DOI:
10.1039/D0SC04434B
(Edge Article)
Chem. Sci., 2020,
11, 11562-11569
Ruthenium-catalyzed cascade C–H activation/annulation of N-alkoxybenzamides: reaction development and mechanistic insight†
Received
12th August 2020
, Accepted 26th September 2020
First published on 28th September 2020
Abstract
A highly selective ruthenium-catalyzed C–H activation/annulation of alkyne-tethered N-alkoxybenzamides has been developed. In this reaction, diverse products from inverse annulation can be obtained in moderate to good yields with high functional group compatibility. Insightful experimental and theoretical studies indicate that the reaction to the inverse annulation follows the Ru(II)–Ru(IV)–Ru(II) pathway involving N–O bond cleavage prior to alkyne insertion. This is highly different compared to the conventional mechanism of transition metal-catalyzed C–H activation/annulation with alkynes, involving alkyne insertion prior to N–O bond cleavage. Via this pathway, the in situ generated acetic acid from the N–H/C–H activation step facilitates the N–O bond cleavage to give the Ru-nitrene species. Besides the conventional mechanism forming the products via standard annulation, an alternative and novel Ru(II)–Ru(IV)–Ru(II) mechanism featuring N–O cleavage preceding alkyne insertion has been proposed, affording a new understanding of transition metal-catalyzed C–H activation/annulation.
Introduction
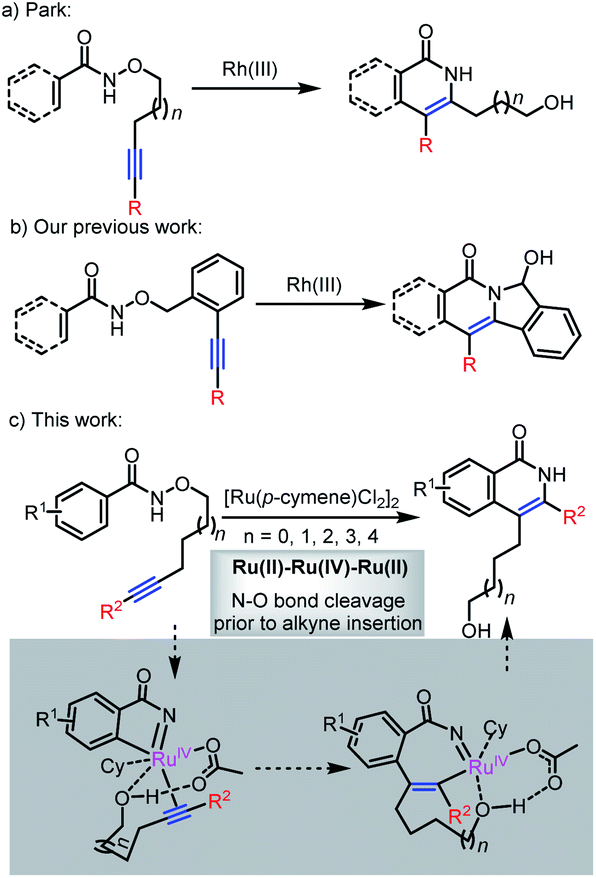
Transition metal-catalyzed C–H activation plays an important role in synthetic organic chemistry.1 In particular, C–H activation followed by annulation with alkynes has emerged as an increasingly effective and attractive strategy for the construction of heterocycles and carbocycles in a step- and atom-economical manner.2 In this regard, Rh(III)3 or Ru(II)4 catalysts have been employed in cascade C–H activation/annulation reactions with various types of alkynes, leading to the synthesis of isoquinolone derivatives through the use of external oxidants.
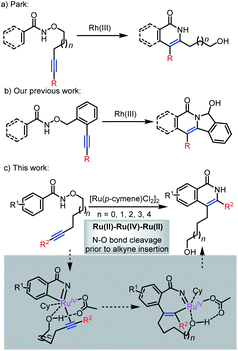
The utilization of internal oxidants exhibits a significant move forward in the area, avoiding the stoichiometric amount of external oxidants and potentially facilitating milder reactions.5,6 For instance, the employment of a N–O bond as an internal oxidant is explored for the construction of isoquinolone analogues in Rh(III)- or Ru(II)-catalyzed C–H activation/annulation reactions and results in a number of examples, which indicated the potential of internal oxidizing directing groups in transition metal-catalyzed C–H activation. One such example was reported by Park,5e who described a rhodium(III)-catalyzed intramolecular annulation reaction of N-alkoxybenzamides to offer a new approach to isoquinolones (Scheme 1a). Subsequently, the internal oxidizing N–O bond has been used to synthesize structurally complex polycyclic compounds.7 In 2014, Lin's group developed a Rh(III)-catalyzed arylative cyclization of N-hydroxybenzamides with cyclohexadienone-containing 1,6-enynes for the synthesis of tetracyclic isoquinolones.7c A Ru(II)-catalyzed regioselective intermolecular annulation of aryl substituted 2-acetylenic ketones with N-methoxybenzamides or acrylamides was reported by Chegondi in 2016.7f Recently, we demonstrated a Rh(III)-catalyzed sequential C(sp2)–H activation and C(sp3)–H amination for the preparation of polycyclic compounds (Scheme 1b).7b,d
 |
| | Scheme 1 Previous works and this approach. | |
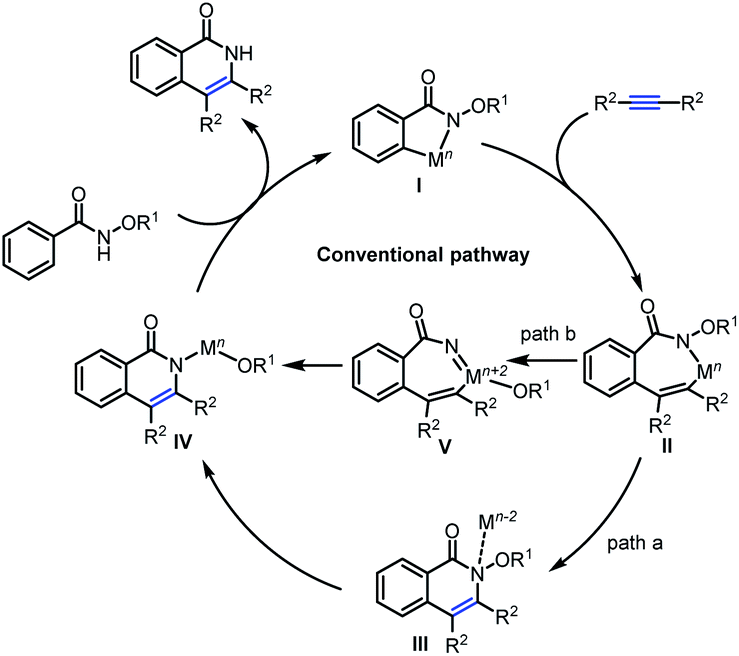
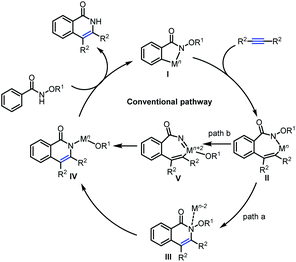
Mechanistically (Scheme 2), these annulations have been explained in the light of an initial C–H activation to generate intermediate I, followed by alkyne insertion into the C–M bond leading to the seven-membered intermediate II, which delivers the isoquinolone products through sequential reductive elimination and oxidative addition into the N–O bond (path a, Mn–Mn−2–Mn pathway). Alternatively, intermediate II possibly undergoes oxidative addition into the N–O bond followed by reductive elimination to deliver the isoquinolones (path b, Mn–Mn+2–Mn pathway).8 However, they are always restricted to alkyne insertion prior to N–O bond cleavage. Mechanistic investigations of the key intermediates in C–H activation/annulation processes are still somewhat limited, so it is not clear whether the mechanistic possibilities in Scheme 2 are sufficient to account for all observed reactivity in these systems. Formation of Rh(V) or Ru(IV) intermediates has been proposed from the experimental results,9 but usually the mechanisms are assumed to involve alkyne insertion prior to N–O bond cleavage (path b). To the best of our knowledge, no unambiguous high-valent Cp*Rh(V) or (p-cymene)Ru(IV) complexes have been isolated to date, but DFT calculations have provided strong support for treating Rh(V) or Ru(IV) species as intermediates in reaction mechanisms.10
 |
| | Scheme 2 Previously proposed catalytic cycle for C–H activation/annulation reactions. | |
In comparison to previous hypotheses, we set out to investigate whether a catalytic cycle involving N–O bond cleavage prior to alkyne insertion might also play a role in some of these reactions.11 The high-valent metal complex generated from N–O bond cleavage should increase the driving force for alkyne insertion and sequential reductive elimination (Scheme 1c). For this purpose, the ruthenium-catalyzed C–H activation/annulation of alkyne-tethered N-alkoxybenzamides is regarded as a platform for verifying this hypothesis.
Results and discussion
Reaction optimization
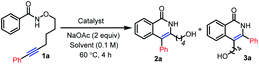
We initiated our studies with the exploration of the ruthenium-catalyzed C–H activation/annulation of alkyne-tethered N-alkoxybenzamide 1a. To our delight, when 1a was treated in the presence of [Ru(p-cymene)Cl2]2 (10 mol%) and NaOAc (2 equiv.) in MeOH at 60 °C for 4 h, the product 3a from inverse annulation was obtained in 81% yield, together with the product 2a from standard annulation in 10% yield (Table 1, entry 1). Next various solvents were screened, such as DCE, CH3CN, THF, acetone, 1,4-dioxane, toluene and DMF, and MeOH was identified as the preferred reaction medium (Table 1, entries 2–8). Adding PivOH (1 equiv.) did not improve the reaction (Table 1, entry 9). Replacing the additive NaOAc with CsOAc (2 equiv.) did not significantly influence the yield (Table 1, entry 10). The employment of AgSbF6 (1 equiv.) instead of NaOAc completely inhibited the reaction (Table 1, entry 11). Decreasing the catalyst loading to 5 mol%, made the reaction much slower (Table 1, entry 12). We also evaluated the performance of other Ru(II)-catalysts, such as [Ru(C6Me6)Cl2]2 and [(C6H5)3P]3RuCl2 (Table 1, entries 13 and 14), but very low conversions were observed.
Table 1 Optimization of the reaction conditionsa
|
|
| Entry |
Catalyst |
Solvent |
2a
(%) |
3a
(%) |
|
Condition: 1a (0.3 mmol), [Ru(p-cymene)Cl2]2, NaOAc (0.6 mmol), solvent (3.0 mL).
Isolated yield.
PivOH (1 equiv.) was added.
CsOAc (2 equiv.) instead of NaOAc.
AgSbF6 (1 equiv.) instead of NaOAc.
|
|
1
|
[Ru(p-cymene)Cl
2
]
2
(10 mol%)
|
MeOH
|
10
|
81
|
| 2 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
DCE |
45 |
39 |
| 3 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
CH3CN |
28 |
16 |
| 4 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
THF |
38 |
32 |
| 5 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
Acetone |
22 |
67 |
| 6 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
1,4-Dioxane |
32 |
19 |
| 7 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
Toluene |
42 |
29 |
| 8 |
[Ru(p-cymene)Cl2]2 (10 mol%) |
DMF |
10 |
21 |
| 9c |
[Ru(p-cymene)Cl2]2 (10 mol%) |
MeOH |
12 |
76 |
| 10d |
[Ru(p-cymene)Cl2]2 (10 mol%) |
MeOH |
11 |
78 |
| 11e |
[Ru(p-cymene)Cl2]2 (10 mol%) |
MeOH |
0 |
0 |
| 12 |
[Ru(p-cymene)Cl2]2 (5 mol%) |
MeOH |
6 |
43 |
| 13 |
[Ru(C6Me6)Cl2]2 (10 mol%) |
MeOH |
<5% |
<5% |
| 14 |
[(C6H5)3P]3RuCl2 (20 mol%) |
MeOH |
<5% |
<5% |
Examination of substrate scope
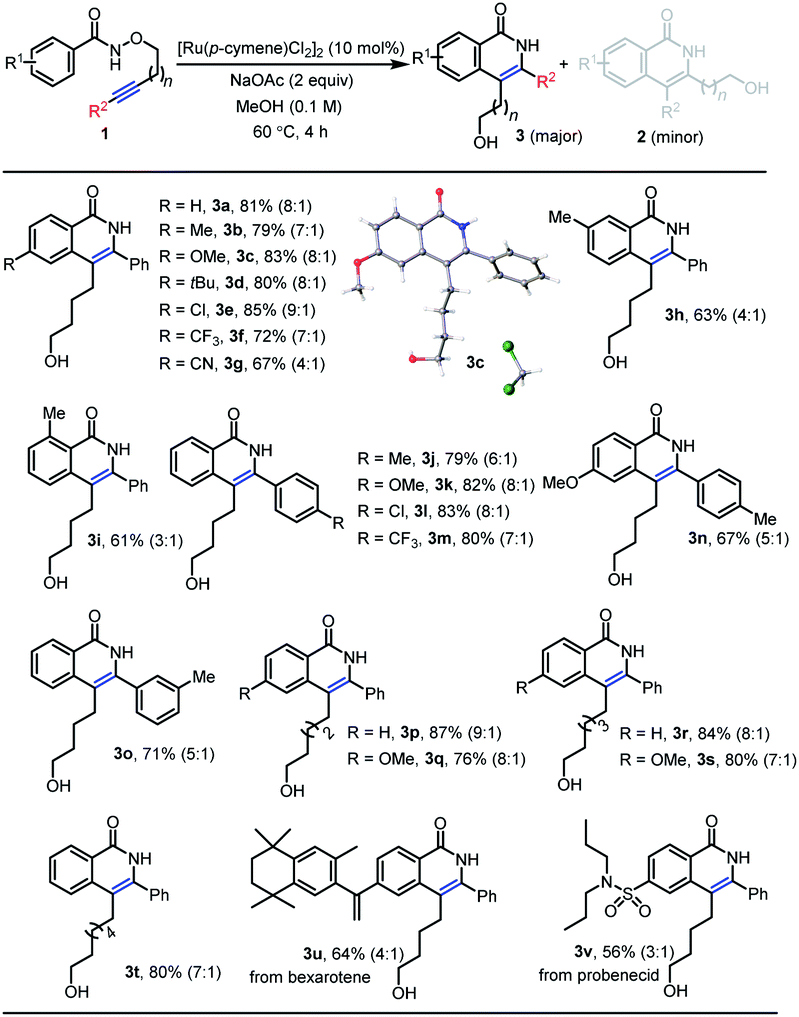
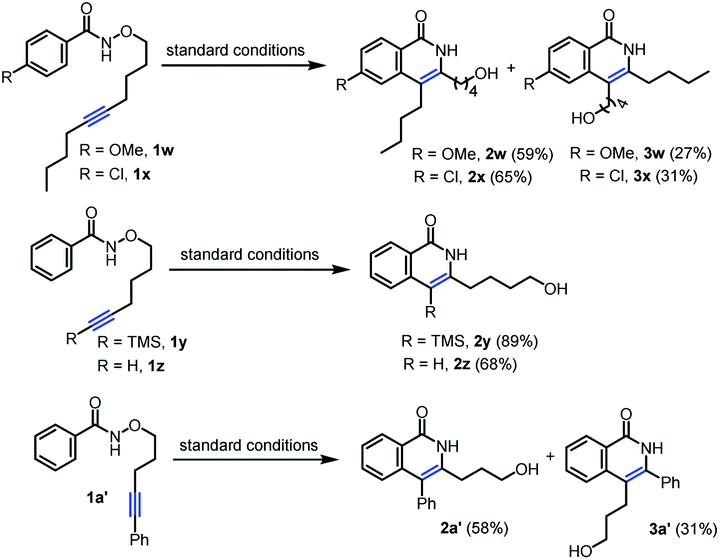
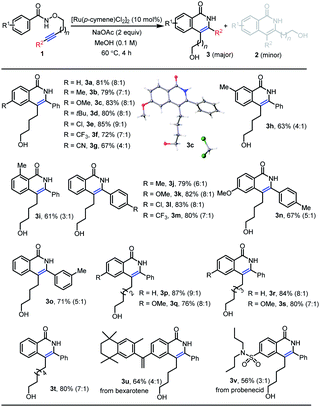
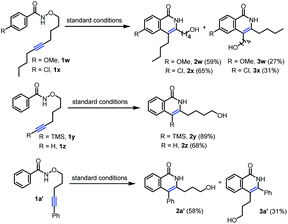
With the optimal reaction conditions in hand, we next investigated diverse substrates to evaluate the scope of the protocol (Table 2 and Scheme 3). Substrates with a wide range of electron-rich and -deficient groups at the para position of the benzamide proceeded smoothly to afford the products 3b–g (67–85%) from inverse annulation (Table 2). The structure of 3c has been confirmed by X-ray diffraction. In particular, the meta-methyl benzamide gave excellent regioselectivity in support of the sterically more accessible C–H bond, delivering product 3h in 63% yield. The ortho-methyl-substituted derivative was also a productive substrate, leading to the product 3i in 61% yield. The annulation reaction also worked well with electron-donating and -withdrawing groups in the aryl substituent of the alkyne, affording the products 3j–o in 67–83% yield. When the aryl group of the alkyne was replaced by n-butyl group (Scheme 3), the products 2w (59%) and 2x (65%) from standard annulation were isolated as the main products, along with concomitant products 3w (27%) and 3x (31%) from inverse annulation. Moreover, only products 2y and 2z from standard annulation were obtained in 89% and 68% yield from the TMS-substituted derivative and the substrate bearing a terminal alkyne respectively (Scheme 3), while previous approaches5e,7b were limited to internal alkynes. Next, we evaluated the length of the tethers between the oxygen atom and the alkyne. Substrates with five-, six- and seven-carbon-atom tethers delivered the products 3p–t from inverse annulation in 76–87% yield. The substrate with a three-carbon-atom tether delivered the product 2a′ (58%) from standard annulation as the main product, together with the product 3a′ from inverse annulation in 31% yield (Scheme 3). To further demonstrate the synthetic utility of this methodology, the annulation reaction was performed using N-alkoxybenzamides derived from the pharmaceuticals bexarotene and probenecid, resulting in the products 3u (64%) and 3v (56%) from inverse annulation.
Table 2 Scope for N-alkoxybenzamide substratesa
Conditions: 1 (0.3 mmol), [Ru(p-cymene)Cl2]2 (0.03 mmol), NaOAc (0.6 mmol) and MeOH (3.0 mL). 3 was isolated by column chromatography. The ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 shown in parenthesis was determined by 1H NMR analysis of the crude reaction mixture. :2 shown in parenthesis was determined by 1H NMR analysis of the crude reaction mixture.
|
|
 |
| | Scheme 3 Ruthenium-catalyzed C–H activation/annulation. | |
Mechanistic studies
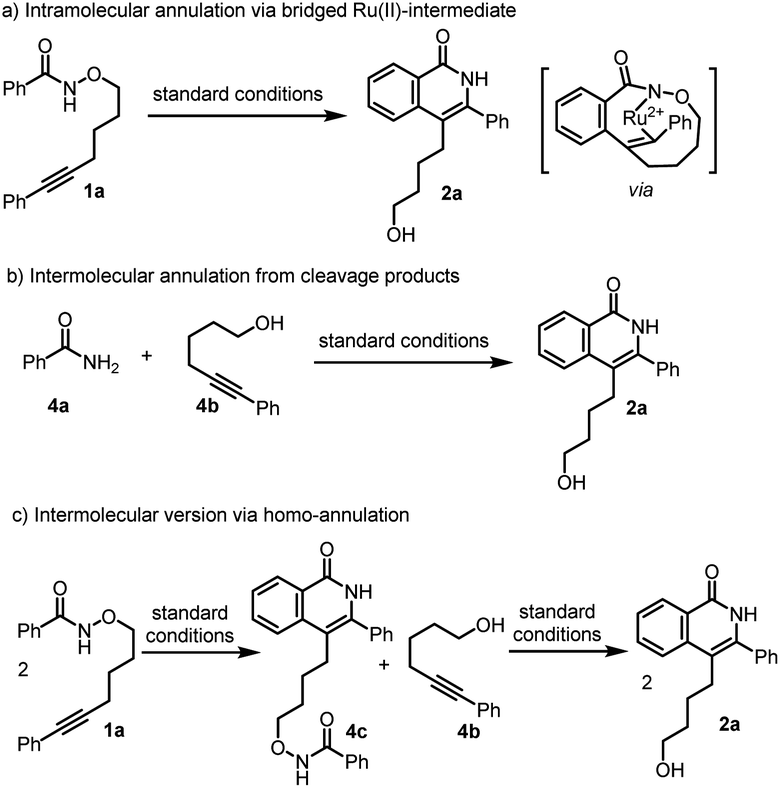
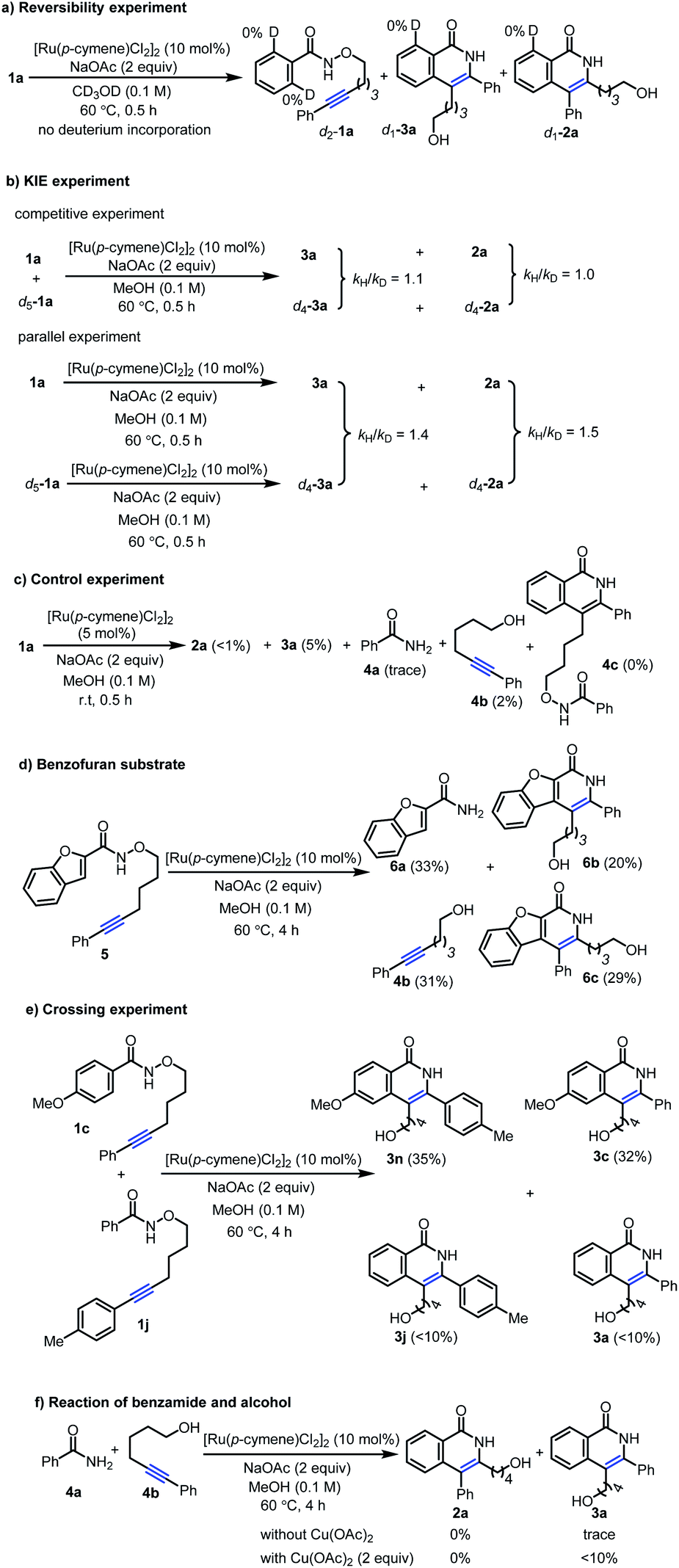
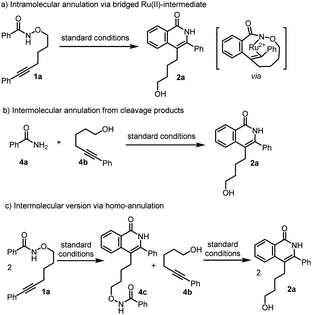
