
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible addition of ethylene to a pincer-based boryl-iridium unit with the formation of a bridging ethylidene†
Yihan
Cao‡
,
Wei-Chun
Shih‡
,
Nattamai
Bhuvanesh
and
Oleg V.
Ozerov
 *
*
Department of Chemistry, Texas A&M University, 3255 TAMU, College Station, Texas 77842, USA. E-mail: ozerov@chem.tamu.edu
First published on 5th October 2020
Abstract
This report examines reactions of a series of Ir complexes supported by the diarylboryl/bis(phosphine) PBP pincer ligand with ethylene: (PBP)IrH4 (1), (PBP)IrH2(CO) (2), and (PBP)Ir(CO)2 (3). The outcomes of these reactions differ from those typical for Ir complexes supported by other pincer ligands and do not give rise to simple ethylene adducts or products of insertion of Ir into the C–H bond of ethylene. Instead, the elements of ethylene are incorporated into the molecules to result in B–C bonds. In the case of 2 and 3, ethylene addition results in the formation of B/Ir bridging ethylidene complexes 5 and 6. For 6, the addition of ethylene (and the analogous addition of 1-hexene) is shown to be partially reversible. Addition of ethylene to 2 and 3 is remarkable because they are saturated at Ir and yet the net outcome is such that ethylene binds without replacing any ligands already present. A mechanistic inquiry suggests that dissociation of CO from 3 or 6 is necessary in order for the addition or loss of ethylene to proceed.
Introduction
The nature of the elementary reactions of binding ethylene and other alkenes to transition metal centers is of importance to many common and impactful catalytic processes that utilize alkenes as feedstocks.1–5 The initial interaction between a suitably unsaturated transition metal complex and an alkene (Scheme 1) typically results in a π-complex (A),6 which may then undergo C–H oxidative addition to yield a vinyl hydride isomer (B),7,8 or isomerization via an initial insertion product C to a metal alkylidene complex (D),9–11 or even a hydrido/alkylidyne isomer (E).12,13 The latter two isomerizations typically require the presence of hydride ligands in the metal fragment and proceed by a combination of an insertion of the olefin into M–H and the subsequent single or double α-H elimination. Moreover, since isomerization of a free hydrocarbon olefin into a free alkylidene is unfavourable by >70 kcal mol−1,14 only the metal centers with an enormous preference for binding an alkylidene vs. olefin are thermodynamically capable of it.9–11 This thermodynamic capacity is more or less restricted to highly electron-rich early-to mid-periodic table metals in low oxidation states and with propensity to form multiple metal–ligand bonds.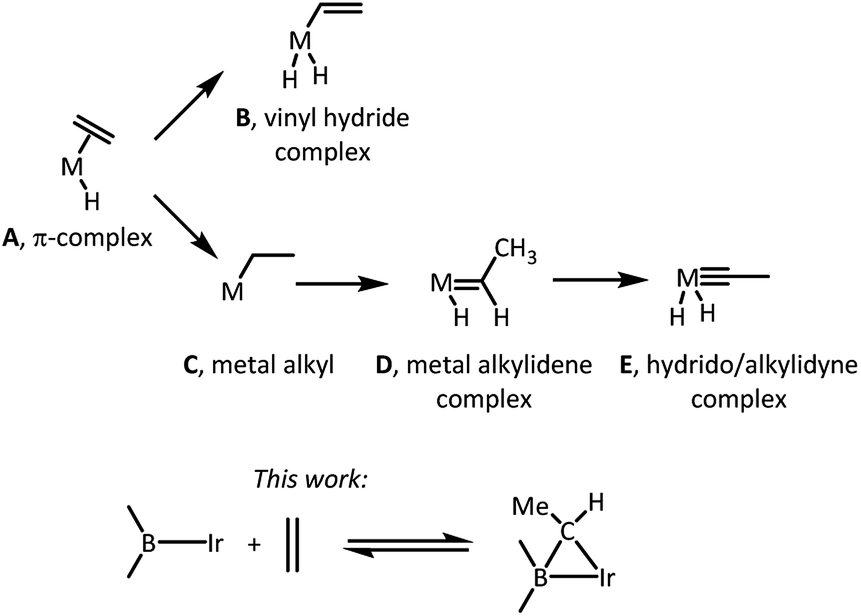 | ||
| Scheme 1 (Top) typical outcomes of a reaction between an olefin (ethylene for simplicity) and a transition metal complex. (Bottom) the new reactivity reported in this work. | ||
In the present work, we report unexpected findings that emerged in the course of our exploration of the reactivity of Ir complexes of a diarylboryl-containing PBP pincer ligand.15–18 In particular, we discovered that ethylene can reversibly add to the boryl-iridium unit as a bridging ethylidene. Complexes of monodentate boryl ligands are well established and important intermediates in such organometallic catalytic processes as hydroboration19 and C–H borylation.20 Some variants of aromatic C–H borylation rely on the presence of sacrificial olefin reagents.20,21 To the best of our knowledge, formation of alkylidenes bridging a boron and a transition metal in C–H borylation of arenes or olefins,20 or in olefin hydroboration,19 has not been documented or considered.
