
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating the ground state, stability and charge transport in OFETs of biradicaloid hexahydro-diindenopyrene derivatives and a proposed method to estimate the biradical character†
Tanguy
Jousselin-Oba
a,
Masashi
Mamada
 *b,
Atsushi
Okazawa
c,
Jérome
Marrot
a,
Takayuki
Ishida
d,
Chihaya
Adachi
be,
Abderrahim
Yassar
f and
Michel
Frigoli
*a
*b,
Atsushi
Okazawa
c,
Jérome
Marrot
a,
Takayuki
Ishida
d,
Chihaya
Adachi
be,
Abderrahim
Yassar
f and
Michel
Frigoli
*a
aUMR CNRS 8180, UVSQ, Institut Lavoisier de Versailles, Université Paris-Saclay, 45 avenue des Etats-Unis, 78035 Versailles Cedex, France. E-mail: michel.frigoli@uvsq.fr
bCenter for Organic Photonics and Electronics Research (OPERA), JST ERATO Adachi Molecular Exciton Engineering Project, Academia-Industry Molecular Systems for Devices Research and Education Center, Kyushu University, Nishi, Fukuoka, 819-0395, Japan. E-mail: mamada@opera.kyushu-u.ac.jp
cDivision of Chemistry, Institute of Liberal Education, Nihon University School of Medicine, Itabashi, Tokyo 173-8610, Japan
dDepartment of Engineering Science, The University of Electro-Communications, Chofu, Tokyo 182-8585, Japan
eInternational Institute for Carbon Neutral Energy Research (WPI-I2CNER), Kyushu University, Nishi, Fukuoka 819-0395, Japan
fEcole Polytechnique, Institut Polytechnique de Paris, LPICM, CNRS, route de Saclay, 91128 Palaiseau, France
First published on 16th September 2020
Abstract
Biradicaloid compounds with an open-shell ground state have been the subject of intense research in the past decade. Although diindenoacenes are one of the most developed families, only a few examples have been reported as active layers in organic field-effect transistors (OFETs) with a charge mobility of around 10−3 cm2 V−1 s−1 due to a steric disadvantage of the mesityl group to kinetically stabilize compounds. Herein, we disclose our efforts to improve the charge transport of the diindenoacene family based on hexahydro-diindenopyrene (HDIP) derivatives with different annelation modes for which the most reactive position has been functionalized with (triisopropylsilyl)ethynyl (TIPS) groups. All the HDIP derivatives show remarkably higher stability than that of TIPS-pentacene, enduring for 2 days to more than 30 days, which depends on the oxidation potential, the contribution of the singlet biradical form in the ground state and the annelation mode. The annelation mode affects not only the band gap and the biradical character (y0) but also the value of the singlet–triplet energy gap (ΔES–T) that does not follow the reverse trend of y0. A method based on comparison between experimental and theoretical bond lengths has been disclosed to estimate y0 and shows that y0 computed at the projected unrestricted Hartree–Fock (PUHF) level is the most relevant among those reported by all other methods. Thanks to their high stability, thin-film OFETs were successfully fabricated. Well balanced ambipolar transport was obtained in the order of 10−3 cm2 V−1 s−1 in the bottom-gate/top-contact configuration, and unipolar transport in the top-gate/bottom-contact configuration was obtained in the order of 10−1 cm2 V−1 s−1 which is the highest value obtained for biradical compounds with a diindenoacene skeleton.
Introduction
In the last ten years, open-shell polycyclic hydrocarbons (PHs) have been the subject of intense research owing to their potential applications in organic electronics and spintronics.1 Several kinds of delocalized open-shell compounds that can be written with a Kekulé resonance form have been prepared based on aromatic six-membered rings as acenes longer than 6 rings,2 anthenes,3 peri-acenes,4 rylene ribbons5 and acenoacenes6 or based on quinoidal compounds comprising only 6-membered rings (6-MR) as zethrene derivatives7 or with the inclusion of two 7-membered rings (7-MRs) as heptalene derivatives8 or 5-membered rings (5-MRs) in the π-conjugated core as bisphenalenyl derivatives9 and diindeno-based PHs.10–16 Among them, diindeno-based PHs are the most developed owing to their rather facile preparation at least for the small members. Since Kekuléan biradicaloid systems are resonance hybrids between the closed- and the open-shell forms, the contribution of each form in the ground state is described by the biradical character index (y0) that can be calculated according to Yamaguchi's scheme.17 The index spans from y0 = 0 for a pure closed-shell to y0 = 1 for a pure open-shell. Most of the biradicaloid PHs with an open-shell nature in the ground state, so-called biradicals in the literature, have singlet ground states owing to the double-spin polarization effect and usually low-lying triplet states.18 An analysis of the reported biradicaloid shows that the emergence of the open-shell form in the ground state can be observed for systems having a relatively large index y0 of ca. 0.6 calculated at the projected unrestricted Hartree–Fock (PUHF) level of theory. So far, the y0 values have been calculated by using different methods and basis sets, making the comparison difficult. The calculated y0 of the reported molecules at the PUHF/6-31+G(d,p) level shows a trend with the threshold of y0 ≥ 0.61 (Chart 1). Two systems with this index value have been reported by Haley's group, which are diindenoanthracene (DIAn)11 and indenoindenodibenzothiophene (anti-IIDBT)13 even though they have different singlet–triplet energy gaps (ΔES–T) of 4.2 and 8.0 kcal mol−1, respectively (Chart 1). This result highlights that the ΔES–T gap does not always follow the reverse trend of the biradical character (y0).13 However, usually, increasing the distance between the two radical centres within a family leads to an increase of y0 and a decrease of the HOMO–LUMO gap and ΔES–T.7,9a,13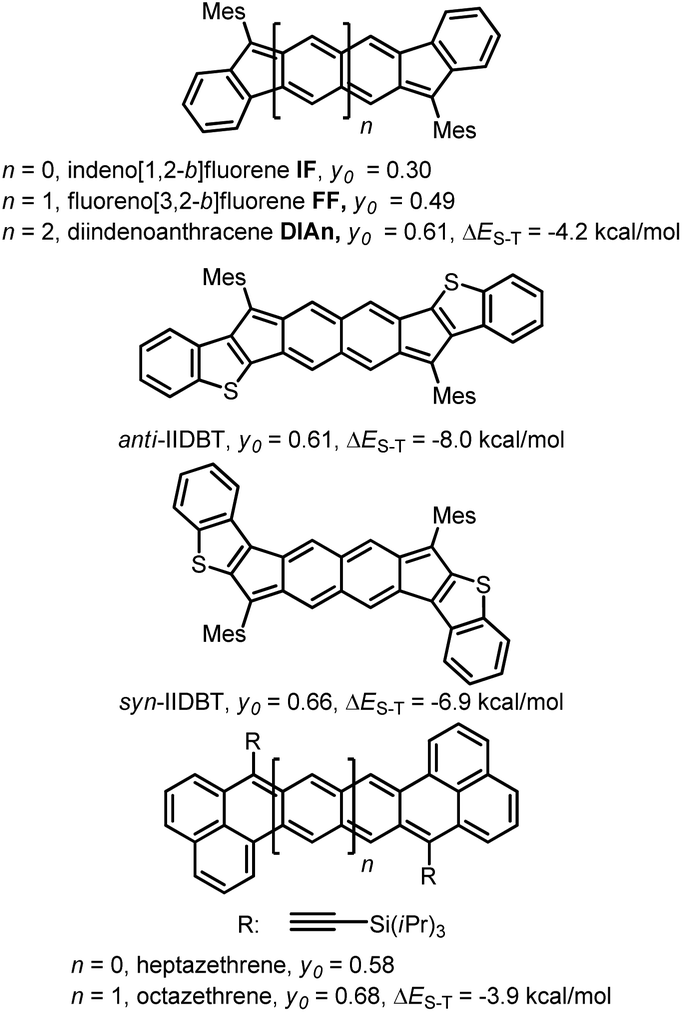 | ||
| Chart 1 Different PHs with their biradical character at the PUHF level of theory and experimental ΔES–T gaps. | ||
Recently, stable singlet biradical systems have emerged as a new class of ambipolar semiconductors in organic field-effect transistors (OFETs). For OFET application, the best singlet biradical system is a pentacenopentacene derivative that shows ambipolar transport >10−2 cm2 V−1 s−1 and hole mobilities over 1 cm2 V−1 s−1.6a Also, three systems belonging to the diindeno-based PH family have been used in OFETs for which ambipolar transport with well-balanced mobilities in a range of 10−3 cm2 V−1 s−1 for holes and electrons has been obtained.11,12,15 These modest mobilities are due to the fact that the apical position of the 5-MR, which is the most reactive position bearing the highest spin density, has to be kinetically blocked against the addition of oxygen to get a stable singlet biradical in the ground state by sterically hindered groups, typically a mesityl group which is not an excellent group to promote good π–π stacking in the solid state. Recently, the functionalization of the 5-MRs in dibenzo-indeno[1,2-b]fluorene derivatives with TIPS-ethynyl groups showing a closed-shell ground state has been proven to be an efficient way to obtain excellent semiconductors in OFETs.19 Consequently, improving the mobilities of diindeno-based PHs with a singlet open-shell ground state in OFETs could be eventually achieved if functionalized with TIPS-ethynyl groups. Since the 9-TIPS-ethynylfluoroenyl radical dimerizes promptly at the apical position of the 5-MR along probably with the oligomerization reaction, the biradical character of open-shell diindeno-based PHs should not be too high in order to get reasonable stability and the structures should be suitably functionalized to avoid dimerization.20 Consequently, we turned our attention to the hexahydro-diindenopyrene derivatives (HDIP) (Chart 2).
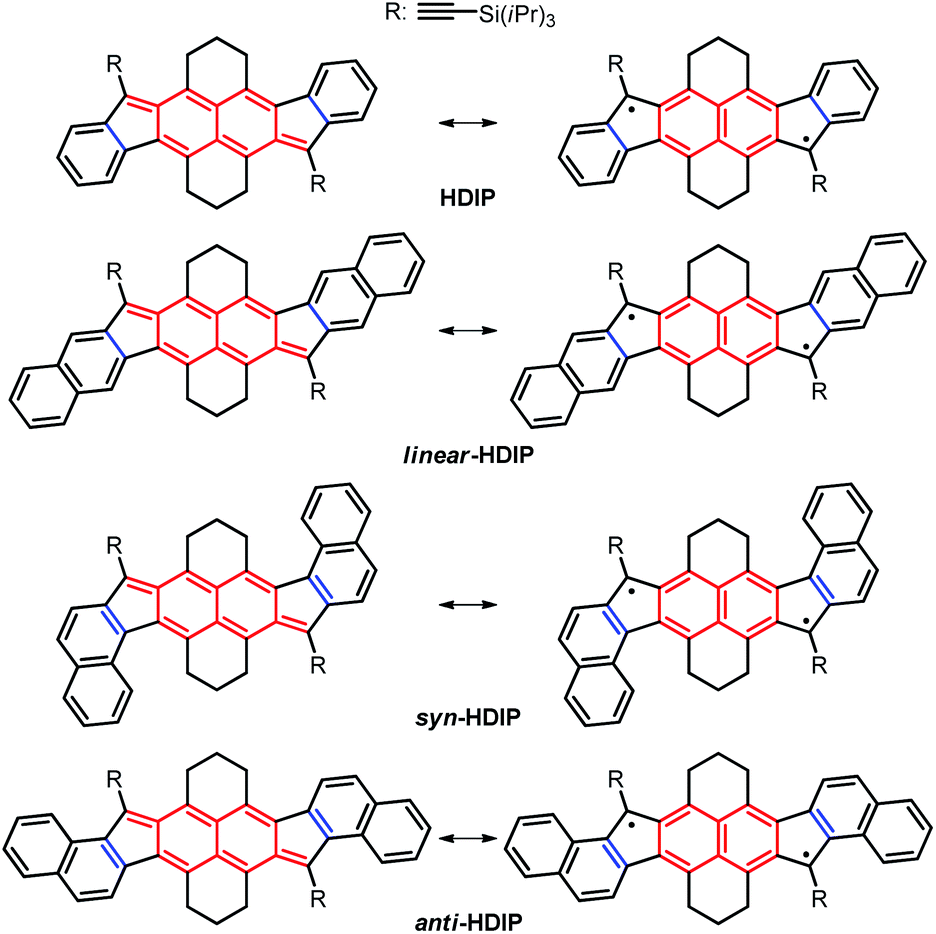 | ||
| Chart 2 Target molecules hexahydro-diindenopyrene (HDIP), linear-dibenzo-HDIP (linear-HDIP), syn-dibenzo-HDIP (syn-HDIP) and anti-dibenzo-HDIP (anti-HDIP). The bond orders at the fusion point (blue lines) are 1.50, 1.33, 1.66 and 1.66 for HDIP, linear-HDIP, syn-HDIP and anti-HDIP, respectively.21 | ||
The naming of the compounds follows the same utilized for dibenzo-indeno[1,2-b]fluorene isomers.21 The hexahydro-diindenopyrene skeleton possesses the same 2,6-naphthoquinodimethane (2,6-NQDM) core as the fluorenofluorene (FF) and IIDBT derivatives disclosed recently (Chart 1).13,22 All of them are functionalized with mesityl groups at the apical positions of the 5-MRs. The functionalization of the 2,6-NQDM core in diindeno-based PHs with TIPS-acetylene is still elusive even though the chosen synthetic path for the preparation of FF could have allowed us to introduce this functionalization.22
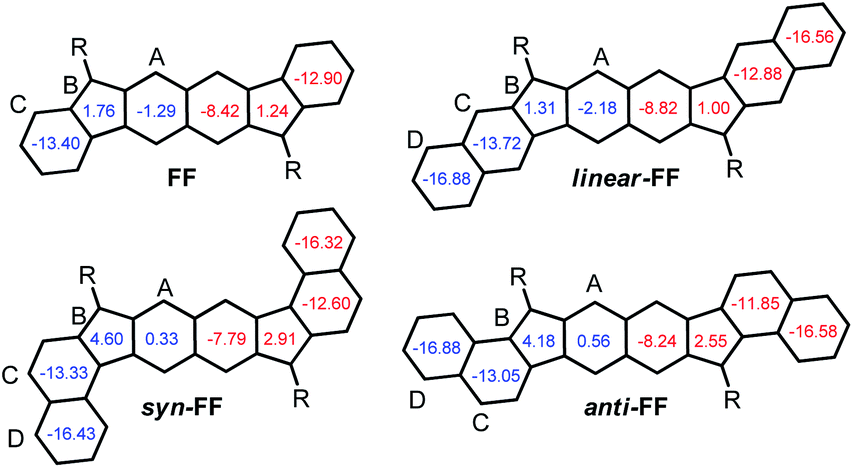
FF has a biradical index y0 of 0.49 and an optical band gap of 1.77 eV and has been categorized as a closed-shell in the ground state.22 Theoretical calculations forecast that the TMS-acetylene-functionalized FF derivative shows an increment of y0 by 0.09 to reach 0.58, which is comparable to that of heptazethrene. Further theoretical calculations on the dibenzo-FF isomers (see ESI†) indicate that dibenzo-fusions increase the biradical character to 0.59 for linear-FF and 0.64 for syn-FF. Note that the latter value exceeds those obtained for DIAn and anti-IIDBT (0.61). Moreover, the expected value of 0.67 for anti-FF approaches that of octazethrene (0.68). Accordingly, across this series of molecules, the contribution of the biradical form in the ground state would be modulated.
Moreover, in their own biradical forms, HDIP derivatives can be seen as the fusion of two benzo-fluorene radicals.23 Since dibenzo-fluorene-based radicals have the unpaired electron mainly delocalized at the fluorenyl moiety at specific positions depending on the annelation mode, the use of the HDIP central core instead of the naphthalene one appeared essential for the preparation of the compounds and very important to increase their stability.23
Herein, we report the synthesis, optoelectronic properties and OFET charge transport behaviour of four HDIP derivatives. Their ground states were discussed and assessed in terms of Variable-Temperature NMR experiments, X-ray structures and calculated and estimated biradical characters. These results were compared with those of other biradicaloid materials reported in the literature. A method to estimate the biradical character is also proposed.
Results and discussion
The synthesis path for the preparation of HDIP derivatives is depicted in Scheme 1. The synthesis starts with a cross-coupling reaction between the known 4,9-dibromo-1,2,3,6,7,8-hexahydropyrene and different boronic ester compounds to furnish the corresponding dicarboxylates 1a–1d in excellent yields. For 1c, two diastereoisomers with the trans/cis configuration could be separated by column chromatography and were obtained in a trans/cis ratio of 59/41 assuming that the trans diastereoisomers are the less polar products. Consequently, a double intramolecular Friedel–Crafts reaction using triflic acid as an activator was performed to yield diketones 2a–2d.24 Then, addition of lithium (triisopropylsilyl)acetylide on diketones 2a–2d followed by a stannous chloride-mediated reduction of the diol intermediates afforded HDIP derivatives with low to moderate yields (13% to 51%).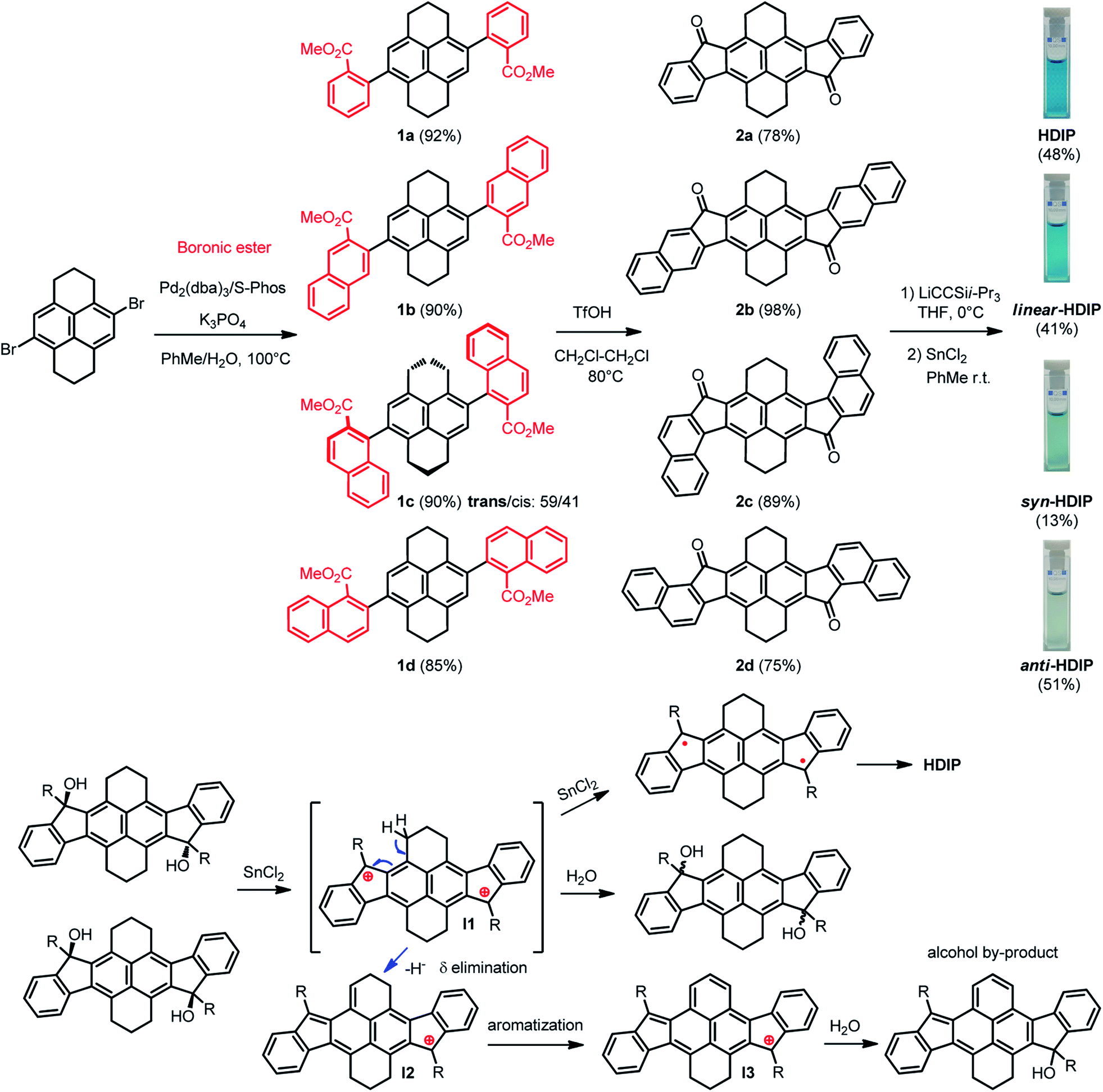 | ||
| Scheme 1 Synthetic path of HDIB derivatives and the proposed mechanism of the formation of an alcohol by-product. | ||
It is worthwhile to note that in the course of the reductive aromatization reaction, for all compounds, a violet spot in thin layer chromatography (TLC) with higher polarity was observed. This by-product was elucidated by means of single-crystal X-ray analysis and the X-ray structure corresponds to a mono-alcohol where one ring of the hexahydropyrene part is oxidized (Scheme 1 and see ESI†). Actually, the diol intermediates with a trans (less polar) or cis (more polar) configuration can be separated by column chromatography using neutral alumina gel.25 Starting from anti- or cis-diols in the presence of stannous chloride does not alter significantly the time and yield of the reaction (see ESI†). Indeed, racemization of the diols occurs in the course of the reaction, which can be easily followed by TLC. This racemization suggests that the key intermediate of the reductive aromatization might be a dication even though its formation is unclear.25 Tykwinski et al. suggest that the presence of water in stannous chloride and/or in solvent might produce a catalytic amount of HCl that would result in the formation of the (di)cation. The dication intermediate (I1) would explain the formation of the alcohol by-product that would begin with a δ elimination of a proton in the benzylic position to give intermediate (I2) followed by a prompt aromatization of the 6-membered ring (I3) and then addition of a water molecule.
Optical properties
The absorption behaviours were investigated in a toluene solution as shown in Fig. 1a. linear-HDIP and HDIP are sky blue in solution and have similar absorption behaviours with an absorbance maximum (λmax) at 698 and 700 nm, respectively. For both, the strong absorption at 698 and 700 nm can be assigned to a symmetry-allowed S0–S1 transition (see ESI†). syn- and anti-HDIP are teal- and light teal-green in solution and have a λmax shifted to the near-infrared (NIR) region of the spectrum with a λmax at 750 nm and 791 nm, respectively, that can be associated with a symmetry-allowed S0–S2 transition because of the symmetry-forbidden S0–S1 transition (see ESI†). Due to the latter, contrary to linear-HDIP, after the λmax, syn-HDIP displays a small transition that extends the absorption up to around 900 nm. anti-HDIP has also a relatively broad absorption band since the difference of 129 nm between the λmax (791 nm) and λedge (920 nm) is observed. Similarly, syn- and anti-IIDBT with the same 2,6-NQDM core have a λmax at 724 and 749 nm, respectively, and absorb up to around 825 nm. Thus, IIDBT and HDIP derivatives should have a similar biradical character since the emergence of the biradical character contribution in the ground state is related to the proaromaticity/aromatic stabilization of the biradical structure, the band gap and the number of π-electrons.14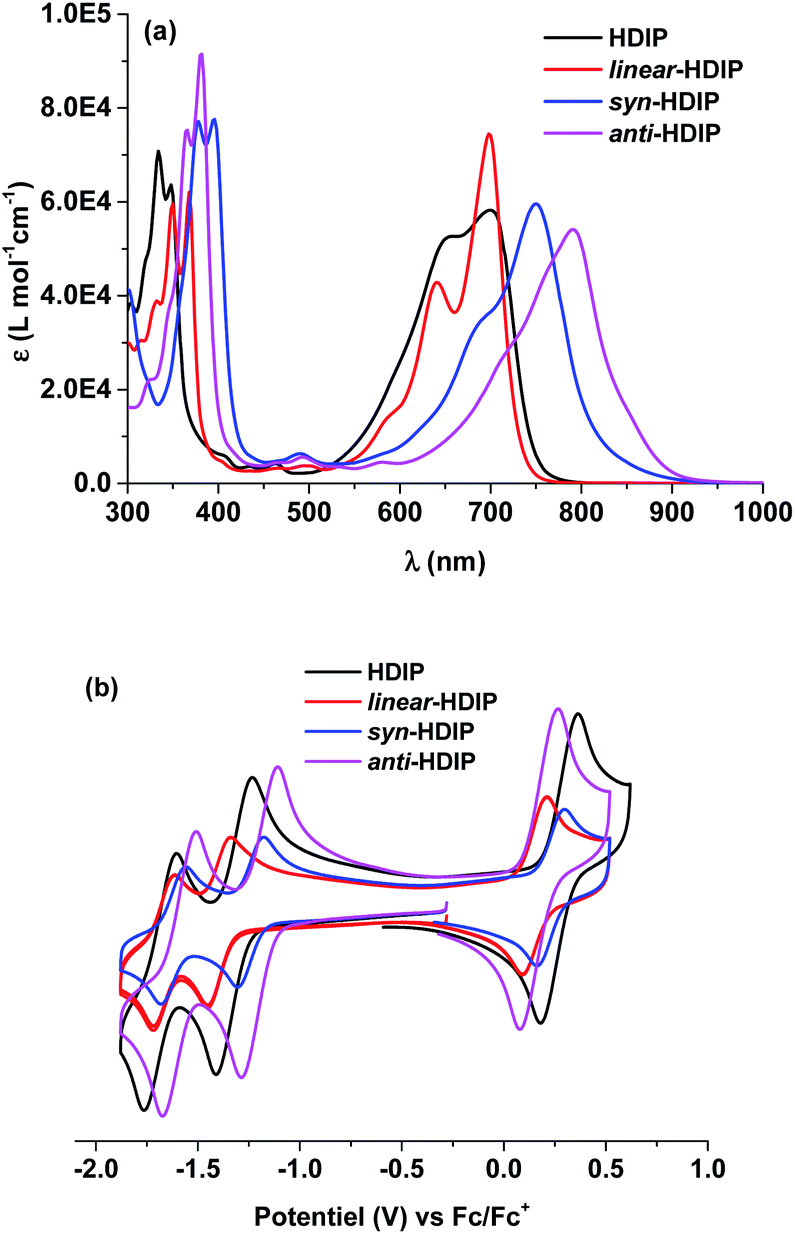 | ||
| Fig. 1 (a) Absorption in toluene and (b) cyclic voltammetry of HDIP derivatives in chlorobenzene. | ||
Electrochemistry
Cyclic voltammetry (CV) was performed to investigate the electrochemical behaviours and probe HOMO/LUMO energy levels of the molecules. For the solubility issue, electrochemical measurements were performed in chlorobenzene and half-wave potentials were determined relative to the ferrocene/ferrocenium (Fc/Fc+) redox couple. CVs of the HDIP molecules are shown in Fig. 1b. All the compounds show amphoteric redox behaviours and exhibit one reversible oxidation wave and two reversible reduction waves. linear- and anti-HDIP show almost the same oxidation peaks with half-peak potential Eox1/2 at 0.15 V and 0.17 V, respectively, while syn-HDIP and HDIP undergo oxidation at higher potentials of Eox1/2 = 0.23 V and 0.27 V, respectively. For the first reduction, the chemical trend is changed. anti-HDIP and syn-HDIP get reduced at lower potentials (Ered1/2) of −1.20 V and −1.26 V, respectively, than those of HDIP and linear-HDIP that get reduced at −1.32 V and −1.40 V, respectively. The HOMO/LUMO energy levels were then estimated to be −4.97 eV/−3.60 eV for anti-HDIP, −5.03 eV/−3.54 eV for syn-HDIP, −4.95 eV/−1.40 eV for linear-HDIP and −5.07 eV/−3.48 eV for HDIP. From the above data, the electrochemical HOMO–LUMO gaps were deduced and are in good agreement with the optical band gap. anti-HDIP has the lowest band gap of 1.37 eV, followed by syn-HDIP with a band gap of 1.49 eV. linear-HDIP and HDIP have a slightly higher band gap than 1.50 eV, which are estimated at 1.55 eV and 1.59 eV, respectively. As reported for indeno[1,2-b]fluorene isomers, the reduction potential is driven by the degree of aromaticity of the cyclopentadienyl anion generated upon reduction which is in turn related to the bond character at the fusion point marked in blue as shown in Chart 2.21 The more that bond has a double character, the more the aromaticity of the cyclopentadienyl anion increases and the more the reduction is facile.Variable-temperature (VT) 1H NMR measurements
Typically, a biradicaloid with a singlet open-shell ground state has usually a low-lying triplet state. Since the triplet state is a paramagnetic species, it can be highlighted by 1H NMR spectroscopy. Accordingly, VT 1H NMR measurements were performed in ortho-dichlorobenzene (o-DCB) from 20 to 140 °C (Fig. 2 and see ESI†). Surprisingly, only anti- and linear-HDIP show signal broadening from 80 °C onwards and the signals do not collapse completely even at 140 °C, suggesting a relatively large energy difference (ΔES–T) between singlet and triplet states. Since the alteration of the signals starts at the same temperature, ΔES–T of anti- and linear-HDIP should be similar. It is worthwhile to note that even the aliphatic signals H1 and H3 in the benzylic positions broaden for anti-HDIP and only H1 for linear-HDIP. The spin density in the α position of H1 is significant enough to alter its signal for both compounds. The alteration of the H3 signal in anti-HDIP is probably due to the concomitant effect of the spin density in the α position of H3 in the naphthalene central core and through-space interaction between H3 and the significant spin bears by carbon C3c since the distance between carbons C3 and C3c is 3.1 Å less than the sum of the van der Waals radius for two carbons (3.4 Å). For HDIP, only a tiny broadening of H1 is observed at 140 °C but not for syn-HDIP for which all the signals remain unchanged (see ESI†), suggesting that the ΔES–T for syn-HDIP is the largest.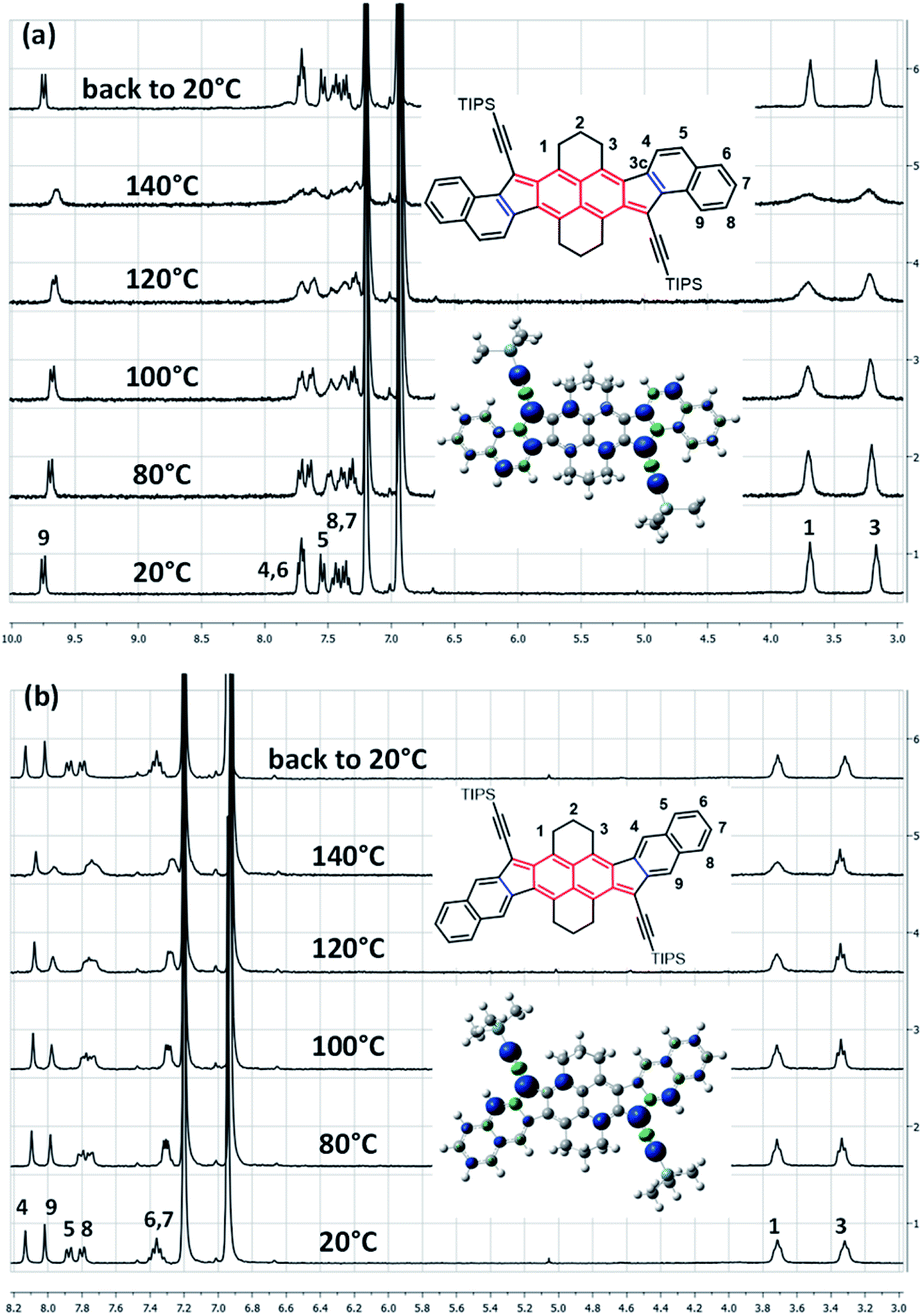 | ||
| Fig. 2 VT 1H NMR spectra in o-DCB-d4 of (a) anti-HDIP and (b) linear-HDIP and the calculated spin density of the triplet state. | ||
ESR and SQUID measurements
ESR and SQUID measurements that give quantitative ΔES–T were unfruitful (see ESI†), because the responses for all the HDIP samples showed anomalous paramagnetic behaviour. All the compounds were ESR active at room temperature and the signals increase with decreasing the temperature. Such anomalous behaviour may originate from the presence of free radicals as observed only in the condensed phase (not in solution) in planar π-conjugated systems.26 Andrew et al. have proposed that the unexpected paramagnetism was derived from a submicromolar concentration of radical cations generated through exposure to an ambient atmosphere (oxygen, water) and light even for extra-purified samples. Due to the presence of free radicals, the magnetic susceptibilities of HDIP derivatives show much higher paramagnetic susceptibilities than that usually obtained from the contribution of the excited triplet state, precluding the estimation of the ΔES–T (see ESI†).7,11,13,16X-ray crystallographic analysis
Fortunately, for all the compounds, single crystals suitable for crystallography analysis were obtained by recrystallization (reliability factor (R) less than 0.06 for linear-HDIP and less than 0.05 for the others). For HDIP, linear- and anti-HDIP, the crystals have a triclinic symmetry with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, whereas for syn-HDIP the crystals have a monoclinic symmetry with the P21/c space group. Note that the crystal of linear-HDIP includes mesitylene solvent molecules, which likely caused a small increase of the R factor. The bond lengths and the packing are disclosed in Fig. 3. For all, the propane groups are up and down to each other. They are almost planar except syn-HDIP that has a helical structure. The strain of the helical structure characterized by ψtot = 40° (the sum of dihedral angles for the atoms of the inner pitch starting from C3 to C4) and the distance d of 3.114 Å between C3 and C4 is lower than that observed for carbo[5]-helicene (ψtot = 67°, d = 2.933 Å).27 HDIP adopts a 2D “bricklayer” arrangement but the π-stacking is only observed in one direction between the molecule in red and the molecules in blue. The short-contacts between the molecule in red and the molecules in green concern only CH–π interactions between propane groups and the outer rings. There are 6 closed contacts between neighbouring molecules (red and blue) in π–π interactions with distances spanning from 3.515 to 3.789 Å. syn- and anti-HDIP exhibit a slipped 1D π-stacking motif. For syn-HDIP, the π–π interactions involved only the two outer rings separated by a distance of 3.492 Å. For linear- and anti-HDIP, 5 rings of adjacent molecules are in intermolecular π–π interactions with an interplanar distance of 3.468 Å and 3.422 Å, respectively.
space group, whereas for syn-HDIP the crystals have a monoclinic symmetry with the P21/c space group. Note that the crystal of linear-HDIP includes mesitylene solvent molecules, which likely caused a small increase of the R factor. The bond lengths and the packing are disclosed in Fig. 3. For all, the propane groups are up and down to each other. They are almost planar except syn-HDIP that has a helical structure. The strain of the helical structure characterized by ψtot = 40° (the sum of dihedral angles for the atoms of the inner pitch starting from C3 to C4) and the distance d of 3.114 Å between C3 and C4 is lower than that observed for carbo[5]-helicene (ψtot = 67°, d = 2.933 Å).27 HDIP adopts a 2D “bricklayer” arrangement but the π-stacking is only observed in one direction between the molecule in red and the molecules in blue. The short-contacts between the molecule in red and the molecules in green concern only CH–π interactions between propane groups and the outer rings. There are 6 closed contacts between neighbouring molecules (red and blue) in π–π interactions with distances spanning from 3.515 to 3.789 Å. syn- and anti-HDIP exhibit a slipped 1D π-stacking motif. For syn-HDIP, the π–π interactions involved only the two outer rings separated by a distance of 3.492 Å. For linear- and anti-HDIP, 5 rings of adjacent molecules are in intermolecular π–π interactions with an interplanar distance of 3.468 Å and 3.422 Å, respectively.
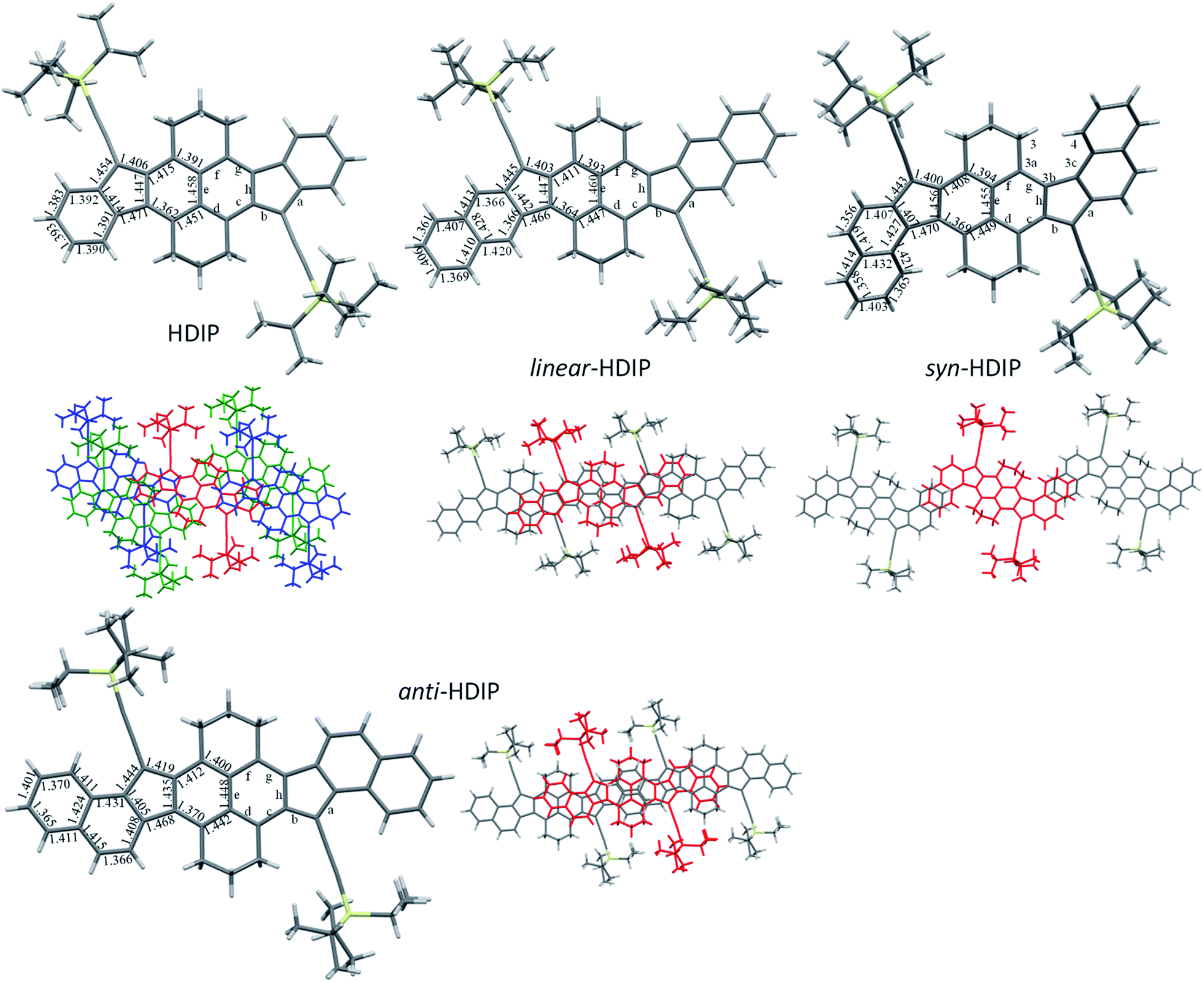 | ||
| Fig. 3 X-ray structures and packing of HDIP derivatives. | ||
Bond length analysis is believed to give insight into the ground state electronic structure of diindeno-based PHs.11 For biradicaloid categorized as a closed-shell in the ground state as for instance in indeno[1,2-b]fluorene derivatives, the bond length a is usually about 1.470 ± 0.05 Å which is a bond length value typically observed between two sp2 carbons and the bond length b considerably increases along with the biradical character and takes a value between 1.380 and 1.401 Å.21,28,29 In the case of biradicaloid systems that have a resonance hybrid between the closed- and open-shell in the ground state, the bond lengths should be intermediate between a quinoidal structure with bond length alternations and the biradical form. In their biradical forms at least for the dibenzo-HDIP derivatives, they can be seen as the fusion of two dibenzo-fluorene radicals. Thankfully, the X-ray structures of linear- and syn-fluorene radicals are known.23 Since they are symmetric molecules, the radical splits in the same manner in the two outer rings leading to identical bond lengths of the 5-MR apical bonds (1.435 Å and 1.438 Å for syn- and linear-fluorene radicals, respectively) and the bond lengths of the two naphthalenes are not much altered. Therefore, at least for the dibenzo-HDIP derivatives, the apical bond lengths a and b should tend to be closer and closer to each other with increasing the biradical character with bond b shorter than bond a due to the closed-shell form contribution in the ground state. Also, bond lengths c and f should decrease whereas bond lengths d and g should increase due to the partial retrieve of the naphthalene central core in the biradical form.
For the dibenzo-HDIP derivatives, the bond lengths a are similar (1.443–5 Å) but the bond lengths b differ from 1.400 Å and 1.403 Å for syn- and linear-HDIP, respectively, to 1.419 Å for anti-HDIP, indicating that the latter has a more important contribution of the biradical form in the ground state, whereas syn- and linear-HDIP have a similar ground state. This statement is also seen with the bond lengths c, d, g and f. For HDIP, the bond length b (1.406 Å) is slightly longer than that of syn- and linear-HDIP. However, bond lengths of c and f are the longest while the bond lengths of d and g are the shortest suggesting that the contribution of the biradical form in the ground state for HDIP is likely to be slightly less important. According to the crystallographic analysis, the biradical character should increase in this order: HDIP < linear-HDIP ≈ syn-HDIP < anti-HDIP.
Theoretical calculations
For a better understanding of the electronic nature of HDIP derivatives, computational calculations were performed using Gaussian 16 except for odd-electron density and nucleus-independent chemical shift (NICS) calculations (see ESI† for details).30 The biradical character was estimated from the electron occupancies of the frontier natural orbitals through Yamaguchi's scheme (y0) at the PUHF/6-31+G(d,p) level of theory and computed from an optimized geometry obtained at the R-B3LYP/6-311G(d,p) level.17,31 This is perfectly coherent since the optimized structures obtained at the R-B3LYP level correspond fairly well to the X-ray HDIP structures and also other biradicaloid systems with an open-shell ground state when the crystal structure is of high quality (see ESI†).7a,16 The results are gathered in Table 1 along with the values obtained for FF derivatives, their very closed counterparts and the calculated apical bond length b. For FF derivatives, the biradical character increases with the increase of the apical bond length b with a little exception for linear-FF that has the lowest bond length. The same trend is obtained for DIAn derivatives.16| Compoundsa | y 0 | Bond bc | ΔES–Td | E g |
|---|---|---|---|---|
| a All the compounds are functionalized with TMS-ethynyl groups and accept native compounds. b Calculated at the PUHF/6-31+G(d,p) level of theory in Å. c At the R-B3LYP/6-311G(d,p) level. d At the B3LYP/6-311G(d,p) level in kcal mol−1. e Electrochemical energy gap in eV. f Experimental value taken from ref. 14. g Optical band gap. h Taken from ref. 14. | ||||
| Native FF | 0.48 | 1.382 | −9.69 | — |
| FF | 0.58 | 1.404 | −6.01 | — |
| linear-FF | 0.59 | 1.403 | −5.73 | — |
| syn-FF | 0.64 | 1.406 | −5.84 | — |
| anti-FF | 0.67 | 1.417 | −5.11 | — |
| Native HDIP | 0.46 | 1.382 | −11.70 | — |
| HDIP | 0.56 | 1.409/1.406(2)exp | −7.26 | 1.59 |
| linear-HDIP | 0.58 | 1.406/1.403(3)exp | −6.52 | 1.55 |
| syn-HDIP | 0.59 | 1.407/1.400(2)exp | −8.49 | 1.49 |
| anti-HDIP | 0.65 | 1.423/1.419(2)exp | −6.49 | 1.37 |
| anti-IIDBT | 0.61 | — | −7.48 (−8.0)f | 1.50g |
| syn-IIDBT | 0.66h | — | −6.84 (−6.9)f | 1.50g |
Compared to FF derivatives, surprisingly, the HDIP derivatives have an index y0 slightly reduced by 0.02 except for syn-HDIP for which the reduction is more important (0.05) even if the bond b is genuinely longer for HDIP derivatives than that obtained for FF derivatives. A longer bond b for HDIP derivatives can be attributed to the steric repulsion between the aliphatic bridge and the ethynyl function. The distance between these two groups is less than 3.1 Å, which is less than the sum of the van der Waals radius between two carbons (3.4 Å). Since native FF and native HDIP have the same bond length b because of no steric repulsion, the reduction of y0 can be attributed to a little hypsochromic shift of the main transition in the vis/NIR region going from FF- to HDIP derivatives (see ESI†). syn-HDIP has a [5]-helicene-like structure that reduces the apical bond length b which becomes lower than that obtained for HDIP and consequently leads to a decrease of y0 more important than expected. Even if syn-HDIP has a lower band gap than HDIP and linear-HDIP, they have similar y0 (0.56–0.59). It has been shown that increasing the twisting of the helical backbone of diindenopicene derivatives leads to a decrease of the biradical character and an increase of ΔES–T gap while the optical behaviours are very similar.12anti-HDIP has the highest biradical character and the lowest band gap.
Regarding the ΔES–T, it does not follow the reverse trend of the biradical character but agrees well with the VT 1H NMR experiments. anti-HDIP and linear-HDIP have the lowest ΔES–T, while syn-HDIP has the highest one. An intermediate ΔES–T is obtained for HDIP. These theoretical results alongside with VT 1H NMR experiments indicate that the ΔES–T gap can be significantly modulated with the annelation mode. These results are in line with the behaviours of anti and syn-IIDBT.13,14 According to our theoretical calculations, syn-IIDBT has a ΔES–T close to those of linear- and anti-HDIP and the signal broadening in 1H NMR of syn-IIDBT starts at 75 °C which is close to the temperature obtained for linear- and anti-HDIP (80 °C).14 In a similar manner, HDIP and anti-IIDBT have similar ΔES–T and the signal broadening of protons in 1H NMR starts at similar temperatures (125 °C and 120–140 °C for anti-IIDBT and HDIP, respectively).13 Even though the results of VT 1H NMR experiments are not quantitative but rather qualitative, there is a clear correlation between the starting temperature of proton broadening and the calculated ΔES–T gap.
The trend of ΔES–T values for HDIP derivatives is different from that observed recently in dibenzo-DIAn derivatives which possess the anthracene central core and the mesityl group at the apical position.16 However, they are not strictly comparable since HDIP derivatives have the TIPS-ethynyl group at the apical position and two aliphatic bridges at the central core. Since the ΔES–T of native FF follows the reverse trend of the biradical character according to the calculation (see ESI†), the functionalization plays an important role in ΔES–T.7b These results emphasize the fact that a lower ΔES–T is not always correlated with a higher biradical character and a lower band gap even within a family.7b,16
So far, systems that are considered as biradicaloids with an open-shell ground state are those for which the thermally activated triplet state is close enough to the ground singlet state in energy. However, due to a strong spin polarization effect, some systems could be biradicals in the ground state even though they have large ΔES–T. For instance, TIPS-heptazethrene7a,b and TIPS-hexacene7b are considered as a closed shell in the ground state while the calculated y0 is 0.58 at the PUHF,6 and this value is very close to that of DIAn and anti-IIDBT (0.61). Also, the calculation of y0 strongly depends on the functional used. TIPS-heptazethrene and a diindenopicene derivative (open-shell) have a y0 of 0.58 and 0.64, respectively, at the PUHF, while the y0 at the UCAM-B3LYP drops to 0.16 and 0.23, respectively.7,12 DIAn (open-shell) has a y0 of 0.62 at the PUHF, while the y0 is only 0.21 at the CASCI.16 Considering systems as biradicals in the ground state while the contribution of the biradical form in the ground state would be less than 50% appears somewhat strange. Therefore, finding a way to estimate the biradical character would constitute a breakthrough in the domain.
In general, in the literature, two functionals such as the B3LYP and the CAM-B3LYP are used with the restricted approach to determine a closed-shell equilibrium structure (CS) and with the unrestricted approach to determine the open-shell broken symmetry equilibrium structure (BS). The latter is possible when a more stable open-shell BS solution is found. However, the optimized geometries using these two functionals give different states of the molecules. The four calculated structures of HDIP are provided in Fig. 4 and the structures of the other derivatives can be seen in the ESI.†
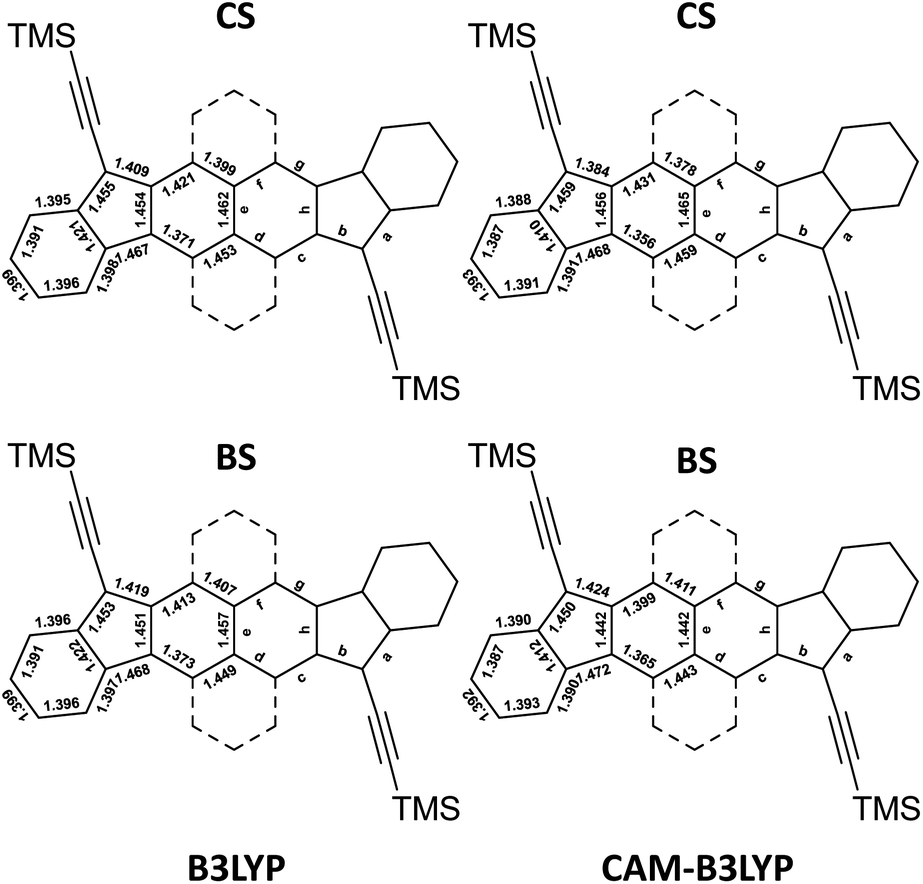 | ||
| Fig. 4 Bond lengths of CS and BS solutions calculated at B3LYP (left) and CAM-B3LYP (right) levels for HDIP. | ||
As mentioned earlier, the CS solution at the B3LYP level gives a fairly good representation of the X-ray structures for HDIP derivatives and other biradicaloid systems when the measurements are made on a good quality crystal.16 Therefore, the structure calculated at the R-B3LYP level is a resonance hybrid between the closed- and open-shell forms in the ground state and not a representation of the quinoidal resonance structure when the BS solution is lower in energy. The CS and BS solutions at the CAM-B3LYP level are more in agreement with expected structures for a quinoidal closed-shell and a biradical form, respectively. Indeed, the CS solution shows a shorter apical bond length b and the bond lengths c and f are much longer than those of d and g. The BS solution at the CAM-B3LYP level shows a longer bond length b and the bond lengths c and g are shorter than those of d and f as expected for the naphthalene moiety. It should be noted that the bond length c is longer than g and the bond length d is shorter than f. This trend is also seen in the X-ray structure of the linear-fluorene free radical and can be ascribed to the spin delocalization which is more important in the bonds c than in g.23 Considering these two canonical forms, it is possible to deduce the biradical character by calculating the contribution of each form from the apical bond length b which is the most altered bond between the two canonical forms as bexp = bCS × (1 − y0) + bBS × y0, where bexp, bCS and bBS are the apical bond lengths b of the X-ray structure and of the CS and BS solution calculated at the CAM-B3LYP level, respectively, and y0 is the biradical character. Applying the found coefficients to all the bond lengths of interest, the resulting hybrid structures are matching very well with the X-ray structures (see ESI†). Accordingly, the biradical character would be in a range of 0.55 ± 0.05, 0.48 ± 0.07, 0.53 ± 0.05 and 0.57 ± 0.05 for HDIP, linear-, syn- and anti-HDIP, respectively, by taking into account the standard deviation of the bond length b found in the X-ray structures. For linear-HDIP, the calculation is slightly less accurate since the quality of the X-ray measurement is slightly less good with the R factor above 0.05. The estimation of y0 by this way is fairly in agreement with the calculated y0 at the PUHF level within the uncertainties of the X-ray structures and the CS and BS solutions at the CAM-B3LYP level. Therefore, the calculated y0 at the PUHF level is the most relevant among those reported by all other methods and indicates that a system considered as a biradical in the ground state should have a contribution of the biradical form more than 50% in the ground state. It could be argued that CS and BS solutions could still not be a pure closed- and open-shell since diindenothienothiophene having a very low biradical character functionalized with TIPS-ethynyl groups has an apical bond length of around 1.370 Å.32 Nevertheless, decreasing the apical bond length of the CS solution and increasing that of the BS solution with the same gap would give rise to the same biradical character. This methodology works well for X-ray structures with an R factor less than 0.05. Indeed, it also works for linear-DIAn (0.65 ± 0.05), TIPS-heptazethrene (0.56 ± 0.05), and octazethrene (0.67 ± 0.05) (see ESI†).
For all the HDIP compounds, the driving force to be a biradical in the ground state is the same going from a proaromatic quinoidal 2,6-NQDM core to the aromatic stabilization of the naphthalene in the biradical form. NICS computations using π-only methods (NICS(1.7)πzz) of the CS (blue) and BS (red) solution at the CAM-B3LYP level support this statement as shown in Fig. 5.30 Since NICS computations were difficult on HDIP derivatives, the calculations were made on FF derivatives with the TMS-ethynyl group at the apical position instead and odd electron density maps for HDIP derivatives were also calculated (Fig. 6).11,16
 | ||
| Fig. 5 NICS (1.7)πzz values in ppm inside the rings for CS (blue) and BS (red) solutions at the CAM-B3LYP level for FF derivatives with the TMS-ethynyl group. | ||
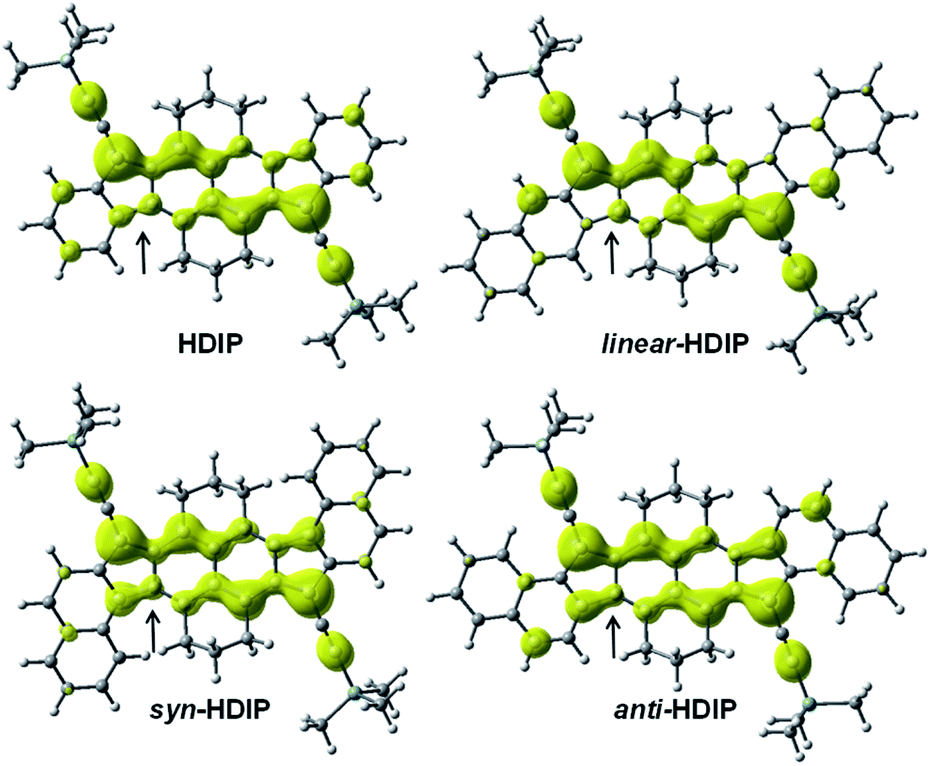 | ||
| Fig. 6 Odd electron density maps for HDIP derivatives, calculated at the LC-UBLYP/6-311+G(d,p) level with a contour value of 0.001 a.u. | ||
For the quinoidal form (blue), the central rings A are nearly atropic for all since the NICS values are close to 0 and should be considered as non-aromatic rings. In contrast, in the biradical form (red), the rings A sustain a diatropic ring current of around −8 ppm indicating a partial retrieve of the aromaticity of the naphthalene ring. Due to the delocalization of the spins on both rings A as seen in odd electron density maps (Fig. 6), the aromaticity is reduced compared to a naphthalene (−18 ppm). The difference of the tropicity values of the rings A for the quinoidal and biradical forms depends somehow on the strength of the paratropicity of the 5-MRs which in turn depends on the bond order at the fusion point. The higher the bond order at the fusion point is, the higher the paratropicity of the 5-MRs is. Furthermore, the paratropicity of the 5-MRs is slightly reduced from the quinoidal to the biradical form. The farer outer rings D for dibenzo-HDIP derivatives show similar values and the diatropic ring current of the rings D is higher than that of C, which indicates that the aromaticity is more localized on the rings D compared to C.23 The aromaticity difference of the rings D between the quinoidal and biradical form is slightly more important for linear- and anti-FF (≈0.30 ppm) than that for syn-FF (0.11 ppm) but the delocalization of the spins in the biradical form is small as seen in odd spin density maps. In contrast, the diatropicity of the rings C is more altered and decreases from the quinoidal to the biradical form. The most difference is observed for the anti-FF derivative (1.20 ppm), which indicates a higher spin delocalization on the rings C followed by linear- (0.84 ppm) and syn-FF (0.73 ppm). Actually, there is a clear correlation between the spin delocalization at the outer rings and the ΔES–T. As can be seen in Table 1 for FF derivatives, the ΔES–T is lower when the delocalization is higher, indicating that increasing the distance between the two radical centers decreases the ΔES–T. However, for this series of molecules for which the driving force to be biradical in the ground state is the same, the biradical character is first governed by the HOMO–LUMO gap and then by the delocalization at the outer rings. The HOMO–LUMO gap primarily depends on the nature of the 5-MR. The more the 5-MR is antiaromatic, the more the molecule absorbs at longer wavelengths since the reduction potential is more altered than the oxidation one. However, due to a higher spin delocalization at the outer rings for anti-compared to syn-HDIP, anti-HDIP gets reduced and oxidized at a lower potential.
Since HDIP derivatives have a higher ΔES–T than FF derivatives, it may be inferred that the presence of the two aliphatic bridges decreases the delocalization at the outer rings by stabilizing the spins at the centre of the molecule by the electronic effect. Due to the helical structure of syn-HDIP, the delocalization of the spins at the outer rings might be even less important leading to an increase of ΔES–T.12
As can be seen from the odd electron density maps (Fig. 6), the spin densities are similar in the central core but different at the outer rings and also at the 5-MRs as indicated by the black arrows due to the bond order at the fusion point (Chart 2, blue bonds) and the conjugation mode. The odd electron density maps show clearly the importance of the TIPS-ethynyl groups for the increase of the biradical character due to an increase of the spin delocalization and an increase of the distances between the two radical centers. They also show the importance of the two aliphatic bridges to protect the central core towards oxidation and to hamper the possible dimerization (oligomerization) at the apical and ethynyl positions.
Stability test
Stability experiments of the molecules in a saturated air–toluene solution under room light conditions were performed by following the decrease of absorbance at their own λmax in the course of time (Fig. 7, see ESI†).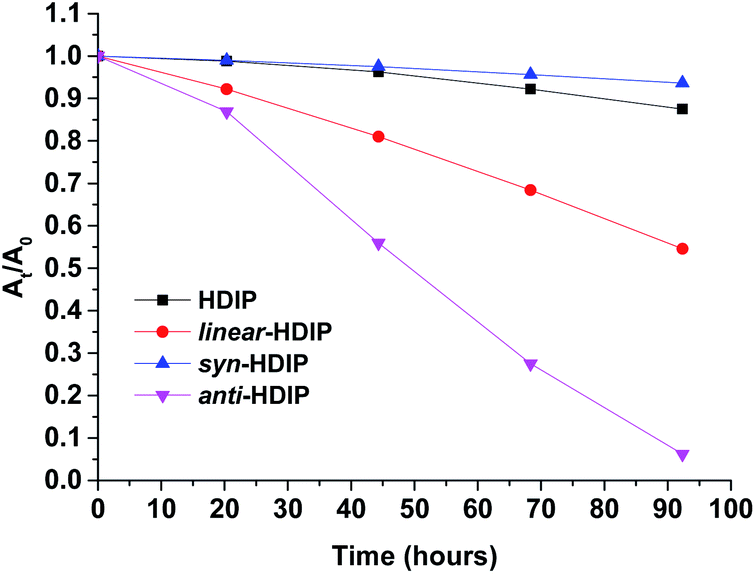 | ||
| Fig. 7 Change of UV/Vis absorption (followed at their own λmax) over time in toluene (26.6 μm) of HDIP derivatives. | ||
The least stable is anti-HDIP with a half-life (τ1/2) of 2.1 days followed by linear-HDIP having a τ1/2 value of 4.3 days. HDIP and syn-HDIP have great stability and lose 13% and 6%, respectively, of the original absorbance after 5 days. Actually, all the compounds are more stable than TIPS-pentacene (τ1/2 = 1.7 days).6a The order of stability is correlated with the first oxidation potential as known for instance in the acene series and dibenzo-fluorene-based radicals.23anti- and linear-HDIP have the lowest oxidation potential of the series and consequently they are the least stable. The difference of stability between the anti- and linear-HDIP can be associated with the biradical character. Since the contribution of the biradical form in the ground state increases from linear- to anti-HDIP, the stability decreases from linear- to anti-HDIP. syn-HDIP is the most stable. This result may be explained by the spin delocalization at the outer rings which is mainly on internal quaternary carbons for syn-HDIP and on tertiary carbons more facile to be oxidized for the others. Indeed, the stability of HDIP derivatives follows the trend observed in dibenzo-fluorene-based radicals.23 It is worthwhile to note that TIPS-heptazethrene and linear-HDIP have the same half-lives of 4 days and have the same calculated biradical character (0.58).7aanti-HDIP is slightly more stable (2.1 days, y0 = 0.65) than TIPS-octazethrene (1.4 days, y0 = 0.68) and has a slightly less biradical character.7a
OFET devices
According to the π–π stacking structures revealed by the single crystal analysis, HDIP derivatives are expected to have excellent charge transport capabilities. The small HOMO–LUMO energy gaps lower than 1.5 eV owing to a moderate biradical character enable the injection of both holes and electrons, and thus biradicaloid compounds are promising for high performance bipolar semiconducting materials. However, the mobilities have remained at a low level so far, probably due to the limited intermolecular π–π interactions with bulky protection groups. Therefore, the potential of charge transport behaviours for biradicaloid compounds such as diindeno-based PHs has not been fully explored. Here, we fabricated OFET devices for HDIP derivatives with bottom-gate/top-contact (BG/TC) and top-gate/bottom-contact (TG/BC) configurations (see ESI†). Thin films of semiconductor layers were formed by drop-casting of an o-DCB solution with polystyrene. The polymer blends are known to lead to better mobility and uniformity.33Interestingly, HDIP and linear-HDIP showed unipolar p-type characteristics under any conditions, whereas syn-HDIP and anti-HDIP exhibited bipolar transport (Fig. 8 and Table 2), clearly related to deeper LUMO levels (Fig. 1b). The hole and electron mobilities of anti-HDIP in the BG/TC device were estimated to be relatively balanced as 4.5 × 10−3 and 1.1 × 10−3 cm2 V−1 s−1, respectively. The TG/BC device with a CYTOP dielectric showed improved hole mobilities higher than 0.1 cm2 V−1 s−1 for HDIP, linear-HDIP and anti-HDIP, which are among the best reported for singlet biradical compounds based on diindeno-based PHs. The n-type characteristics were not clear, although the current increase at VG = 0 V in the output scans indicates an electron injection possibility. Thus, it is attributed to largely unbalanced charge transport behaviours. Although the mobilities of syn-HDIP were slightly improved in the TG/BC device, those values of 10−3 cm2 V−1 s−1 are in the same range as those of DIAn and other compounds, indicating that a small region of orbital overlaps had limited the charge transport.
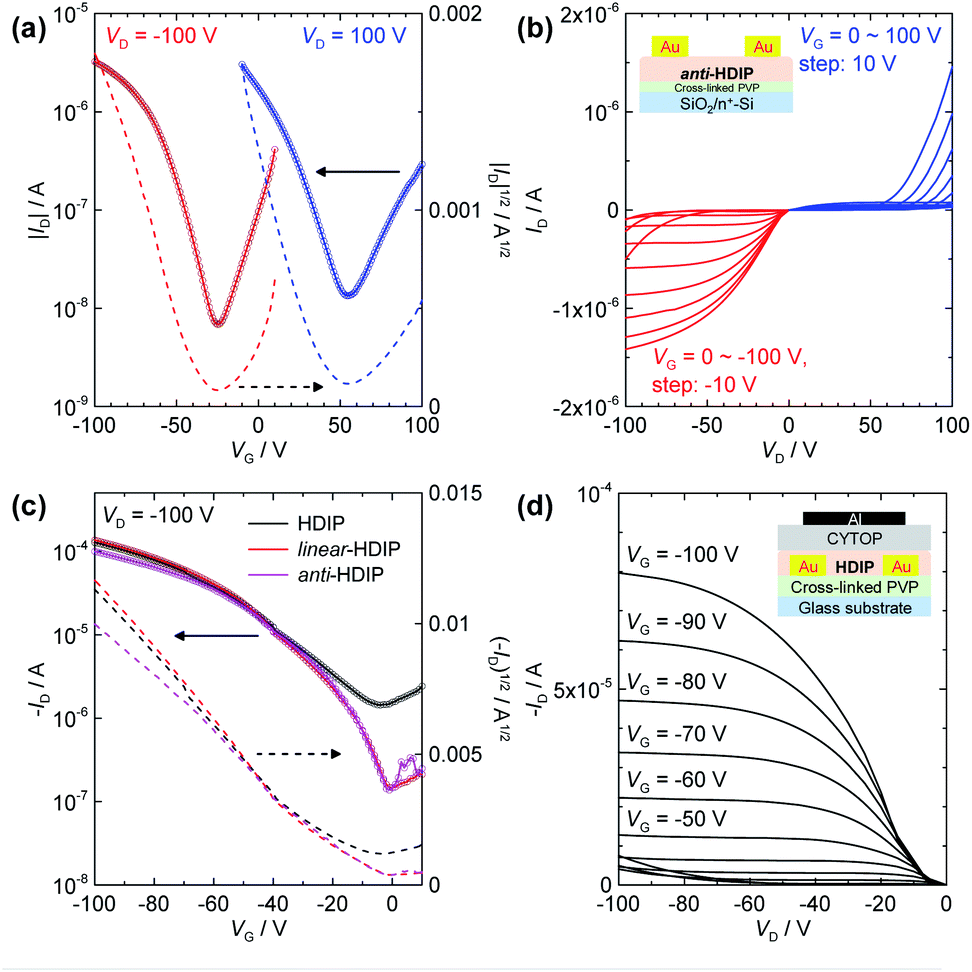 | ||
| Fig. 8 OFET devices of HDIP derivatives. (a) Transfer and (b) output characteristics of a BG/TC device with a drop-cast anti-HDIP. (c) Transfer and (d) output characteristics of TG/BC devices with a drop-cast HDIP (black lines), linear-HDIP (red lines), and anti-HDIP (magenta lines). L/W = ca. 50/2000 μm. | ||
| Compd | Struct.a | μ h (cm2 V−1 s−1) | V th (V) | μ e (cm2 V−1 s−1) | V th (V) |
|---|---|---|---|---|---|
| a BG/TC is bottom-gate/top-contact, TG/BC is top-gate/bottom-contact. | |||||
| HDIP | BG/TC | 0.041 | −17 | — | |
| linear-HDIP | BG/TC | 1.9 × 10−3 | −32 | — | |
| syn-HDIP | BG/TC | 1.5 × 10−4 | −51 | 4.0 × 10−5 | 53 |
| anti-HDIP | BG/TC | 4.5 × 10−3 | −38 | 1.1 × 10−3 | 67 |
| HDIP | TG/BC | 0.40 | −20 | — | |
| linear-HDIP | TG/BC | 0.30 | −18 | — | |
| syn-HDIP | TG/BC | 1.3 × 10−3 | −34 | 8.6 × 10−4 | 70 |
| anti-HDIP | TG/BC | 0.20 | −14 | — | |
Conclusions
We have reported the synthesis of four HDIP derivatives, biradicaloid compounds with the open-shell ground state functionalized at the most reactive site with the TIPS-ethynyl groups. All the HDIP derivatives show remarkable stability higher than TIPS-pentacene going from 2 days to more than 30 days depending on the oxidation potential, the contribution of the singlet biradical form in the ground state and the annelation mode. The annelation mode also plays an important role in the modulation of the band gap going from 1.59 eV to 1.37 eV and also of the ΔES–T gap and the packing. The value of ΔES–T is not entirely correlated with the band gap and the biradical character but rather with the spin delocalization at the outer rings. A method to estimate the biradical character has been proposed and shows that the biradical character computed at the PUHF level is the most relevant among those reported by all other methods. The presence of TIPS-groups at the apical position of the 5-MRs is very important for the emergence of the singlet biradical in the ground state by reducing the band gap and increasing the distance between the two radical centres. The highest stability was found for the syn-annelation providing a guideline for molecular design to get stable biradicals. Thanks to their high stability, thin film OFETs were fabricated. Well balanced ambipolar transport was obtained in the order of 10−3 cm2 V−1 s−1 in the BG/TC configuration and unipolar transport in the order of 10−1 cm2 V−1 s−1 in the TG/BG configuration which is the highest value obtained for biradicals based on the diindenoacene skeleton.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr T. Matsumoto for the measurements of X-ray single crystal analysis. M. F. and A. Y. acknowledge financial support from the Agence Nationale de la Recherche ANR-16-CE07-0024 (GATE). T. J.-O. acknowledges CNRS for PhD grant. C. A. and M. M. acknowledge financial support from JST ERATO Grant Number JPMJER1305 and JSPS KAKENHI Grant Number 19H02790.Notes and references
- (a) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC; (b) T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC; (c) X. Hu, W. Wang, D. Wang and Y. J. Zheng, J. Mater. Chem. C, 2018, 6, 11232–11242 RSC.
- (a) M. Bendikov, H. M. Duong, K. Starkey, K. N. Houk, E. A. Carter and F. Wudl, J. Am. Chem. Soc., 2004, 126, 7416–7417 CrossRef CAS; (b) C. Tonshoff and H. F. Bettinger, Top. Curr. Chem., 2014, 349, 1–30 Search PubMed; (c) K. J. Thorley and J. E. Anthony, Isr. J. Chem., 2014, 54, 642–649 CrossRef CAS; (d) D. Chun, Y. Cheng and F. Wudl, Angew. Chem., Int. Ed., 2008, 47, 8380 CrossRef CAS; (e) E. Qu and C. Chi, Org. Lett., 2010, 12, 3360–3363 CrossRef.
- (a) A. Konishi, Y. Hirao, M. Nakano, A. Shimizu, E. Botek, B. T. Champagne, D. Shiomi, K. Sato, T. Takui, K. Matsumoto, H. Kurata and T. Kubo, J. Am. Chem. Soc., 2010, 132, 11021–11023 CrossRef CAS; (b) A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, R. Kishi, Y. Shigeta, M. Nakano, K. Tokunaga, K. Kamada and T. Kubo, J. Am. Chem. Soc., 2013, 135, 1430–1437 CrossRef CAS.
- (a) J. Liu, P. Ravat, M. Wagner, M. Baumgarten, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 12442–12446 CrossRef CAS; (b) M. R. Ajayakumar, Y. Fu, J. Ma, F. Hennersdorf, H. Komber, J. J. Weigand, A. Alfonsov, A. Popov, R. Berger, J. Liu, K. Müllen and X. Feng, J. Am. Chem. Soc., 2018, 140, 6240–6244 CrossRef CAS; (c) Y. Ni, T. Y. Gopalakrishna, H. Phan, T. S. Herng, S. Wu, Y. Han, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 9697–9701 CrossRef CAS.
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Chem, 2017, 2, 81–92 CrossRef CAS.
- (a) T. Jousselin-Oba, M. Mamada, J. Marrot, A. Maignan, C. Adachi, A. Yassar and M. Frigoli, J. Am. Chem. Soc., 2019, 141, 9373–9381 CrossRef; (b) Y. Gu, Y. G. Tullimilli, J. Feng, H. Phan, W. Zeng and J. Wu, Chem. Commun., 2019, 55, 5567–5570 RSC.
- (a) Y. Li, W.-K. Heng, B. S. Lee, N. Aratani, J. L. Zafra, N. Bao, R. Lee, Y. M. Sung, Z. Sun, K.-W. Huang, R. D. Webster, J. T. López Navarrete, D. Kim, A. Osuka, J. Casado, J. Ding and J. Wu, J. Am. Chem. Soc., 2012, 134, 14913–14922 CrossRef CAS; (b) R. Huang, H. Phan, T. S. Herng, P. Hu, W. Zeng, S.-Q. Dong, S. Das, Y. Shen, J. Ding, D. Casanova and J. Wu, J. Am. Chem. Soc., 2016, 138, 10323–10330 CrossRef CAS; (c) W. Zeng, Z. Sun, T. S. Herng, T. P. Gonçalves, T. Y. Gopalakrishna, K.-W. Huang, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 8615–8619 CrossRef CAS; (d) W. Zeng, T. Y. Gopalakrishna, H. Phan, T. Tanaka, T. S. Herng, J. Ding, A. Osuka and J. Wu, J. Am. Chem. Soc., 2018, 140, 14054–14058 CrossRef CAS.
- A. Konishi, K. Horii, D. Shiomi, K. Sato, T. Takui and M. Yasuda, J. Am. Chem. Soc., 2019, 141, 10165–10170 CrossRef CAS.
- (a) A. Shimizu, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, M. Uruichi and K. Yakushi, Chem. Commun., 2012, 48, 5629–5631 RSC; (b) H. Koike, M. Chikamatsu, R. Azumi, J. y. Tsutsumi, K. Ogawa, W. Yamane, T. Nishiuchi, T. Kubo, T. Hasegawa and K. Kanai, Adv. Funct. Mater., 2016, 26, 277–283 CrossRef CAS.
- (a) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS; (b) H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2014, 5, 163–168 RSC; (c) X. Shi, E. Quintero, S. Lee, J. Jing, T. S. Herng, B. Zheng, K.-W. Huang, J. T. López Navarrete, J. Ding, D. Kim, J. Casado and C. Chi, Chem. Sci., 2014, 5, 4490–4503 RSC; (d) K. Sbargoud, M. Mamada, J. Marrot, S. Tokito, A. Yassar and M. Frigoli, Chem. Sci., 2015, 6, 3402–3409 RSC; (e) T. Maekawa, H. Ueno, Y. Segawa, M. M. Haley and K. Itami, Chem. Sci., 2016, 7, 650–654 RSC; (f) J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y.-Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280–3284 CrossRef CAS; (g) R.-Q. Lu, S. Wu, L.-L. Yang, W.-B. Gao, H. Qu, X.-Y. Wang, J.-B. Chen, C. Tang, H.-Y. Shi and X.-Y. Cao, Angew. Chem., Int. Ed., 2019, 58, 7600–7605 CrossRef CAS; (h) J. Ma, K. Zhang, K. S. Schellhammer, Y. Fu, H. Komber, C. Xu, A. A. Popov, F. Hennersdorf, J. J. Weigand, S. Zhou, W. Pisula, F. Ortmann, R. Berger, J. Liu and X. Feng, Chem. Sci., 2019, 10, 4025–4031 RSC; (i) A. S. Hacker, M. Pavano, J. E. Wood, H. Hashimoto, K. M. D'Ambrosio, C. K. Frederickson, J. Zafra, C. J. Gomez Garcia, V. Postils, A. Ringer McDonald, D. Casanova, D. K. Frantz and J. Casado, Chem. Commun., 2019, 55, 14186–14189 RSC; (j) M. A Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stepien, Chem. Sci., 2019, 10, 3413–3420 RSC; (k) S. Mori, M. Akita, S. Suzuki, M. S. Asano, M. Murata, T. Akiyama, T. Matsumoto, C. Kitamura and S.-i. Kato, Chem. Commun., 2020, 56, 5881–5884 RSC.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. Ponce Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS.
- Y.-C. Hsieh, C.-F. Wu, Y.-T. Chen, C.-T. Fang, C.-S. Wang, C.-H. Li, C.-H. L.-Y. Chen, M.-J. Cheng, C.-C. Chueh, P.-T. Chou and Y.-T. Wu, J. Am. Chem. Soc., 2018, 140, 14357–14366 CrossRef CAS.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS.
- J. E. Barker, J. J. Dressler, A. Ca¡rdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2020, 142, 1548–1555 CrossRef CAS.
- J. Melidonie, E. Dmitrieva, K. Zhang, Y. Fu, A. A. Popov, W. Pisula, R. Berger, J. Liu and X. Feng, J. Org. Chem., 2020, 85, 215–223 CrossRef CAS.
- J. J. Dressler, A. Ca¡rdenas Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Chem, 2020, 6, 1–16 Search PubMed ; only compound 10 (linear-DIAn) has an R factor lower than 0.05.
- (a) D. Doehnert and J. Koutecky, J. Am. Chem. Soc., 1980, 102, 1789–1796 CrossRef CAS; (b) K. Yamaguchi, T. Kawakami, Y. Takano, Y. Kitagawa, Y. Yamashita and H. Fujita, Int. J. Quantum Chem., 2002, 90, 370–385 CrossRef CAS.
- P. J. Karafiloglou, J. Chem. Educ., 1989, 66, 816 CrossRef CAS.
- A. M. Zeidell, L. Jennings, C. K. Frederickson, Q. Ai, J. J. Dressler, L. N. Zakharov, C. Risko, M. M. Haley and O. D. Jurchescu, Chem. Mater., 2019, 31, 6962–6970 CrossRef CAS.
- S. Qiu, Y. Zhang, X. Huang, L. Bao, Y. Hong, Z. Zeng and J. Wu, Org. Lett., 2016, 18, 6018–6021 CrossRef CAS.
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS.
- J. E. Barker, C. K. Frederickson, M. H. Jones, L. N. Zakharov and M. M. Haley, Org. Lett., 2017, 19, 5312–5315 CrossRef CAS.
- Y. Tian, K. Uchida, H. Kurata, Y. Hirao, T. Nishiuchi and T. Kubo, J. Am. Chem. Soc., 2014, 136, 12784–12793 CrossRef CAS.
- (a) T. Jousselin-Oba, K. Sbargoud, G. Vaccaro, F. Meinardi, A. Yassar and M. Frigoli, Chem.–Eur. J., 2017, 23, 16184–16188 CrossRef CAS; (b) K. Sbargoud, M. Mamada, T. Jousselin-Oba, Y. Takeda, S. Tokito, A. Yassar, J. Marrot and M. Frigoli, Chem.–Eur. J., 2017, 23, 5076–5080 CrossRef CAS.
- J. L. Marshall, D. Lehnherr, B. D. Lindner and R. R. Tykwinski, ChemPlusChem, 2017, 82, 967–1001 CrossRef CAS.
- G. P. Eyer, K. R. Kittilstved and T. L. Andrew, J. Phys. Chem. C, 2017, 121, 24929–24935 CrossRef CAS.
- M. Frigoli, J. Marrot, P. L. Gentili, D. Jacquemin, M. Vagnini, D. Pannacci and F. Ortica, ChemPhysChem, 2015, 16, 2447–2458 CrossRef CAS.
- J.-I. Nishida, S. Tsukaguchi and Y. Yamashita, Chem.–Eur. J., 2012, 18, 8964–8970 CrossRef CAS.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS.
- R. Gershoni-Poranne and A. Stanger, Chem.–Eur. J., 2014, 20, 5673–5688 CrossRef CAS.
- S. Canola, J. Casado and F. Negri, Phys. Chem. Chem. Phys., 2018, 20, 24227–24238 RSC.
- G. E. Rudebusch, A. G. Fix, H. A. Henthorn, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 3627–3633 RSC.
- (a) J. Kang, N. Shin, D. Y. Jang, V. M. Prabhu and D. Y. Yoon, J. Am. Chem. Soc., 2008, 130, 12273–12275 CrossRef CAS; (b) T. Ohe, M. Kuribayashi, R. Yasuda, A. Tsuboi, K. Nomoto, K. Satori, M. Itabashi and J. Kasahara, Appl. Phys. Lett., 2008, 93, 053303 CrossRef; (c) J. Smith, R. Hamilton, M. Heeney, D. M. de Leeuw, E. Cantatore, J. E. Anthony, I. McCulloch, D. D. C. Bradley and T. D. Anthopoulos, Appl. Phys. Lett., 2008, 93, 253301 CrossRef; (d) M. Mamada, H. Shima, Y. Yoneda, T. Shimano, N. Yamada, K. Kakita, T. Machida, Y. Tanaka, S. Aotsuka, D. Kumaki and S. Tokito, Chem. Mater., 2015, 27, 141–147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1987607–1987611. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04583g |
| This journal is © The Royal Society of Chemistry 2020 |