
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganoborohydride-catalyzed Chichibabin-type C4-position alkylation of pyridines with alkenes assisted by organoboranes†
Ying
Wang‡
a,
Runhan
Li‡
 b,
Wei
Guan
b,
Yanfei
Li
a,
Xiaohong
Li
a,
Jianjun
Yin
a,
Ge
Zhang
a,
Qian
Zhang
a,
Tao
Xiong
*a and
Qian
Zhang
*ac
b,
Wei
Guan
b,
Yanfei
Li
a,
Xiaohong
Li
a,
Jianjun
Yin
a,
Ge
Zhang
a,
Qian
Zhang
a,
Tao
Xiong
*a and
Qian
Zhang
*ac
aJilin Province Key Laboratory of Organic Functional Molecular Design & Synthesis, Department of Chemistry, Northeast Normal University, Changchun 130024, China. E-mail: xiongt626@nenu.edu.cn; zhangq651@nenu.edu.cn
bInstitute of Functional Material Chemistry, Department of Chemistry, Northeast Normal University, Changchun 130024, China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China
First published on 29th September 2020
Abstract
The first NaBEt3H-catalyzed intermolecular Chichibabin-type alkylation of pyridine and its derivatives with alkenes as the latent nucleophiles is presented with the assistance of BEt3, and a series of branched C4-alkylation pyridines, even highly congested all-carbon quaternary center-containing triarylmethanes can be obtained in a regiospecific manner. Therefore, the conventional reliance on high cost and low availability transition metal catalysts, prior formation of N-activated pyridines, organometallic reagents, and extra oxidation operation for the construction of a C–C bond at the C4-position of the pyridines in previous methods are not required. The corresponding mechanism and the key roles of the organoborane were elaborated by the combination of H/D scrambling experiments, 11B NMR studies, intermediate trapping experiments and computational studies. This straightforward and mechanistically distinct organocatalytic technology not only opens a new door for the classical but still far less well-developed Chichibabin-type reaction, but also sets up a new platform for the development of novel C–C bond-forming methods.
Introduction
The development of direct C–C bond-forming reactions of pyridine and its derivatives is of paramount importance because these motifs are among the most important heterocyclic structural skeletons and exist diffusely in a large number of natural products, agrochemicals, FDA approved drugs and functional materials.1 Therefore, numerous methods,2 including traditional electrophilic aromatic substitution (SEAr), nucleophilic aromatic substitution of organometallic reagents (SNAr),3 the metalation-trapping strategy with a strong base,2a,d,f radical-based Minisci-type reactions,4 and transition-metal-catalyzed C–H bond activation reactions,5,6 have been well established to predominantly access a wide range of C2- and C3-position functionalized pyridine-containing molecules. By comparison, the direct C–C bond-formation at the C4-position of pyridines is far less developed.Until recently, several highly atom-economical and cost-effective protocols, including the pioneering bimetallic Ni/Al catalysis7 and subsequent mono-transition-metal catalysis (e.g., Co, Cr, and Y),8 which enabled C4-position selective alkylation and alkenylation of pyridines have been established (Scheme 1A). More recently, Buchwald and co-workers have elegantly disclosed a Cu-catalyzed asymmetric C4-position reductive coupling reaction of pyridines and pyridazines with aryl alkenes through a successive dearomatization/reoxidation process.9a In addition, Shi et al. also reported an unprecedented Ni-catalyzed intramolecular asymmetric C–H cyclization of pyridines with alkenes.10 In spite of these significant advances, some limitations, such as the absence of methods enabling the generation of all-carbon quaternary centers so far, ineluctable reliance on the undesirable in pharmaceutical industry and non-environmentally benign transition metal catalysts and some frequently accompanying undesired side reactions such as β-hydride elimination, indicated that the development of a transition metal free C4-position C–C bond-forming reaction of pyridines is in high demand.
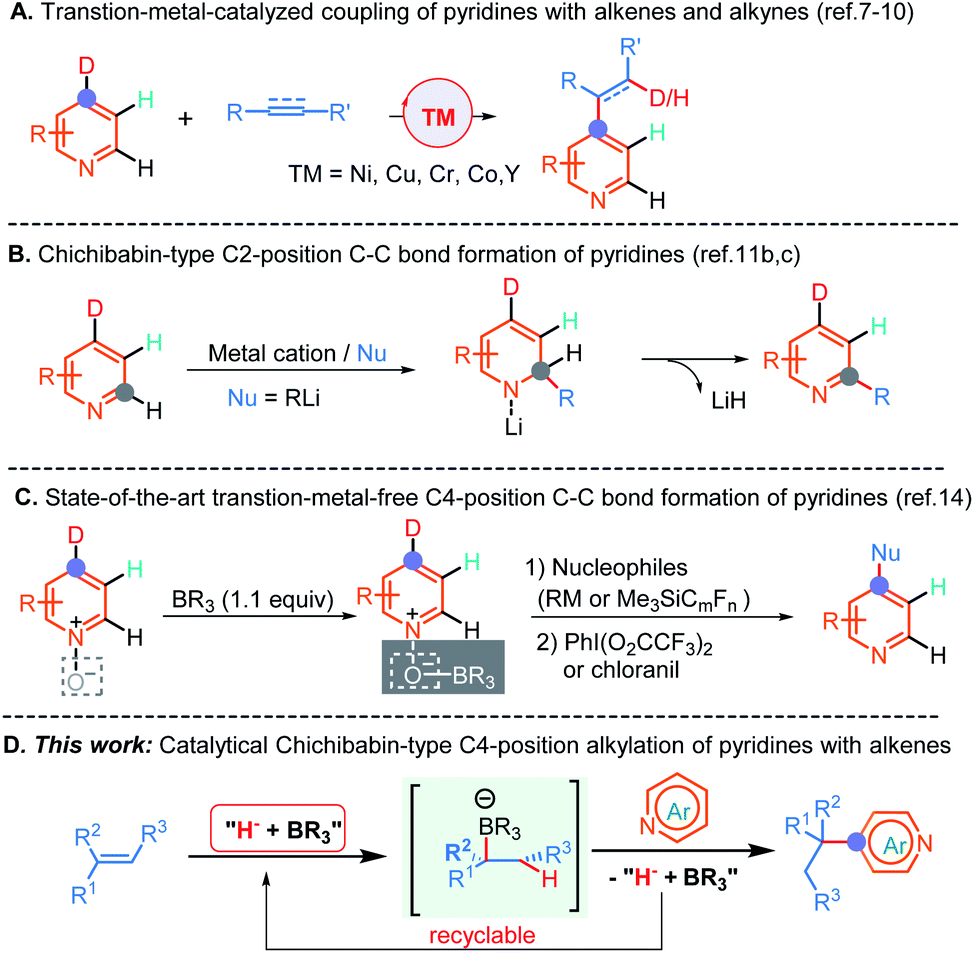 | ||
| Scheme 1 Direct C4-position C–C bond-forming of pyridines and Chichibabin-type transformations. | ||
The Chichibabin amination reaction,11a which involves a C2-position addition of the amine anion to pyridine with the aid of the coordination of pyridine to the sodium cation, followed by an elimination of NaH to form 2-aminopyridine, provided a straightforward and alternative strategy for the synthesis of functionalized pyridines under transition metal free conditions. Although this approach has been established for more than one hundred years, analogous examples have rarely been reported to date, and moreover, strongly basic organolithium reagents as nucleophiles are indispensable in these conversions (Scheme 1B).11b,c Further, the more challenging direct construction of the C–C bond at the C4-position by this method is still unexplored to date. The major issues for the sluggish progress in