
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis and immunological evaluation of entirely carbohydrate conjugate Globo H-PS A1†
Samir
Ghosh‡
,
Kevin R.
Trabbic‡
,
Mengchao
Shi
,
Sharmeen
Nishat
,
Pradheep
Eradi
,
Kristopher A.
Kleski
and
Peter R.
Andreana
 *
*
The University of Toledo, Department of Chemistry and Biochemistry, 2801 West Bancroft Street, Toledo, Ohio, USA 43606. E-mail: peter.andreana@utoledo.edu
First published on 19th October 2020
Abstract
An anticancer, entirely carbohydrate conjugate, Globo H-polysaccharide A1 (Globo H-PS A1), was chemically prepared and immunologically evaluated in C57BL/6 mice. Tumor associated carbohydrate antigen Globo H hexasaccharide was synthesized in an overall 7.8% yield employing a convergent [3 + 3] strategy that revealed an anomeric aminooxy group used for conjugation to oxidized PS A1 via an oxime linkage. Globo H-PS A1, formulated with adjuvants monophosphoryl lipid A and TiterMax® Gold. After immunization an antigen specific immune response was observed in ELISA with anti-Globo H IgG/IgM antibodies. Specificity of the corresponding antibodies was determined by FACS showing cell surface binding to Globo H-positive cancer cell lines MCF-7 and OVCAR-5. The anti-Globo H antibodies also exhibited complement-dependent cellular cytotoxicity against MCF-7 and OVCAR-5 cells.
Introduction
Aberrant glycosylation is one of the characteristic events observed during carcinogenesis.1–3 It is the result of drastic alterations of glycotransferases and glycosidases eventually leading to the overexpression of antigenic oligomers on tumor cell membranes that are termed tumor associated carbohydrate antigens (TACAs). TACAs have been found on many different types of cancer cells and malignant tissues, however, negligible amounts are detected on healthy tissue. This phenotypical difference opens a therapeutic window for the development of anticancer vaccines through harnessing the power of human immunity to induce TACA-specific immune responses and hence generate antibodies and effector cells to potentially eradicate cancer cells and/or halt tumor progression.4 Many studies have shown that properly presented TACAs in an immunogenic construct can induce specific T cell dependent immune responses and give rise to high affinity anti-TACA antibodies.5–10 Generally, TACAs are incapable of triggering adaptive immunity alone due to their weak immunogenic nature. In order to circumvent this obstacle, TACAs have been covalently conjugated to immunogenic carriers, thus, the conjugates as a whole, can be processed by antigen presenting cells (APCs) and induce the desired immune response.11,12 To date, there are several types of carriers that have been applied toward developing cancer vaccines;13,14 the most prevalent carriers have been proteinaceous. For example, KLH,15–17 CRM197 (ref. 18, 19) and TT20 TACA-conjugates have been extensively studied and proven capable of TACA-specific antibody (Ab) production and tumor cell cytotoxicity.Several anti-TACA monoclonal antibodies (mAbs) were developed and applied for clinical practice. The anti-GD2 Ab, unituxin, was granted FDA approval in 2015 for the treatment of neuroblastomas.21,22 There are several anti-TACA mAbs in pre-clinical/clinical trial for example mAbs to GD3,23,24 GM2,25 fucosyl-GM1,26 Lewis Y,27,28 Tn,29,30 STn31,32 and TF.33 The TACAs are viable targets for immunotherapy, however, developing a qualified therapeutic anti-TACA mAb for specific types of cancer can be extremely challenging. First, generation of strong immune responses against the TACA component of conjugates is essential for the design of TACA vaccines, thus selection of carriers and conjugation methods are critical. Second, after vaccination a pool of polyclonal antibodies are obtained which can bind various epitopes; for example, the carbohydrate antigens and/or the fragments of antigen carrier.34 Some antibodies lack the ability to bind to natural TACAs that display on the surface of tumor cells; only limited to recognize clustered/multivalent display of glycans.30 Therefore, identifying a mAb that possess both high specificity and affinity against TACAs can be especially difficult. Furthermore, therapeutic mAbs should be capable of inducing effector functions, which induce antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). In some case good affinity and specificity do not render good effector functions, which are resultant of the lack of interactions with the Fc with Fc receptors, or the failed recruitment of complement.35
However, the imperfections of TACA-protein conjugates have also been unmasked and hindered success in clinical trials as a direct result of the “epitope suppression” effect which negatively affects immunization efficacy.36–39 Additionally, less chemically well-defined conjugation methods can lead to inconsistencies in chemical composition from batch-to-batch samples.40 The non-protein carrier approach, therefore, can be an attractive alternative towards cancer vaccine development and to this end, our group has been dedicated to developing TACA specific immunity employing an entirely carbohydrate conjugate strategy utilizing bacterial polysaccharide PS A1 from Bacteroides fragilis (ATCC 25285/NCTC 9343) as the immune stimulating “carrier” (Fig. 1).41,42 PS A1 possesses unique immunological properties in comparison to protein counterparts. This capsular zwitterionic polysaccharide (ZPS) has been involved in immunological studies that have indicated processing by APCs and activated CD4+ T cells through the major histocompatibility complex II (MHCII) dependent pathway.43–45 The efficacy of PS A1 as an immunogenic “carrier” was first evaluated and validated through our studies with TACA-PS A1 constructs including Tn-PS A1,46,47 TF-PS A1,48,49 STn-PS A1 (ref. 50) and GM3-PS A1 (ref. 51) (Fig. 1a) in which all conjugates were able to induce specific adaptive immunity (target ID), production of high affinity IgG antibodies (target specificity) and tumor cell lysis (function).29
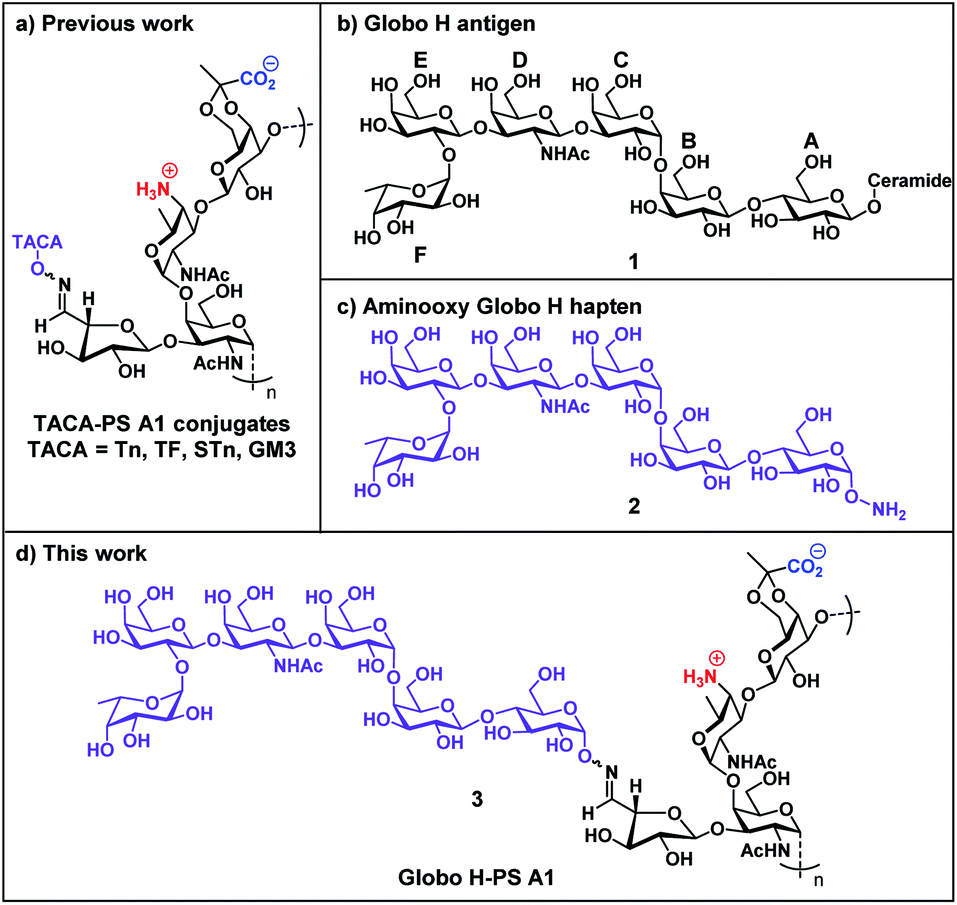 | ||
| Fig. 1 (a) TACA-PS A1 glycoconjugate strategy; (b) chemical structure of native Globo H (1); (c) chemical structure of aminooxy Globo H (2); (d) Globo H-PS A1 conjugate (3). | ||
Globo H (1 and 2) is a TACA overexpressed on a variety of tumor cells but scarcely found on healthy tissue.52 The Globo H-PS A1 (3) construct is a continuing effort in our research group aimed at generating TACA-specific adaptive immunity and by-passing immunological challenges with protein carriers. Globo H (1 and 2) is a hexasaccharide glycan with a ceramide lipid attached at the reducing end and it is anchored endogenously on the outer membrane of tumor cells.53 A murine anti-Globo H mAb MBr1 was developed, identified Globo H as a biomarker for human breast cancer54 and subsequent immunohistology studies revealed that abnormally high levels of Globo H were detected on various tumor specimens, including small-cell lung, prostate, pancreas, gastric, and ovarian cancers.55–57 In clinical practice, overexpression of Globo H is correlated to a poor prognosis of cancer patients.58,59 One possible reason is the binding of Globo H to translin-associated factor X (TRAX) promotes angiogenesis, which is a key factor for the rapid progression of cancer.60 Globo H has therefore become one of the most attractive carbohydrate-based targets for cancer vaccine development.61–63 Previous epitope mapping studies of the anti-Globo H mAb MBr1 indicated that the terminal tetrasaccharide (residue CDEF) is the minimal antigenic structure for Globo H recognition;64,65 the structural rigidity is crucial for MBr1 binding.66,67 The pursuit of higher Ag epitope specificity encouraged us to develop a chemically well-defined immunogenic glycoconjugate that carries Globo H hexasaccharide glycan with the specific aim of generating highly specific Abs that can bind to Globo H antigens on tumor cells. Our previous studies of TACA-PS A1 conjugates have driven our interests to synthesize the important aminooxy Globo H (2) motif, which when conjugated to oxidized PS A1, renders a semisynthetic Globo H-PS A1 (3) construct. Furthermore, we have carefully designed immunological evaluation studies of Globo H-PS A1 as a vaccine candidate in combatting breast and ovarian cancers.
Results and discussion
Chemical synthesis of aminooxy-Globo H antigen
The hexasaccharide structure of Globo-H antigen (Fucα(1→2)Galβ(1→3)GalNAcβ(1→3)Galα(1→4)Galβ(1→4)Glcβ(1)) has garnered a substantial amount of interest from the synthetic community over the years. The first chemical synthesis of Globo-H was reported in 1995 where a convergent [3 + 3] strategy was employed.68 To date, several other synthetic routes and strategies have been developed, even for analogs, which have led to improved overall yields, large scale production and improved stereoselectivity.69 For example, linear pre-activation one-pot strategy,70 two-directional strategy,71 solid-phase synthesis72 and chemo-enzymatic synthesis73 have all rendered Globo H for further biological analysis.Based on literature precedents, we designed a strategy for aminooxy Globo H (2) as illustrated in Scheme 1. The [3 + 3] (5 (DEF) + 6 (ABC)) glycosylation, as one of the most critical steps in constructing the glycosidic skeleton, would be accomplished using the trichloroacetimidate Schmidt strategy and give rise to fully protected hexasaccharide 4. The trisaccharide donor 5 and acceptor 6, were chosen because of their ease of preparation. Building block 10 (AB) would incorporate an O-succinimidyl group at the reducing end to later reveal an aminooxy group that would allow for the formation of the stable conjugate via an oxime link. The involvement of an O-succinimidyl group plays a key role for the successful implementation of this strategy because the O-succinimidyl can withstand the requisite strong acidic conditions of the [3 + 3] and AB + C glycosylation as noted from our previous work.49–51 In the later stages of synthesis, the O-succinimidyl can be selectively removed by simple treatment with hydrazine hydrate in the form of a Gabriel reaction.
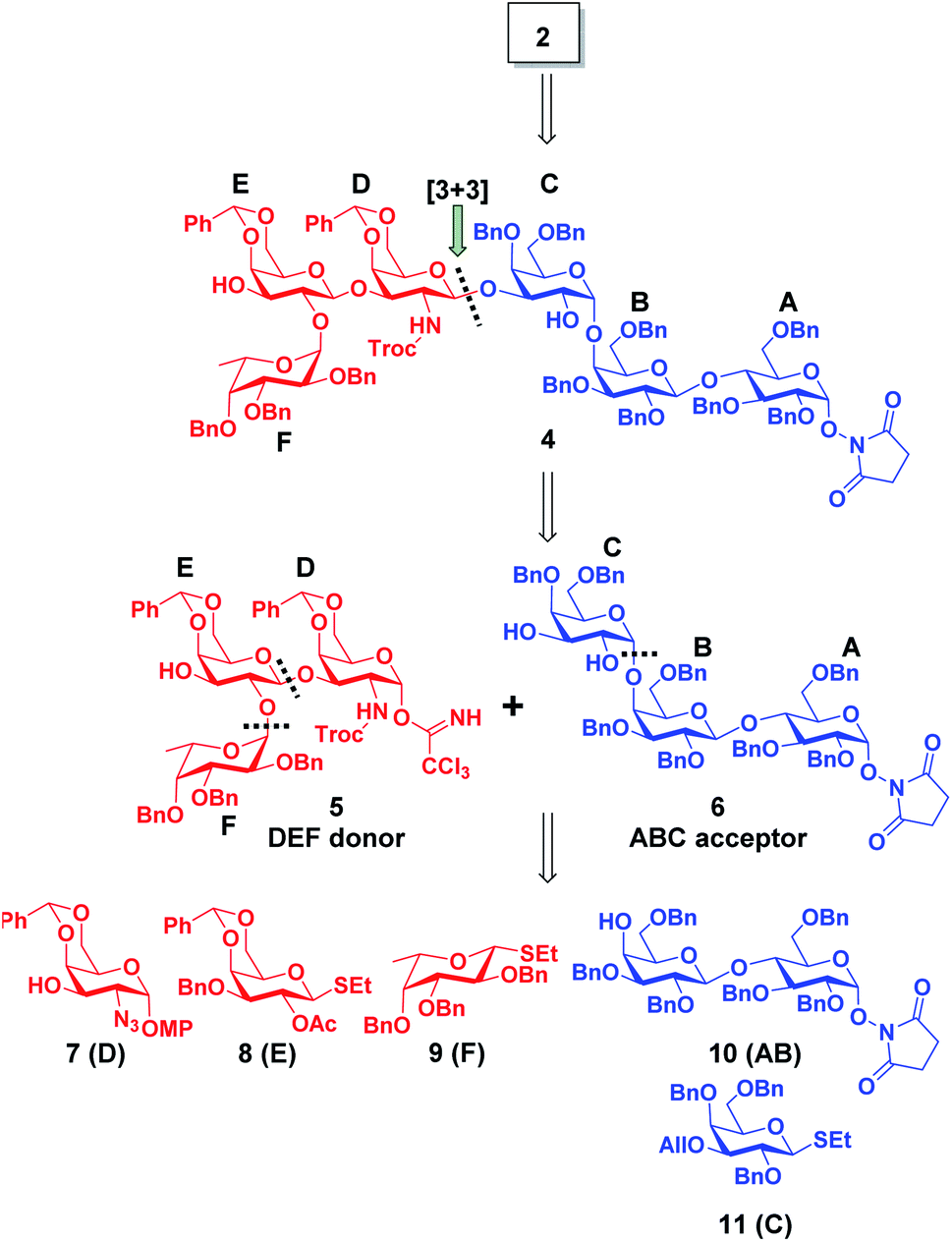 | ||
| Scheme 1 Retrosynthetic analysis of aminooxy Globo H (2). | ||
Therefore, our synthesis commenced with preparing trisaccharide donor 5 using monosaccharide intermediates 7,748,75 and 971,76 (Scheme 1). They were assembled in a linear sequence given rise to trisaccharide 13 (Scheme 2). The synthesis of trisaccharide 5 was quite concise and efficient, requiring only five transformations starting from the coupling of acceptor 7 and donor 8. Therefore, the glycosylation of D and E was accomplished under a NIS/TMSOTf promoter system and the designed 2′-OAc group was set in place to exert a neighboring group participation effect that ensured exclusive formation of the β-anomer. When subjected to Zemplén deacetylation conditions, disaccharide 12 was afforded with a single hydroxyl group at the C2′ position. The DEF trisaccharide 13 was then prepared by selective fucosylation using fucosyl donor 9 under the same NIS/TMSOTf activation conditions mentioned previously in 70% yield with an α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β ratio of 20:1. The choice of solvents, for the fucosylation, can affect the stereoselective outcome of the reaction; use of dichloromethane (DCM) gave us only a 1:1 mixture of α/β anomers, however, use of a DCM/diethyl ether (1:1, v/v) mixed solvent system dramatically improved the α selectivity and subsequent yield. The α selectivity was confirmed by examining the coupling constant of the fucosyl anomeric proton (J = 3.8 Hz at δ = 5.68). One reason for the high selectivity of this reaction might be due to diethyl ether exhibiting a stabilization effect on the oxocarbenium ion in such a way where the β-face is hindered and nucleophilic attack on the α-face is favorable.77–82 With the trisaccharide in hand, the C2′ azido group was converted to a primary amine in the presence of 1,3-propanedithiol and subsequently capped with a Troc protecting group giving rise to 13. The Troc group was chosen to act as a directing moiety for neighboring group participation in anticipation for the 1,3 glycosylation. The p-methoxyphenyl (PMP) group at the reducing end of 13 was then selectively removed by oxidation with ceric ammonium nitrate (CAN) to yield a hemiacetal intermediate, which was subsequently converted to the Schmidt trichloroacetimidate donor 5 (Scheme S3†) in preparation for the convergent [3 + 3] coupling.83,84
:β ratio of 20:1. The choice of solvents, for the fucosylation, can affect the stereoselective outcome of the reaction; use of dichloromethane (DCM) gave us only a 1:1 mixture of α/β anomers, however, use of a DCM/diethyl ether (1:1, v/v) mixed solvent system dramatically improved the α selectivity and subsequent yield. The α selectivity was confirmed by examining the coupling constant of the fucosyl anomeric proton (J = 3.8 Hz at δ = 5.68). One reason for the high selectivity of this reaction might be due to diethyl ether exhibiting a stabilization effect on the oxocarbenium ion in such a way where the β-face is hindered and nucleophilic attack on the α-face is favorable.77–82 With the trisaccharide in hand, the C2′ azido group was converted to a primary amine in the presence of 1,3-propanedithiol and subsequently capped with a Troc protecting group giving rise to 13. The Troc group was chosen to act as a directing moiety for neighboring group participation in anticipation for the 1,3 glycosylation. The p-methoxyphenyl (PMP) group at the reducing end of 13 was then selectively removed by oxidation with ceric ammonium nitrate (CAN) to yield a hemiacetal intermediate, which was subsequently converted to the Schmidt trichloroacetimidate donor 5 (Scheme S3†) in preparation for the convergent [3 + 3] coupling.83,84
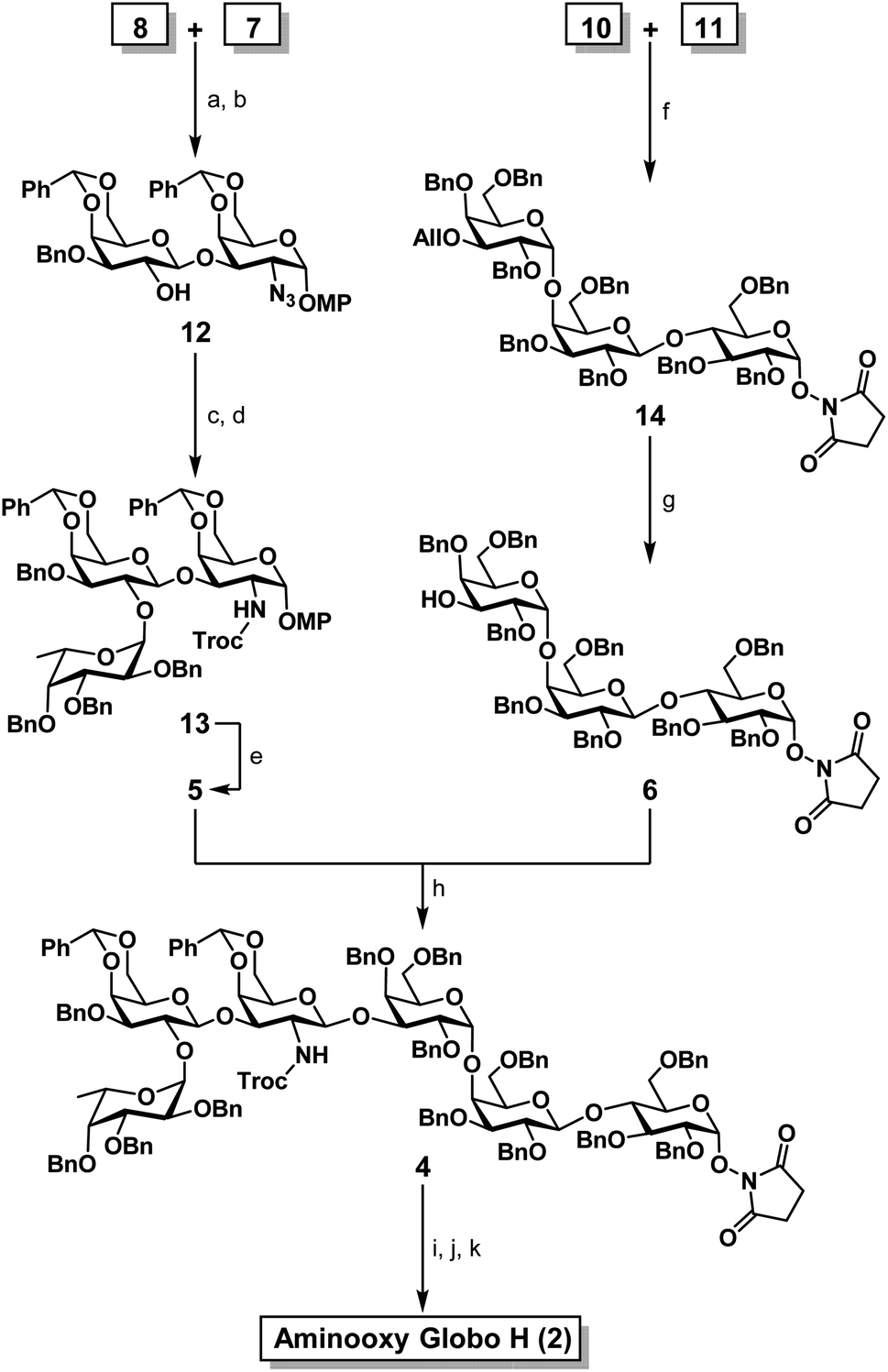 | ||
| Scheme 2 Synthesis of aminooxy Globo H (2). (a) NIS, TMSOTf, CH2Cl2, 4 Å molecular sieves, −30 °C, 30 min, 73%; (b) NaOMe/MeOH, 40 °C, 12 h, 81%; (c) 9, NIS, TMSOTf, CH2Cl2/Et2O (1:1), 4 Å molecular sieves, −30 °C, 30 min, 70% (α anomer), α:β = 20:1; (d) (1) 1,3-propanedithiol, Et3N, CH2Cl2/MeOH (1:1), reflux 12 h; (2) TrocCl, NaHCO3, THF, 3 h (92% over two steps); (e) (1) CAN, CH3CN/H2O (4:1), rt, 3 h; (2) TCA, DBU, CH2Cl2, 0 °C, 1 h, (92% over two steps); (f) NIS, TMSOTf, CH2Cl2/Et2O (4:1), 4 Å molecular sieves, 0 °C, 1 h, 66% (α only); (g) PdCl2, CH2Cl2:MeOH (4:1), 3 h, rt, 86%; (h) TMSOTf, CH2Cl2, 4 Å molecular sieves, −30 °C, 45 min, 39% (β only); (i) Zn, THF:AcOH:Ac2O (3:2:1), 0 °C to rt; (j) 10% Pd/C, H2, MeOH, rt, 3 h; (k) NH2NH2·H2O, H2O/MeOH (1:1), rt, 12 h (57% over three steps). | ||
One of the major hurdles in our synthetic efforts was to produce the ABC acceptor 6 (Scheme 2). This trisaccharide component can be retro-synthetically analyzed from a lactose building block 10 containing the desired O-succinimidyl group and a hydroxyl at the C4′ position reacting with building block 11 (C). Therefore, building blocks 1085 and 1186 were synthesized as per literature protocols. The glycosyl acceptor 10 and donor 11 were assembled using NIS/TMSOTf activation to afford the desired trisaccharide 14 in quantitative α-selectivity and in 66% yield. The formation of an α(1→4) glycosidic bond was confirmed by the anomeric proton coupling constant (J = 3.2 Hz δ = 5.04). The allyl ether in compound 14 was then readily removed with palladium(II) chloride to furnish the desired trisaccharide acceptor 6. With trisaccharide moieties 5 and 6 in hand, we focused our synthetic efforts toward the decisive [3 + 3] glycosylation (Scheme 2). Schmidt donor 5 was mixed with acceptor 6 in the presence of TMSOTf, which, led to the formation of the anticipated hexasaccharide 4 in exclusive β-selectivity in ∼40% yield. Global deprotection of 4 commenced with in situ N-Troc deprotection/acetylation utilizing activated zinc in a mixture of THF:AcOH:Ac2O (v/v/v = 3:2:1) to remove the N-Troc group and concomitantly introduce the N-acetyl group.87 The remaining benzyl and benzylidene groups were then removed with palladium catalyzed hydrogenolysis at room temperature.88 Upon the completion of hydrogenolysis, the resulting residue was directly subjected to a Gabriel-type hydrazine hydrate reaction for the removal of the succinimide giving rise to the aminooxy motif51 (Scheme S4†) in 57% over three steps. Fully deprotected and purified aminooxy Globo-H (2) was obtained after conducting a size exclusion column quickly followed by lyophilization. The overall calculated yield of 2 was 7.8% starting from intermediate 7 and achieved in a combined 11 steps. By implementing this synthetic route, within a relative short period of time, a sufficient amount of fully protected hexasaccharide 4 can be readily prepared and provide Globo H analogs and other probes for furthering various preclinical studies.
Preparation of Globo H-PS A1 conjugate through oxime bond
Toward the goal of evaluating our Globo H-PS A1 concept, we conjugated compound 2 to “carrier” polysaccharide PS A1 giving rise to an entirely carbohydrate construct 3 as our immunogen. The first step of the conjugation protocol involved a sodium periodate mediated selective vicinal diol oxidation of naturally isolated PS A1 giving an aldehyde functional handle (Scheme 3). The aldehyde was in turn reacted with aminooxy 2 under a mild acidic sodium acetate buffer (pH = 5.0) to form the oxime bond and yield Globo H-PS A1 (3). Purified 3 was obtained by filtration through a 10 kDa molecular weight cutoff centrifugal concentrator and the purified product was lyophilized rendering a white porous solid.50,51,84 Globo H-PS A1 was characterized using 1H NMR spectrum at 60 °C. The indicative signals were inclusive of an oxime moiety (δ = 7.46–7.47, doublet), an N-acetyl peak (δ = 2.32), and the presence of the fucosyl C5′ methyl (δ = 1.49–1.50, doublet). The relative ratio of Globo H:PS A1 was determined to be 1:2.75 based on the integration of the fucosyl C5′ methyl and PS A1 methyl peaks, which gave us an approximate 40 Globo H molecules to every PS A1 molecule used.
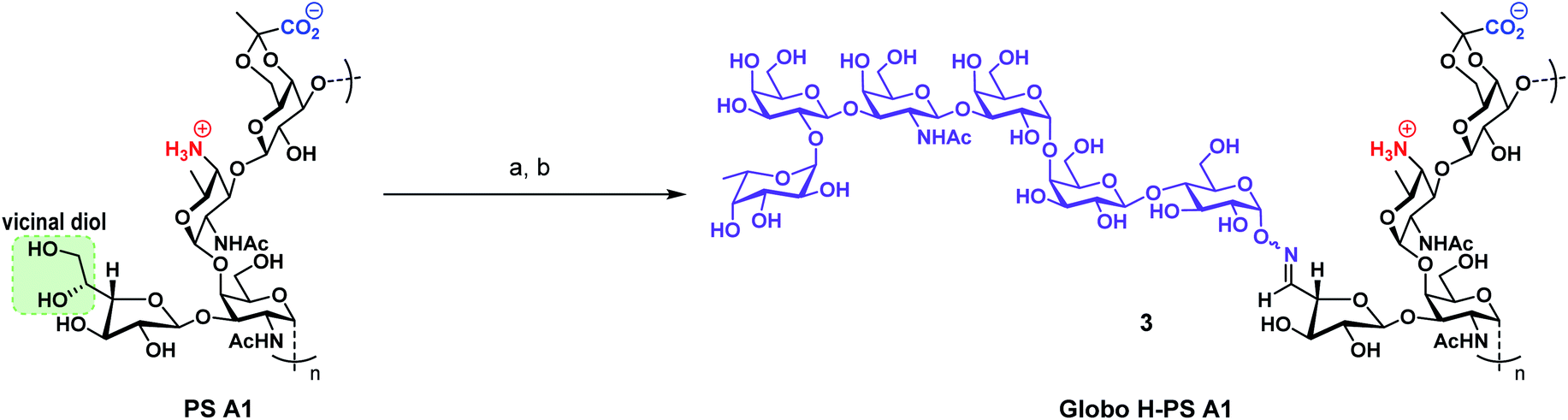 | ||
| Scheme 3 Preparation of Globo H-PS A1 (3) immunogen from PS A1 and aminooxy Globo H (2). (a) NaIO4, 0.1 M NaOAc buffer pH = 5.0, dark, rt, 1.5 h; (b) 2, 0.1 M NaOAc buffer pH = 5.0, rt, 18 h. | ||
Immunological evaluation: Ab response against Globo H-PS A1 conjugate
The immunological evaluation of Globo H-PS A1 (3) commenced with the immunization of Jax C57BL/6 mice. As shown in Fig. 2, the production and specificity of anti-Globo H antibodies was determined by serological assays. One formulation consisted of Globo H-PS A1 plus Sigma Adjuvant System® (SAS). SAS can prime the system for Ab production and particularly aides in a robust IgG response that was observed in our studies.89 Another group of mice was concomitantly vaccinated with Globo H-PS A1 plus TiterMax® Gold (TMG) and showed a moderate level of Ab production. A control group of mice, with no administered adjuvant, gave an immune response albeit at lower levels of Ab production against Globo H given the same incubation period in the mouse. Based on the serological assay results, the IgG/IgM ratio suggests that immunoglobulin isotype switching occurred during the trials and demonstrates that T cells play a role in generating anti-Globo H antibodies.90–93 | ||
| Fig. 2 Determining IgG/IgM antibody titers induced by Globo H-PS A1 + SAS (red), Globo H-PS A1 + TMG (blue) and Globo H-PS A1 (green). The error bars represent standard deviation (SD) of two triplicate tests. | ||
The serological assays also revealed an interesting finding in that the anti-serum had rather high titers of anti-Gb3 antibodies (Fig. S1†). Since Gb3 is a biosynthetic precursor of Globo H,94,95 it is plausible that Globo H might exhibit cross reactivity toward Gb3. Additionally, in the antigen presenting cells, the depolymerization of Globo H-PS A1 might possibly trigger the release of a large amount of reactive oxygen species (ROS)96 potentially leading to fragmented Globo H as Gb3-like glycans.
The presence of the Fucα(1→2)Gal glycosidic bond and L-fucose in Globo H-PS A1 are possible conventions for generating undesirable cross reactivity against blood group A (BGA) and blood group B (BGB) antigens. Anti-serum from Globo-H-PS A1 vaccinations were screened for binding to BGA and BGB antigens at 1:200 dilution (Fig. S2†). It was observed that anti-sera from Globo H-PS A1 (both SAS and TMG groups) showed negligible IgG/IgM binding to either BGA or BGB. This result suggests that vaccination using the Globo H-PS A1 construct would be unlikely to cause cytotoxicity towards red blood cells. The cross-reactivity test results clearly suggested the specificity of antigenic epitope recognition of the raised Ab was indeed achieved.
Antibody binding to the Globo H positive cancer cell surface
Fluorescence-activated cell sorting (FACS) was then utilized to study the immunological potential of Globo H-PS A1 as a vaccine designed to target the Globo H antigens on human tumor cell membranes (Fig. 3). Human breast cancer cell line MCF-7 and ovarian cancer cell line OVCAR-5 are known to overexpress Globo H on their surface.2 The anti-sera from Globo H-PS A1 plus SAS/TMG groups were selected for the IgG surface binding experiments. Cancer cell lines treated with anti-PS A1 serum were used as serological controls, and cells treated exclusively with secondary FITC-labeled anti-IgG were used as Ab isotype controls. The FACS study indicated both anti-sera (Globo H-PS A1 + SAS and Globo H-PS A1 + TMG) exhibited very strong binding to MCF-7 and OVCAR-5 lines. For the anti-serum obtained from Globo H-PS A1 + TMG group, 84% and 83% positive shifts were observed on MCF-7 and OVCAR-5 respectively. The Globo H-PS A1 + SAS group showed 94% positive for MCF-7, as well as 81% positive for the OVACR-5 cell lines.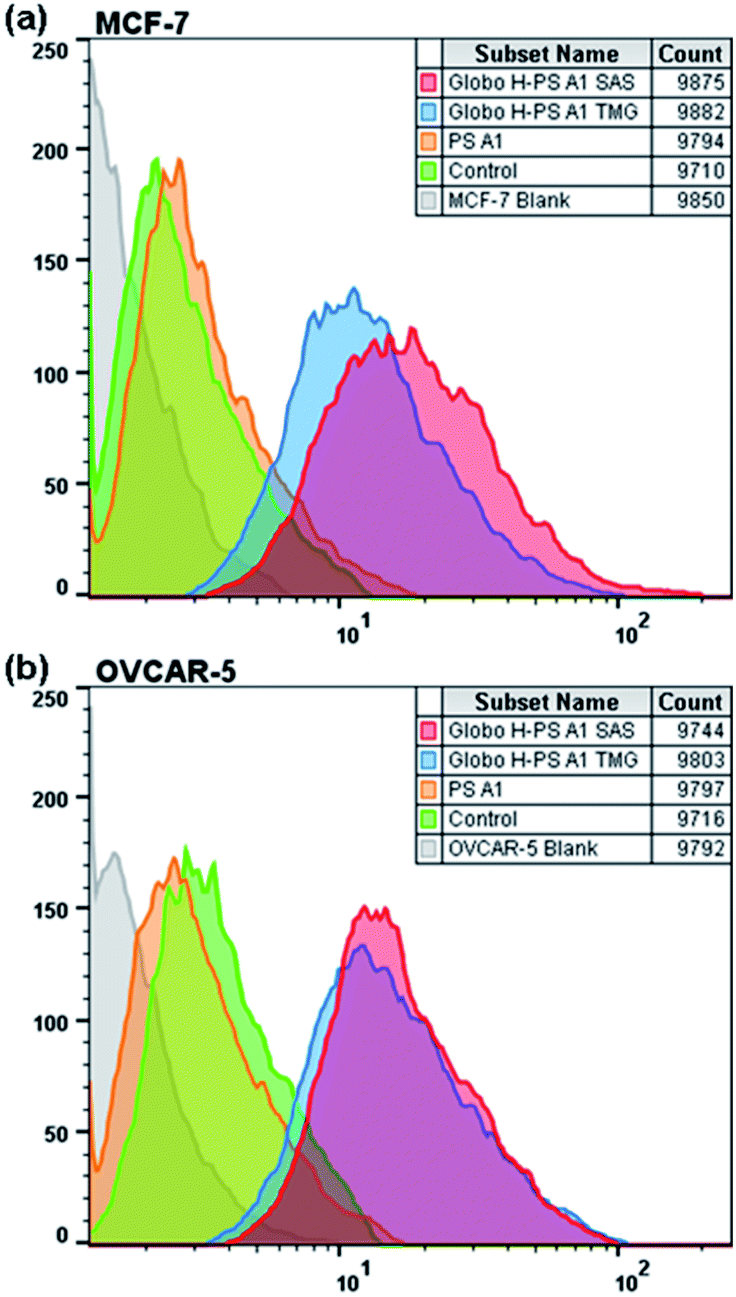 | ||
| Fig. 3 FACS analysis of IgG binding against Globo H positive tumor cells, (a) MCF-7 and (b) OVCAR-5. | ||
Production of highly specific anti-Globo H IgGs as noted from the FACS results in combination with serological assays. These data, when combined, offer critical evidence for isotype switching and T cell dependent immune responses. There is a distinct difference of Ab titers between Globo H-PS A1 + SAS group and Globo H-PS A1 + TMG group, however the cancer cell surface reactivity of anti-Globo H IgGs that was raised from both formulations compare favorably to each other. This interesting titer observation, to a large extent, is most likely because of different adjuvants; the monophosphoryl lipid A is the active component of SAS and has been proven to act as an agonist for TLR-4 as well as an efficient immunostimulant. On the other hand, TMG, which is noted to be a benign adjuvant and efficient on the delivery of biomacromolecules, is less toxic and less potent compared to SAS. The difference on the Ab titers only reflects the potency difference of these two adjuvants.89,97 More importantly both adjuvants can assist the Globo H-PS A1 immunogen to induce adaptive immunity and generate high affinity IgGs; studies were done in C57BL/6 mice.
Antibody-mediated complement-dependent cytotoxicity (CDC)
The antibodies raised from Globo H-PS A1 + SAS group immunizations exhibited strong binding to the naturally occurring Globo H. As a result, anti-sera obtained from this group was selected for further testing using a complement dependent cytotoxicity (CDC) assay.98,99 Antibody-mediated cytolysis is of great significance when determining the potential therapeutic value of Globo H-PS A1 conjugate and CDC was measured against cancer cell lines MCF-7 and OVCAR-5 as well as non-tumorigenic cell line MCF-10A as the control. The substance control was setup with the exclusive treatment of rabbit complement. The percent of cell lysis was determined by a lactate dehydrogenase (LDH) assay (Roche Applied Science) without further optimization (Fig. 4). LDH assay results revealed there was significant cytotoxicity against MCF-7 and OVCAR-5 cells upon the treatment with anti-sera Globo H-PS A1 + SAS. For MCF-7, a reasonably good CDC effect was observed with a 47% net cell lysis rate in comparision with the control, whereas for OVCAR-5 only a moderate CDC effect was found with a 24% net cell lysis rate recorded. There was no statistically significant cytotoxicity observed with MCF-10A cells due to the absence of surface Globo H antigens. Collectively, it was noted that anti-Globo-H-PS A1 sera exhibited good to moderate cytotoxicity against human breast and ovarian tumor cell lines and no cytotoxicity to the normal cell line was observed.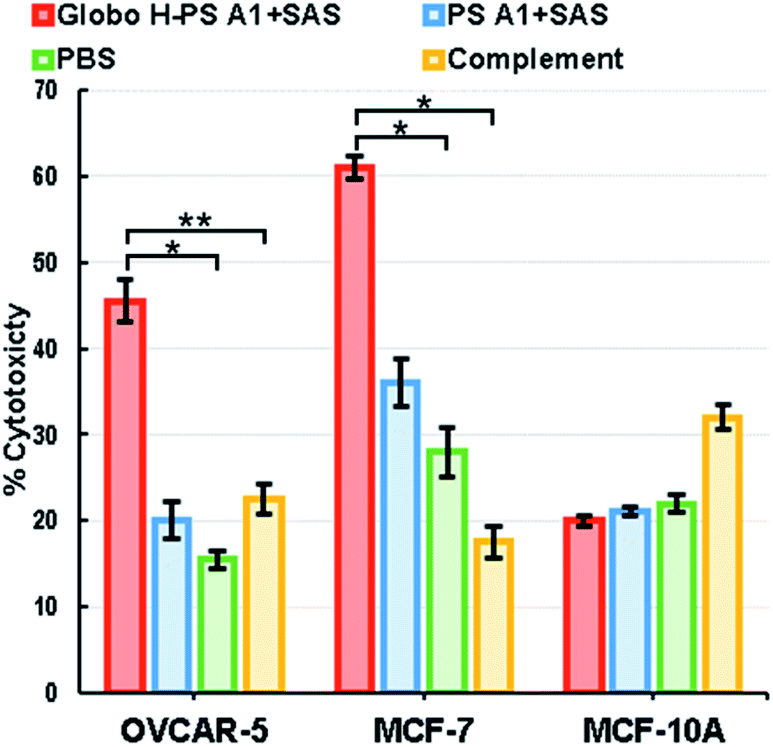 | ||
| Fig. 4 Lysis of Globo H positive cancer cells through antibody mediated complement-dependent cytotoxicity (CDC). The cytotoxicity was determined using an LDH assay. Data shown are mean values of two parallel triplicate tests, where *P < 0.05 and **P < 0.1 were obtained using a Student's t-test. The error bars represent standard deviation (SD) of two triplicate tests. | ||
Conclusions
Herein, we have described the chemical synthesis of aminooxy Globo H (2), the preparation of Globo H-PS A1 conjugate (3) and the corresponding immunological evaluation as a potential cancer vaccine candidate. A convergent [3 + 3] synthetic strategy was developed for 2, which started from building block 7 and took 12 steps to obtain 2 in an overall 7.8% yield. The aminooxy glycan 2 was conjugated to oxidized PS A1 with an oxime linkage to afford Globo H-PS A1 (3). The formulations of Globo H-PS A1 + SAS and Globo H-PS A1 + TMG were able to induce high affinity anti-Globo H antibodies when C57BL/6 mice were immunized as indicated by ELISA. FACS studies further confirmed binding events with Globo H expressing MCF-7 and OVCAR-5 cancer cell lines. The combined results of ELISA and FACS demonstrated excellent specificity and fidelity of antibodies raised against the Globo H-PS A1 immunogen using SAS or TMG as the adjuvant for targeting the native Globo H antigen even when BGA and BGB glycans were used in the study. Additionally, an in vitro LDH assay indicated that anti-Globo H antibodies were able to generate complement-dependent cytotoxicity and lead to tumor cell lysis, meanwhile showing negligible toxicity toward normal cells. The test results from the mentioned experiments suggest the efficacy and potential of Globo H-PS A1 as cancer vaccine candidate. Further studies are currently underway to investigate tumor challenge mouse models and the immunological mechanisms behind it.Ethical statement
Animal study was performed in strict compliance with protocols approved by the by the Institutional Animal Care and Use Committee of the University of Toledo (Toledo, Ohio, United States), NIH guidelines for the care and use of laboratory animals (NIH publication no. 85-23, rev. 1985) were observed.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is dedicated to Professor David Crich on the occasion of his 60th birthday. Financial Support for this work was provided by the National Institutes of Health (NIH NCI R01 CA156661 and NIH NIGMS U01 GM125271). The authors thank Dr Kandace Williams of the University of Toledo College of Medicine and Life Sciences for providing the MCF-10A cell line and Dr Andrea Kalinoski of the Department of Surgery and the University of Toledo Advanced Microscopy and Imaging Center for assistance with FACS. Dr Frederic Valeriote of Henry Ford Health Systems and Halina Pietraszkiewicz of 21st Century Therapeutics provided MCF-7 and OVCAR-5 cell lines and expertise with tissue cell culture. Dr Yong-Wah Kim in the Department of Chemistry and Biochemistry at the University of Toledo provided assistance with some NMR experiments.Notes and references
- T. Feizi, Cancer Surv., 1985, 4, 245–269 CAS.
- S.-I. Hakomori, Cancer Res., 1985, 45, 2405–2414 CAS.
- K. O. Lloyd, Semin. Cancer Biol., 1991, 2, 421–431 CAS.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477 CrossRef CAS.
- P. O. Livingston, Curr. Opin. Immunol., 1992, 4, 624–629 CrossRef CAS.
- T. Boon, Int. J. Cancer, 1993, 54, 177–180 CrossRef CAS.
- A. Lanzavecchia, Science, 1993, 260, 937–944 CrossRef CAS.
- D. M. Pardoll, Curr. Opin. Immunol., 1993, 5, 719–725 CrossRef CAS.
- M.-H. Tao and R. Levy, Nature, 1993, 362, 755–758 CrossRef CAS.
- P. O. Livingston, S. Zhang and K. O. Lloyd, Cancer Immunol. Immunother., 1997, 45, 1–9 CrossRef CAS.
- S. Stevanovic, Nat. Rev. Cancer, 2002, 2, 514 CrossRef CAS.
- T. Freire, S. Bay, S. Vichier-Guerre, R. Lo-Man and C. Leclerc, Mini-Rev. Med. Chem., 2006, 6, 1357–1373 CrossRef CAS.
- S. Lang and X. Huang, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.00284.
- R. Mettu, C.-Y. Chen and C.-Y. Wu, J. Biomed. Sci., 2020, 27, 9 CrossRef CAS.
- B. M. Longenecker, M. Reddish, R. Koganty and G. D. MacLean, Adv. Exp. Med. Biol., 1994, 353, 105–124 CrossRef CAS.
- B. M. Longenecker, M. Reddish, R. Koganty and G. D. MacLean, Ann. N. Y. Acad. Sci., 1993, 690, 276–291 CrossRef CAS.
- R. Jennemann, C. Gnewuch, S. Bosslet, B. L. Bauer and H. Wiegandt, J. Biol. Chem., 1994, 115, 1047–1052 CAS.
- H. Y. Lee, C. Y. Chen, T. I. Tsai, S. T. Li, K. H. Lin, Y. Y. Cheng, C. T. Ren, T. J. Cheng, C. Y. Wu and C. H. Wong, J. Am. Chem. Soc., 2014, 136, 16844–16853 CrossRef CAS.
- Y. L. Huang, J. T. Hung, S. K. Cheung, H. Y. Lee, K. C. Chu, S. T. Li, Y. C. Lin, C. T. Ren, T. J. Cheng, T. L. Hsu, A. L. Yu, C. Y. Wu and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2517–2522 CrossRef CAS.
- D. J. Lefeber, J. P. Kamerling and J. F. Vliegenthart, Chemistry, 2001, 7, 4411–4421 CrossRef CAS.
- N.-K. V. Cheung, I. Y. Cheung, K. Kramer, S. Modak, D. Kuk, N. Pandit-Taskar, E. Chamberlain, I. Ostrovnaya and B. H. Kushner, Int. J. Cancer, 2014, 135, 2199–2205 CrossRef CAS.
- A. L. Yu, A. L. Gilman, M. F. Ozkaynak, W. B. London, S. G. Kreissman, H. X. Chen, M. Smith, B. Anderson, J. G. Villablanca, K. K. Matthay, H. Shimada, S. A. Grupp, R. Seeger, C. P. Reynolds, A. Buxton, R. A. Reisfeld, S. D. Gillies, S. L. Cohn, J. M. Maris and P. M. Sondel, N. Engl. J. Med., 2010, 363, 1324–1334 CrossRef CAS.
- A. N. Houghton, D. Mintzer, C. Cordon-Cardo, S. Welt, B. Fliegel, S. Vadhan, E. Carswell, M. R. Melamed, H. F. Oettgen and L. J. Old, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 1242–1246 CrossRef CAS.
- A. M. Scott, F.-T. Lee, W. Hopkins, J. S. Cebon, J. M. Wheatley, Z. Liu, F. E. Smyth, C. Murone, S. Sturrock, D. MacGregor, N. Hanai, K. Inoue, M. Yamasaki, M. W. Brechbiel, I. D. Davis, R. Murphy, A. Hannah, M. Lim-Joon, T. Chan, G. Chong, G. Ritter, E. W. Hoffman, A. W. Burgess and L. J. Old, J. Clin. Oncol., 2001, 19, 3976–3987 CrossRef CAS.
- P. O. Livingston, C. Hood, L. M. Krug, N. Warren, M. G. Kris, T. Brezicka and G. Ragupathi, Cancer Immunol. Immunother., 2005, 54, 1018–1025 CrossRef CAS.
- O. Nilsson, F. T. Brezicka, J. Holmgren, S. Sörenson, L. Svennerholm, F. Yngvason and L. Lindholm, Cancer Res., 1986, 46, 1403–1407 CAS.
- A. M. Scott, N. Tebbutt, F.-T. Lee, T. Cavicchiolo, Z. Liu, S. Gill, A. M. T. Poon, W. Hopkins, F. E. Smyth, C. Murone, D. MacGregor, A. T. Papenfuss, B. Chappell, T. H. Saunder, M. W. Brechbiel, I. D. Davis, R. Murphy, G. Chong, E. W. Hoffman and L. J. Old, Clin. Cancer Res., 2007, 13, 3286–3292 CrossRef CAS.
- J. A. Posey, M. B. Khazaeli, M. A. Bookman, A. Nowrouzi, W. E. Grizzle, J. Thornton, D. E. Carey, J. M. Lorenz, A. P. Sing, C. B. Siegall, A. F. LoBuglio and M. N. Saleh, Clin. Cancer Res., 2002, 8, 3092–3099 CAS.
- K. R. Trabbic, K. A. Kleski, M. Shi, J.-P. Bourgault, J. M. Prendergast, D. T. Dransfield and P. R. Andreana, Cancer Immunol. Immunother., 2018, 67, 1437–1447 CrossRef CAS.
- J. C. Manimala, T. A. Roach, Z. Li and J. C. Gildersleeve, Glycobiology, 2007, 17, 17C–23C CrossRef CAS.
- J. M. Prendergast, A. P. Galvao da Silva, D. A. Eavarone, D. Ghaderi, M. Zhang, D. Brady, J. Wicks, J. DeSander, J. Behrens and B. R. Rueda, mAbs, 2017, 9, 615–627 CrossRef CAS.
- M. A. Reddish, L. Jackson, R. Rao Koganty, D. Qiu, W. Hong and B. Michael Longenecker, Glycoconjugate J., 1997, 14, 549–560 CrossRef CAS.
- K. R. Trabbic, K. Whalen, K. Abarca-Heideman, L. Xia, J. S. Temme, E. F. Edmondson, J. C. Gildersleeve and J. J. Barchi, Sci. Rep., 2019, 9, 5662 CrossRef.
- E. Sterner, N. Flanagan and J. C. Gildersleeve, ACS Chem. Biol., 2016, 11, 1773–1783 CrossRef CAS.
- A. Natsume, R. Niwa and M. Satoh, Drug Des., Dev. Ther., 2008, 3, 7–16 CrossRef.
- L. A. Herzenberg, T. Tokuhisa and L. A. Herzenberg, Nature, 1980, 285, 664–667 CrossRef CAS.
- M. C. Woodruff, E. H. Kim, W. Luo and B. Pulendran, Cell Rep., 2018, 25, 321–327 CrossRef CAS.
- T. Buskas, Y. H. Li and G. J. Boons, Chem.–Eur. J., 2004, 10, 3517–3524 CrossRef CAS.
- N. R. Bennett, C. M. Jarvis, M. M. Alam, D. B. Zwick, J. M. Olson, H. V. T. Nguyen, J. A. Johnson, M. E. Cook and L. L. Kiessling, Biomacromolecules, 2019, 20, 4370–4379 CrossRef CAS.
- T. Buskas, P. Thompson and G.-J. Boons, Chem. Commun., 2009, 5335–5349, 10.1039/B908664C.
- S. Nishat and P. R. Andreana, Vaccines, 2016, 4 DOI:10.3390/vaccines4020019.
- F. Hossain and P. R. Andreana, Pharmaceuticals, 2019, 12, 84–102 CrossRef CAS.
- W. M. Kalka-Moll, A. O. Tzianabos, P. W. Bryant, M. Niemeyer, H. L. Ploegh and D. L. Kasper, J. Immunol., 2002, 169, 6149–6153 CrossRef CAS.
- J. L. Johnson, M. B. Jones and B. A. Cobb, J. Biol. Chem., 2015, 290, 5007–5014 CrossRef CAS.
- K. Bloem, J. J. Garcia-Vallejo, I. M. Vuist, B. A. Cobb, S. J. van Vliet and Y. van Kooyk, Front. Immunol., 2013, 4, 103 Search PubMed.
- R. A. De Silva, Q. Wang, T. Chidley, D. K. Appulage and P. R. Andreana, J. Am. Chem. Soc., 2009, 131, 9622–9623 CrossRef CAS.
- R. A. De Silva, D. K. Appulage, H. Pietraszkiewicz, K. R. Bobbitt, J. Media, J. Shaw, F. A. Valeriote and P. R. Andreana, Cancer Immunol. Immunother., 2012, 61, 581–585 CrossRef CAS.
- K. A. Kleski, K. R. Trabbic, M. Shi, J. P. Bourgault and P. R. Andreana, Molecules, 2020, 25, 1319–1333 CrossRef CAS.
- J. P. Bourgault, K. R. Trabbic, M. Shi and P. R. Andreana, Org. Biomol. Chem., 2014, 12, 1699–1702 RSC.
- M. Shi, K. A. Kleski, K. R. Trabbic, J.-P. Bourgault and P. R. Andreana, J. Am. Chem. Soc., 2016, 138, 14264–14272 CrossRef CAS.
- K. A. Kleski, M. Shi, M. Lohman, G. T. Hymel, V. K. Gattoji and P. R. Andreana, J. Org. Chem., 2020 DOI:10.1021/acs.joc.0c00320.
- E. Tagliabue, G. Porro, P. Barbanti, G. Della Torre, S. Menard, F. Rilke, S. Cerasoli and M. I. Colnaghi, Hybridoma, 1986, 5, 107–115 CrossRef CAS.
- E. G. Bremer, S. B. Levery, S. Sonnino, R. Ghidoni, S. Canevari, R. Kannagi and S. Hakomori, J. Biol. Chem., 1984, 259, 14773–14777 CAS.
- S. Canevari, G. Fossati, A. Balsari, S. Sonnino and M. I. Colnaghi, Cancer Res., 1983, 43, 1301–1305 CAS.
- R. Mariani-Costantini, M. I. Colnaghi, F. Leoni, S. Menard, S. Cerasoli and F. Rilke, Virchows Arch. A: Pathol. Anat. Histopathol., 1984, 402, 389–404 CrossRef CAS.
- R. Mariani-Costantini, M. I. Colnaghi, F. Leoni, S. Mènard, S. Cerasoli and F. Rilke, Virchows Arch. A: Pathol. Anat. Histopathol., 1984, 402, 389–404 CrossRef CAS.
- S. Zhang, C. Cordon-Cardo, H. S. Zhang, V. E. Reuter, S. Adluri, W. B. Hamilton, K. O. Lloyd and P. O. Livingston, Int. J. Cancer, 1997, 73, 42–49 CrossRef CAS.
- S. Martignone, S. Ménard, A. Bedini, A. Paccagnella, S. Fasolato, R. Veggian and M. I. Colnaghi, Eur. J. Cancer, 1993, 29, 2020–2025 CrossRef.
- F. Perrone, S. Ménard, S. Canevari, M. Calabrese, P. Boracchi, R. Bufalino, S. Testori, M. Baldini and M. I. Colnaghi, Eur. J. Cancer, 1993, 29, 2113–2117 CrossRef.
- J.-Y. Cheng, S.-H. Wang, J. Lin, Y.-C. Tsai, J. Yu, J.-C. Wu, J.-T. Hung, J.-J. Lin, Y.-Y. Wu, K.-T. Yeh and A. L. Yu, Cancer Res., 2014, 74, 6856–6866 CrossRef CAS.
- S. J. Danishefsky, Y.-K. Shue, M. N. Chang and C.-H. Wong, Acc. Chem. Res., 2015, 48, 643–652 CrossRef CAS.
- O. Ouerfelli, J. D. Warren, R. M. Wilson and S. J. Danishefsky, Expert Rev. Vaccines, 2005, 4, 677–685 CAS.
- J. Zhu, Q. Wan, D. Lee, G. Yang, M. K. Spassova, O. Ouerfelli, G. Ragupathi, P. Damani, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 9298–9303 CAS.
- M. E. Perico, D. Mezzanzanica, E. Luison, P. Alberti, L. Panza, G. Russo and S. Canevari, Cancer Immunol. Immunother., 2000, 49, 296–304 CAS.
- E. Adobati, L. Panza, G. Russo, M. I. Colnaghi and S. Canevari, Glycobiology, 1997, 7, 173–178 CrossRef CAS.
- L. Lay, F. Nicotra, L. Panza, G. Russo and E. Adobati, Helv. Chim. Acta, 1994, 77, 509–514 CrossRef CAS.
- L. Lay, L. Panza, G. Russo, D. Colombo, F. Ronchetti, E. Adobati and S. Canevari, Helv. Chim. Acta, 1995, 78, 533–538 CrossRef CAS.
- M. T. Bilodeau, T. K. Park, S. Hu, J. T. Randolph, S. J. Danishefsky, P. O. Livingston and S. Zhang, J. Am. Chem. Soc., 1995, 117, 7840–7841 CrossRef CAS.
- J. R. Allen, J. G. Allen, X. F. Zhang, L. J. Williams, A. Zatorski, G. Ragupathi, P. O. Livingston and S. J. Danishefsky, Chem.–Eur. J., 2000, 6, 1366–1375 CrossRef CAS.
- Z. Wang, L. Zhou, K. El-Boubbou, X.-s. Ye and X. Huang, J. Org. Chem., 2007, 72, 6409–6420 CrossRef CAS.
- T. Zhu and G.-J. Boons, Angew. Chem., Int. Ed., 1999, 38, 3495–3497 CrossRef CAS.
- D. B. Werz, B. Castagner and P. H. Seeberger, J. Am. Chem. Soc., 2007, 129, 2770–2771 CrossRef CAS.
- T.-I. Tsai, H.-Y. Lee, S.-H. Chang, C.-H. Wang, Y.-C. Tu, Y.-C. Lin, D.-R. Hwang, C.-Y. Wu and C.-H. Wong, J. Am. Chem. Soc., 2013, 135, 14831–14839 CrossRef CAS.
- C. Mukherjee and A. K. Misra, Tetrahedron: Asymmetry, 2008, 19, 2746–2751 CrossRef CAS.
- A. A. Sherman, O. N. Yudina, A. S. Shashkov, V. M. Menshov and N. E. Nifant'ev, Carbohydr. Res., 2001, 330, 445–458 CrossRef CAS.
- S. S. Mandal, G. Liao and Z. Guo, RSC Adv., 2015, 5, 23311–23319 RSC.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS.
- A. Demchenko, T. Stauch and G.-J. Boons, Synlett, 1997, 1997, 818–820 CrossRef.
- L. Huang and X. Huang, Chem.–Eur. J., 2007, 13, 529–540 CrossRef CAS.
- H. Satoh, H. S. Hansen, S. Manabe, W. F. van Gunsteren and P. H. Hünenberger, J. Chem. Theory Comput., 2010, 6, 1783–1797 CrossRef CAS.
- H. Tokimoto, Y. Fujimoto, K. Fukase and S. Kusumoto, Tetrahedron: Asymmetry, 2005, 16, 441–447 CrossRef CAS.
- D. Crich, J. Org. Chem., 2011, 76, 9193–9209 CrossRef CAS.
- P. Eradi, S. Ghosh and P. R. Andreana, Org. Lett., 2018, 20, 4526–4530 CrossRef CAS.
- S. Ghosh, S. Nishat and P. R. Andreana, J. Org. Chem., 2016, 81, 4475–4484 CrossRef CAS.
- S. Ghosh and P. R. Andreana, J. Carbohydr. Chem., 2014, 33, 381–394 CrossRef CAS.
- A. Si and A. K. Misra, Synthesis, 2015, 47, 83–88 CAS.
- M. R. Pratt and C. R. Bertozzi, Org. Lett., 2004, 6, 2345–2348 CrossRef CAS.
- A. R. Mandhapati, G. Yang, T. Kato, D. Shcherbakov, S. N. Hobbie, A. Vasella, E. C. Böttger and D. Crich, J. Am. Chem. Soc., 2017, 139, 14611–14619 CrossRef CAS.
- A. L. Gavin, K. Hoebe, B. Duong, T. Ota, C. Martin, B. Beutler and D. Nemazee, Science, 2006, 314, 1936–1938 CrossRef CAS.
- P. J. Gearhart and J. J. Cebra, J. Immunol., 1981, 127, 1030–1034 CAS.
- F. Nimmerjahn and J. V. Ravetch, Immunol. Rev., 2010, 236, 265–275 CrossRef CAS.
- K. D. Pandya, I. Palomo-Caturla, J. A. Walker, V. K. Sandilya, Z. Zhong and K. R. Alugupalli, J. Immunol., 2018, 200, 4078–4084 CrossRef CAS.
- C. Zhu, V. Lee, A. Finn, K. Senger, A. A. Zarrin, L. Du Pasquier and E. Hsu, Curr. Biol., 2012, 22, 872–880 CrossRef CAS.
- S. Alam, A. Fedier, R. S. Kohler and F. Jacob, Glycobiology, 2015, 25, 351–356 CrossRef CAS.
- K. Hamamura, K. Hamajima, S. Yo, Y. Mishima, K. Furukawa, M. Uchikawa, Y. Kondo, H. Mori, H. Kondo, K. Tanaka, K. Miyazawa, S. Goto and A. Togari, Int. J. Mol. Sci., 2019, 20, 2825–2839 CrossRef.
- B. A. Cobb, Q. Wang, A. O. Tzianabos and D. L. Kasper, Cell, 2004, 117, 677–687 CrossRef CAS.
- I. Sarkar, R. Garg and S. van Drunen Littel-van den Hurk, Expert Rev. Vaccines, 2019, 18, 505–521 CrossRef CAS.
- P. O. Livingston, C. Hood, L. M. Krug, N. Warren, M. G. Kris, T. Brezicka and G. Ragupathi, Cancer Immunol. Immunother., 2005, 54, 1018–1025 CrossRef CAS.
- G. Ragupathi, N. X. Liu, C. Musselli, S. Powell, K. Lloyd and P. O. Livingston, J. Immunol., 2005, 174, 5706–5712 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures of chemical synthesis, biological test and spectroscopy data for new compounds. See DOI: 10.1039/d0sc04595k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |