 15 in 91% yield (eqn (c)). Finally, 3b underwent Boc deprotection to give amino ester 16 in 74% yield (eqn (d)).
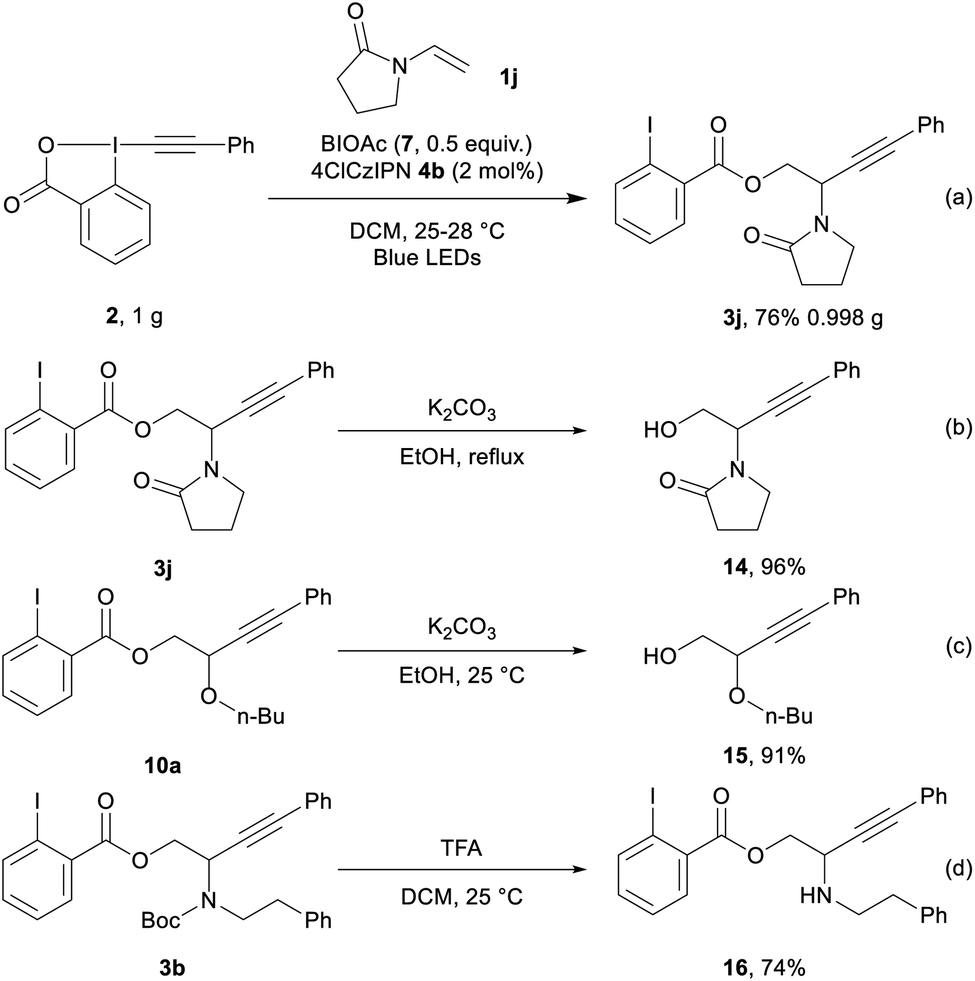 | ||
| Scheme 5 Gram-scale reaction and post-functionalization. | ||
3. Conclusions
In conclusion, we have developed a photocatalytic 1,2-oxy-alkynylation of ene-carbamates based on Umpolung of the reactivity. The transformation proceeds in an atom-economical fashion with EBXs acting both as alkynylating and carboxylating reagents. The reaction occurs at room temperature under blue LED irradiation using 4-ClCzIPN (4b) as an organic photocatalyst and does not require the use of highly reactive electrophilic radicals. The methodology could be extended to enamides and enolethers. The method shows good chemoselectivity for nitrogen or oxygen-substituted olefins over aliphatic alkenes. Based on preliminary mechanistic studies, we propose that an ene-carbamate radical cation is the key intermediate that ensures the anti-Markovnikov regioselectivity initiated by nucleophile addition, contrasting with the classical ATRA mechanism usually invoked for the functionalization of alkenes with hypervalent iodine reagents. This reaction allows quick access to protected 1-alkynyl-1,2-amino alcohols and 1-alkynyl-1,2-diols, which are important building blocks in agrochemical, pharmaceutical and material sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPFL for financial support. This publication was created as part of NCCR Catalysis, a National Centre of Competence in Research funded by the Swiss National Science Foundation.Notes and references
- S. A. Lawrence, Amines: synthesis, properties, and applications, Cambridge University Press, New York, 2004 Search PubMed.