this field might ascribe to: (1) the inherently low electrophilicity of pyridines; (2) the requirement for strongly basic organometallics as nucleophiles that could result in competitive deprotonation/metalation; (3) and the reluctant elimination of hydride from the resulting σH adducts to provide pyridine cores in these processes. Indeed, almost all of these nucleophilic aromatic substitutions of hydrogen (SNH) proceeded through departure of a proton via an oxidative or eliminative pathway rather than departure of a hydride anion so far.12 To surmount these challenges, oxidative Chichibabin-type two-step strategies (nucleophilic addition followed by oxidative aromatization) for mainly accessing C2-position functionalized pyridine derivatives have been developed in recent years.13 For example, by introduction of the sterically bulky Lewis acid to precisely control the regioselectivity, two examples for direct C4-position C–C bond construction with perfluoroalkylsilanes, Grignard, and organozinc reagents as nucleophiles have been successfully disclosed by Kanai, and Knochel, respectively (Scheme 1C).14 Despite these advances, the presynthesis of activated pyridine salts, organometallics and extra oxidation processes are generally necessary. Furthermore, the transition metal free catalytic version of the Chichibabin-type reaction has hitherto never been demonstrated but is highly desirable, especially for directly employing pyridines and readily available and bench-stable alkenes as the feedstocks.
As a result of our continued interested in hydrofunctionalization of alkenes and alkynes,15 herein, we report an unprecedented NaBEt3H-catalyzed Chichibabin-type C4-position alkylation of pyridines with alkenes as the latent nucleophiles under the assistance of BEt3, wherein conventional transition metal catalysts,4–10 prior formation of N-activated pyridines,2e,h,13,14 organometallic regents,2a–d,h,3,11a–d,14 and extra oxidation operation9,14 were not required. Therefore, this method provides a convenient and straightforward strategy to access an array of synthetically important diarylmethanes and more challenging all-carbon quaternary carbon center-containing triarylmethanes in a perfect atom-economical manner (Scheme 1D).
Results and discussion
Optimization of reaction conditions
We initially selected styrene 1a and pyridine 2a as the pilot substrates. After some trials, we delightedly found that the reductive cross-coupling reaction in the presence of NaBEt3H as the hydride source with BEt3 as an additive indeed occurred, and provided the C4-position selective branched product 3a in 90% yield (Table 1, entry 1). Other hydride sources, such as KBEt3H, LiBEt3H, LiBsBu3H, NaH and LiAlH4, were also effective “H−” suppliers, providing 3a in the range of 34 to 90% yields (Table 1, entries 2–6). Considering the reported significant effect of Lewis acids in the reactivity profile and regioselectivity control of six-membered heteroaromatic compounds,7,10,14 we subsequently evaluated various Lewis acids. The experimental results showed that B(n-Bu)3 exhibited comparable reactivity to BEt3, while other Lewis acids including B(OiPr)3, Al(OiPr)3 and AlMe3 led to the substantial decrease of the yields (Table 1, entries 7–10). Likewise, lowering the loading of either the “H−” supplier or BEt3 also resulted in markedly decreased yield (Table 1, entries 11–12). In addition, control experiments revealed that both NaBEt3H and BEt3 were necessary for this transformation to occur (Table 1, entries 13–14). Remarkably, combination of catalytic amounts of LiBsBu3H and BsBu3 instead of NaBEt3H and BEt3 also showed excellent reactivity profiles and the branched coupling product 3a was afforded in excellent yield (Table 1, entry 15). Unfortunately, we found that this reaction condition exhibited narrow substrate scope. It is worth noting that all the reactions occurred at the 4-position of pyridine and 2-position alkylated pyridine was not observed.|
|
|||
|---|---|---|---|
| Entry | “H−” source | Additive | Yieldb (%) |
| a Reaction conditions: 1a (0.75 mmol, 1.5 equiv), 2a (0.5 mmol), NaBEt3H (0.2 mmol), BEt3 (1.0 mmol) in dry THF (1 mL) at 100 °C under a N2 atmosphere. b Yields were determined by 1H NMR spectroscopy of the crude mixture, using CH2Br2 as the internal standard. c 30 mol% NaBEt3H was used. d 1.0 equiv. BEt3 was used. e 20 mol% LiBsBu3H and 10 mol% BsBu3 were used. | |||
| 1 | NaBEt3H | BEt3 | 90 |
| 2 | KBEt3H | BEt3 | 79 |
| 3 | LiBEt3H | BEt3 | 80 |
| 4 | LiBsBu3H | BEt3 | 64 |
| 5 | NaH | BEt3 | 90 |
| 6 | LiAlH4 | BEt3 | 34 |
| 7 | NaBEt3H | B(n-Bu)3 | 89 |
| 8 | NaBEt3H | B(OiPr)3 | 20 |
| 9 | NaBEt3H | Al(OiPr)3 | 15 |
| 10 | NaBEt3H | AlMe3 | 35 |
| 11c | NaBEt3H | BEt3 | 73 |
| 12d | NaBEt3H | BEt3 | 15 |
| 13 | — | BEt3 | 0 |
| 14 | NaBEt3H | — | 0 |
| 15e | LiBsBu3H | BsBu3 | 95 |
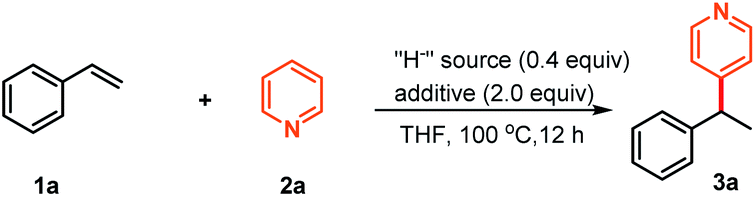
Substrate scope