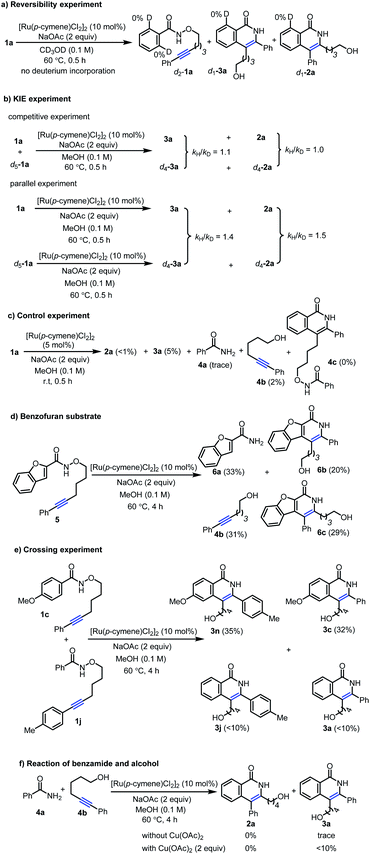
Based on above experimental results, three possible reaction pathways from conventional mechanism for the inverse annulation were firstly proposed. That is, intramolecular annulation via bridged Ru(II)-intermediate (Scheme 4a), or intermolecular annulation from cleavage products (Scheme 4b), or intermolecular version via homo-annulation (Scheme 4c). To gain more mechanistic insight of the present reaction, a series of control experiments were performed. Reversibility experiments concerning the C–H activation step, using 1a in the presence of deuterated methanol (Scheme 5a), showed that there is no deuterium incorporation in the recovered substrate and products, which indicates that the C–H activation step is irreversible. To further investigate the C–H activation process, kinetic isotope effect (KIE) experiments were performed (Scheme 5b). The competitive experiment gave kH/kD = 1.1 for the formation of 3a and kH/kD = 1.0 for the formation of 2a, while the parallel experiment delivered kH/kD = 1.4 for the formation of 3a and kH/kD = 1.5 for the formation of 2a, clearly indicating that the C–H bond cleavage process is not involved in the rate-limiting step, which is different from Park's work.5e When 1a was treated under [Ru(p-cymene)Cl2]2 (5 mol%) at r.t. for 0.5 h (Scheme 5c), the product 3a and alcohol 4b were obtained in 5% and 2% yields respectively, while only <1% product 2a and trace amount of benzamide 4a were detected. We did not observe the formation of compound 4c. When benzofuran substrate 5 was employed under the standard conditions, the product 6b from inverse annulation was isolated in 20%, together with the product 6c (29%) from standard annulation, benzamide 6a (33%) and alcohol 4b (31%) (Scheme 5d). The isolation of cleavage products amide and alcohol as well as no compound 4c formed, suggest that the N–O bond cleavage occurs prior to the alkyne insertion. Therefore, pathways in Scheme 4a and c can be ruled out. Also, from DFT calculation results, pathway in Scheme 4a is less favorable (see ESI†).12d We also did the crossing experiment of 1c and 1j under the standard conditions (Scheme 5e), resulting in four annulation products. This result further indicates that the N–O bond cleavage occurs prior to the alkyne insertion. Under the standard conditions, the reaction of 4a and 4b could not deliver the annulation products (Scheme 5f), and only minor amount of product 3a was obtained when Cu(OAc)2 (2 equiv.) was added. This indicates that the formation of 2a and 3a does not follow the pathway involving 4a and 4b from the cleavage of 1a. Thus pathway in Scheme 4b can be ruled out.
 |
| | Scheme 4 Possible mechanisms from initial assumption. | |
 |
| | Scheme 5 Control experiments and deuterium-labeling experiments. | |
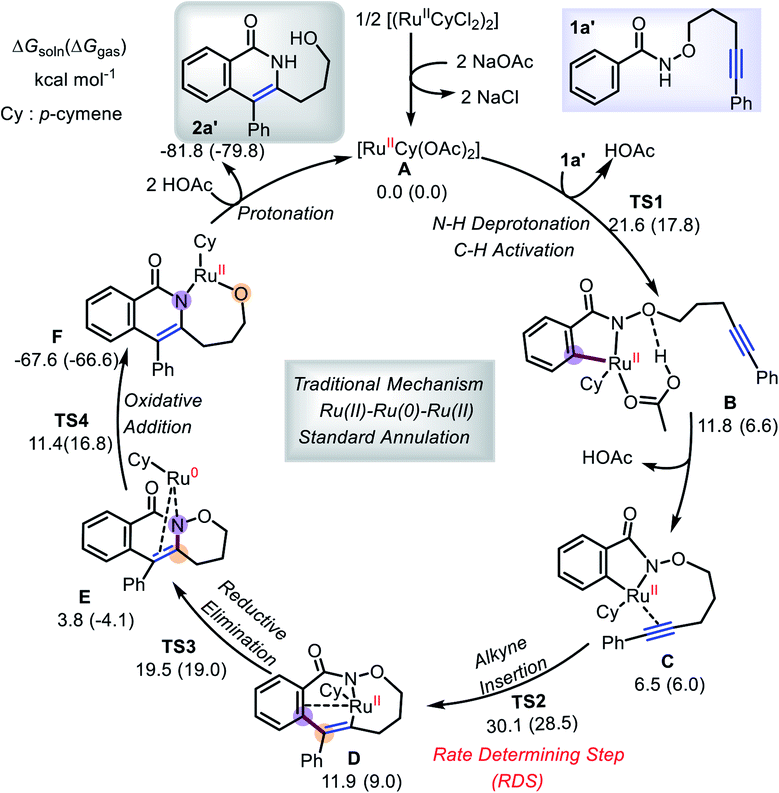
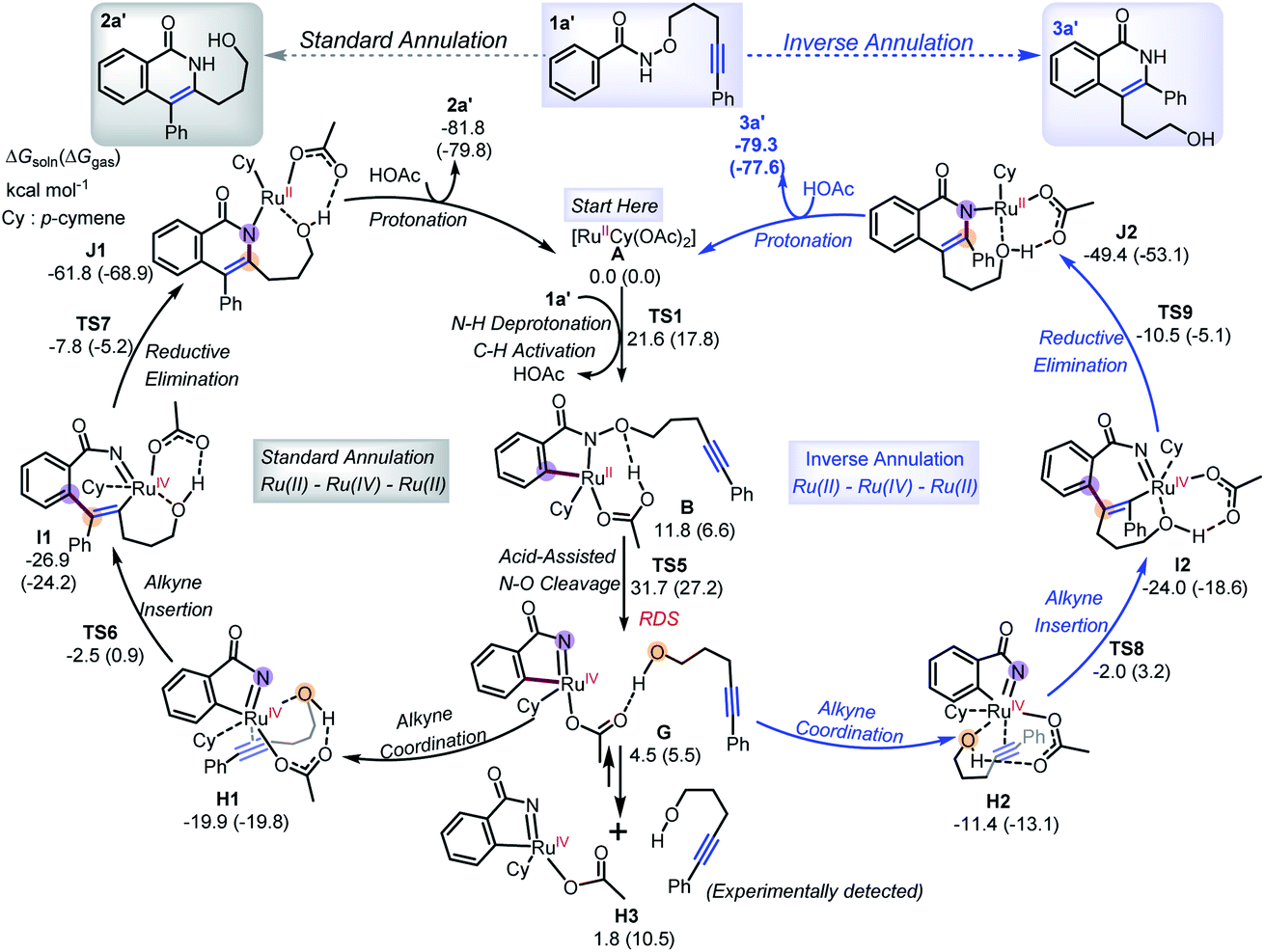
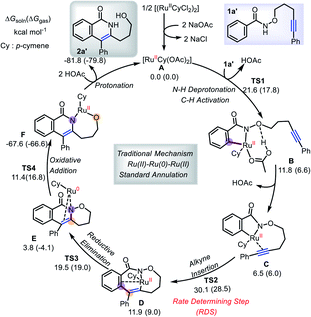
To provide more light on the formation of annulation products, DFT calculations employing B3LYP-D3/def2-TZVP method were performed with the Ru(OAc)2(p-cymene) catalyst A and the three-carbon-atom (1a′) tether substrate. We first consider the previously proposed Ru(II)–Ru(0)–Ru(II) mechanism (Scheme 6),5e,7,10 which involves an initial two-step C–H activation (TS1), followed by the rate-determining step (RDS), alkyne insertion through TS2. These steps are followed by reductive elimination (TS3) and N–O oxidative cleavage (TS4). The overall free energy barrier is 28.5–30.1 kcal mol−1, and only the product 2a′ from the standard annulation reaction can be formed.12
 |
| | Scheme 6 Traditional mechanistic cycle leading to the standard annulation product. Computed relative free energies (kcal mol−1, B3LYP-D3/def2-TZVP (SMD:methanol)). | |
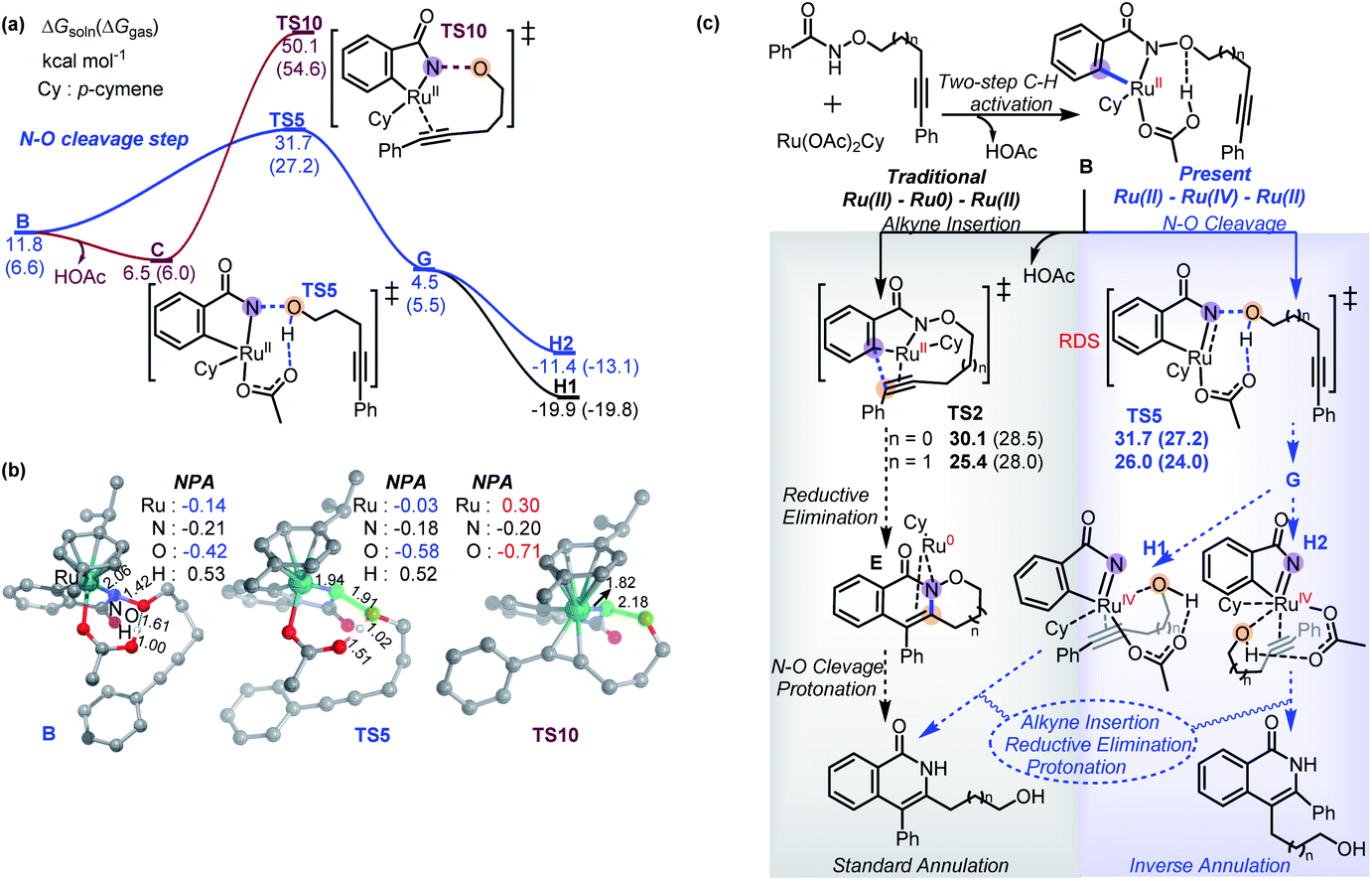
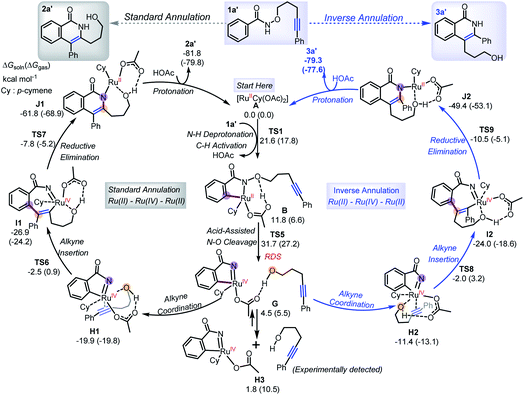
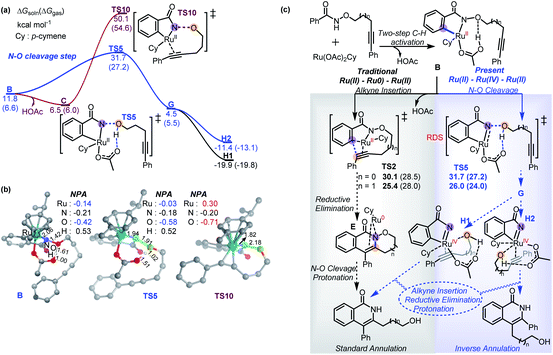
Based on the experimental observations and DFT calculations, we then propose an alternative competitive Ru(II)–Ru(IV)–Ru(II) mechanism (Scheme 7) featuring N–O cleavage preceding alkyne insertion, which can lead to formation of both the standard 2a′ and inverse 3a′ annulation products. As in the previous mechanism, the first steps are N–H deprotonation and C–H activation (TS1, 17.8–21.6 kcal mol−1) forming metallacycle B. At this point, as well as the previously suggested alkyne insertion, the system can undergo an acid-assisted N–O cleavage process (TS5, 27.2–31.7 kcal mol−1) to afford the Ru(IV)-nitrene species G (4.5–5.5 kcal mol−1), in which the forming alcohol (5-phenylpent-4-yn-1-ol) is H-bonded to the acetate ligand. Dissociation of alcohol from G is weakly endergonic by 1.8 kcal mol−1 in solution