- Amino group chemistry: from synthesis to the life sciences, ed. A. Ricci, John Wiley & Sons, Inc, Weinheim, 2008 Search PubMed.
- Modern alkyne chemistry: catalytic and atom-economic transformations, ed. B. M. Trost and C.-J. Li, Wiley-VCH, Weinheim, 2015 Search PubMed.
- D. Clark and D. A. Travis, Bioorg. Med. Chem., 2001, 9, 2857–2862 Search PubMed.
- H. Fukumoto, K. Takahashi, J. Ishihara and S. Hatakeyama, Angew. Chem., Int. Ed., 2006, 45, 2731–2734 Search PubMed.
- Y. He and R. L. Funk, Org. Lett., 2006, 8, 3689–3692 Search PubMed.
- D. R. Cohen and C. A. Townsend, Nat. Chem., 2017, 10, 231 Search PubMed.
- K. C. Nicolaou, D. Das, Y. Lu, S. Rout, E. N. Pitsinos, J. Lyssikatos, A. Schammel, J. Sandoval, M. Hammond, M. Aujay and J. Gavrilyuk, J. Am. Chem. Soc., 2020, 142, 2549–2561 Search PubMed.
- P. Gangadhar, A. Sathish Reddy and P. Srihari, Tetrahedron, 2016, 72, 5807–5817 Search PubMed.
- K. Horikawa, T. Yagyu, Y. Yoshioka, T. Fujiwara, A. Kanamoto, T. Okamoto and M. Ojika, Tetrahedron, 2013, 69, 101–106 Search PubMed.
- R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292–301 Search PubMed.
- K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 Search PubMed.
- G. Bernadat and G. Masson, Synlett, 2014, 25, 2842–2867 Search PubMed.
- N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Chem. - Eur. J., 2014, 20, 7548–7564 Search PubMed.
- T. Courant, G. Dagousset and G. Masson, Synthesis, 2015, 47, 1799–1856 Search PubMed.
- X. Cai, M. Yang and H. Guo, Curr. Org. Synth., 2019, 16, 70–97 Search PubMed.
- T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945–6952 Search PubMed.
- C. Poittevin, V. Liautard, R. Beniazza, F. Robert and Y. Landais, Org. Lett., 2013, 15, 2814–2817 Search PubMed.
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240–1243 Search PubMed.
- E. L. S. de Souza, C. Wiethan and C. R. D. Correia, ACS Omega, 2019, 4, 18918–18929 Search PubMed.
- P. Kramer, M. Halaczkiewicz, Y. Sun, H. Kelm and G. Manolikakes, J. Org. Chem., 2020, 85, 3617–3637 Search PubMed.
- Q. Fu, Z.-Y. Bo, J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Nat. Commun., 2019, 10, 3592 Search PubMed.
- C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224–8229 Search PubMed.
- K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 Search PubMed.
- D. Nicewicz and D. Hamilton, Synlett, 2014, 25, 1191–1196 Search PubMed.
- N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 Search PubMed.
- D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 Search PubMed.
- T. M. Nguyen, N. Manohar and D. A. Nicewicz, Angew. Chem., Int. Ed., 2014, 53, 6198–6201 Search PubMed.
- A. J. Perkowski and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 10334–10337 Search PubMed.
- T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 Search PubMed.
- D. J. Wilger, N. J. Gesmundo and D. A. Nicewicz, Chem. Sci., 2013, 4, 3160–3165 Search PubMed.
- D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 Search PubMed.
- D. P. Hari, S. Nicolai and J. Waser, in PATAI’S Chemistry of Functional Groups, 2018, pp. 1–58 Search PubMed.
- X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 Search PubMed.
- Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L. Lu and W. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196–11199 Search PubMed.
- F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 Search PubMed.
- F. Le Vaillant and J. Waser, Chem. Sci., 2019, 10, 8909–8923 Search PubMed.
- K. Shen and Q. Wang, Chem. Sci., 2017, 8, 8265–8270 Search PubMed.
- W.-J. Han, Y.-R. Wang, J.-W. Zhang, F. Chen, B. Zhou and B. Han, Org. Lett., 2018, 20, 2960–2963 Search PubMed.
- Y. Li, R. Lu, S. Sun and L. Liu, Org. Lett., 2018, 20, 6836–6839 Search PubMed.
- H. Jiang and A. Studer, Chem. - Eur. J., 2019, 25, 516–520 Search PubMed.
- X. Yang and G. C. Tsui, Org. Lett., 2019, 21, 8625–8629 Search PubMed.
- D. P. Hari and J. Waser, J. Am. Chem. Soc., 2016, 138, 2190–2193 Search PubMed.
- D. P. Hari and J. Waser, J. Am. Chem. Soc., 2017, 139, 8420–8423 Search PubMed.
- J. Borrel, G. Pisella and J. Waser, Org. Lett., 2020, 22, 422–427 Search PubMed.
- A. Boelke, P. Finkbeiner and B. J. Nachtsheim, Beilstein J. Org. Chem., 2018, 14, 1263–1280 Search PubMed.
- S. Bertho, R. Rey-Rodriguez, C. Colas, P. Retailleau and I. Gillaizeau, Chem. - Eur. J., 2017, 23, 17674–17677 Search PubMed.
- For an example of atom-economical functionalization of other types of olefins with Togni reagent, see: H. Egami, R. Shimizu, Y. Usui and M. Sodeoka, J. Fluorine Chem., 2014, 167, 172–178 Search PubMed.
- S. Banerjee, B. Senthilkumar and N. T. Patil, Org. Lett., 2019, 21, 180–184 Search PubMed.
- For the interception of radical cation generated from non-activated alkenes and strong stoichiometric oxidants by external oxygen nucleophiles and EBX reagents, see ref. 40.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 Search PubMed.
- F. Le Vaillant, M. Garreau, S. Nicolai, G. Gryn’ova, C. Corminboeuf and J. Waser, Chem. Sci., 2018, 9, 5883–5889 Search PubMed.
- M. Garreau, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2019, 58, 8182–8186 Search PubMed.
- J. L. Brice, J. E. Meerdink and S. S. Stahl, Org. Lett., 2004, 6, 1845–1848 Search PubMed.
- J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. - Eur. J., 2012, 18, 5655–5666 Search PubMed.
- Following previous reports, 2 is purified by trituration at 80°C in MeCN, but impurities remain. Recrystallization from EtOAc:MeOH 2:1 or column chromatography afforded the compound in greater purity, ESI.‡.
- H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881–1884 Search PubMed.
- 0.1 equiv. was also tested and gave similar results. However, to ensure a general procedure for all substrates, the use of 0.5 equiv. was favored.
- Other solvents tested such as THF, Tol, DME, EtOAc or MeCN gave lower yields.
- Reaction at the non-activated alkene was not observed and the low yield was mostly due to decomposition upon isolation.
- The anti-Markovnikov product was also observed as a mixture of diastereoisomers in 10% yield, but could not be isolated in pure form. See ESI‡ for details.
- N-Boc 3,4-dihydro-2H-pyridine was also tested in the reaction condition, but led to a very complex mixture.
- 10ha and 10hb were obtained as an inseparable mixture. Based on 1H NMR analysis, both compounds are speculated to have been obtained as single diastereoisomers in cis configuration.
- Alkyl-EBX reagents were not tolerated under our reaction conditions.
- M. Ochiai, T. Ito, H. Takahashi, A. Nakanishi, M. Toyonari, T. Sueda, S. Goto and M. Shiro, J. Am. Chem. Soc., 1996, 118, 7716–7730 Search PubMed.
- V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192–5197 Search PubMed.
- J. Barluenga, E. Campos-Gómez, D. Rodríguez, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2005, 44, 5851–5854 Search PubMed.
- Surprisingly, fluorescence quenching, albeit weaker than 1a, was also observed with substrate 1k, but no reaction was obtained with this substrate.
- F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790–1800 Search PubMed.
Footnotes |
| † Raw data for NMR, MS and IR is available at: DOI: 10.5281/zenodo.4043189 |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03655b |
| This journal is © The Royal Society of Chemistry 2020 |