With these optimized reaction conditions in hand, we then examined the substrate scope of this NaBEt3H-catalyzed Chichibabin-type alkylation of pyridines. As illustrated in Scheme 2A, an array of styrenes bearing a variety of functional groups, including -alkyl, -aryl, -OCH3, -OSiR3, -SCH3, -F, -Cl, -Br, -OCF3, and -CF3, could be effectively cross-coupled with pyridine and delivered corresponding products 3a–3v in generally good to excellent yields with exclusive regioselectivities. The styrenes 1l and 1m containing a silyl ether functional group and a relatively strong acidic benzylic C–H bond, were well-tolerated under these reaction conditions, providing corresponding products 3l and 3m in good yields. The accommodation of silyl ether and aryl halides provided more opportunities for further elaborations. In addition, lower reactivities of electron-donating functional groups, such as para-OMe and -Me substituted styrenes than styrenes bearing electron-withdrawing functional groups (e.g. -F, -Cl, -Br, and -OCF3) were also observed, as demonstrated by 3f and 3iversus3p–3t, 3v. A heteroaromatic substituted alkene, a common scaffold in bioactive relevant targets, was also coupled with pyridine efficiently, furnishing expected product 3w in 75% yield. 1,4-Divinylbenzene and (−)-menthol-derived styrene could also be converted into corresponding mono-pyridation products 3x and 3y in moderate yields. The more steric hindrance α-methyl styrene, which can't be used as the intermolecular coupling partner under the metal catalysis conditions,7–10 to our delight, was found to be a suitable substrate to afford a quaternary carbon center-containing product 3z. In addition to terminal alkenes, aryl substituted internal alkenes were also compatible and the respective products 3aa and 3ab were obtained in good yields. Subsequently, a further study was initiated to explore the substrate scope regarding pyridines. Various alkyl-, aryl substituted pyridine derivatives could also be effectively alkylated with styrene, giving desired products 3ac–3ah in the range of 50–89% yields. Thienyl, methoxyl and synthetically versatile -B(pin) (pin = pinacolate) substituted pyridines, as well as fused pyridine derivatives were also found to be valid substrates for this transformation, delivering the corresponding products 3ai–3am. For ortho-substituted pyridines, we noticed that these reactions became sluggish and required increased reaction temperature, which might be attributed to the steric hindrance. | ||
| Scheme 2 Substrate scope of alkenes and pyridines.a,b aReaction conditions for (A) styrenes (0.75 mmol, 1.5 equiv.), pyridines (0.5 mmol), NaBEt3H (0.2 mmol), BEt3 (1.0 mmol) in dry THF (1.0 mL) at 100 °C for 12 hours under a N2 atmosphere; yield was determined by 1H NMR spectroscopy of the crude mixture, using CH2Br2 as the internal standard. bReaction conditions for (B) 1,1-diaryl alkenes (0.75 mmol, 1.5 equiv.), pyridine (0.5 mmol), NaBEt3H (0.15 mmol), BEt3 (1.0 mmol) in dry THF (1.0 mL) at 70 °C for 12 hours under a N2 atmosphere; isolated yields. cUsing NaH instead of NaBEt3H. d24% alkene was recovered. e57% alkene was recovered. fThese reactions were carried out at 140 °C. g50% alkene was recovered. | ||
The success with sterically hindered α-methyl styrene as the substrate encouraged us to examine whether 1,1-diaryl alkenes could be used as the valid coupling components to synthesize structurally more intriguing all-carbon quaternary carbon center-containing triarylmethanes. These compounds are well-known substructures in photochromic agents,16 leuco dye precursors,17 and materials and medicinal chemistry,18 and could otherwise be difficult to access.19 After briefly screening the reaction conditions, to our delight, the alkylation reaction of pyridine with 1,1-dibenzylethene could proceed with a relatively lower loading of organoborohydride and milder conditions (30 mol% NaBEt3H, 70 °C). The corresponding coupling product 4a was afforded in 93% yield in a regiospecific manner. Additionally, this reaction could also be performed at room temperature, despite the relatively low yield and requirement for prolonged reaction times (80% yield, 48 h). As illustrated in Scheme 2B, various diversely functionalized 1,1-diarylethylenes, including these aryl groups having alkyl, methoxyl, methylthio, siloxy, silyl, alkenyl, phenyl, boronate, fluoro-, chloro-, naphthyl- and heteroaryl, were found to be valid substrates to furnish the corresponding products 4b–4o in moderate to excellent yields. In addition, some representative examples using NaH as the hydride supplier were also reported and the corresponding expected products were indeed obtained with similar reactivities compared with those of NaBEt3H, as demonstrated by 3a–3d and 4a–4c. Additionally, to demonstrate the practicality of this approach, we carried out gram-scale synthesis of products 3a and 4a under the optimal reaction conditions (see the ESI† for details). There were negligible changes in the chemical yields (88% and 79% yields for 3a and 4a, respectively), suggesting that large-scale chemical production might be possible.
Mechanistic investigation
To gain preliminary insight into the mechanism, a series of control experiments were performed. As illustrated in Scheme 3, a series of isotope labeling experiments were carried out firstly. By the reaction of 2a-d5 (>99% D) instead of 2a with styrene under the standard reaction conditions, 0.66 D incorporation at the β-position methyl of the product 5 was observed, which means the C4-position D atom of the pyridine indeed participated in the catalytic cycle (eqn (1)). In addition, when commercially available LiAlD4 was used as the hydride supplier instead of NaBEt3H, the coupling product 6 with 0.48 D at the β-position was obtained and this result suggests that the addition of the deuterium anion to the alkene occurred (eqn (2)). Simultaneously, 0.36 D at the ortho-position of the pyridine core in 6 was observed. To explain this phenomenon, a control experiment with 4-phenylpyridine as the substrate in the presence of LiAlD4 was performed, and 0.32 D incorporation at the ortho-position of 4-phenylpyridine was also observed (see the ESI† for details), indicating that a hydrogen/deuterium exchange could proceed directly between pyridines and LiAlD4. Moreover, when we conducted the reaction under optimal conditions with LiAlD4 as the hydride source and 2a-d5 as the substrate, the corresponding deuterated product 7 containing 1.0 D in the methyl group was observed, which clearly reveals again that the newly introduced D atom in the methyl group of the product entirely comes from the hydride supplier and the para-position D atom of pyridine (eqn (3)). Collectively, these isotopic labeling experimental results indicate that the C4-position D atom of the pyridine departed as a hydride anion and could then participate in the catalytic cycle. Moreover, several 11B NMR experiments were also conducted to elucidate the possible intermediates in this transformation. As shown in Fig. 1A, two peaks were observed from the commercially available NaBEt3H solution (1.0 M in THF) and confirmed as BEt3 (δ = 86.5 ppm) and the tetraorganoborate anion [BEt3H]− (δ = −16.8 ppm), respectively. This experiment indicates that a dissociation equilibrium existed in the NaBEt3H solution. In addition, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of pyridine and BEt3 in THF showed a new peak at 2.8 ppm, assigned to their complex,20 which means BEt3-activated pyridine could readily generate and might be involved in this reaction. Moreover, the peak of BEt3 completely disappeared which suggests their strong interaction, which might provide the potential clue of using stoichiometric BEt3 in this Chichibabin-type alkylation reaction. Pleasingly, either with the mixture of NaBEt3H and 4-fluorostyrene (1:1) or the reaction of pyridine with 4-fluorostyrene under the standard conditions, the same new signal was formed at −15.0 ppm, and was confirmed as tetradentate benzylorganoborate anion species,21 which means the tetradentate benzylorganoborate anion intermediate is likely involved in this catalytic reaction. Besides, other signals, including tetradentate [BEt3H]− and the complex of BEt3 with pyridine were also observed in the last 11B NMR experiment. In addition, the peaks at 55 and 53.5 in these 11B NMR spectra were likely to be Et2BOR analogues,21a which indicates that the decomposition of BEt3 might occur. To further confirm the intermediate of this transformation, we proposed that the tetraorganoborate anion intermediate might be trapped by an oxidation step to form corresponding alcohol (Fig. 1B, top). Indeed, by the reaction of 4-phenylstyrene with a stoichiometric quantity of LiBEt3H in THF at 70 °C, followed by the treatment with an equimolar amount of methanesulfonic acid and oxidation with NaOH/H2O2, 56% 1-phenylethanol 8 was isolated. This experimental result also provided positive evidence that the tetradentate benzylorganoborate anion species is generated in this NaBEt3H-catalyzed Chichibabin-type alkylation reaction. Moreover, we also investigated the possible intermediate of the coupling between 1,1-dibenzylethene and pyridine, but the signal of the analogous tetradentate benzylorganoborate anion species was not detected by the 11B NMR experiment. Instead, a deep red color was formed immediately when 1.1 equiv. NaBEt3H was added to the solution of 1,1-dibenzylethene, which suggested 1-sodium-1,1-diphenylethane was likely generated rather than its corresponding organoborate.22 This probable intermediate was further supported by a trapping experiment with MeOH, providing 1,1-diphenylethane 9 in 95% yield (Fig. 1B, bottom).