Results and discussion
Reaction of ethylene with (PBP)Ir complexes
For the reactions with ethylene, we chose the recently disclosed complexes 1, 2, and 3.17b Solutions of 1, 2, and 3 in toluene or benzene were blanketed with an atmosphere of ethylene and allowed to react overnight (Scheme 2). Reactions with 1 and 2 proceeded at ambient temperature, while reaction with 3 required thermolysis at 50 °C for 36 h. Upon removal of the volatiles and recrystallization, the new complexes 4, 5, and 6 were obtained in pure form. NMR spectroscopic analysis of 4 indicated that it possesses an intact PBP ligand with apparent Cs symmetry, an sp3-hybridized boron center (11B NMR: δ 20.3 ppm), a single hydride, an Ir-bound ethylene, and a vinyl group. These data point to the structure depicted in Scheme 2, which was confirmed by an X-ray diffraction study (vide infra). 4 is the product of addition of two equivalents of ethylene to the (PBP)Ir fragment. This is atypical for reactions of (pincer)IrH2 or (pincer)IrH4 with an olefin, which normally yield either a (pincer)Ir(η2-olefin) complex or a (pincer)Ir(H)(alkenyl) isomer in cases of high steric congestion,7 both being products of the reaction of the pincer complex with only one molecule of alkene.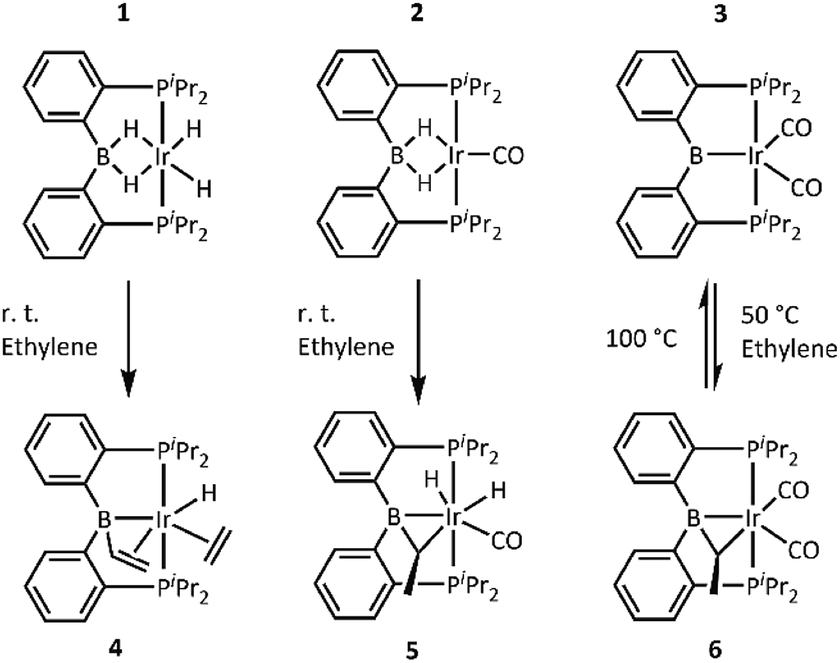 | ||
| Scheme 2 Reactions of 1, 2, and 3 with ethylene. | ||
Complexes 5 and 6 both contained all the expected 1H NMR resonances for the PBP ligand and their 11B NMR chemical shifts (17.8 and 20.4 ppm, respectively) also indicated sp3 hybridization at boron. However, there were no resonances in either 5 or 6 that could be ascribed to a π-bound ethylene or a vinyl group. Instead, 5 and 6 each possessed a pair of 1H NMR resonances in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 integral ratio consistent with a CHCH3 fragment (5: δ 3.37 (m, 1H) and 1.75 (d, JH–H = 7.0 Hz, 3H) ppm; 6: δ 2.72 (m, 1H) and 1.39 (dd, JH–H = 7.0, JP–H = 0.7 Hz, 3H) ppm). 5 displayed resonances for two inequivalent hydrides (−11.10 and −12.88 ppm), whereas 6 displayed none. These data were consistent with the presence of an ethylidene (CHCH3) unit bridging B and Ir. Furthermore, the observed C1 symmetry in the NMR spectra of 5 and 6 was consistent with the presence of a carbon center with four different substituents (the methine carbon of the bridging alkylidene). IR spectroscopic observations suggested the presence of a single CO ligand in 5 (νCO = 1977 cm−1) and two CO ligands in 6 (νCO = 1987 and 1942 cm−1).
:3 integral ratio consistent with a CHCH3 fragment (5: δ 3.37 (m, 1H) and 1.75 (d, JH–H = 7.0 Hz, 3H) ppm; 6: δ 2.72 (m, 1H) and 1.39 (dd, JH–H = 7.0, JP–H = 0.7 Hz, 3H) ppm). 5 displayed resonances for two inequivalent hydrides (−11.10 and −12.88 ppm), whereas 6 displayed none. These data were consistent with the presence of an ethylidene (CHCH3) unit bridging B and Ir. Furthermore, the observed C1 symmetry in the NMR spectra of 5 and 6 was consistent with the presence of a carbon center with four different substituents (the methine carbon of the bridging alkylidene). IR spectroscopic observations suggested the presence of a single CO ligand in 5 (νCO = 1977 cm−1) and two CO ligands in 6 (νCO = 1987 and 1942 cm−1).
Interestingly, addition of ethylene to 3 is partially reversible. Thermolysis of a solution of 6 at 90 °C led to the appearance of signals for 3 and free ethylene. Traces of free ethylene were also observed upon thermolysis of 5 at 80 °C for 10 min.
Structural characterization
Solid-state structures of 4, 5, and 6 were determined by single-crystal X-ray diffractometry (Fig. 1). The structure of 5 contained two independent molecules in the asymmetric unit. Both molecules displayed disorder, which was successfully modelled through varying the positions of the atoms in the Ir–CHMe unit, as well as CO for one of the molecules. The disorder likely involves other atoms in the molecules, but it was not possible to improve the model via consideration of the other atom positions. Because of this, while the connectivity of the non-hydrogen atoms was unambiguously established, the metrics associated with the immediate coordination sphere of Ir could not be reliably interpreted.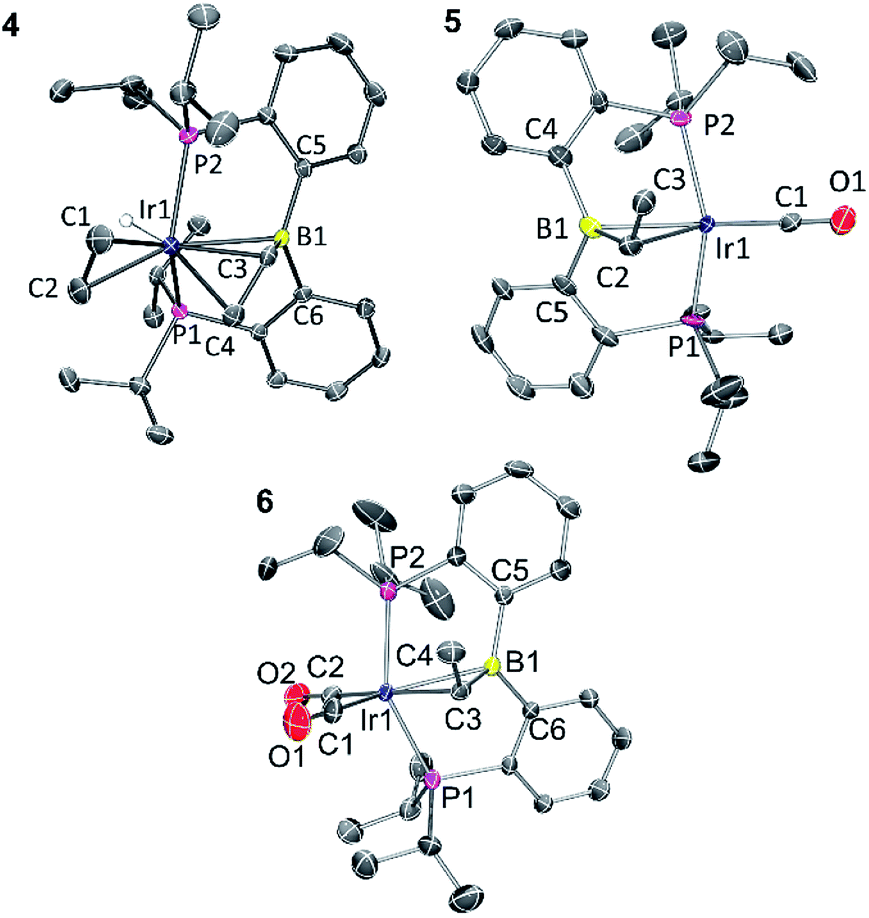 | ||