relative to A, consistent with the experimental observation of small amounts of alcohol side-product. The N–O cleavage is strongly acid-assisted: in the absence of HOAc, the barrier to this process is much higher (TS10, 50.1–54.6 kcal mol−1 in Scheme 8a), while TS2 is acid-insensitive.12a Analysis of the electronic structure of the TS indicates that this is because this step is a direct heterolytic cleavage between two atoms with small electronegativity difference (Scheme 8b). Natural population analysis (NPA) shows that the alkoxyl oxygen atom in TS10 has a large negative charge (−0.71e), which can be stabilized by proton transfer in TS5, and the resulting acetate base could delocalize the negative charge.13 These results suggest that the in situ generated acetic acid from N–H/C–H activation step could serve to facilitate the N–O bond cleavage.
 |
| | Scheme 7 New suggested mechanism. Computed relative free energies (kcal mol−1, B3LYP-D3/def2-TZVP (SMD:methanol)). | |
 |
| | Scheme 8 Role of acetic acid in the N–O cleavage. (a) Energy profiles (kcal mol−1) for the N–O cleavage step in the Ru(II)–Ru(IV)–Ru(II) pathway. (b) Structures of the N–O cleavage transition states, including NPA charges of key atoms and selected interatomic distances (in Å). (c) Schematics pathways for Ru-catalyzed annulation reaction. | |
Further coordination of the alkyne fragment in 5-phenylpent-4-yn-1-ol to the metal from intermediate G is very exothermic and can form two different Ru(IV) intermediates (Scheme 7), H1 and H2, which differ based on the orientation of the phenyl substituent of 5-phenylpent-4-yn-1-ol. They thereby lead to different annulation products, with H1 forming 2a′, while H2 yields 3a′, in each case through a sequence of steps involving alkyne insertion (TS6/TS8), reductive elimination (TS7/TS9) and protonation. The corresponding barriers are much smaller than that of the N–O cleavage process,14 and similar in the standard annulation case, consistent with the observation of both types of annulation products.
The TS of the RDS step (acid-assisted N–O cleavage process, TS5) in the present Ru(II)–Ru(IV)–Ru(II) mechanism (Scheme 7) is only slightly different in free-energy to that of the key TS (for alkyne insertion, TS2) in the traditional Ru(II)–Ru(0)–Ru(II) mechanism (Scheme 6), which makes it difficult to determine solely from computation which mechanism should be dominant, especially as the relative free energies are somewhat sensitive to the nature of the density functional used in the computations (see ESI†). In additional calculations with the four-carbon-atom tethered substrate, the two mechanisms are also found to have similar activation free energies (Scheme 8c). For both the three- (1a′) and four-carbon-atom (1a) tethered substrates, TS5 is lower (by 1.3 kcal mol−1, and 4.0 kcal mol−1, respectively) in the gas phase free energy, but relatively higher (by 1.6 kcal mol−1, and 0.6 kcal mol−1, respectively) in the solution phase free energy than TS2. In experiments, reaction with 1a′ yields more standard annulation product 2a′ (58%) than the inverse annulation product 3a′ (31%), while reaction with 1a leads to more inverse annulation product 3a (81%) and less standard annulation product 2a (10%). The calculated slightly lower solution phase free energies for TS2 than for TS5 should make the traditional mechanism, and thereby the standard annulation product, dominant in both cases. However, given the small calculated energy difference and the expected error bars (the two TSs differ significantly in electronic structure, so favorable error compensation cannot be expected), our results are consistent with the new mechanism viaTS5 playing an important role. Therefore, our DFT calculations combined with experimental observations suggest that formation of the inverse annulation products follows the Ru(II)–Ru(IV)–Ru(II) cycle, while the “standard” annulation products could be formed by either the Ru(II)–Ru(0)–Ru(II) or Ru(II)–Ru(IV)–Ru(II) mechanism, or both.
Conclusions
In summary, we have developed a ruthenium-catalyzed C–H activation/annulation process of alkyne-tethered N-alkoxybenzamides with high selectivity. This methodology is compatible with various kinds of N-alkoxybenzamides including bexarotene and probenecid derivatives, providing a wide range of products from inverse annulation. Detailed experimental investigations and DFT calculations reveal that the inverse annulation follows a Ru(II)–Ru(IV)–Ru(II) mechanism featuring N–O cleavage preceding alkyne insertion. Formation of the putative Ru-nitrene intermediate is assisted by the in situ generated acetic acid from N–H/C–H activation step. Formation of the standard annulation products might follow either the traditional Ru(II)–Ru(0)–Ru(II) pathway involving alkyne insertion prior to N–O bond cleavage, or the newly proposed Ru(II)–Ru(IV)–Ru(II) mechanism featuring N–O cleavage preceding alkyne insertion. The balance between the two mechanisms and between the inverse and standard routes will depend on the catalyst (e.g. Rh(III) vs. Ru(II)), the nature of the substrate (alkyl chain or benzylidene linker) and other factors (e.g., solvents, additives). Kinetic studies and theoretical studies suggest that C–H bond cleavage process is not involved in the rate-limiting step.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Liangliang Song appreciates the Postdoctoral Mandate (PDM) of KU Leuven. Xiaoyong Zhang appreciates the China Scholarship Council (CSC) for providing a doctoral scholarship. LVM thanks the Hercules Foundation for supporting the purchase of the diffractometer through project AKUL/09/0035. We acknowledge the FWO [Fund for Scientific Research-Flanders (Belgium)] and the Research Council of KU Leuven (BOF) for financial support. We acknowledge the support of RUDN University Program 5-100. The computational resources and services used in this work are provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation-Flanders (FWO) and the Flemish Government-department EWI.
Notes and references
- For selected reviews on catalytic C–H bond activation, see:
(a) J. He, M. Wasa, K. S. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2016, 117, 8754 CrossRef;
(b) K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302 CrossRef CAS;
(c) J. C. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS;
(d) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2018, 119, 2192 CrossRef;
(e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS;
(f) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS;
(g) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC;
(h) N. Kuhl, N. Schroeder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS.
- For reviews, see:
(a) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC;
(b) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS;
(c) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS;
(d) M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000 CrossRef;
(e) Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872 RSC;
(f) S. Prakash, R. Kuppusamy and C.-H. Cheng, ChemCatChem, 2018, 10, 683 CrossRef CAS.
- For rhodium-catalyzed annulation with external oxidants, see:
(a) S. Mochida, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2010, 39, 744 CrossRef CAS;
(b) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS;
(c) G. Song, D. Chen, C.-L. Pan, R. H. Crabtree and X. Li, J. Org. Chem., 2010, 75, 7487 CrossRef CAS;
(d) N. S. Upadhyay, V. H. Thorat, R. Sato, P. Annamalai, S.-C. Chuang and C.-H. Cheng, Green Chem., 2017, 19, 3219 RSC;
(e) L. Song, G. Tian, Y. He and E. V. Van der Eycken, Chem. Commun., 2017, 53, 12394 RSC;
(f) P. Tao and Y. Jia, Chem. Commun., 2014, 50, 7367 RSC;
(g) J. Zheng, S. B. Wang, C. Zheng and S. L. You, Angew. Chem., 2017, 129, 4611 CrossRef;
(h) J. Li, J. Liu, J. Yin, Y. Zhang, W. Han, J. Lan, D. Wu, Z. Bin and J. You, J. Org. Chem., 2019, 84, 15697 CrossRef CAS;
(i) L. Song, G. Tian, A. Blanpain, L. Van Meervelt and E. V. Van der Eycken, Adv. Synth. Catal., 2019, 361, 4442 CrossRef CAS.
- For ruthenium-catalyzed annulation with external oxidants, see:
(a) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379 CrossRef CAS;
(b) L. Ackermann, A. V. Lygin and N. Hofmann, Org. Lett., 2011, 13, 3278 CrossRef CAS;
(c) R. P. Tulichala, M. Shankar and K. K. Swamy, J. Org. Chem., 2017, 82, 5068 CrossRef CAS;
(d) H. Lin, S.-S. Li and L. Dong, Org. Biomol. Chem., 2015, 13, 11228 RSC;
(e) D. N. Garad and S. B. Mhaske, J. Org. Chem., 2019, 84, 1863 CrossRef CAS.
- For rhodium-catalyzed annulation with internal oxidants, see:
(a) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS;
(b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS;
(c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS;
(d) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592 CrossRef CAS;
(e) X. Xu, Y. Liu and C. M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS;
(f) L. Song, G. Tian and E. V. Van der Eycken, Mol. Catal., 2018, 459, 129 CrossRef CAS;
(g) X. Wu, B. Wang, Y. Zhou and H. Liu, Org. Lett., 2017, 19, 1294 CrossRef CAS;
(h) S. Lee, N. Semakul and T. Rovis, Angew. Chem., Int. Ed., 2020, 59, 4965 CrossRef CAS;
(i) M. Xu, C. Wang, W. Jiang and B. Xiao, Adv. Synth. Catal., 2020, 362, 1706 CrossRef CAS;
(j) T. J. Saiegh, H. Chédotal, C. Meyer and J. Cossy, Org. Lett., 2019, 21, 8364 CrossRef CAS;
(k) E. Tan, O. Quinonero, M. Elena de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166 CrossRef CAS;
(l) H. Gao, M. Sun, H. Zhang, M. Bian, M. Wu, G. Zhu, Z. Zhou and W. Yi, Org. Lett., 2019, 21, 5229 CrossRef CAS;
(m) T. Yamada, Y. Shibata, S. Kawauchi, S. Yoshizaki and K. Tanaka, Chem.–Eur. J., 2018, 24, 5723 CrossRef CAS;
(n) J. F. Tan, C. T. Bormann, K. Severin and N. Cramer, ACS Catal., 2020, 10, 3790 CrossRef CAS;
(o) W. J. Cui, Z. J. Wu, Q. Gu and S. L. You, J. Am. Chem. Soc., 2020, 142, 7379 CrossRef CAS.
- For ruthenium-catalyzed annulation with internal oxidants, see:
(a) B. Li, H. Feng, S. Xu and B. Wang, Chem.–Eur. J., 2011, 17, 12573 CrossRef CAS;
(b) L. Ackermann and S. Fenner, Org. Lett., 2011, 13, 6548 CrossRef CAS;
(c) H. Huang, S. Nakanowatari and L. Ackermann, Org. Lett., 2017, 19, 4620 CrossRef CAS;
(d) X. Wu, B. Wang, S. Zhou, Y. Zhou and H. Liu, ACS Catal., 2017, 7, 2494 CrossRef CAS;
(e) F. Yang and L. Ackermann, J. Org. Chem., 2014, 79, 12070 CrossRef CAS;
(f) E. Tan, A. I. Konovalov, G. A. Fernández, R. Dorel and A. M. Echavarren, Org. Lett., 2017, 19, 5561 CrossRef CAS.
-
(a) S. Y. Hong, J. Jeong and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 2408 CrossRef CAS;
(b) L. Song, X. Zhang, G. Tian, K. Robeyns, L. Van Meervelt, J. N. Harvey and E. V. Van der Eycken, Mol. Catal., 2019, 463, 30 CrossRef CAS;
(c) Y. Fukui, P. Liu, Q. Liu, Z.-T. He, N.-Y. Wu, P. Tian and G.-Q. Lin, J. Am. Chem. Soc., 2014, 136, 15607 CrossRef CAS;
(d) L. Song, G. Tian, J. Van der Eycken and E. V. Van der Eycken, Beilstein J. Org. Chem., 2019, 15, 571 CrossRef CAS;
(e) J.-P. Krieger, D. Lesuisse, G. Ricci, M.-A. Perrin, C. Meyer and J. Cossy, Org. Lett., 2017, 19, 2706 CrossRef CAS;
(f) K. K. Gollapelli, S. Kallepu, N. Govindappa, J. B. Nanubolu and R. Chegondi, Chem. Sci., 2016, 7, 4748 RSC;
(g) M. Shankar, K. Ghosh, K. Mukherjee, R. K. Rit and A. K. Sahoo, Org. Lett., 2018, 20, 5144 CrossRef CAS.
-
(a) J.-Q. Wu, S.-S. Zhang, H. Gao, Z. Qi, C.-J. Zhou, W.-W. Ji, Y. Liu, Y. Chen, Q. Li, X. Li and H. Wang, J. Am. Chem. Soc., 2017, 139, 3537 CrossRef CAS;
(b) X. Wang, T. Gensch, A. Lerchen, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 6506 CrossRef CAS;
(c) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS;
(d) Y. F. Liang, L. Yang, T. Rogge and L. Ackermann, Chem.–Eur. J., 2018, 24, 16548 CrossRef CAS;
(e) M. Simonetti, D. M. Cannas, X. Just-Baringo, I. J. Vitorica-Yrezabal and I. Larrosa, Nat. Chem., 2018, 10, 724 CrossRef CAS.
-
(a) C. Q. Wang, Y. Zhang and C. Feng, Angew. Chem., Int. Ed., 2017, 56, 14918 CrossRef CAS;
(b) Z. Zhou, G. Liu, Y. Shen and X. Lu, Org. Chem. Front., 2014, 1, 1161 RSC.
-
(a) S. Vasquez-Cespedes, X. Wang and F. Glorius, ACS Catal., 2018, 8, 242 CrossRef CAS;
(b) Z. Wang, P. Xie and Y. Xia, Chin. Chem. Lett., 2018, 29, 47 CrossRef CAS;
(c) G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS;
(d) L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. Xia, J. Org. Chem., 2012, 77, 3017 CrossRef CAS;
(e) B. Ling, Y. Liu, Y.-Y. Jiang, P. Liu and S. Bi, Organometallics, 2019, 38, 1877 CrossRef CAS;
(f) Y.-F. Yang, K. N. Houk and Y.-D. Wu, J. Am. Chem. Soc., 2016, 138, 6861 CrossRef CAS.
-
(a) S. Huh, S. Y. Hong and S. Chang, Org. Lett., 2019, 21, 2808 CrossRef CAS;
(b) E. Azek, M. Khalifa, J. Bartholoméüs, M. Ernzerhof and H. Lebel, Chem. Sci., 2019, 10, 718 RSC.
-
(a) In the traditional mechanism, the acetic acid has a negligible
effect on the barrier height of the rate determining alkyne insertion step, since TS2 in the presence of acid (Scheme S2 in ESI†) is calculated to be 30.7 kcal mol−1, comparable to the one (30.1 kcal mol−1) in the absence of acid;
(b) The pathway with alkyne insertion into the Ru–N bond prior to C–C reductive elimination is less favorable with a barrier of 35.4 kcal mol−1;
(c) After alkyne insertion into the Ru–C bond, the pathway with N–O oxidative cleavage prior to reductive elimination involves TS13 and TS14 (>23.2 kcal mol−1), higher in energy than TS3 (19.5 kcal mol−1) and TS4;
(d) Alkyne insertion following the intramolecular annulation to inverse annulation product is calculated to involve a transition state more than 10.0 kcal mol−1 (Scheme S13†) higher than that to standard annulation product. Thus, the formation of inverse annulation product cannot be explained by the tradition mechanism. More details can be found in ESI.†.
- The effect of methanol was explicitly considered in the N–O cleavage step. In absence of acetic acid, the methanol could also transfer its hydrogen to neutralize alkoxyl oxygen atom of the substrate, and result into a negative methoxyl group. However, the calculated barrier is much higher (>46.9 kcal mol−1) than that of the acetic acid assisted mechanism (Scheme S9†), which could be explained by the stronger acid of acetic acid to promote proton transfer, and the more stabilization of negative charge in the carboxylate group.
- In the new suggested mechanism, once N–O cleavage completes, the barriers of alkyne insertion and reductive elimination in the absence of the acetic acid are at least 6.0 kcal mol−1 higher in energy (ESI, Scheme S11†), than that of the route (H2 to J2 in Scheme 6) in the presence of the acetic acid.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1959807. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04434b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Xiao
Tang
c,
Luc
Van Meervelt
d,
Johan
Van der Eycken
e,
Jeremy N.
Harvey
b,
Xiao
Tang
c,
Luc
Van Meervelt
d,
Johan
Van der Eycken
e,
Jeremy N.
Harvey