:1 ratio of pyridine and BEt3 in THF showed a new peak at 2.8 ppm, assigned to their complex,20 which means BEt3-activated pyridine could readily generate and might be involved in this reaction. Moreover, the peak of BEt3 completely disappeared which suggests their strong interaction, which might provide the potential clue of using stoichiometric BEt3 in this Chichibabin-type alkylation reaction. Pleasingly, either with the mixture of NaBEt3H and 4-fluorostyrene (1:1) or the reaction of pyridine with 4-fluorostyrene under the standard conditions, the same new signal was formed at −15.0 ppm, and was confirmed as tetradentate benzylorganoborate anion species,21 which means the tetradentate benzylorganoborate anion intermediate is likely involved in this catalytic reaction. Besides, other signals, including tetradentate [BEt3H]− and the complex of BEt3 with pyridine were also observed in the last 11B NMR experiment. In addition, the peaks at 55 and 53.5 in these 11B NMR spectra were likely to be Et2BOR analogues,21a which indicates that the decomposition of BEt3 might occur. To further confirm the intermediate of this transformation, we proposed that the tetraorganoborate anion intermediate might be trapped by an oxidation step to form corresponding alcohol (Fig. 1B, top). Indeed, by the reaction of 4-phenylstyrene with a stoichiometric quantity of LiBEt3H in THF at 70 °C, followed by the treatment with an equimolar amount of methanesulfonic acid and oxidation with NaOH/H2O2, 56% 1-phenylethanol 8 was isolated. This experimental result also provided positive evidence that the tetradentate benzylorganoborate anion species is generated in this NaBEt3H-catalyzed Chichibabin-type alkylation reaction. Moreover, we also investigated the possible intermediate of the coupling between 1,1-dibenzylethene and pyridine, but the signal of the analogous tetradentate benzylorganoborate anion species was not detected by the 11B NMR experiment. Instead, a deep red color was formed immediately when 1.1 equiv. NaBEt3H was added to the solution of 1,1-dibenzylethene, which suggested 1-sodium-1,1-diphenylethane was likely generated rather than its corresponding organoborate.22 This probable intermediate was further supported by a trapping experiment with MeOH, providing 1,1-diphenylethane 9 in 95% yield (Fig. 1B, bottom).
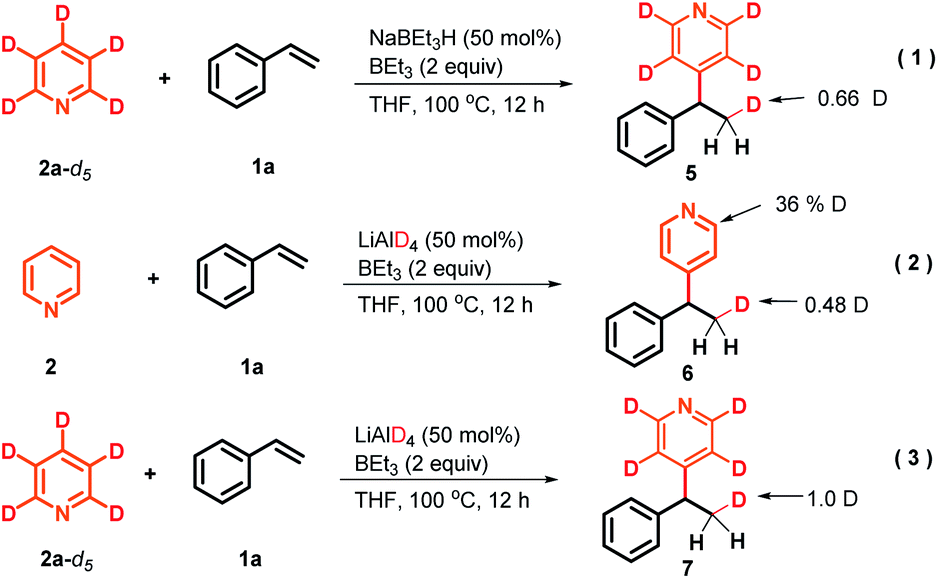 | ||
| Scheme 3 H/D scrambling experiments. | ||
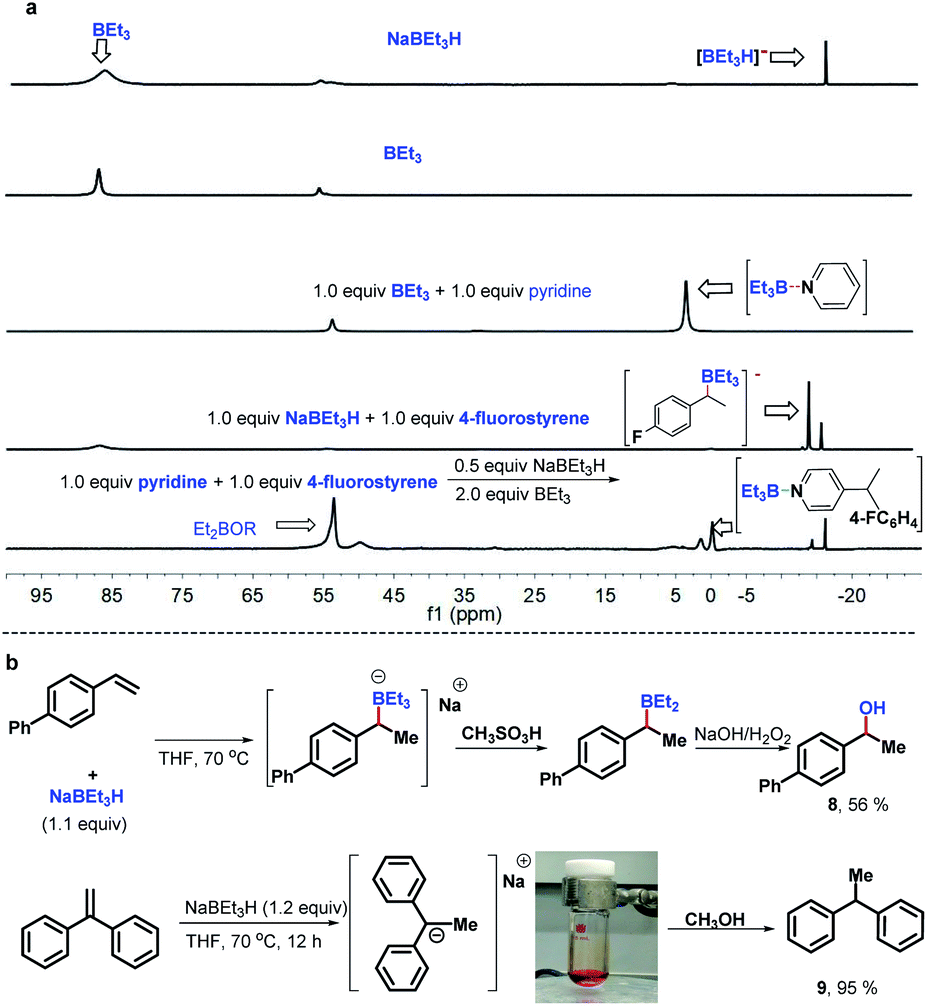 | ||
| Fig. 1 Intermediate investigation experiments. | ||
To further shed light on the details of this transformation, we conducted density functional theory (DFT) calculations. The calculated free energy profile showed that the lone pair of the N atom in pyridine interacts with the empty orbital of the B atom of BEt3 to produce a relatively stable complex Sub2 with a Gibbs free energy change (ΔG°) of −4.1 kcal mol−1 (Fig. 2A). Thus, Sub2 directly participates in the reaction. As shown in Fig. 2B (yellow line), NaBEt3H is initially combined with styrene through the electrostatic interaction to form intermediate 1′ (5.1 kcal mol−1 exergonic), followed by an insertion of the hydride ion into the C–C double bond process assisted by BEt3 to generate intermediate 2′ and release the BEt3 (5.3 kcal mol−1, viaTS-1). Then, 2′ could isomerize to a more stable intermediate 3′ (4.3 kcal mol−1 exergonic). The following nucleophilic addition of 3′ to Sub2 occurs viaTS2 to afford intermediate 4′ (20.0 kcal mol−1). By contrast, the nucleophilic addition to the C2-position of Sub2 is less favorable with a higher energy barrier of 22.3 kcal mol−1 (Fig. S18†). Finally, as a Lewis acid the BEt3 is again involved in the hydride ion transfer to provide the target product 5′ (viaTS-3) and simultaneously regenerate the NaBEt3H. The hydride ion transfer is the rate-determining step of the whole reaction, which requires a moderate Gibbs activation energy (ΔG°‡) value of 25.0 kcal