| Fig. 1 ORTEP drawings showing selected atom labeling of 4, 5 and 6. Hydrogen atoms (except Ir–H) are omitted for clarity. 4: Ir1–B1, 2.396(3) Å; Ir1–C1, 2.200(3) Å; Ir1–C2, 2.223(3) Å; Ir1–C3, 2.213(3) Å; Ir1–C4, 2.248(3) Å; C1–C2, 1.402(4) Å; C3–C4, 1.405(4) Å; C3–B1, 1.556(4) Å; C5–B1–C6, 120.4(2)°; C6–B1–C3, 117.5(2)°; C3–B1–C5, 115.6(2)°. 5: the structure contains two independent molecules and each is disordered, including the Ir position. One of the independent molecules is drawn. See ESI† for additional information. 6: Ir1–B1, 2.475(4) Å; Ir1–C3, 2.262(4) Å; C3–C4, 1.523(6) Å; C3–B1, 1.530(6) Å; C1–O1, 1.160(5) Å; C2–O2, 1.136(6) Å; C1–Ir1–C3, 175.7(2)°; P1–Ir1–P2 140.36(4)°; C5–B1–C6, 122.0(3)°; C6–B1–C3, 118.2(3)°; C3–B1–C5, 114.6(3)°. | ||
In the structure of 4, the coordination sphere of Ir contains a hydride, two phosphines, two olefin donors, and a relatively distant interaction with a boron center (2.396(3) Å). If the latter were ignored, the molecule could be viewed as a monovalent, five-coordinate IrXL4 center with a geometry intermediate between square pyramidal and trigonal planar (τ = 0.43).22 This geometry probably results from a combination of innate electronic preferences and the constraint imposed by the chelating ligand. The Ir–B distance in 4 is ca. 0.1 Å longer than those recorded for Ir and Rh complexes of BP3 and PB(Ph)P where the central borane site functioned as a Z-type ligand; 23,24 and it is ca. 0.25 Å longer than the Ir–B(boryl) bond distance in 3.17b The sum of C–B–C angles about boron (ca. 354°) indicates only modest pyramidalization. The proximity of B to Ir is also dictated by the vinyl–Ir interaction. Furthermore, the BCHCH2 unit could alternatively be viewed as a η3-borataallyl25,26 fragment bound to Ir (Fig. 2). The related η3-binding of a B–Ph group in boranes coordinating to transition metals has also been reported.27–31 The B1–C3 distance of 1.556(4) Å is shorter than the B–Caryl distances in 4 or 6 (ca. 1.59–1.61 Å).
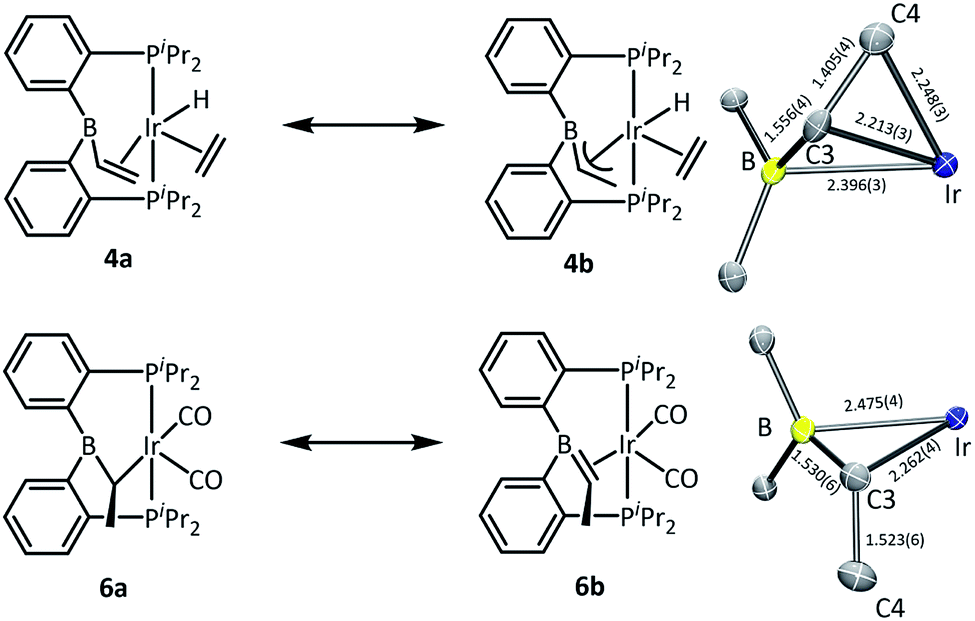 | ||
| Fig. 2 ChemDraw interpretations and POV-Ray rendition of the ORTEP drawing (50% thermal ellipsoids, truncated molecules with boron center and atoms around boron) of 4 and 6. | ||
The structure of 6 contains an even more distant interaction between Ir and B (2.475(4) Å) and the boron center is also only slightly pyramidalized (sum of C–B–C angles ca. 355°). If the Ir–B interaction were discounted, the molecule could be viewed as an IrXL4 five-coordinate (τ = 0.60),22 where X is the boryl-substituted alkyl ligand connected to Ir via C3. However, the Ir–C3 distance of 2.261(4) Å is considerably longer than the sum of Ir and C covalent radii (2.17 Å).32 In fact, it is even slightly longer than the Ir–C distances (2.20–2.25 Å) to the π-bound olefins in 4. In addition, the B1–C3 distance (1.530(6) Å) is shorter than is expected for a single B–C bond (cf. the B–Caryl distances in 4 and 6). These metrics point to an alternative view of this structure as an η2-borataalkene33 complex of monovalent Ir (Fig. 2). It is important to emphasize that the alternative descriptions of the observed structures for 4 (vinylborane vs. borataallyl complex) and 6 (boryl-substituted alkyl or borataalkene complex) are not possible isomers but rather idealized or extreme descriptions of the same molecule.
Mechanistic analysis
We recently reported that 3 can selectively activate the ortho-C–H bonds in pyridine derivatives. 3 is an 18-electron complex at Ir and thus cannot undergo direct oxidative addition. Thus, the mechanism of C–H activation of pyridines was proposed whereby one of the CO ligands in 3 has to dissociate to allow for the C–H oxidative addition to take place at the monocarbonyl intermediate. It was observed that the C–H activation of pyridines was retarded by the presence of free CO.We surmised that the activation of ethylene by 3 may proceed by a related mechanism34 (Scheme 3). We propose that dissociation of CO creates unsaturation and permits coordination of the olefin to give intermediate 8 and then oxidative addition of the vinylic C–H bond to give 9. The resultant vinyl group may then migrate from Ir to B to give 10, and then insertion into the Ir–H produces 11 and after recapturing CO, 6, with the observed bridging ethylidene structure. We previously observed facile migration of phenyl between B and Ir (or Rh) in complexes of this PBP ligand.17a The proposed migration of a vinyl is reasonable by analogy. Alternatively, one could envision that 8 is converted to 10via a 2,1-olefin insertion into the Ir–B bond and the subsequent β-hydrogen elimination from the resultant boroalkyl 12. Although insertions of olefins into M–B bonds are well precedented,35–39 it is not clear that the chelate constraint here would enable 1,2-insertion or that it should proceed with the regioselectivity needed for the eventual production of 6′.
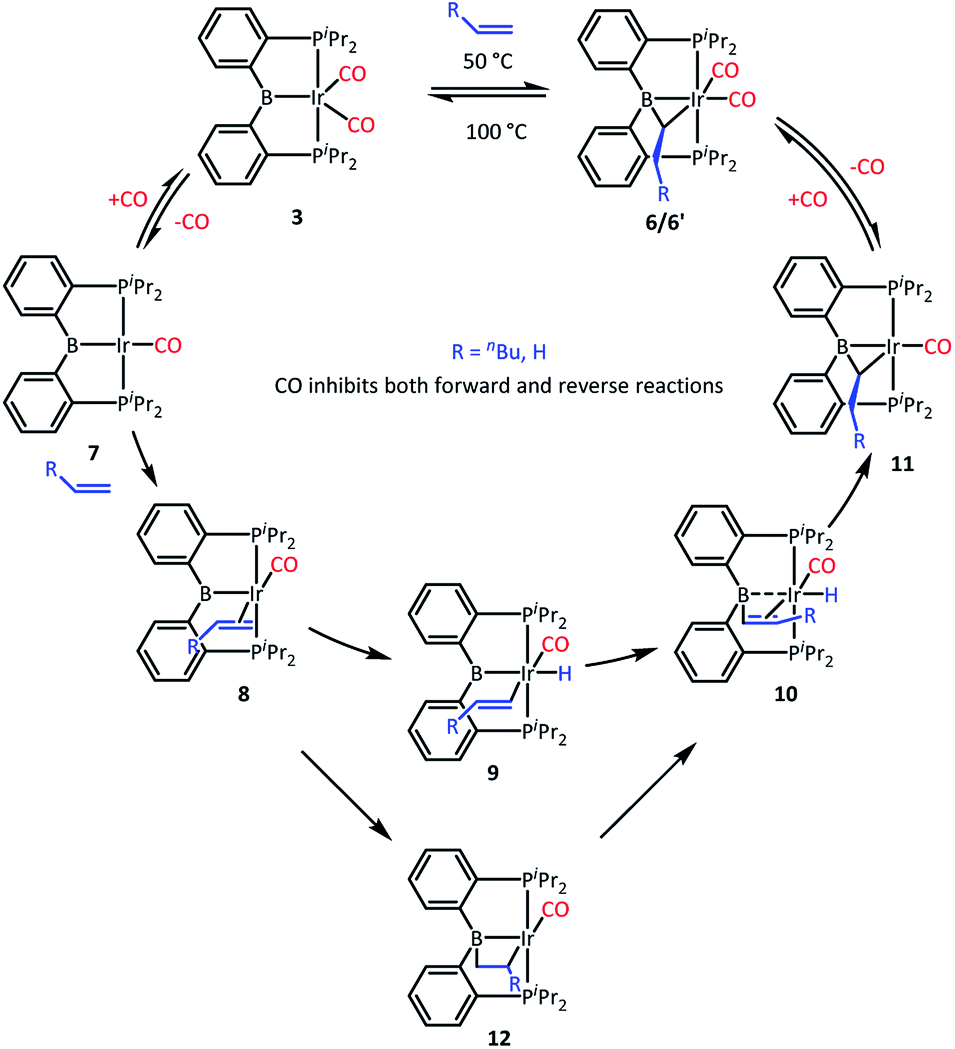 | ||
| Scheme 3 Proposed mechanism. | ||
We tested our mechanistic proposal by examining whether the rate of ethylene addition to 3 was affected by the presence of free CO. Owing to the practical challenges in varying the pressures or concentrations of two gaseous reagents (C2H4 and CO) in NMR tube experiments, we elected to carry out test reactions with 1-hexene instead. First, we established that 1-hexene indeed formed the analogous product (Scheme 4). Treatment 3 with 80 equivalents of 1-hexene, after thermolysis at 50 °C for 16 h, resulted in the formation of a product 6′, whose NMR spectroscopic features closely matched those of 6, except for the presence of a pentyl group in place of a methyl in the bridging alkylidene (See Table S1†). Interestingly, the reaction of 3 with trans-2-hexene also gave 6c as the major product after 5 d at 100 °C (see ESI†). We then examined the progress of reactions of 3 with 1-hexene under the atmosphere of Ar vs. CO but otherwise identical conditions (C6D6 solution 50 °C, 90 h) and concentrations. NMR analysis revealed 73% consumption of 3 (all converted to 6′) under Ar. Under CO, only a trace of 3 was consumed to form a water adduct we previously described,34 with no evidence for the formation of 6′. Higher concentrations of 1-hexene correlated with faster conversion of 3 to 6′. Thus, the reaction displays positive dependence on [1-hexene] and apparent inverse dependence on [CO], indicating that reversible dissociative displacement of CO with 1-hexene constitutes the rate-determining sequence.
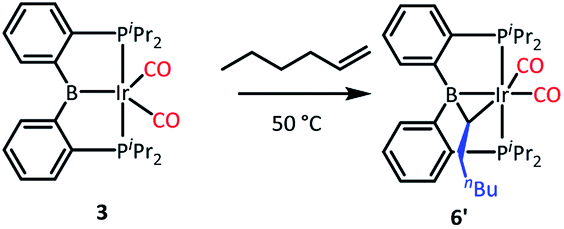 | ||
| Scheme 4 Reaction of 3 with 1-hexene. | ||
The mechanism proposed in Scheme 3 suggested, by the principle of microscopic reversibility, that loss of ethylene from 6 should also be retarded by the presence of free CO. Indeed, thermolysis (100 °C, 2 h) of two identically constituted C6D6 solutions of 6 resulted in diminished conversion to 3 (20% vs. 40% after 2 h, see details in the ESI†) in the reaction carried out under 1 atm of CO as opposed to 1 atm of Ar. This difference is not dramatic, and may indicate that rate of the back-reaction of 11 with CO is competitive with the loss of ethylene from 11, or even that multiple mechanisms for ethylene loss may be operative. With that qualifier, the CO inhibition experiments are consistent with the proposed mechanism (Scheme 3) for the reactions of 3. It is also reasonable to think that the reaction of 2 with ethylene proceeds by a similar mechanism, initiated by the dissociation of CO or H2 from 2. The product of the reaction of 3 with ethylene can be viewed as analogous to the intermediate 10 in Scheme 3 (with η2-ethylene in place of CO).
Conclusion
In summary, we have described a series of unusual outcomes in the reactions of ethylene with simple pincer complexes of Ir. These reactions demonstrate that the boryl of the PBP ligand is not merely an electronically special ligand or even a potential Lewis acid to Lewis basic sites. The presence of the boryl donor in the pincer can also significantly alter the preferred reactions products when contrasted with pincer complexes with different central heteroatoms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the US National Science Foundation (grant CHE-1565923 to O. V. O.) for support of this research.Notes and references
- J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS.
- (a) C. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS; (b) J. F. Hartwig, Organotransition Metal Chemistry From Bonding to Catalysis, University Science Books, Sausalito, CA, 2009, pp. 1047–1100 Search PubMed.
- (a) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS; (b) O. M. Ogba, N. C. Warner, D. J. O. Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS.
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry From Bonding to Catalysis, University Science Books, Sausalito, CA, 2009, pp. 47–51 Search PubMed.
- K. B. Renkema, Y. V. Kissin and A. S. Goldman, J. Am. Chem. Soc., 2003, 125, 7770–7771 CrossRef CAS.
- Formation of an olefin π-complex as an intermediate prior to the insertion into the vinylic C–H bond is not always necessary: P. O. Stoutland and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 4581–4582 CrossRef CAS.
- R. R. Schrock, K.-Y. Shih, D. A. Dobbs and W. M. Davis, J. Am. Chem. Soc., 1995, 117, 6609–6610 CrossRef CAS.
- K. F. Hirsekorn, A. S. Veige, M. P. Marshak, Y. Koldobskaya, P. T. Wolczanski, T. R. Cundari and E. B. Lobkovsky, J. Am. Chem. Soc., 2005, 127, 4809–4830 CrossRef CAS.
- O. V. Ozerov, L. A. Watson, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2003, 125, 9604–9605 CrossRef CAS.
- O. V. Ozerov, J. C. Huffman, L. A. Watson and K. G. Caulton, Organometallics, 2003, 22, 2539–2541 CrossRef CAS.
- O. V. Ozerov, L. A. Watson, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2004, 126, 6363–6378 CrossRef CAS.
- J. N. Coalter, G. J. Spivak, H. Gérard, E. Clot, E. R. Davidson, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1998, 120, 9388–9389 CrossRef CAS.
- For examples of the chemistry of diaminoboryl centered PBP ligands originally developed by Yamashita and Nozaki, see: (a) Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201–9203 CrossRef CAS; (b) E. H. Kwan, Y. J. Kawai, S. Kamakura and M. Yamashita, Dalton Trans., 2016, 45, 15931–15941 RSC; (c) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS; (d) A. F. Hill and C. M. A. McQueen, Organometallics, 2014, 33, 1977–1985 CrossRef CAS.
- For the chemistry of boryl pincer complexes containing the meta-carborane core, see: (a) A. M. Spokoyny, M. G. Reuter, C. L. Stern, M. A. Ratner, T. Seideman and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 9482–9483 CrossRef CAS; (b) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS; (c) B. J. Eleazer, M. D. Smith, A. A. Popov and D. V. Peryshkov, J. Am. Chem. Soc., 2016, 138, 10531–10538 CrossRef CAS.
- For the chemistry of the diarylboryl PBP complexes of Ir, see: (a) W.-C. Shih, W. Gu, M. C. MacInnis, S. D. Timpa, N. Bhuvanesh, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 2086–2089 CrossRef CAS; (b) W.-C. Shih and O. V. Ozerov, Organometallics, 2017, 36, 228–233 CrossRef CAS; (c) W.-C. Shih and O. V. Ozerov, J. Am. Chem. Soc., 2017, 139, 17297–17300 CrossRef CAS.
- For the diarylboryl PBP complexes of Pd, see: D. Schuhknecht, F. Ritter and M. E. Tauchert, Chem. Commun., 2016, 52, 11823–11826 RSC.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef CAS.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS.
- L. P. Press, A. J. Kosanovich, B. J. McCulloch and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 9487–9497 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) S. Bontemps, H. Gornitzka, G. Bouhadir, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2006, 45, 1611–1614 CrossRef CAS; (b) C. M. Conifer, D. J. Law, G. J. Sunley, A. J. P. White and G. J. P. Britovsek, Organometallics, 2011, 30, 4060–4066 CrossRef CAS; (c) W.-C. Shih, W. Gu, M. C. MacInnis, D. E. Herbert and O. V. Ozerov, Organometallics, 2017, 36, 1718–1726 CrossRef CAS.
- (a) H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 4251–4258 CrossRef CAS; (b) H. Kameo and H. Nakazawa, Organometallics, 2012, 31, 7476–7484 CrossRef CAS.
- Z. Hui, T. Watanabe and H. Tobita, Organometallics, 2017, 36, 4816–4824 CrossRef CAS.
- K. B. Kolpin and D. J. H. Emslie, Angew. Chem., Int. Ed., 2010, 49, 2716–2719 CrossRef CAS.
- M. Sircoglou, S. Bontemps, M. Mercy, K. Miqueu, S. Ladeira, N. Saffon, L. Maron, G. Bouhadir and D. Bourissou, Inorg. Chem., 2010, 49, 3983–3990 CrossRef CAS.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS.
- D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938–4941 CrossRef CAS.
- M. A. Nesbit, D. L. M. Suess and J. C. Peters, Organometallics, 2015, 34, 4741–4752 CrossRef CAS.
- D. J. H. Emslie, B. E. Cowie and K. B. Kolpin, Dalton Trans., 2012, 41, 1101–1117 RSC.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 21, 2832 RSC.
- K. S. Cook, W. E. Piers, P. G. Hayes and M. Parvez, Organometallics, 2002, 21, 2422–2425 CrossRef CAS.
- We also recently described addition N–H, O–H, and F–H bonds across the B–Ir moiety in 3. These reactions appear to proceed via protonation of the Ir center either directly by the sufficiently acidic reagent, or after binding of the heteroatom to the Lewis acidic boron site. This mechanism does not appear to be applicable to a substrate such as ethylene, possessing neither the necessary Brønsted acidity in the free form nor the affinity for binding to a σ-Lewis acid. Y. Cao, W.-C. Shih and O. V. Ozerov, Organometallics, 2019, 38, 4076–4081 CrossRef CAS.
- R. T. Baker, J. C. Calabrese, S. A. Westcott, P. Nguyen and T. B. Marder, J. Am. Chem. Soc., 1993, 115, 4367–4368 CrossRef CAS.
- L. Dang, H. Zhao, Z. Lin and T. B. Marder, Organometallics, 2007, 26, 2824–2832 CrossRef CAS.
- D. S. Laitar, E. Y. Tsui and J. P. Sadighi, Organometallics, 2006, 25, 2405–2408 CrossRef CAS.
- G. R. Clark, G. J. Irvine, W. R. Roper and L. J. Wright, Organometallics, 1997, 16, 5499–5505 CrossRef CAS.
- C. N. Iverson and M. R. Smith III, Organometallics, 1996, 15, 5155–5165 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and pictorial NMR spectra, details of the computational studies and the coordinate files. CCDC 1858843–1858845. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04748a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |