
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic redox-neutral hydroxyalkylation of N-heteroaromatics with aldehydes†
Hiromu
Fuse
a,
Hiroyasu
Nakao
a,
Yutaka
Saga
b,
Arisa
Fukatsu
c,
Mio
Kondo
 b,
Shigeyuki
Masaoka
b,
Harunobu
Mitsunuma
*a and
Motomu
Kanai
*a
b,
Shigeyuki
Masaoka
b,
Harunobu
Mitsunuma
*a and
Motomu
Kanai
*a
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: h-mitsunuma@mol.f.u-tokyo.ac.jp; kanai@mol.f.u-tokyo.ac.jp
bDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
cDepartment of Life and Coordination-Complex Molecular Science, Institute for Molecular Science (IMS), 5-1 Higashiyama, Myodaiji, Okazaki, Aichi 444-8787, Japan
First published on 12th October 2020
Abstract
Hydroxyalkylation of N-heteroaromatics with aldehydes was achieved using a binary hybrid catalyst system comprising an acridinium photoredox catalyst and a thiophosphoric acid organocatalyst. The reaction proceeded through the following sequence: (1) photoredox-catalyzed single-electron oxidation of a thiophosphoric acid catalyst to generate a thiyl radical, (2) cleavage of the formyl C–H bond of the aldehyde substrates by a thiyl radical acting as a hydrogen atom transfer catalyst to generate acyl radicals, (3) Minisci-type addition of the resulting acyl radicals to N-heteroaromatics, and (4) a spin-center shift, photoredox-catalyzed single-electron reduction, and protonation to produce secondary alcohol products. This metal-free hybrid catalysis proceeded under mild conditions for a wide range of substrates, including isoquinolines, quinolines, and pyridines as N-heteroaromatics, as well as both aromatic and aliphatic aldehydes, and tolerated various functional groups. The reaction was applicable to late-stage derivatization of drugs and their leads.
Introduction
Hydroxyalkylated N-heteroaromatics are ubiquitously present in bioactive compounds and are versatile intermediates in the synthesis of pharmaceuticals/agrochemicals. Although transition metal-catalyzed addition reactions of aromatic compounds to various electrophiles through C(sp2)–H bond activation are reported,1 the use of carbonyl groups as electrophiles has remained difficult due to the reversibility of the C–C bond-forming step.1,2 Specifically, the catalyzed direct N-heteroarylation of aldehydes has been limited to a C3-selective dehydrogenative reaction in the presence of silanes, which proceeded at a high temperature (135 °C) and has a limited substrate scope.3 Consequently, N-heteroarylation of aldehydes mainly relies on stoichiometric carbanion chemistry through the deprotonation of a C(sp2)–H bond with pKa greater than 40 using strong bases (Fig. 1A(a)).4 Regio- and chemoselective C–H metalation and functionalization reactions of N-heteroaromatics using less nucleophilic Brønsted bases are reported.5 Several types of functional groups, however, are not compatible with these reaction conditions.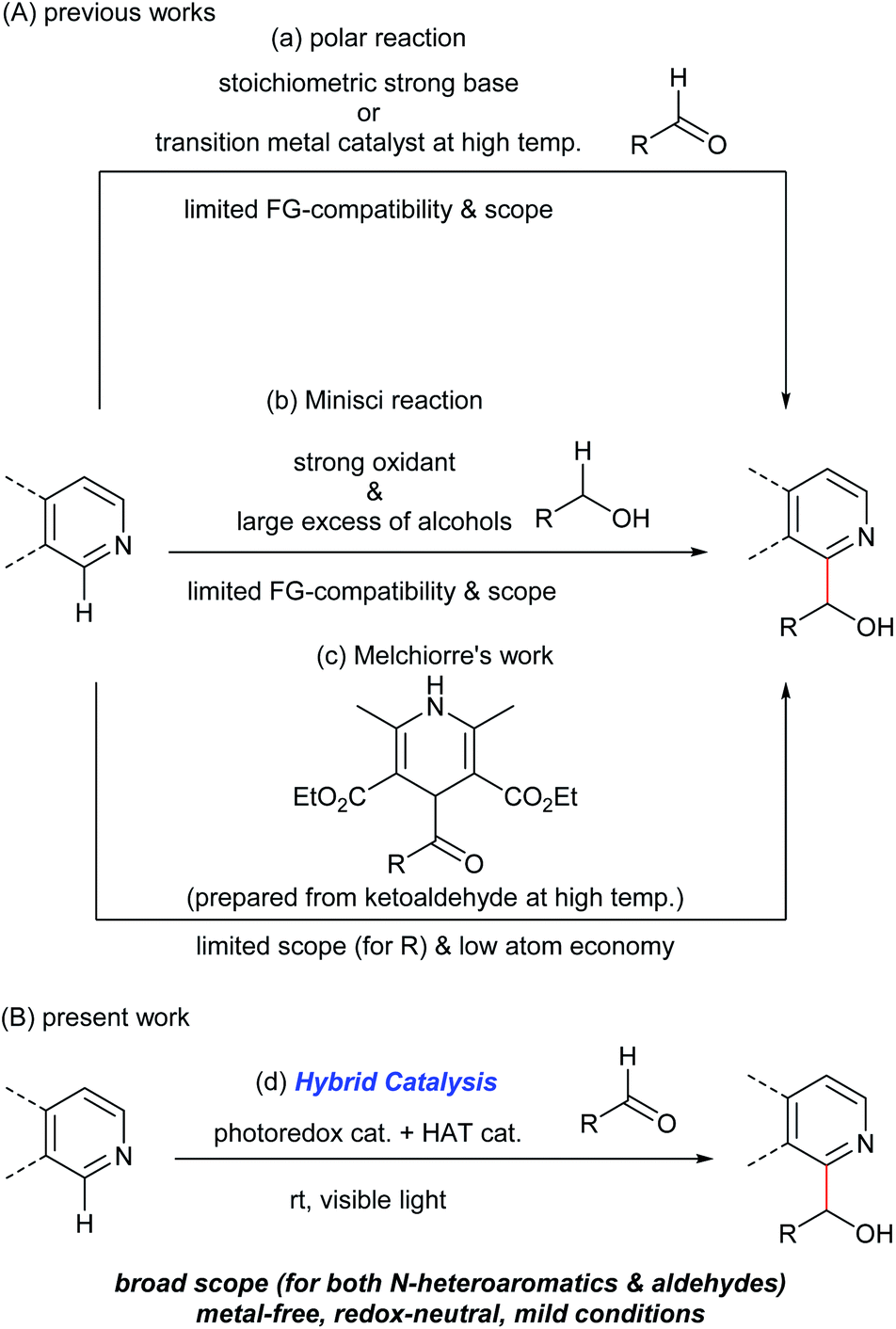 | ||
| Fig. 1 Overview of C–H hydroxyalkylation of N-heteroaromatics. (A) Previous studies: (a) using a stoichiometric strong base or a transition metal catalyst. (b) Oxidative Minisci reaction. (c) Melchiorre's work using 4-acyl-1,4-dihydropyridines. (B) Present work: (d) binary hybrid catalysis comprising a photoredox catalyst and an organo-HAT catalyst. | ||
An alternative pathway to hydroxyalkylated N-heteroaromatics is an oxidative Minisci reaction (Fig. 1A(b)).6–8 Due to the inertness of the α-oxy C–H bond of alcohols, however, this process requires a strong oxidant and large excess of alcohols resulting in low generality. On the other hand, Melchiorre recently reported the photochemical hydroxyalkylation of quinolines and isoquinolines by a novel spin-center shift (SCS) process (Fig. 1A(c)).9,10 Although the reaction proceeded under mild conditions, the acyl radical sources, 4-acyl-1,4-dihydropyridines, were prepared from the corresponding ketoaldehydes under harsh conditions (i.e. heating neat at 120–130 °C). This limited substrate generality of hydroxyalkyl groups made the overall process less atom economical. Herein, we report a new catalytic method for synthesizing hydroxyalkylated N-heteroaromatics through a Minisci-type reaction between N-heteroaromatics and aldehydes, mediated by a hybrid catalyst system comprising an acridinium photoredox catalyst and a thiophosphoric acid (TPA) organo-hydrogen atom transfer (HAT) catalyst (Fig. 1B).11 Notably, this reaction proceeded under redox-neutral conditions with high atom economy. This is in contrast to previously reported Minisci reactions between N-heteroaromatics and aldehydes, which required stoichiometric strong oxidants and produced acylated products.12
Results and discussion
Optimization of reaction conditions
Because the bond dissociation energy of a formyl C–H bond of aldehydes (88.7 kcal mol−1)13a is much smaller than that of a C(sp2)–H bond of N-heteroaromatics (105 kcal mol−1),13b we planned to incorporate a SCS process in the overall catalytic cycle to produce alcohol products (see below). There are preceding examples in which the SCS process was combined with the photoredox-catalyzed Minisci-type reaction of N-heteroaromatics.14 In these examples, formal alkylations of N-heteroaromatics were achieved using alcohols, ethers, or carbonyl compounds as an alkyl source. Specifically, MacMillan14a and Huang14c,d achieved the incorporation of HAT and SCS processes. Despite the versatile roles of hydroxy groups in organic synthesis, however, they were eliminated under their reaction conditions. Moreover, their mechanistic studies implied that hydroxyalkylated N-heteroaromatics were unstable under their reaction conditions.14a,dOur working hypothesis for the catalytic cycle is shown in Fig. 2. A photoredox catalyst Mes-Acr+ in the excited state (*Mes-Acr+) oxidizes an organocatalyst (RSH) to produce a radical (RS˙) acting as a HAT catalyst. This radical cleaves the formyl C–H bond of aldehyde 2. The resulting acyl radical 4 reacts with protonated N-heteroaromatics 5 through a Minisci reaction, giving radical cation 6. Deprotonation of 6 affords benzylic carbon-centered radical 7, which is converted to the α-oxy radical 8via SCS. Finally, 8 is reduced by the reduced photoredox catalyst (Mes-Acr), and the subsequent protonation affords target compound 3. On the basis of our previous findings, we envisioned that the combination of an acridinium photoredox catalyst (Mes-Acr+) and TPA organocatalyst would realize the designed hybrid catalysis.15,16
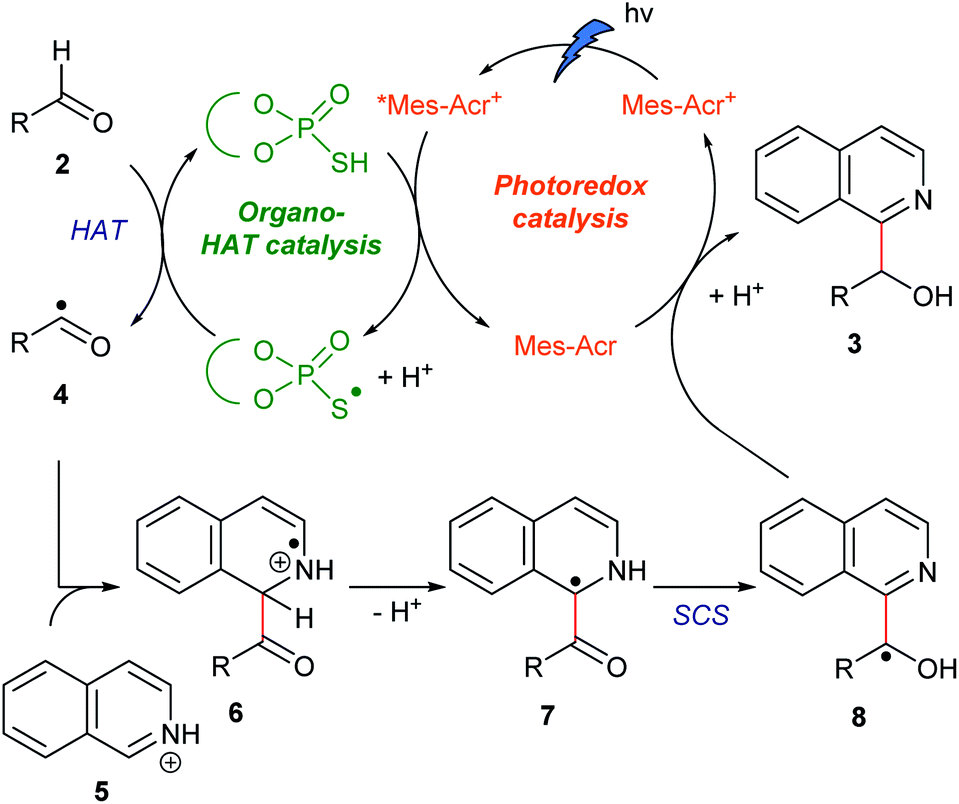 | ||
| Fig. 2 Proposed catalytic cycle. | ||
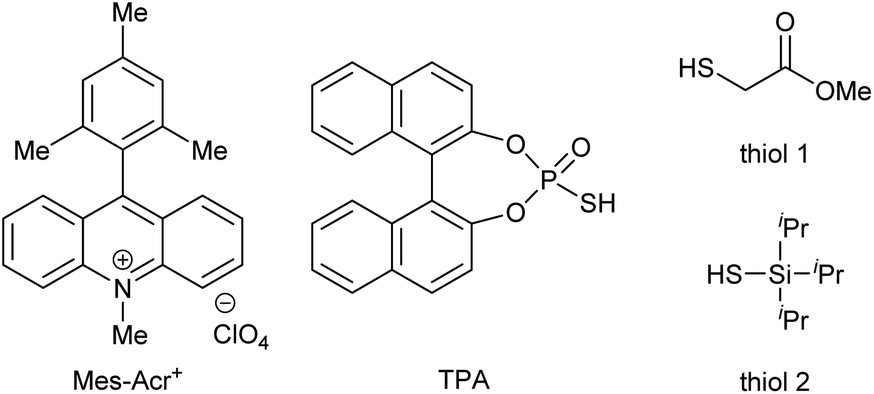
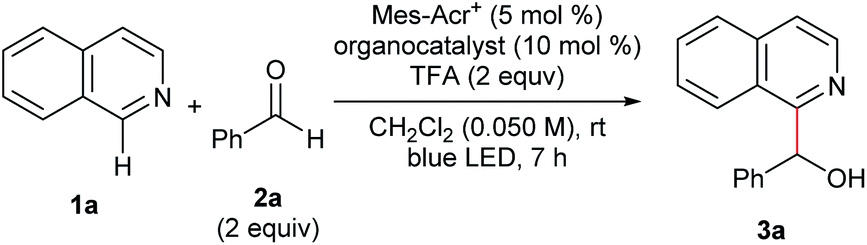
We conducted optimization studies using isoquinoline (1a) and benzaldehyde (2a) (Table 1). Despite several possible side reactions such as reductive deoxygenation of 3a and reduction of 2a,14 we identified the optimized reaction conditions comprising 5 mol% Mes-Acr+ photoredox catalyst and 10 mol% TPA organocatalyst in the presence of 2 equiv. TFA, affording 3a in 94% yield (entry 1). Other organocatalysts for HAT, such as thiols,14a,17a quinuclidine,17b and benzoic acid17c did not afford the product (entries 2–5). Control experiments revealed that Mes-Acr+, TPA, TFA, and visible light irradiation were indispensable for efficient reaction progress (entries 6–9). The addition of 1 equiv. TEMPO inhibited the reaction (entry 10), indicating the presence of radical intermediates.
|
|
||
|---|---|---|
| Entry | Organocatalyst | 3a (%) |
| a General reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), Mes-Acr+ (0.005 mmol), TPA (0.010 mmol), and TFA (0.20 mmol) were reacted in dichloromethane (DCM; 2.0 mL) at room temperature under blue LED irradiation for 7 h. b Yield was determined by 1H NMR analysis of the crude mixture using 1,1,2,2-tetrachloroethane as an internal standard. c Isolated yield in parentheses. d Without Mes-Acr+. e Without TFA. f Without photoirradiation. g 1 equiv. TEMPO was added. | ||
| 1 | TPA | 94 (89)c |
| 2 | Thiol 1 | ND |
| 3 | Thiol 2 | ND |
| 4 | Quinuclidine | 3 |
| 5 | Benzoic acid | ND |
| 6d | TPA | 29 |
| 7 | — | ND |
| 8e | TPA | 1 |
| 9f | TPA | ND |
| 10g | TPA | ND |
|
||
Substrate scope
Under the optimized conditions, we investigated the substrate scope (Fig. 3). We first studied the aldehyde side (3a–l) using isoquinoline (1a) as an N-heteroaromatic substrate. Aromatic aldehydes containing various functional groups such as halogen (3b–d), ether (3e), trifluoromethyl (3f), and alkyl (3g) groups tolerated the reaction conditions. The electron density of the aromatic ring did not significantly affect the results. Moreover, steric hindrance at the ortho-position of the phenyl ring of aldehydes did not interfere with the reaction (3g). Aliphatic aldehydes, which are challenging substrates due to the presence of acidic α-C–H bonds and low electrophilicity,3a were a competent class of substrates in this reaction (3h–l). As a result, several hydroxy-alkylated isoquinolines containing ester (3i), phthalimide (3j), ketone (3k), and halogen (3l) groups were obtained in high yield. We then examined the scope of N-heteroaromatics. Various brominated isoquinolines exhibited good reactivity (3m–p). The reactions of isoquinolines containing amide (3q) and ester (3r) groups proceeded in good yield. This catalyst system was also applicable to the hydroxyalkylation of quinolines (3s–z). Quinolines containing ester (3u), halogen (3v–x), ether (3y), and phenyl (3z) groups were competent substrates. Moreover, pyridine derivatives were successfully converted to the corresponding products (3aa–ad). Ketone and ester functionalities were tolerated. Steric hindrance did not interrupt reaction progress and substituted pyridines were synthesized (3ab–ac).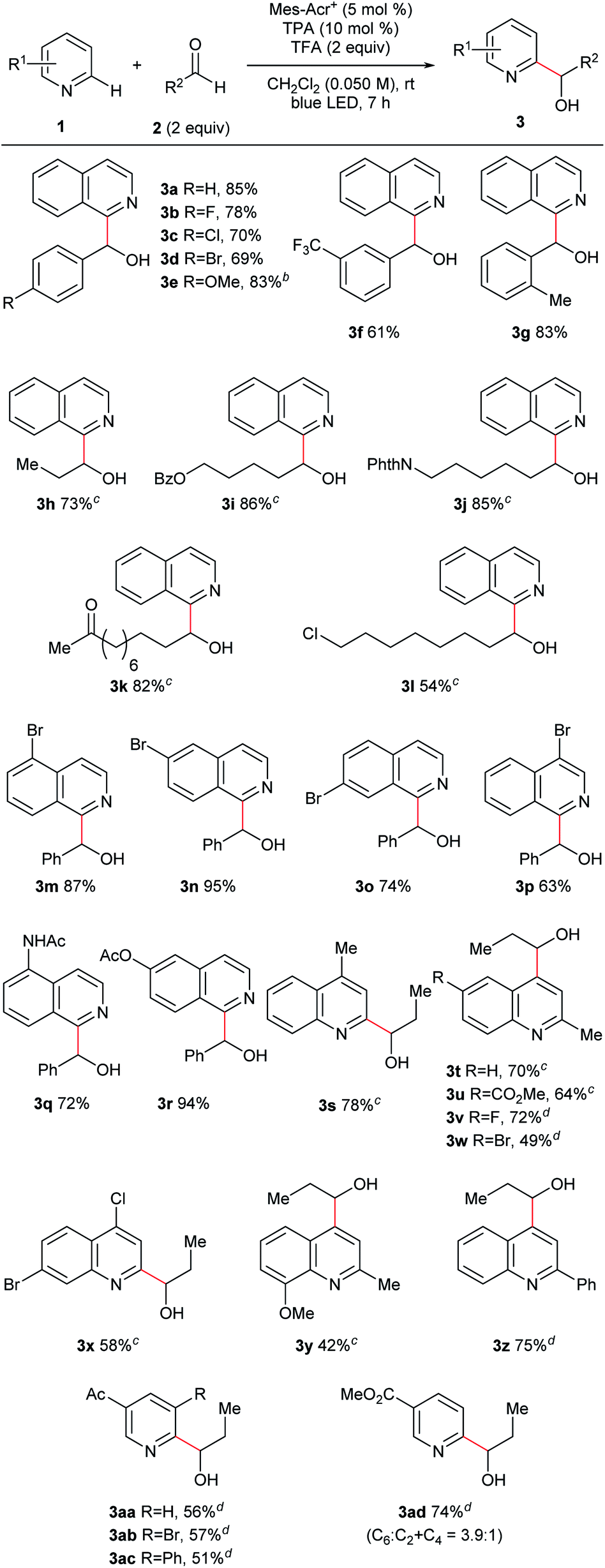 | ||
| Fig. 3 Substrate scope. aGeneral reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), Mes-Acr+ (0.005 mmol), TPA (0.010 mmol), and TFA (0.20 mmol) were reacted in dichloromethane (DCM; 2.0 mL) at room temperature under blue LED irradiation for 7 h. Yield is isolated yield unless otherwise noted. b5 equiv. of the aldehyde was used with the reaction time 13 h. cReaction time was 3 h. dReaction time was 5 h. | ||
Furthermore, this reaction was applicable to late-stage modifications of complex molecules because of the mild conditions (Fig. 4). Thus, camptothecin, which has anti-cancer activity, was transformed to the corresponding C7-hydroxyalkylated derivative (3ae). Introduction of functional groups to the C7-position of camptothecin could improve its pharmacological properties.18 A fasudil (ROCK2 inhibitor) derivative also reacted with propionaldehyde with excellent yield (3af). Functionalized hydroxyalkyl groups, which were difficult to use in the previous radical-mediated reactions, were also introduced to N-heterocycles under the present conditions (3ag–3ak). Thus, aldehydes derived from oxaprozin, loxoprofen, dehydrocholic acid, an amino acid derivative, and a sugar derivative afforded the corresponding N-heteroarylated products in good yields. It is noteworthy that substrates containing an oxazole ring which is susceptible to oxidative conditions (3ag)19 or a relatively weak benzylic C–H bond (3ah) tolerated our reaction conditions.
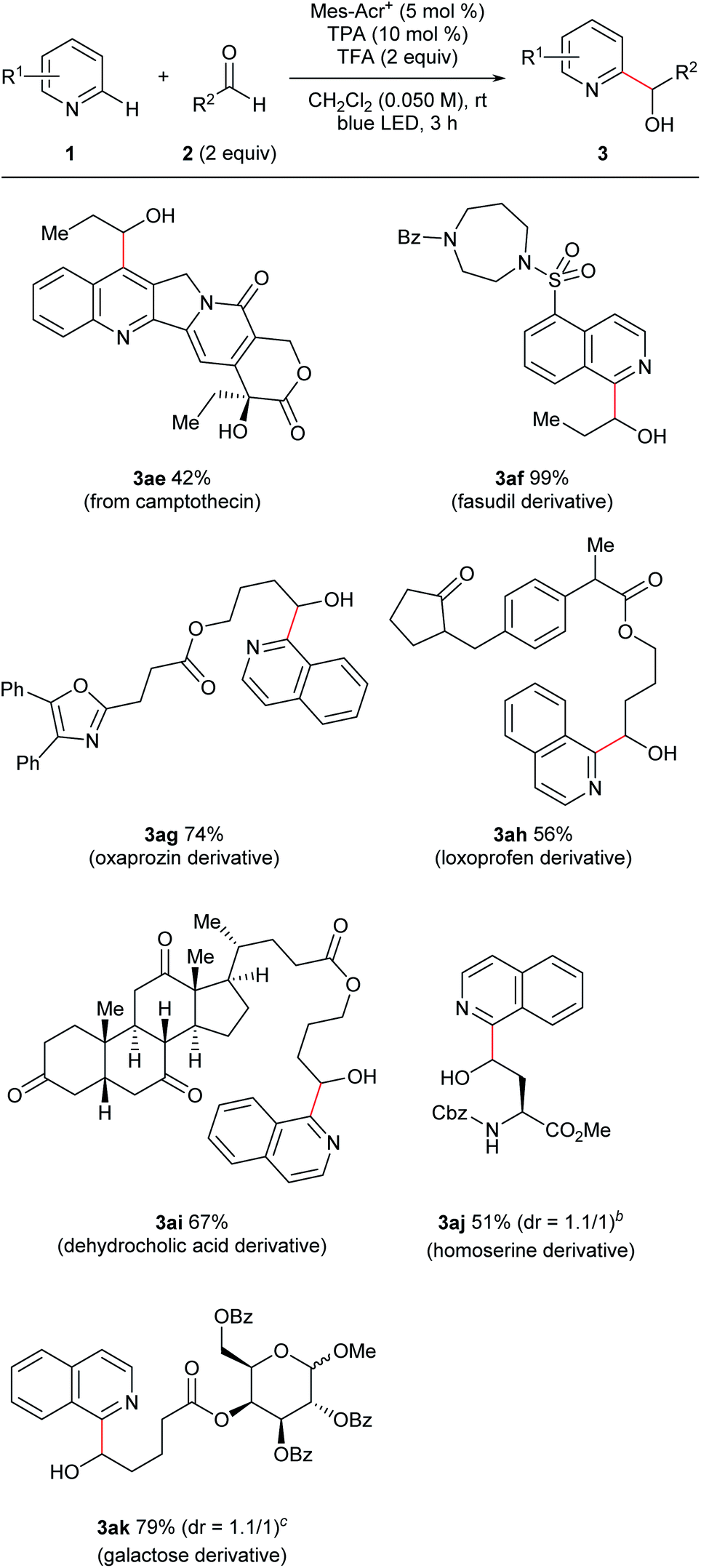 | ||
| Fig. 4 Late-stage modification of multifunctional substrates. aGeneral reaction conditions: 1 (0.10 mmol), 2 (0.20 mmol), Mes-Acr+ (0.005 mmol), TPA (0.010 mmol), and TFA (0.20 mmol) were reacted in dichloromethane (DCM; 2.0 mL) at room temperature under blue LED irradiation for 3 h. Yield is isolated yield unless otherwise noted. b1.2 equiv. of the aldehyde was used. The target compound was isolated as the lactone. cThe reaction was conducted at the 0.035 mmol scale. | ||
To gain preliminary insight into the mechanism, reactions were conducted using deuterated compounds (Fig. 5). When benzaldehyde-d (2a-d) was exposed to the reaction conditions, target compound 3a contained 85%-H at the α-position of the hydroxy group (Fig. 5a). Thus, the formyl C–H bond of the aldehyde was cleaved in the overall catalytic cycle. When TFA-d was utilized as an acid additive, however, 62%-H was still incorporated at the α-position (Fig. 5b). This result was contradictory to our hypothesis (Fig. 2), and could be due to contamination by H2O. Therefore, we assessed the H/D ratio incorporated in 3a in mixed solvents containing variable amounts of D2O. As a result, incorporation of D increased up to 95% according to the D2O concentration (Fig. 5b). This result indicates that the hydrogen atom at the α-position of the hydroxy group is introduced via protonation. These deuteration experiments support the feasibility of our mechanistic hypothesis shown in Fig. 2. The quantum yield was determined to be 0.046, which supports that the reaction proceeded through a closed catalytic cycle, not a radical chain pathway (see the ESI† for details).
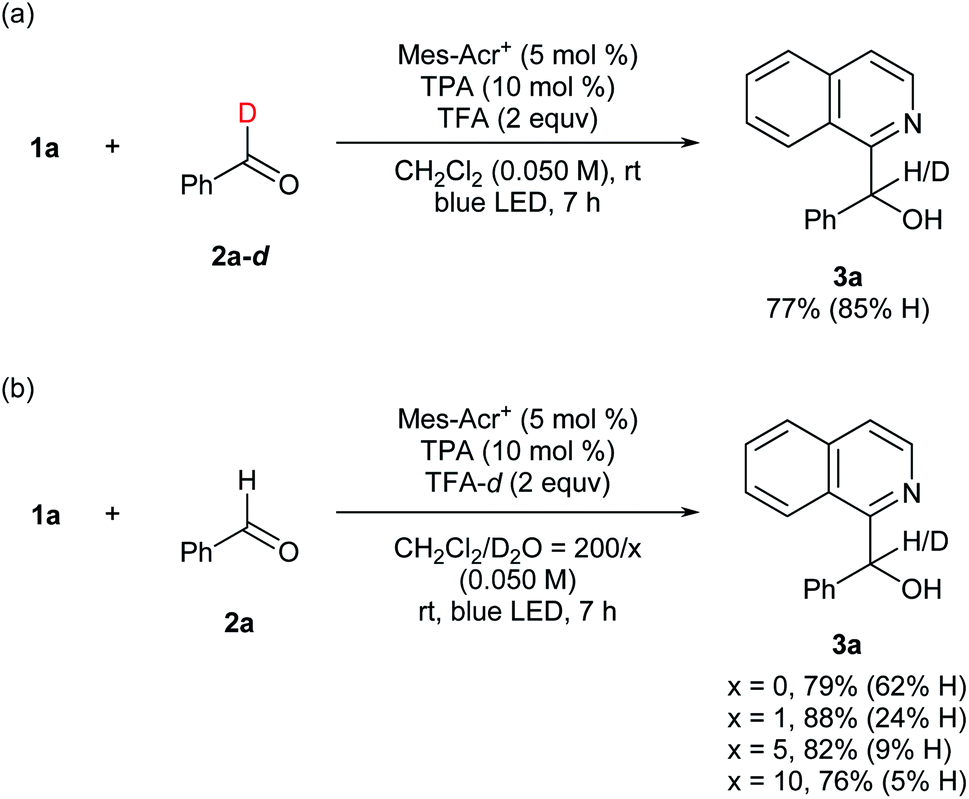 | ||
| Fig. 5 Mechanistic information for deuterium incorporation. aYield was determined by 1H NMR analysis of the crude mixture using 1,1,2,2-tetrachloroethane as an internal standard. | ||
Conclusions
In conclusion, we developed a binary hybrid catalyst system to achieve a one-step, redox-neutral hydroxyalkylation of N-heteroaromatic compounds with aldehydes without using a metal species. The reaction proceeded under mild conditions and high atom economy, enabling a broad substrate scope and application to late-stage modifications of drug-related molecules. Keys to the success were the sequential HAT, Minisci, and SCS processes. Further mechanistic studies are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Numbers JP17H06442 (M. Ka.) (Hybrid Catalysis), 18H05969 (H. M.), 19J23073 (H. F.), 17H06444 (S. M.) (Hybrid Catalysis), 20H02754 (M. Ko.), and 20K15955 (Y. S.). We thank Professor Masahiro Terada and Dr Jun Kikuchi in Tohoku University for sending us an enantiomerically pure TPA derivative. We also thank Professor Yasuteru Urano, Dr Tasuku Ueno, and Dr Toru Komatsu at the University of Tokyo for helpful discussion on mechanistic studies.Notes and references
- (a) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC; (b) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS; (c) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS.
- Previously reported transition metal-catalyzed C(sp2)–H addition to aldehydes depended on the use of directing groups, and required electron-deficient aldehydes or stoichiometric metal species. For representative examples, see: (a) L. Yang, C. A. Correia and C. J. Li, Adv. Synth. Catal., 2011, 353, 1269 CrossRef CAS; (b) Y. Li, X. S. Zhang, K. Chen, K. H. He, F. Pan, B. J. Li and Z. J. Shi, Org. Lett., 2012, 14, 636 CrossRef CAS; (c) Y. Li, X. S. Zhang, Q. L. Zhu and Z. J. Shi, Org. Lett., 2012, 14, 4498 CrossRef CAS; (d) B. W. Zhou, Y. Y. Hu and C. Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13659 CrossRef CAS; (e) H. Jo, J. Park, M. Choi, S. Sharma, M. Jeon, N. K. Mishra, T. Jeong, S. Han and I. S. Kim, Adv. Synth. Catal., 2016, 358, 2714 CrossRef CAS.
- Only aromatic aldehydes were applicable: (a) B. J. Li and Z. J. Shi, Chem. Sci., 2011, 2, 488 RSC. For a related transformation with imidazoles, see: (b) Y. Fukumoto, K. Sawada, M. Hagihara, N. Chatani and S. Murai, Angew. Chem., Int. Ed., 2002, 41, 2779 CrossRef CAS.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568 CrossRef CAS.
- For a review, see: (a) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS. For representative examples, see: (b) Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539 CrossRef CAS; (c) T. Imahori and Y. Kondo, J. Am. Chem. Soc., 2003, 125, 8082 CrossRef CAS; (d) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS.
- (a) C. A. Correia, L. Yang and C.-J. Li, Org. Lett., 2011, 13, 4581 CrossRef CAS; (b) C. A. Huff, R. D. Cohen, K. D. Dykstra, E. Streckfuss, D. A. DiRocco and S. W. Krska, J. Org. Chem., 2016, 81, 6980 CrossRef CAS; (c) L. Zhou, N. Okugawa and H. Togo, Eur. J. Org. Chem., 2017, 6239 CrossRef CAS; (d) L. Niu, J. Jiamei Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef; (e) S. Wang, S. Xing, Y. Zhang, Y. Fan, H. Zhao, J. Wang, S. Zhang and W. Wang, RSC Adv., 2019, 9, 41847 RSC.
- (a) M. A. J. Duncton, MedChemComm, 2011, 2, 1135 RSC; (b) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666 CrossRef CAS. For recent representative examples about the Minisci reaction, see: (c) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS; (d) Á. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251 CrossRef; (e) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS; (f) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS.
- Early examples of radical reactions were known as the Emmert reaction. Use of HgCl2–Mg, HgCl2–Al, Li or SmI2 resulted in low generality. (a) B. Emmert and A. Asendorf, Ber. Dtsch. Chem. Ges. B, 1939, 72, 1188 CrossRef; (b) C. E. Crawforth, C. A. Russell and O. Meth-Cohn, J. Chem. Soc. D, 1970, 1406 RSC; (c) D. J. O'Neill and P. Helquist, Org. Lett., 1999, 1, 1659 CrossRef; (d) J. A. Weitgenant, J. D. Mortison, D. J. O'Neill, B. Mowery, A. Puranen and P. Helquist, J. Org. Chem., 2004, 69, 2809 CrossRef CAS.
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219 CrossRef CAS.
- B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16878 CrossRef CAS.
- After a preliminary version of this manuscript was posted in ChemRxiv (DOI: 10.26434/chemrxiv.12085875), Ji, Wang, and Huang reported a similar photocatalytic transformation promoted by 4CzlPN–LiBr. The scope of their method, however, is limited to reactions of simple aromatic aldehydes with quinoline derivatives and an isoquinoline derivative. See: X. Ji, Q. Liu, Z. Wang, P. Wang, G.-J. Deng and H. Huang, Green Chem., 2020 10.1039/d0gc01872d.
- (a) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082 CrossRef CAS; (b) Y. Siddaraju, M. Lamani and K. R. Prabhu, J. Org. Chem., 2014, 79, 3856 CrossRef CAS; (c) P. Cheng, Z. Qing, S. Liu, W. Liu, H. Xie and J. Zeng, Tetrahedron Lett., 2014, 55, 6647 CrossRef CAS; (d) J. Chen, M. Wan, J. Hua, Y. Sun, Z. Lv, W. Li and L. Liu, Org. Biomol. Chem., 2015, 13, 11561 RSC; (e) Y. Siddaraju and K. R. Prabhu, Tetrahedron, 2016, 72, 959 CrossRef CAS.
- (a) L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020 CrossRef CAS; (b) J. H. Kiefer, Q. Zhang, R. D. Kern, J. Yao and B. Jursic, J. Phys. Chem. A, 1997, 101, 7061 CrossRef CAS.
- (a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS; (b) J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Sci. Adv., 2019, 5, eaax9955 CrossRef CAS; (c) Z. Wang, X. Ji, T. Han, G. J. Deng and H. Huang, Adv. Synth. Catal., 2019, 361, 5643 CrossRef CAS; (d) Z. Wang, Q. Liu, X. Ji, G.-L. Deng and H. Huang, ACS Catal., 2020, 10, 154 CrossRef CAS. For recent reports using SCS for reactions other than the Minisci reaction under photoirradiation, see: (e) E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322 CrossRef CAS; (f) S. Barata-Vallejo, C. Ferreri, B. T. Golding and C. Chatgilialoglu, Org. Lett., 2018, 20, 4290 CrossRef CAS; (g) Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 2755 CrossRef CAS.
- (a) S. Kato, Y. Saga, M. Kojima, H. Fuse, S. Matsunaga, A. Fukatsu, M. Kondo, S. Masaoka and M. Kanai, J. Am. Chem. Soc., 2017, 139, 2204 CrossRef CAS; (b) H. Fuse, M. Kojima, H. Mitsunuma and M. Kanai, Org. Lett., 2018, 20, 2042 CrossRef CAS; (c) H. Fuse, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 4493 CrossRef CAS; (d) S. Tanabe, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2020, 142, 12374 CrossRef CAS.
- Electron transfer between *Mes-Acr+ and TPA is feasible on the basis of the following experimental results and data: (a) Stern–Volmer quenching was observed between Mes-Acr+ and TPA, but not between Mes-Acr+ and aldehyde 2a. See the ESI.†; (b) The transient absorption spectra supported electron transfer from TPA to *Mes-Acr+. See ref. 15a; (c) The oxidation potential of TPA (+1.18 V vs. SCE, see ref. 15a) is smaller than the reduction potential of *Mes-Acr+ (1.88 V vs. SCE, see S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS.
- (a) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74 CrossRef CAS; (b) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532 CrossRef CAS; (c) S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200 CrossRef CAS.
- D. J. Adams, M. L. Wahl, J. L. Flowers, B. Sen, M. Colvin, M. W. Dewhirst, G. Manikumar and M. C. Wani, Cancer Chemother. Pharmacol., 2006, 57, 145 CrossRef CAS.
- (a) D. A. Evans, P. Nagorny and R. Xu, Org. Lett., 2006, 8, 5669 CrossRef CAS; (b) H. H. Wasserman and M. B. Floyd, Tetrahedron, 1966, 22, 441 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04114a |
| This journal is © The Royal Society of Chemistry 2020 |