mol−1 relative to 3′. The present calculations are consistent with the experimental results. This reaction can be achieved at a temperature of 373 K. Moreover, we also investigated the hydride ion insertion process without the extra BEt3 (Fig. 2B, black line). Obviously, these results clearly show that the participation of BEt3 dramatically accelerates the insertion process and following reaction steps. Actually, one of the key roles of BEt3 is generation of the stable intermediates 3′ and 4′, which are more stable than the corresponding intermediates 7′ and 8′ and then significantly stabilizes the potential energy surface and lowers the energy barrier of the rate-determining step. In addition, we have also presented the molecular orbital distributions and energy levels of 3′, 4′, 7′ and 8′ in Fig. 2b, respectively. The BEt3 coordination stabilizes the HOMO levels of 3′ and 4′, respectively, leading to the increases in their HOMO–LUMO energy gaps, which make the intermediates 3′ and 4′ more stable (Fig. S17†). On the other hand, the experimental phenomenon with 1,1-diaryl alkenes as the alkylating reagents suggested that the tetradentate benzylorganoborate anion species might not be involved in this transformation (Fig. 1B, bottom). This observation is further supported by DFT calculations. The formation of the C–B bond during this process is energetically disfavored (Fig. 2C, bottom, 10′ → 10′′, 3.0 kcal mol−1 endergonic). However the C–B bond could significantly stabilize similar intermediate 6′ in the alkylation of pyridine with styrene (Fig. 2C, top, 6′ → 3′, 15.3 kcal mol−1 exergonic). Moreover, the Gibbs energy profiles of the alkylation between 1,1-diphenylethene and pyridine were also provided (Fig. 2D), and the calculated results showed that the Gibbs activation energy of the rate-determining step is 20.7 kcal mol−1 relative to 9′, lower than that of styrene (25 kcal mol−1). This is also consistent with the experimental results.
 | ||
| Fig. 2 Calculated energy profiles of the NaBEt3H-catalyzed alkylation reaction of pyridine with the alkene. (A) Calculated energy profile of pyridine with BEt3. (B) DFT-computed reaction pathway for NaBEt3H-catalysed alkylation of styrene with pyridine. (C) The relative Gibbs energies and structures of different C–B interactions between styrene and 1,1-diphenylethene. (D) DFT-computed reaction pathway for NaBEt3H-catalysed alkylation of diphenylethene with pyridine. | ||
On the basis of the above experimental observations and computational studies, a plausible mechanism of this NaBEt3H-catalyzed Chichibabin-type alkylation reaction is depicted in Scheme 4. An addition of the hydride catalyst to the alkene could occur firstly in the presence of the organoborane, furnishing “organoborate” intermediate I,22 which has been recognized as a class of particularly versatile synthetic intermediates in organic synthesis.23 Intermediate I could then undergo an intermolecular regioselective addition to organoborane-activated heteroarenes II leading to σH-adduct intermediates III and IV. Finally, by an elimination of the hydride anion, a Chichibabin-type-like process,11 could occur with the assistance of the organoborane to furnish the expected alkylation product simultaneously regenerating the hydride catalyst to participate in the next catalytic cycle. For the mechanism of the alkylation of pyridine with 1,1-diaryl alkenes, according to the experimental phenomenon and computational studies, a free 1,1-diphenyl stabilized carbon anion intermediate rather than its tetradentate benzylorganoborate species might react directly with intermediate II to provide the product 4 and regenerate the hydride catalyst. The relatively high loading of NaBEt3H and BEt3 under the present catalytic conditions might be ascribed to the ineluctable trace amount of water in the reaction, the strong interaction of BEt3 with pyridine cores and the decomposition of BEt3.
