
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiple rotational rates in a guest-loaded, amphidynamic zirconia metal–organic framework†
Aaron
Torres-Huerta
 a,
Dazaet
Galicia-Badillo
a,
Andrés
Aguilar-Granda‡
a,
Jacob T.
Bryant
b,
Fernando J.
Uribe-Romo
*b and
Braulio
Rodríguez-Molina
*a
a,
Dazaet
Galicia-Badillo
a,
Andrés
Aguilar-Granda‡
a,
Jacob T.
Bryant
b,
Fernando J.
Uribe-Romo
*b and
Braulio
Rodríguez-Molina
*a
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, 04510, Ciudad de México, Mexico. E-mail: brodriguez@iquimica.unam.mx
bDepartment of Chemistry and Renewable Energy and Chemical Transformations Cluster, University of Central Florida, 4111 Libra Drive, Room 251 PSB, 32816-2366, Orlando, Florida, USA. E-mail: fernando@ucf.edu
First published on 24th September 2020
Abstract
Amphidynamic motion in metal–organic frameworks (MOFs) is an intriguing emergent property, characterized by high rotational motion of the phenylene rings that are embedded within an open, rigid framework. Here, we show how the phenylene rings in the organic linkers of the water stable MOF PEPEP-PIZOF-2 exhibit multiple rotational rates as a result of the electronic structure of the linker, with and without the presence of highly interacting molecular guests. By selective 2H enrichment, we prepared isotopologues PIZOF-2d4 and PIZOF-2d8 and utilized solid-state 13C and 2H NMR to differentiate the dynamic behavior of specific phenylenes in the linker at room temperature. A difference of at least one order of magnitude was observed between the rates of rotation of the central and outer rings at room temperature, with the central phenylene ring, surrounded by ethynyl groups, undergoing ultrafast 180° jumps with frequencies higher than 10 MHz. Moreover, loading tetracyanoquinodimethane (TCNQ) within the pores produced significant changes in the MOF's electronic structure, but very small changes were observed in the rotational rates, providing an unprecedented insight into the effects that internal dynamics have on guest diffusion. These findings would help elucidate the in-pore guest dynamics that affect transport phenomena in these highly used MOFs.
Amphidynamic crystals are an emerging class of materials made of molecular components that exhibit fast internal motion within a rigid lattice.1,2 Metal–organic frameworks (MOFs) can be considered as intrinsically amphidynamic materials, because they are formed by the assembly of organic molecules that carry high degrees of freedom linked to inorganic clusters that form an extended solid matrix.3 This assembly allows for the organic components to behave like rotators, while the solid matrix/framework acts as a stator, with gyroscope-type motion enabled by the open architecture of the MOF with motion modulated by the molecular structure of the linker.4–6 In order to create materials with targeted dynamic properties for real-life applications, like molecular machines, it is important to determine whether the chemical environment of the linkers can produce dynamics at multiple rates and how the presence of molecular guests affect such dynamics. To do so, it is important to use MOFs that are chemically stable to water and humidity, because this robustness increases the reproducibility of the results and the applicability of the MOF. The interplay between guest diffusion, linker dynamics and the overall framework flexibility has been actively investigated in recent years.7
Here, we prepared a water-stable MOF, PEPEP-PIZOF-2 (Fig. 1a), strategically labelled with deuterium atoms to probe the multiple segmental motion in the pristine and guest-loaded materials. Utilizing solid-state NMR techniques, we elucidated that this MOF exhibits bimodal rotational rates, with the central ring of the linker having free rotation above the 10 MHz limit of quantitation, and with the outer rings having slower rotation. This double-rate internal dynamics is preserved even in the presence of a very “sticky,” electron-deficient guest such as tetracyanoquinodimethane, TCNQ. Studying the molecular dynamics of this class of MOFs helps in accelerating their use as applied materials and for the fundamental studies of transfer phenomena that occur in MOFs such as mass, heat, and momentum transfer.
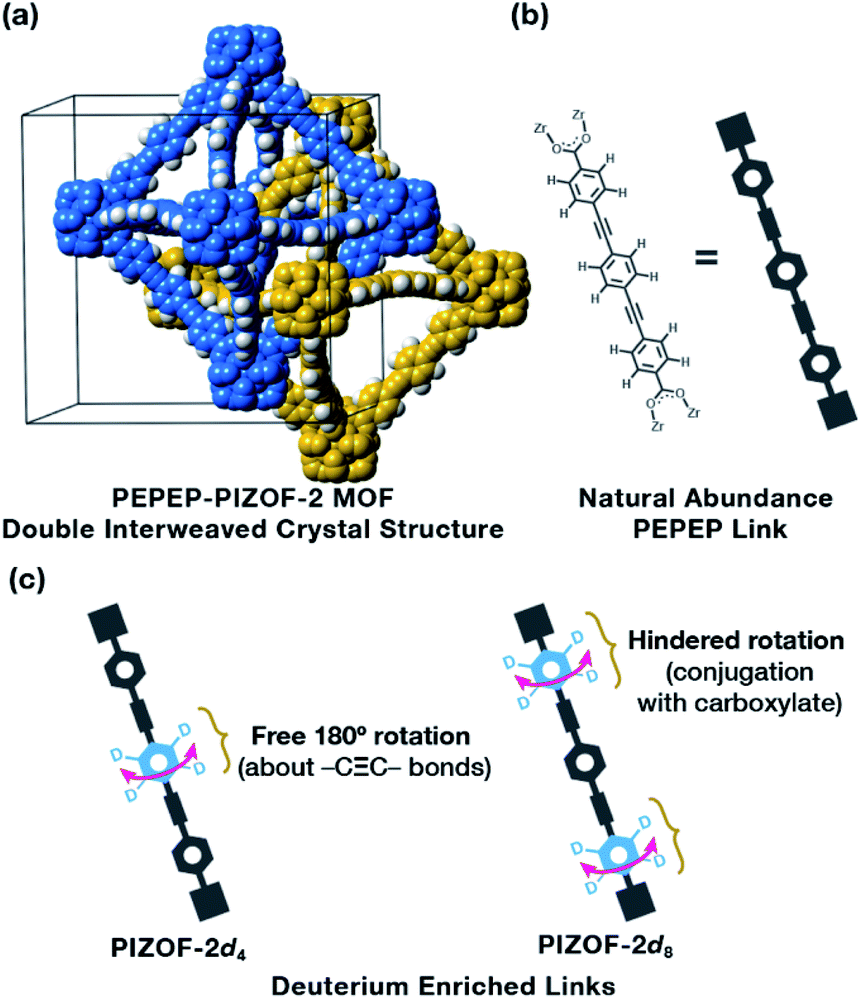 | ||
| Fig. 1 (a) Crystal structure of the double interweaved MOF PIZOF-2, showing each framework in separate colors. (b) Molecular structure of the PEPEP link. (c) Deuterium enriched linkers used in this study emphasizing the deuterium location in the link and the different chemical environments. | ||
Zirconia MOFs have been shown to exhibit unprecedented chemical stability, of which the family of Porous Interpenetrated Zirconia Organic Frameworks (PIZOFs) features superior stability combined with a unique molecular composition of their linkers.8,9 The linkers in PIZOF MOFs are linear and made by a combination of phenylene rings and ethynylene groups, where multiple chemical environments can be created around the phenylenes, thus altering their rotational behavior. Of the series, PEPEP-PIZOF-2 (hereafter PIZOF-2) is a high symmetry interweaved MOF (interweaved = interpenetrated with minimally displaced frameworks10,11) made with linkers that contain three phenylenes (P) and two ethynylenes (E) in an alternating form (hence PEPEP, Fig. 1b), creating two different types of chemical and crystallographic environments around the rotor moieties: the central phenylene ring is surrounded by two alkyne groups that provide a negligible electronic barrier for rotation and two outer phenylene rings surrounded by an alkyne and a carboxylate. So, we expect to observe significant differences in dynamics for each component of the linker.12 To properly observe the gyroscope-like rotation, protons were replaced with deuterons either in the inner ring (PIZOF-2d4, Fig. 1c) or in the outer rings (PIZOF-2d8, Fig. 1c). These two modes of isotopic labeling allowed the isolation of each ring to study of their dynamics by 2H NMR.
Samples of the PIZOF-2 MOF containing natural and isotopically enriched PEPEP links were prepared from adapted published procedures (ESI†).13 The MOFs were prepared via solvothermal crystallization of the respective linkers in DMF in the presence of ZrCl4 and proline-HCl at 120 °C for 24 h, resulting in crystalline powder samples of formula Zr6O4(OH)4[PEPEP]6, Zr6O4(OH)4[PEPEP-d4]6, and Zr6O4(OH)4[PEPEP-d8]6. Powder X-ray diffraction (PXRD) patterns of all three isotopologues exhibited sharp diffraction lines starting at 3.84° 2θ (CuKα radiation) characteristic of the cubic PIZOF-2 MOF phase (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group symmetry) (Fig. 2a).14 Phase purity was assessed using Rietveld refinement of the experimental patterns using the single crystal unit cell data resulting in phase pure samples with low residuals (Fig. S1–S4†).
m space group symmetry) (Fig. 2a).14 Phase purity was assessed using Rietveld refinement of the experimental patterns using the single crystal unit cell data resulting in phase pure samples with low residuals (Fig. S1–S4†).
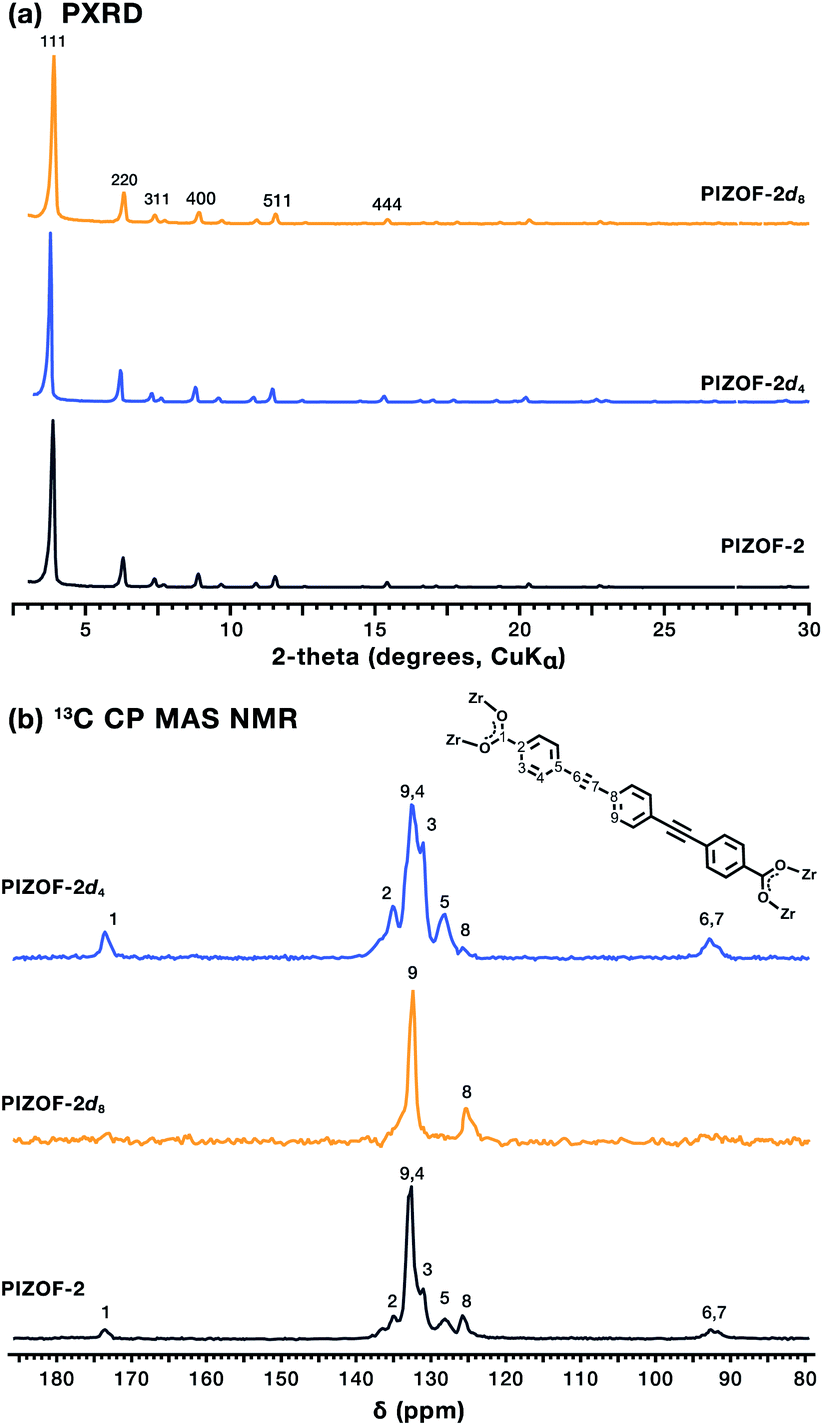 | ||
| Fig. 2 (a) Powder X-ray diffraction of the natural and isotopically enriched PIZOF-2 MOFs demonstrating their isoreticular nature. Miller indices of the most intense peaks are indicated. (b) 13C CPMAS NMR spectra of natural and isotopically enriched PIZOF-2 MOFs. | ||
The internal structure of the MOFs was analyzed using 13C Cross-Polarization with Magic Angle Spinning (CP MAS) NMR spectroscopy, where the intensities of 13C signals varied according to the level of deuteration of the linker in each MOF (Fig. 2b). PIZOF-2 exhibits a 13C spectrum with signals at around 92 ppm, corresponding to the internal ethynyl, signals between 120 and 140 ppm corresponding to the phenylene carbons, and signals at 173 ppm that correspond to carboxylates, consistent with the expected structure. In PIZOF-2d4 the signals that correspond to the central phenylene ring are attenuated (Fig. 2b, signal 8) compared to the natural material, whereas in PIZOF-2d8, the only visible signals are those of the central ring, due to the absence of vicinal protons required for CP. Peaks associated with solvents and other reagents were not observed indicating a successfully evacuated framework, which in addition to high crystallinity and the magnetic field produces changes in the spectral line shape that can be associated with different types of motion.15 In the case of the PIZOF MOFs, the differences in the molecular substructure and porosity ensured having optimal samples for dynamic studies. Despite being double interweaved, the distances between centroids of the aromatic rings of the interpenetrating frameworks have values in the range of 6.23 Å to 8.04 Å (Fig. S7†). Considering that the volume of revolution of the phenylene is ca. 6 Å, significant changes in the internal rotational dynamics caused by interpenetration were ruled out. Besides, it is expected that the phenylene rings have sufficient space to undergo fast rotational displacement, as it has been observed in other MOFs.16 To determine this, the deuterated samples were studied using solid-state quadrupolar echo 2H NMR spectroscopy. The reorientation of the C–2H bond vectors with respect to the external between outer and inner rings is expected to afford different rotational rates.
The 2H NMR line shape at room temperature of PIZOF-2d8 displays signals characteristic of motions in the intermediate exchange regime. A successful fitting of the spectrum using NMRweblab17 was obtained using a model that assumed two-fold flip jumps, indicating a rotational rate at room temperature of the outer rings of krot = 2.10 MHz (Fig. 3 top). The rate of rotation of the deuterated outer rings is similar to that reported in UiO-66(Zr)18 (krot = 2.3 MHz at rt) and much larger than that of other simple MOFs like MOF-5(Zn),12MIL-47(V),19 and MIL-53(Cr)19 (krot < 0.001 MHz at rt). The rotation of the outer rings could be then regulated by the electronic conjugation of the phenylene with the carboxylates and/or affected by the interactions with the metal oxide clusters.
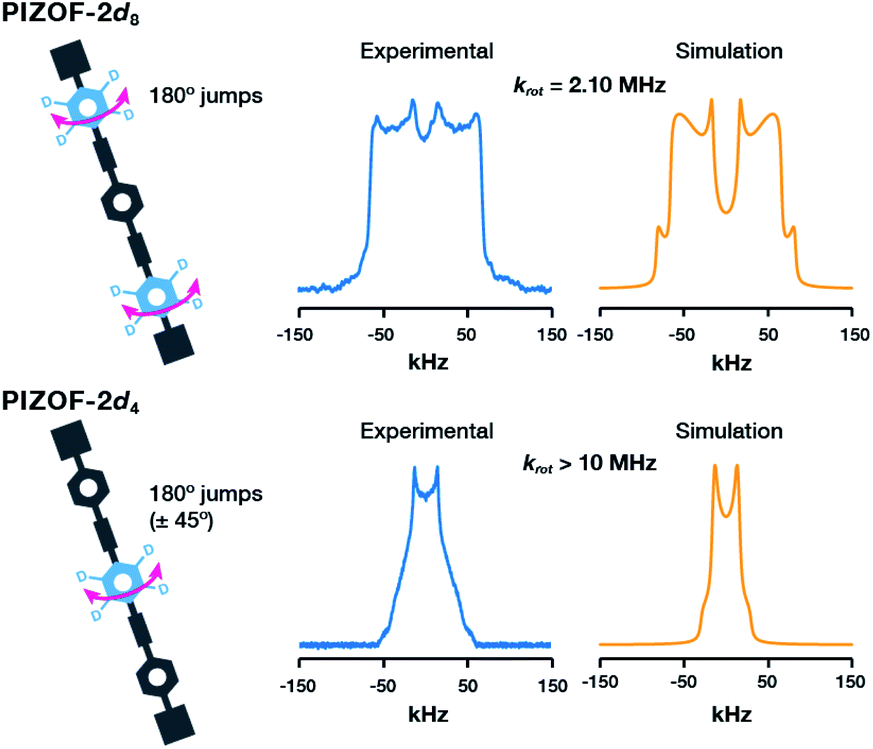 | ||
| Fig. 3 Experimental (blue) and calculated (orange) deuterium line shapes of PIZOF-2 at 295 K: (top) PIZOF-2d8 and (bottom) PIZOF-2d4. | ||
Conversely, in the case of PIZOF-2d4 (Fig. 3 bottom) the narrow 2H NMR spectrum is characteristic of ultrafast reorientations about the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– axis. A fitting of the spectrum was carried out assuming fast 180° jumps and large amplitude vibrations, indicating a rate of rotation of krot > 10 MHz, the upper limit of the 2H NMR sensitivity, so at 295 K the inner rings are rotating freely. This rate correlates with the minimal electronic barrier given by the flanking alkynes as has also been observed in a Zn-pyrazolate MOF that contains the same diethynyl-phenylene-diethynyl moiety.16 To date, this is the first time a MOF exhibits multiple rotational rates of their phenylene rings, which has implications for understanding and improving guest-diffusion related phenomena such as guest storage, catalysis, and separations.
C– axis. A fitting of the spectrum was carried out assuming fast 180° jumps and large amplitude vibrations, indicating a rate of rotation of krot > 10 MHz, the upper limit of the 2H NMR sensitivity, so at 295 K the inner rings are rotating freely. This rate correlates with the minimal electronic barrier given by the flanking alkynes as has also been observed in a Zn-pyrazolate MOF that contains the same diethynyl-phenylene-diethynyl moiety.16 To date, this is the first time a MOF exhibits multiple rotational rates of their phenylene rings, which has implications for understanding and improving guest-diffusion related phenomena such as guest storage, catalysis, and separations.
As the transport of guests throughout the MOF would be affected by the interactions between the guest and the static and dynamic components of the framework, we impregnated deuterated PIZOF-2 samples with tetracyanoquinodimethane (TCNQ). Given the electron-rich nature of the linker, electron deficient TCNQ was selected because it fits into the pores and has a high propensity to form strong π–π stacking bonds, often in the form of charge transfer complexes.20 In other words, TCNQ is a very sticky molecule known to affect the electronic structure of MOFs and has been used as an additive to enhance their charge conduction properties for device applications.21,22
The incorporation of TCNQ into the MOF was performed by immersing MOF powder samples in CH2Cl2 solutions for a minimum of 6 h at 295 K followed by rinsing, resulting in a loading capacity of 28.6 ± 0.2 TCNQ molecules per unit cell. At this saturated state, the white crystals changed to a green color and showed a strong EPR signal with g = 2.0025 (Fig. S8c†), compared to the pristine MOF. This could be attributed to a charge transfer event that produces organic radicals which overshadows the intrinsic paramagnetism of the zirconia oxoclusters.23 We also observed a quench of the emission, with a significant change in the quantum yield from ΦF = 8.5% to ΦF < 0.1% (Fig. S8d†). Fluorescence quenching was expected due to the interaction of electron deficient molecules with the conjugated oligo-phenylene-ethynylene linkers that make the MOF emissive.24
The 13C CPMAS spectrum of TCNQ loaded PIZOF-2d4 not only confirmed the guest within the pores (Fig. S12†), but it also revealed the changes in the chemical environment around the linkers: the appearance of a second carboxylate signal around δ = 174 ppm and a second quaternary carbon signal around δ = 128 ppm, with higher intensities with an increased loading time (Fig. 4a), attributable to the interaction of TCNQ with the outer rings of the PEPEP links, closer to the Zr cluster. Surprisingly, despite the evidence of the diffusion of TCNQ into the MOF, the solid-state 2H NMR spectrum of PIZOF-2d4 loaded with TCNQ for 6 h remained unaltered (Fig. 4b). Increasing the impregnation time to 72 h or increasing the temperature to 60 °C resulted in similar line shapes. These results suggest that the guest may have adsorbed near the outer phenylene rings of the linker. To demonstrate this, PIZOF-2d8 loaded with TCNQ for 6 h (Fig. 4c) was studied by 2H NMR. Interestingly, the fitting of the 2H line shape indicated slightly faster rotational rates compared to pristine PIZOF-2d8, changing from krot = 2.1 MHz to krot = 3.3 MHz. Only rising the impregnation temperature to 60 °C for 24 h allowed faster adsorption equilibration, decreasing the rotational rate to krot = 1.2 MHz. This indicates that the diffusion of TCNQ is slow and may require longer equilibration times at higher temperature to reach an equilibrium. Furthermore, considering the changes in the chemical shift of the carboxylate peak observed by 13C NMR CP MAS upon the diffusion of the guest (Fig. 4a, pink mark), as well as the minor changes in the rotational dynamics of the aromatic rings, we postulate that the TCNQ is located closer to the metal cluster, which agrees well with previously observed guest-loaded Zr-based MOFs.25,26
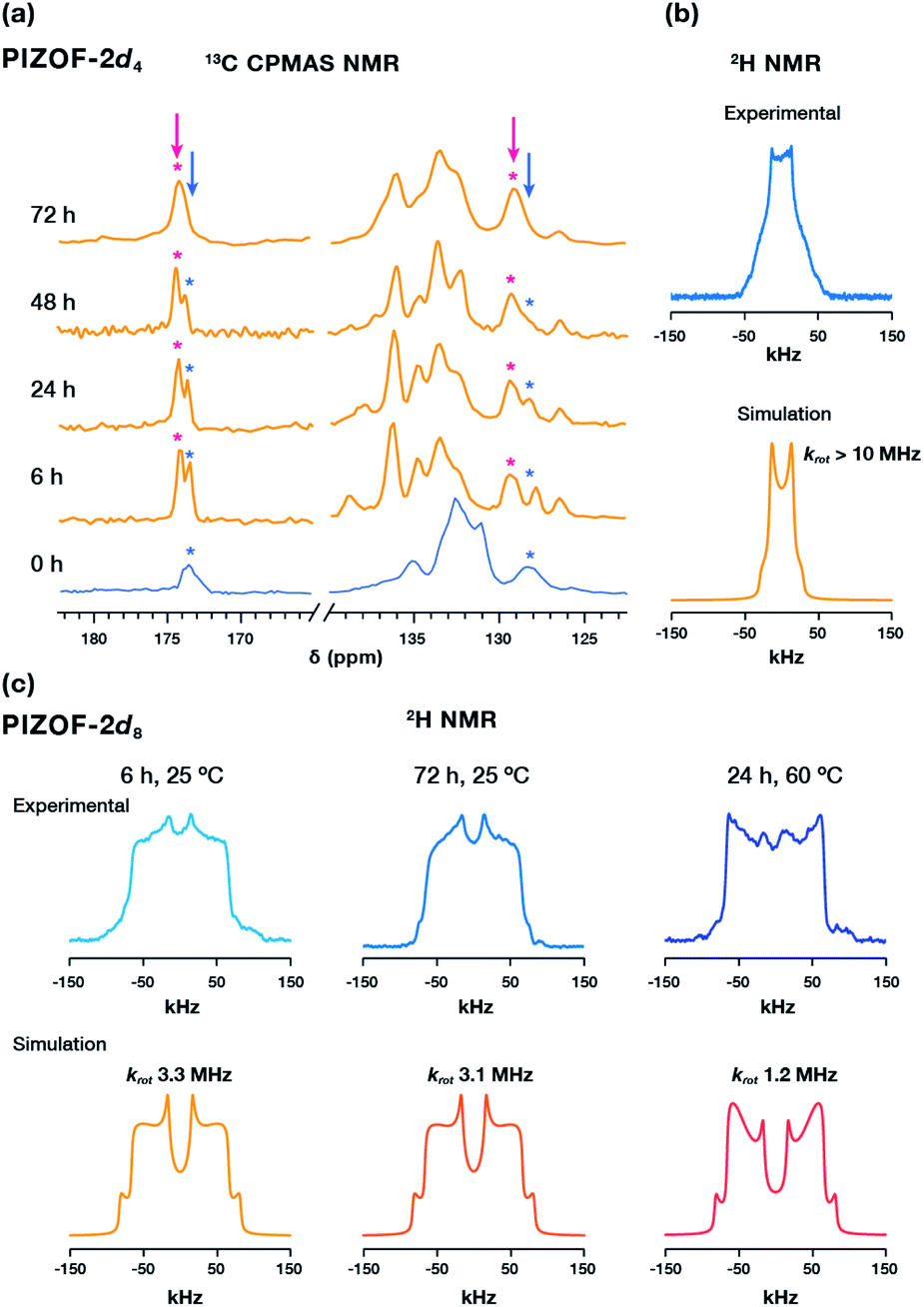 | ||
| Fig. 4 (a) 13C CPMAS of PIZOF-2d4 at different TCNQ loading times. (b) Experimental and simulated 2H NMR spectra of PIZOF-2d4 revealing that the signal from the central phenylene remains unaffected. (c) Experimental and simulated 2H NMR of PIZOF-2d8 under different TCNQ loading conditions. | ||
This work highlights that our approach can tackle one of the challenges in guest-loaded MOFs, which is the understanding of the interactions between the guest and the framework, a problem often exacerbated by the difficulty of acquiring high-quality single crystals. Furthermore, even after obtaining suitable crystals, X-ray diffraction studies provide only averaged space and time information. Conversely, solid-state NMR, as it is time-resolved, is ideal to analyze guest loaded MOFs in bulk samples, providing kinetic information such as transient π-interaction sites,27 gas-absorption diffusional rates,28 internal rotational dynamics,6 and other kinetic details.29,30
Conclusions
In summary, we studied for the first time the amphidynamic behavior of the ultra-stable PIZOF-2 MOF in the absence and presence of the TCNQ guest through a combined material characterization and solid-state NMR technique using isotopically enriched linkers. We found that the phenylene rings in the linkers of this MOF exhibit two different rotational rates and that they are not equally affected by the inclusion of the guest. This study offers insights into the complex molecular dynamics within MOFs aiming for tunable properties of synthetic molecular machines. Further work exploring the dynamics of the MOF PIZOF-2 at different temperatures and the effect of the guest on the rotation under those conditions is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from CONACYT A1-S-32820 and PAPIIT IN209119. We thank R. A. Toscano, M. C. García González, M. A. Peña, E. Huerta, V. Gómez-Vidales, U. Hernández-Balderas and A. Nuñez Pineda for technical support. The authors thank UNAM for support related to UNAM's BGSI node. We thank Mr A. Colin-Molina for help with measurements and discussion.Notes and references
- C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892–1910 RSC.
- A. Colin-Molina, D. P. Karothu, M. J. Jellen, R. A. Toscano, M. A. Garcia-Garibay, P. Naumov and B. Rodríguez-Molina, Matter, 2019, 1, 1033–1046 CrossRef.
- C. S. Vogelsberg, F. J. Uribe-Romo, A. S. Lipton, S. Yang, K. N. Hou, S. Brown and M. A. Garcia-Garibay, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13613–13618 CrossRef CAS.
- J. Gonzalez, R. Nandini Devi, D. P. Tunstall, P. A. Cox and P. A. Wright, Microporous Mesoporous Mater., 2005, 84, 97–104 CrossRef CAS.
- G. Bastien, C. Lemouchi, P. Wzietek, S. Simonov, L. Zorina, A. Rodríguez-Fortea, E. Canadell and P. Batail, Z. Anorg. Allg. Chem., 2014, 640, 1127–1133 CrossRef CAS.
- X. Jiang, H. B. Duan, S. I. Khan and M. A. Garcia-Garibay, ACS Cent. Sci., 2016, 2, 608–613 CrossRef CAS.
- A. Gonzalez-Nelson, F. X. Coudert and M. A. van der Veen, Nanomaterials, 2019, 9, 330–336 CrossRef CAS.
- A. Schaate, P. Roy, T. Preuße, S. J. Lohmeier, A. Godt and P. Behrens, Chem.–Eur. J., 2011, 17, 9320–9325 CrossRef CAS.
- R. J. Marshall, Y. Kalinovskyy, S. L. Griffin, C. Wilson, B. A. Blight and R. S. Forgan, J. Am. Chem. Soc., 2017, 139, 6253–6260 CrossRef CAS.
- B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- S. L. Gould, D. Tranchemontagne, O. M. Yaghi and M. a Garcia-garibay, J. Am. Chem. Soc., 2008, 130, 3246–3247 CrossRef CAS.
- R. J. Marshall, S. L. Griffin, C. Wilson and R. S. Forgan, Chem.–Eur. J., 2016, 22, 4870–4877 CrossRef CAS.
- T. L. H. Doan, H. L. Nguyen, H. Q. Pham, N. N. Pham-Tran, T. N. Le and K. E. Cordova, Chem.–Asian J., 2015, 10, 2660–2668 CrossRef CAS.
- G. E. Pake, J. Chem. Phys., 1948, 16, 327–336 CrossRef CAS.
- S. Bracco, F. Castiglioni, A. Comotti, S. Galli, M. Negroni, A. Maspero and P. Sozzani, Chem.–Eur. J., 2017, 23, 11210–11215 CrossRef CAS.
- V. Macho, L. Brombacher and H. W. Spiess, Appl. Magn. Reson., 2001, 20, 405–432 CrossRef CAS.
- D. I. Kolokolov, A. G. Stepanov, V. Guillerm, C. Serre, B. Frick and H. Jobic, J. Phys. Chem. C, 2012, 116, 12131–12136 CrossRef CAS.
- D. I. Kolokolov, H. Jobic, A. G. Stepanov, V. Guillerm, T. Devic, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 4791–4794 CrossRef CAS.
- H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 CrossRef CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS.
- M. D. Allendorf, M. E. Foster, F. Léonard, V. Stavila, P. L. Feng, F. P. Doty, K. Leong, E. Y. Ma, S. R. Johnston and A. A. Talin, J. Phys. Chem. Lett., 2015, 6, 1182–1195 CrossRef CAS.
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wanga, Chem. Commun., 2012, 48, 11656–11658 RSC.
- D. C. Santra, M. K. Bera, P. K. Sukul and S. Malik, Chem.–Eur. J., 2016, 22, 2012–2019 CrossRef CAS.
- A. Van Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS.
- A. R. Muguruza, R. F. de Luis, N. Iglesias, B. Bazán, M. K. Urtiaga, E. S. Larrea, A. Fidalgo-Marijuan and G. Barandika, J. Inorg. Biochem., 2020, 205, 110977 CrossRef CAS.
- X. Xu, S. Li, Q. Liu, Z. Liu, W. Yan, L. Zhao, W. Zhang, L. Zhang, F. Deng, H. Cong and H. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 973–981 CrossRef CAS.
- B. E. G. Lucier, S. Chen and Y. Huang, Acc. Chem. Res., 2018, 51, 319–330 CrossRef CAS.
- W. Yan, S. Li, T. Yang, Y. Xia, X. Zhang, C. Wang, Z. Yan, F. Deng, Q. Zhou and H. Deng, J. Am. Chem. Soc., 2020, 142(38), 16182–16187 CrossRef CAS.
- E. Brunner and M. Rauche, Chem. Sci., 2020, 11, 4297–4304 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis procedures, powder X-ray diffractograms, solid-state NMR spectra, EPR, X-ray, and fluorescence analyses. See DOI: 10.1039/d0sc04432f |
| ‡ Current address: Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. |
| This journal is © The Royal Society of Chemistry 2020 |