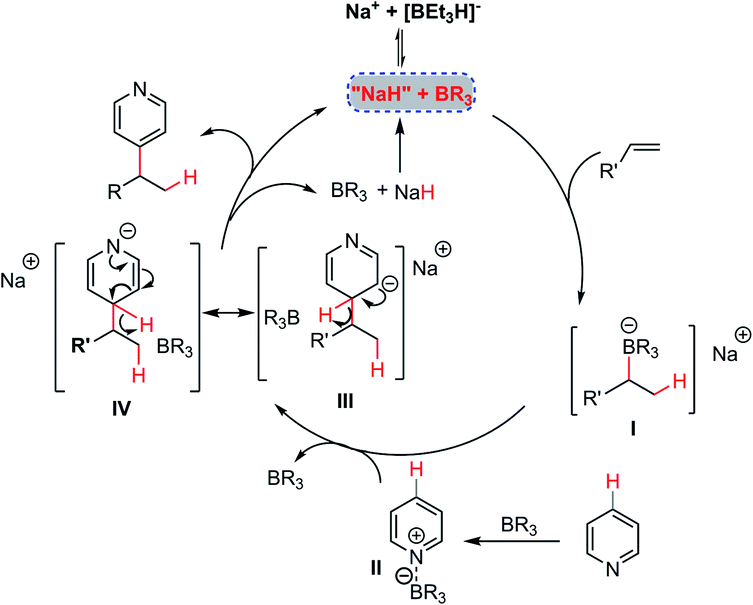 | ||
| Scheme 4 Plausible mechanism. | ||
Conclusions
In summary, we have developed the first NaBEt3H-catalyzed Chichibabin-type alkylation of pyridines with alkenes in the presence of BEt3 in a perfect atom-economical and regiospecific fashion. This method allows for facile access to an array of branched C4-alkylation pyridines, without the requirement for conventional transition metal catalysts, prior formation of N-activated pyridines, organometallic reagents, and oxidation processes. Moreover, highly congested all-carbon quaternary center-containing triarylmethanes could also be efficiently synthesized. The corresponding mechanism and the key roles of the organoborane were also elaborated by the combination of H/D scrambling experiments, 11B NMR studies, intermediate trapping experiments and computational studies. This novel and mechanically complementary methodology could not only open a new door for the classical but still far less well-developed Chichibabin-type reaction, but could also set up a new platform for the development of novel C–C bond-forming methods. Explorations of the potential of this organoborohydride catalysis for C–C bond formations are currently underway in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the NSFC (21672033, 21801039, and 21831002), Jilin Educational Committee (JJKH20190269KJ), Fundamental Research Funds for the Central Universities, and Ten Thousand Talents Program for generous financial support.Notes and references
- (a) J. W. Daly, H. M. Garraffo and T. F. Spande, in Alkaloids: Chemical and Biological Perspectives, ed. W. W. Pelletier, Elsevier, New York, 1999, vol. 13, pp. 92–104 Search PubMed; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS; (c) Y. G. Skrypink and T. F. Doroshenko, Mater. Sci., 1996, 32, 537–544 CrossRef; (d) N. Leclerc, S. Sanaur, L. Galmiche, F. Mathevet, A.-J. Attias, J.-L. Fave, J. Roussel, P. Hapiot, N. Lemaître and B. Geffroy, Chem. Mater., 2005, 17, 502–513 CrossRef CAS.
- For leading reviews, see: (a) V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS; (b) P. Gros and Y. Fort, Eur. J. Org. Chem., 2002, 3375–3383 CrossRef CAS; (c) M. Schlosser and F. Mongin, Chem. Soc. Rev., 2007, 36, 1161–1172 RSC; (d) P. C. Gros and Y. Fort, Eur. J. Org. Chem., 2009, 4199–4209 CrossRef CAS; (e) M. Ahamed and M. H. Todd, Eur. J. Org. Chem., 2010, 5935–5942 CrossRef CAS; (f) Y. Nakao, Synthesis, 2011, 3209–3219 CrossRef CAS; (g) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef; (h) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS; (i) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807–4815 RSC; (j) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS.
- (a) J. L. Jeffrey and R. Sarpong, Org. Lett., 2012, 14, 5400–5403 CrossRef CAS; (b) F.-F. Zhuo, W.-W. Xie, Y.-X. Yang, L. Zhang, P. Wang, R. Yuan and C.-S. Da, J. Org. Chem., 2013, 78, 3243–3249 CrossRef CAS.
- For recent reviews on the Minisci reaction, see: (a) M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC; (b) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS; For selected examples, see: (c) F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS; (d) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. D. Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292–3295 CrossRef CAS; (e) G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS; (f) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS; (g) Y. Fujiwara, J. A. Dixon, F. O. Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herle, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS; (h) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS; (i) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorious, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS; (j) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907–911 CrossRef CAS; (k) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS.
- Selected literature of C2-selectivity, see: (a) R. F. Jordan and D. F. Taylor, J. Am. Chem. Soc., 1989, 111, 778–779 CrossRef CAS; (b) S. Rodewald and R. F. Jordan, J. Am. Chem. Soc., 1994, 116, 4491–4492 CrossRef CAS; (c) M. Murakami and S. Hori, J. Am. Chem. Soc., 2003, 125, 4720–4721 CrossRef CAS; (d) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332–5333 CrossRef CAS; (e) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 CrossRef CAS; (f) A. M. Berman, J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 14926–14927 CrossRef CAS; (g) B.-T. Guan and Z. Hou, J. Am. Chem. Soc., 2011, 133, 18086–18089 CrossRef CAS; (h) H. Kaneko, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2011, 133, 19626–19629 CrossRef CAS; (i) B. Liu, Y. Huang, J. Lan, F. Song and J. You, Chem. Sci., 2013, 4, 2163–2167 RSC; (j) D. G. Johnson, J. M. Lynam, N. S. Mistry, J. M. Slattery, R. J. Thatcher and A. C. Whitwood, J. Am. Chem. Soc., 2013, 135, 2222–2234 CrossRef CAS; (k) G. Song, W. N. O. Wylie and Z. Hou, J. Am. Chem. Soc., 2014, 136, 12209–12212 CrossRef CAS; (l) H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2015, 137, 640–643 CrossRef CAS; (m) A. Kundu, M. Inoue, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2018, 140, 7332–7342 CrossRef CAS.
- Selected literature of C3-selectivity, see: (a) M. Ye, G.-L. Gao and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 6964–6967 CrossRef CAS; (b) M. Ye, G.-L. Gao, A. J. F. Edmunds, P. A. Worthington, J. A. Morris and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19090–19093 CrossRef CAS; (c) P. Guo, J. M. Joo, S. Rakshit and D. Sames, J. Am. Chem. Soc., 2011, 133, 16338–116341 CrossRef CAS.
- (a) C.-C. Tsai, W.-C. Shih, C.-H. Fang, C.-Y. Li, T.-G. Ong and A. G. P. Yap, J. Am. Chem. Soc., 2010, 132, 11887–11889 CrossRef CAS; (b) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS; (c) W.-C. Lee, C.-H. Chen, C.-Y. Liu, M.-S. Yu, Y.-H. Lin and T. G. Ong, Chem. Commun., 2015, 51, 17104–17107 RSC.
- (a) T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213–3216 CrossRef CAS; (b) Y. Li, G. Deng and X. Zeng, Organometallics, 2016, 35, 747–750 CrossRef CAS; transition-metal-mediated alkylation with additional hydride sources, see: (c) T. Mizumori, T. Hata and H. Urabe, Chem.–Eur. J., 2015, 21, 422–426 CrossRef CAS.
- (a) M. W. Gribble Jr, S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057–5060 CrossRef; (b) M. W. Gribble Jr, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 11252–11269 CrossRef.
- W.-B. Zhang, X.-T. Yang, J.-B. Ma, Z.-M. Su and S.-L. Shi, J. Am. Chem. Soc., 2019, 141, 5628–5634 CrossRef CAS.
- (a) A. E. Chichibabin and O. A. Zeide, J. Russ. Phys.-Chem. Soc., 1914, 46, 1216 CAS; (b) J. C. W. Evans and C. F. H. Allen, Org. Synth., 1938, 18, 70 CrossRef CAS; (c) J. L. Jeffrey and R. Sarpong, Org. Lett., 2012, 14, 5400–5403 CrossRef CAS; transition-metal-catalysed Chichibabin-type C2-position functionalization, see: (d) M. Tobisu, I. Hyodo and N. Chatani, J. Am. Chem. Soc., 2009, 131, 12070 CrossRef CAS, and also see ref. 8a. Related fluorination reactions, see: (e) P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956–960 CrossRef CAS.
- (a) M. Mąkosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666 CrossRef; (b) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855–2868 RSC; (c) O. N. Chupakhin and V. N. Charushin, Tetrahedron Lett., 2016, 57, 2665–2672 CrossRef CAS.
- For reviews, see: (a) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS; (b) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141–1156 RSC; (c) H. Andersson, R. Olsson and F. Almqvist, Org. Biomol. Chem., 2011, 9, 337–346 RSC; (d) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef CAS.
- (a) Q. Chen, X. M. du Jourdin and P. Knochel, J. Am. Chem. Soc., 2013, 135, 4958–4961 CrossRef CAS; (b) M. Nagase, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 6103–6106 CrossRef CAS.
- (a) G. Xu, H. Zhao, B. Fu, A. Cang, G. Zhang, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13130–13134 CrossRef CAS; (b) G. Xu, B. Fu, H. Zhao, Y. Li, G. Zhang, Y. Wang, T. Xiong and Q. Zhang, Chem. Sci., 2019, 10, 1802–1806 RSC; (c) B. Fu, X. Yuan, Y. Li, Y. Wang, Q. Zhang, T. Xiong and Q. Zhang, Org. Lett., 2019, 21, 3576–3580 CrossRef CAS; (d) G. Zhang, Y. Liang, T. Qin, T. Xiong, S. Liu, W. Guan and Q. Zhang, CCS Chem., 2020, 2, 1737–1745 CrossRef.
- D. F. Duxbury, Chem. Rev., 1993, 93, 381–433 CrossRef CAS.
- P. Ryss and H. Zollinger, Fundamentals of the chemistry and application of dyes, Wiley VCH, 1972 Search PubMed.
- M. S. Shchepinov and V. A. Korshun, Chem. Soc. Rev., 2003, 32, 170–180 RSC.
- M. Nambo and C. M. Crudden, ACS Catal., 2015, 5, 4734–4742 CrossRef CAS.
- A. V. Fedorov, A. A. Ermoshkin, A. Mejiritski and D. C. Neckers, Macromolecules, 2007, 40, 3554–3560 CrossRef CAS.
- (a) R. B. Bedford, N. J. Gower, M. F. Haddow, J. N. Harvey, J. Nunn, R. A. Okopie and R. F. Sankey, Angew. Chem., Int. Ed., 2012, 51, 5435–5438 CrossRef CAS; (b) M. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061–9065 CrossRef CAS.
- H. C. Brown and S.-C. Kim, J. Org. Chem., 1984, 49, 1064–1071 CrossRef CAS.
- C. G. Watson, P. J. Unsworth, D. Leonori and V. K. Aggarwal, Lithium-Boron Chemistry: A Synergistic Strategy in Modern Synthesis, in Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley, 2014, pp. 397–422 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04808a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |