
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycoside hydrolase stabilization of transition state charge: new directions for inhibitor design†
Weiwu
Ren‡
 a,
Marco
Farren-Dai
a,
Natalia
Sannikova
a,
Katarzyna
Świderek
b,
Yang
Wang‡
a,
Oluwafemi
Akintola
a,
Robert
Britton
*a,
Vicent
Moliner
*b and
Andrew J.
Bennet
*a
a,
Marco
Farren-Dai
a,
Natalia
Sannikova
a,
Katarzyna
Świderek
b,
Yang
Wang‡
a,
Oluwafemi
Akintola
a,
Robert
Britton
*a,
Vicent
Moliner
*b and
Andrew J.
Bennet
*a
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada. E-mail: bennet@sfu.ca; rbritton@sfu.ca; Tel: +1-778-782-8814
bDepartament de Química Física i Analítica, Universitat Jaume I, 12560 Castellón, Spain. E-mail: moliner@uji.es
First published on 16th September 2020
Abstract
Carbasugars are structural mimics of naturally occurring carbohydrates that can interact with and inhibit enzymes involved in carbohydrate processing. In particular, carbasugars have attracted attention as inhibitors of glycoside hydrolases (GHs) and as therapeutic leads in several disease areas. However, it is unclear how the carbasugars are recognized and processed by GHs. Here, we report the synthesis of three carbasugar isotopologues and provide a detailed transition state (TS) analysis for the formation of the initial GH-carbasugar covalent intermediate, as well as for hydrolysis of this intermediate, using a combination of experimentally measured kinetic isotope effects and hybrid QM/MM calculations. We find that the α-galactosidase from Thermotoga maritima effectively stabilizes TS charge development on a remote C5-allylic center acting in concert with the reacting carbasugar, and catalysis proceeds via an exploded, or loose, SN2 transition state with no discrete enzyme-bound cationic intermediate. We conclude that, in complement to what we know about the TS structures of enzyme-natural substrate complexes, knowledge of the TS structures of enzymes reacting with non-natural carbasugar substrates shows that GHs can stabilize a wider range of positively charged TS structures than previously thought. Furthermore, this enhanced understanding will enable the design of new carbasugar GH transition state analogues to be used as, for example, chemical biology tools and pharmaceutical lead compounds.
Introduction
Biological catalysts that remove carbohydrate residues from complex biomolecules are found throughout nature; within that functional group, glycoside hydrolases (GHs; glycosidases) are the main family of enzymes responsible for hydrolytically cleaving a glycosidic bond, the covalent link between a sugar residue and another molecule. Glycoside hydrolases are classified into more than 160 families based on sequence and structure.1,2 Further grouping of some GH families into “clans” occurs for those that have similar active site amino acids3 and identical structural motifs, and intraclan GH families tend to share the same catalytic mechanism of action.4 Glycoside hydrolases cleave glycosidic bonds to give a hemiacetal product that has one of two stereochemical outcomes: a product with either the same (retention) or opposite (inversion) configuration at the anomeric center relative to the substrate.5 In the case of most retaining enzymes, general acid catalysis facilitates the departure of the leaving group prior to or simultaneous with the nucleophilic attack by an active site residue on the anomeric center of the carbohydrate substrate, which traverses a pyranosylium ion-like transition state (TS) to give a covalently bound glycosyl enzyme intermediate (Fig. 1A) that is subsequently hydrolyzed.6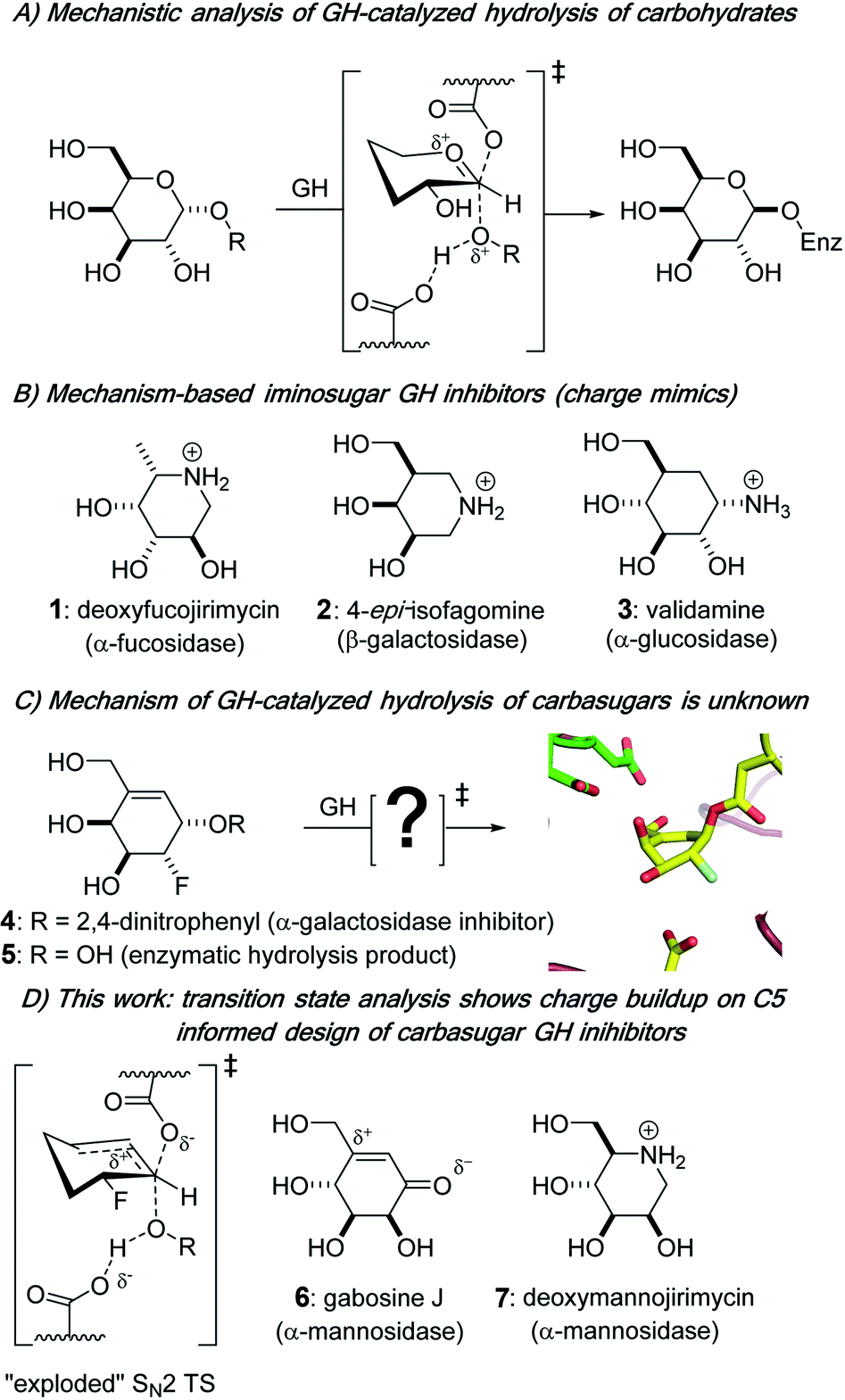 | ||
| Fig. 1 Panel (A): currently accepted mechanism for the formation of the covalently bound glycosyl–enzyme intermediate in retaining glycoside hydrolases. Panel (B): structures of the iminosugar and carbasugar competitive inhibitors of glycoside hydrolases, deoxyfucojirimycin (1), 4-epi-isofagomine (2), and validamine (3). Panel (C): GH covalent inhibitor 4, its hydrolysis product 5, and the structure of the enzyme-intermediate with the T. maritima α-galactosidase. Panel (D): depiction of the TS for covalent labeling of TmGalA, and the structures of GH inhibitors gabosine J (6) and deoxymannojirimycin (7). | ||
Glycoside hydrolases are among the most catalytically proficient of enzymes7 and play critical roles in diverse biological processes, from viral pathogenesis to the body's immune response and maintenance of cellular homeostasis.8,9 Aberrant activity of GHs is often associated with disease states; from a pharmaceutical angle, interest is growing in the potential of controlling the activity of these enzymes with small molecule inhibitors.10–12 The design of small molecule GH inhibitors is informed by nature as well as an understanding of the unique mechanism of action of the specific enzyme being targeted. Three common classes of GH competitive inhibitors, all of which bind to the active site of the enzyme via non-covalent interactions and have weakly basic amines that are protonated at physiological pH values (Fig. 1B), are: (i) the natural product nojirimycin-based inhibitors,13 which include the α-fucosidase inhibitor deoxyfucojirimycin 1,14 (ii) the synthetic isofagomine family of inhibitors, e.g.2, where the positive charge is located on the pseudo-anomeric center of the six-membered ring,15,16 and (iii) validamine 3,17,18 a natural carbasugar structural component of validamycin A,19,20 which is a bacterially produced antibiotic. Of note, iminosugar- and carbasugar-based GH inhibitors have proven to be effective in the treatment of various GH-dependent disorders, including viral infections,21 diabetes,22 and lysosomal storage disease.23 Mechanistically, tight-binding iminosugars and weakly basic carbasugars are often presumed to be transition state analogs (TSAs) by virtue of their assumed ability to be good charge mimics of pyranosylium ion-like enzymatic transition states (Fig. 1A).5,24–26 However, despite the tight binding that many carbasugars exhibit towards GHs, it remains controversial as to whether they are indeed TSA inhibitors.24,27–29 A more theoretically based approach to the design of TSAs has been used and incorporates the measurement of kinetic isotope effects (KIEs) with computational methods to determine the geometric and electronic structure of enzymatic transition states for the reactions involving natural substrates,30–32 and to use this structural information to design of TSA inhibitors.33–35
In contrast to previous work on non-covalent inhibition of GHs,36,37 we presented studies of small molecule alkenyl, e.g., 4,38,39 and cyclopropyl39,40 carbasugar probes that both covalently label GHs and we provided structural insights into the conformations of the enzyme bound and covalent intermediates that are present during GH inactivation (Fig. 1C).38,40 Despite these and other efforts37,41 it has yet to be determined how the developing charge at the enzymatic TSs is delocalized and stabilized during GH catalysis with non-natural carbasugar substrates, an information void that is a barrier to improving the design of GH-based therapeutics. Herein, we report a detailed TS analysis of a carbasugar GH covalent inhibitor. We combine laboratory-based KIE measurements and hybrid quantum mechanics/molecular mechanics (QM/MM) calculations to show that pseudo-glycosylation of the enzymatic nucleophile in the GH36 α-galactosidase from Thermotoga maritima (TmGalA) proceeds via an exploded, or loose, SN2 TS with no discrete enzyme-bound cationic intermediate (Fig. 1D). This result highlights the ability of GHs to stabilize positive charge development at positions remote from the pseudo-anomeric center and provides a clear rationale for the improved potency of some carbasugar GH inhibitors (e.g., gabosine J 6)42,43 over their iminosugar counterparts (e.g., deoxymannojirimycin 7).44
Results and discussion
We measured three KIEs for isotopic substitutions at the pseudo-anomeric carbon in order to “solve” the TS for covalent labeling by our allylic carbasugar inhibitor. Specifically, we targeted (i) the α-secondary deuterium effect, which reports on rehybridization at the reaction centre, and (ii) the two primary heavy-atom effects, in particular, the reaction center 13C and the leaving group 18O, which report on bonding changes between the ground state (GS) and TS.Synthesis of isotopically labeled carbasugars
The required carbasugars (1-2H)-, (1-13C)-, and (1-13C,1-18O)-4 for the measurement of KIEs were synthesized using a de novo approach recently reported by us.38 Here, the required deuterium-labeled aldehyde (3-2H)-8 was accessed through LiAlD4 reduction of a dienone followed by oxidative cleavage of the remote alkene function. The corresponding 13C-labeled aldehyde (3-13C)-8 was synthesized from K13CN through displacement of bromide from allyl bromide followed by nitrile hydrolysis and a sequence of reactions including reduction/oxidation and addition of vinyl magnesium bromide. With the 2H aldehyde in hand, a D-proline catalyzed α-fluorination–aldol reaction using ketone 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 proceeded in good yield to provide a mixture of unstable fluorohydrin aldol adducts that were directly treated with the lithium anion derived from methylsulfonyl phenyltetrazole to provide a separable mixture of dienes to give the required stereoisomer 10 (91.8% ee).
38 proceeded in good yield to provide a mixture of unstable fluorohydrin aldol adducts that were directly treated with the lithium anion derived from methylsulfonyl phenyltetrazole to provide a separable mixture of dienes to give the required stereoisomer 10 (91.8% ee).
Likewise, Julia–Kocienski olefination of the fluorohydrin aldol adduct derived from 13C-labeled aldehyde (3-13C)-8 provided dienes 12 (92.3% ee) and 13 (93.8% ee). Acetylation of the free alcohol function, followed by removal of the silyl protecting group and ring closing metathesis (Grubbs' II) then provided the series of cyclohexenes 14, 15, and 16, arrived at by migration of the acetyl group during the deprotection step. For the 2H- and 13C-labeled carbasugars, arylation of 14 or 15 using 1-fluoro-2,4-dinitrobenzene and deprotection afforded (1-2H)-4 and (1-13C)-4, respectively. Finally, to introduce an 18O-label we removed the pseudo-anomeric acetate from 16 and used a Mitsunobu reaction with 4-nitro[18O1]benzoate to give an approximate 60:40 mixture of (1-13C)-4 and (1-13C,1-18O)-4 after standard debenzoylation, arylation, and deprotection (Fig. 2).
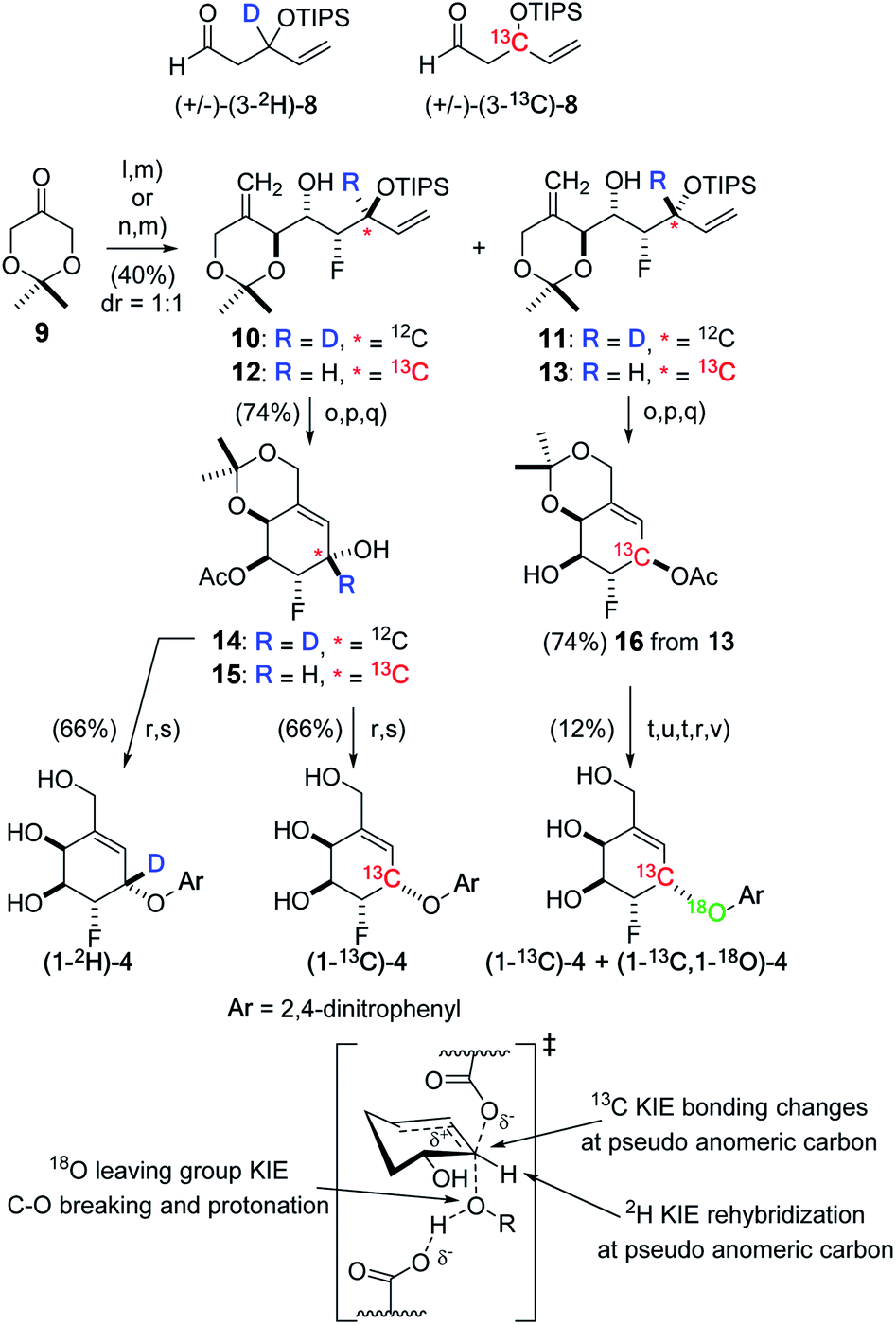 | ||
| Fig. 2 Synthesis of (2H)-, (13C)-, and (13C,18O)-4. (a) PCC, CH2Cl2, rt; (b) LiAlD4, Et2O, 0 °C; (c) TIPSCl, imidazole, CH2Cl2, rt (26% from hexan-1,5-dien-3-ol); (d) AD-mix, tBuOH-H2O, rt; (e) NaIO4, THF, H2O, rt (38% over two steps); (f) bis(triphenylphosphine)iminium chloride (5 mol%), CH2Cl2, H2O (0.5 M), 0 °C; (g) HCl (conc.), reflux (60% from allyl bromide); (h) LiAlH4, Et2O, 0 °C to rt; (i) DMP, NaHCO3, CH2Cl2, 0 °C; (j) CH2CHMgBr, CH2Cl2, Et2O, 0 °C; (k) TIPSCl, imidazole, CH2Cl2, rt [30% from (1-13C)but-3-enoic acid]; (l) (±)-(3-2H)-8, D-proline, Selectfluor, DMF, 0 °C then 9, D-proline, CH2Cl2; (m) methylsulfonyl phenyltetrazole, LiHMDS, THF, −78 °C (40% from 8, 10:11 or 12:13 = 1:1); (n) (±)-(3-13C)-8, D-proline, Selectfluor, DMF, 0 °C then 9, D-proline, CH2Cl2; (o) Ac2O, Et3N, DMAP, CH2Cl2, rt (84%); (p) TBAF, AcOH, THF, 0 °C to rt; (q) Grubbs' II catalyst, CH2Cl2, 40 °C; (r) 1-fluoro-2,4-dinitrobenzene, quinuclidine, DMF, rt; (s) K2CO3, MeOH, 0 °C then HCl (1 N) (66% from 12 or 14); (t) K2CO3, MeOH, 0 °C (88%); (u) 4-nitro[18O1]benzoic acid, PPh3, DIAD, THF, rt (58%); (v) TFA, CH2Cl2, rt. | ||
Measurement of kinetic isotope effects on GH covalent labeling
With the (2H)-, (13C)-, and (13C,18O)-alkenyl carbasugars in-hand, we turned our attention to measuring KIEs on the TmGalA-catalyzed hydrolysis of these compounds. For the enzyme-catalyzed reaction, kinact/Ki is determined by the differences in free energy between the ground state (free substrate/inhibitor and enzyme in solution) and the first irreversible step (cleavage of the glycosidic bond).24,38,45,46 Therefore, we decided to use 19F NMR spectroscopy to measure isotopologue ratios (R, the ratio of heavy-to-light isotopologues for the remaining starting material) as the reaction progressed (F, the fraction of reaction for the light isotopologue) to evaluate the secondary deuterium and 13C-KIEs.47,48 We note that because our syntheses involved enantioselective catalysis our covalent inhibitors are not enantiopure, as also would be the case with chiral pool starting materials, such as galactose. We corrected our R and F values, assuming that the enzyme would have no reaction with the small quantity of L-carbasugar in the reaction mixture (details in the Experimental section and Tables S1 and S2 in ESI†). Nonlinear least squares fitting of the corrected R and F values to eqn (1) gave values for two of the three competitive KIEs (Table 1) resulting from isotopic substitution at the pseudo-anomeric center.45,49,50 To measure the leaving group 18O-KIE we introduced a 13C probe nucleus at the pseudo-anomeric center. However, because 13C is a less sensitive probe nucleus than 19F, the concentration of covalent inhibitor was increased for NMR spectroscopic analysis. After checking that inclusion of ethanol-d6 (10% v/v) into the buffer had no effect on the activity of TmGalA, we measured the 18O-KIE at 50 °C—an increase in temperature from 37 °C to improve the solubility of 4—by following by following standard procedures.50–52 We note that the expected effect of the higher temperature on the measured 18O-KIE is smaller than the experimental error in the value (Table 1). Also, as the two enzyme-catalyzed chemical steps (kinact and kreact, Scheme 1) have drastically different first-order rate constants for the reaction of 4 with TmGalA,38 we were able to measure the α-secondary deuterium KIE on the turnover of 4 by measuring individual rate constants for kcat, or turnover, of 4 and (1-2H)-4.49 In addition, to show that the ratio of the rate constants for C–O bond cleavage to the rate constant for dissociation of the inhibitor from the Michaelis complex (E·4) (i.e., the forward commitment factor)53,54 is very small, we made the corresponding 4-nitrophenyl covalent inhibitor S-6 (ESI†), and found that it reacts over 100-fold less rapidly than 4 (ESI†). In other words, the enzyme-catalyzed cleavage of the allylic C–O bond in 4 is the first irreversible step and the measured competitive KIEs are intrinsic values.55| R = R0(1 − F1)(1/KIE − 1) | (1) |
| KIE (kinact/Ki) | k H/kD | k 12/k13b | k 16/k18c | k H/kD (kreact)d |
|---|---|---|---|---|
| a Conditions 25 mM HEPES, pH 7.4, 10% v/v D2O T = 37 °C. b Calculated from weighted averages of kH/kD × kD/k13 (Table S2, ESI). c T = 50 °C, value calculated at 37 °C, assuming T × ln(KIE) = constant is 1.045. d Measured by UV/vis spectroscopy for turnover of 4, conditions 50 mM HEPES, pH 7.4, 1 mg mL−1 BSA T = 37 °C. | ||||
| Experimental | 1.172 ± 0.011 | 1.029 ± 0.013 | 1.043 ± 0.006 | 1.147 ± 0.006 |
| QM/MM | 1.246 ± 0.022 | 1.041 ± 0.004 | 1.056 ± 0.005 | 1.107 ± 0.078 |
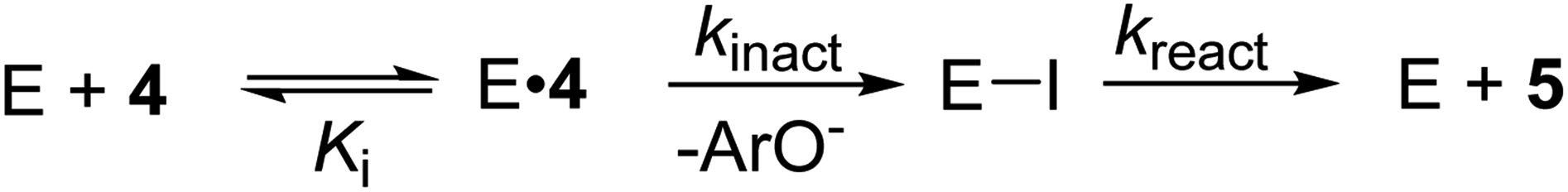 | ||
| Scheme 1 Kinetic scheme for the covalent inhibition of GH36 α-galactosidase by carbasugar analogue 4 to give, after hydrolysis of the covalent intermediate (E–I) the hydrolyzed product 5. | ||
Free energy surfaces for the covalent labeling of TmGalA by carbasugar 4
To understand how TmGalA stabilizes the TS for its covalent labeling by 4, we computationally studied the two chemical steps in the enzyme-catalyzed turnover of the covalent inhibitor (kinact and kreact, Scheme 1). To do this, we computed the free energy surface (FES) for the covalent labeling event (kinact, Scheme 1) at a DFT/MM level, after setting up the molecular model of the full E·4 system solvated in a box of water molecules (see ESI†). Our results show that nucleophilic attack of D327 and proton transfer to the departing dinitrophenolate leaving group take place in a stepwise manner (Fig. 3A). Here, we found that the covalent labeling event avoids formation of a high energy enzyme-bound allylic cation (top right corner Fig. 3A). Surprisingly, though little to no nucleophilic attack has occurred as the reaction transverses TS1 (Fig. 3A), this process still proceeds via a SN2 reaction, because after traversing the TS the covalent intermediate's C–O bond forms without crossing any other energy barriers. The computed inter-atomic distances of the forming and breaking bonds at the TSalk, 2.925 ± 0.683 and 2.212 ± 0.060 Å, respectively, as reported in Table S4 (ESI†), values that confirms its exploded or loose character.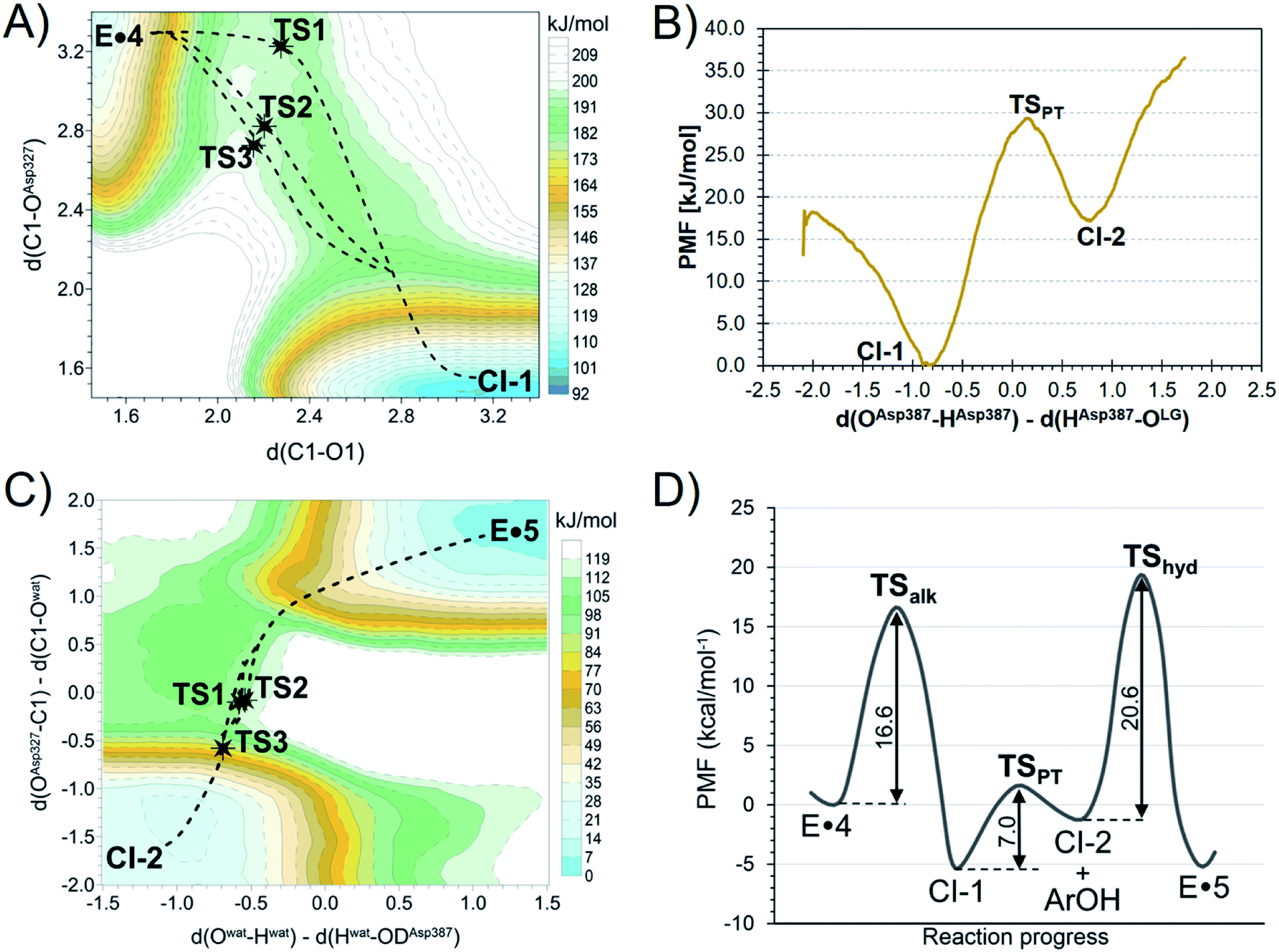 | ||
| Fig. 3 M06-2X:AM1/MM free energy surfaces: Panel (A): transformation of the Michaelis complex E·4 to the first covalent intermediate CI-1. The positions of three TS structures (TSalk) optimized at M06-2X/MM are indicated as stars, while dashed lines indicate the intrinsic reaction coordinate (IRC) paths. Panel (B): transformation of CI-1 to CI-2 by deprotonation of Asp387 by 2,4-dinitrophenolate. Panel (C): transformation of the second covalent intermediate CI-2 to the enzyme product complex (E·5). The positions of three TS structures (TShyd) optimized at M06-2X/MM are indicated as stars, while dashed lines indicate the intrinsic reaction coordinate (IRC) paths. Panel (D): overall reaction coordinate free energy for the transformation of the E·4 complex via the TSs for covalent labeling (TSalk, kinact), proton transfer (TSPT) and hydrolysis of the covalent intermediate (TShyd, kreact) and two covalent intermediates (CI-1 & CI-2) to give the enzyme complex of the product (E·5). | ||
Notably, directly after cleavage of the pseudo-glycosidic bond in 4, the covalent intermediate (CI-1) has a bound 2,4-dinitrophenolate while the acid catalyst (D387) remains protonated. The interconversion of the first covalent intermediate CI-1 to the second intermediate CI-2 corresponds to the proton transfer from D387 to the leaving group (Fig. 3B). Then, as described in detail in the ESI,† the leaving group is released from the active site and hydrolysis of the carbasugar covalent intermediate (CI-2) renders the enzyme complex of the hydrolyzed inhibitor 5, a process that is assisted by the deprotonated D387 (Fig. 3C). Again, we note that this reaction step avoids formation of an enzyme bound allylic cation. The overall free energy profile for the interconversion of the Michaelis complex to the product-bound species is shown in Fig. 3D. Our calculations are in accord with the experimental observation that hydrolysis of the covalent-enzyme intermediate is rate-limiting for inhibitor turnover (kinact ≫ kreact, Scheme 1).
To confirm our simulations, KIEs for this α-galactosidase-catalyzed reaction were computed (Table 1) by evaluating 3 × 3 combinations of three TS structures of the alkylation step (see Fig. 3A and their overlay in Fig. 4) and three GS structures of 4 solvated in a box of water molecules, optimized at DFT/MM level. We find, relative to the GS, that at the TSs for covalent labeling, positive charge develops on the remote allylic carbon atom C5 (Table S7, ESI†). This result clearly differentiates this class of inhibitor from pyranosides where positive charge development occurs mainly on the two adjacent positions to the anomeric carbon atom within the six-membered ring: the ring oxygen in the pyranosylium ion (C+–O ↔ C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+) and carbon-2 (via hyperconjugation).56,57 In addition, we note that the computed FES is relatively flat in the vicinity of the quadratic region of the TS (Fig. 3A), a conclusion that suggests that the enzyme is able to stabilize positive charge development on C5 via a plethora of different TS structures.
O+) and carbon-2 (via hyperconjugation).56,57 In addition, we note that the computed FES is relatively flat in the vicinity of the quadratic region of the TS (Fig. 3A), a conclusion that suggests that the enzyme is able to stabilize positive charge development on C5 via a plethora of different TS structures.
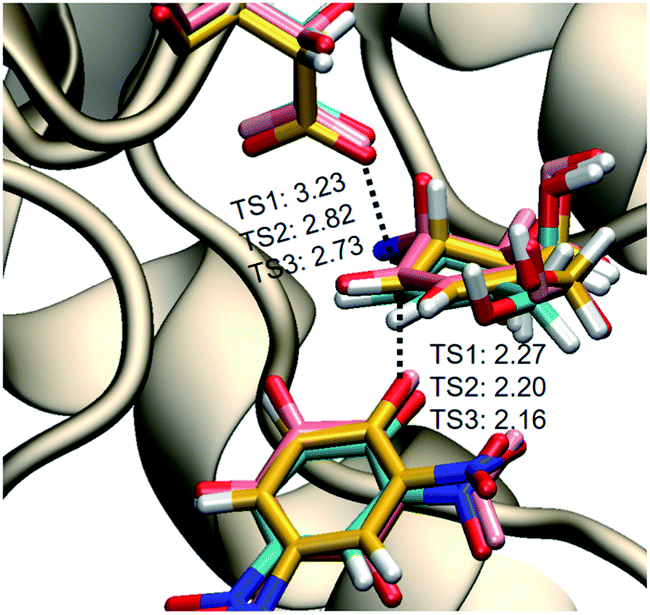 | ||
| Fig. 4 Overlay of the three TSs for the covalent labeling of TmGalA by 4, TSalk, optimized at M06-2X/MM level. | ||
We suggest that catalysis involves the GH's intrinsic ability to support positive charge development further removed from the anomeric center, and that this is clearly the origin of our carbasugar 4 being an excellent α-galactosidase covalent inhibitor. Fig. 5A shows the map of electrostatic potential computed on the reacting system at the TSalk under the effect of the protein environment, while Fig. 5B shows the electrostatic potential (V) generated by each protein residue on C5 of the carbasugar ring (Table S8, ESI†). The spatial location of the four aspartic acid residues, which provide the majority of the stabilization at the TS are shown in Fig. 5C (in blue) along with the two main destabilizing residues (in red). Moreover, as shown in Fig. 5B, the two largest contributors to stabilization of the positive charge on C5 are the two carboxylate residues (Asp 220 and Asp 221, top left Fig. 5C) that are positioned for H-bonding with the C4 and C6 hydroxyl groups; such interactions are common in GHs. As a result, we suggest that new designs of competitive, as well as covalent inhibitors, for GHs should incorporate design elements that allow positive charge incorporation, or development, at sites other than those that correspond to the anomeric carbon and the ring oxygen atoms.
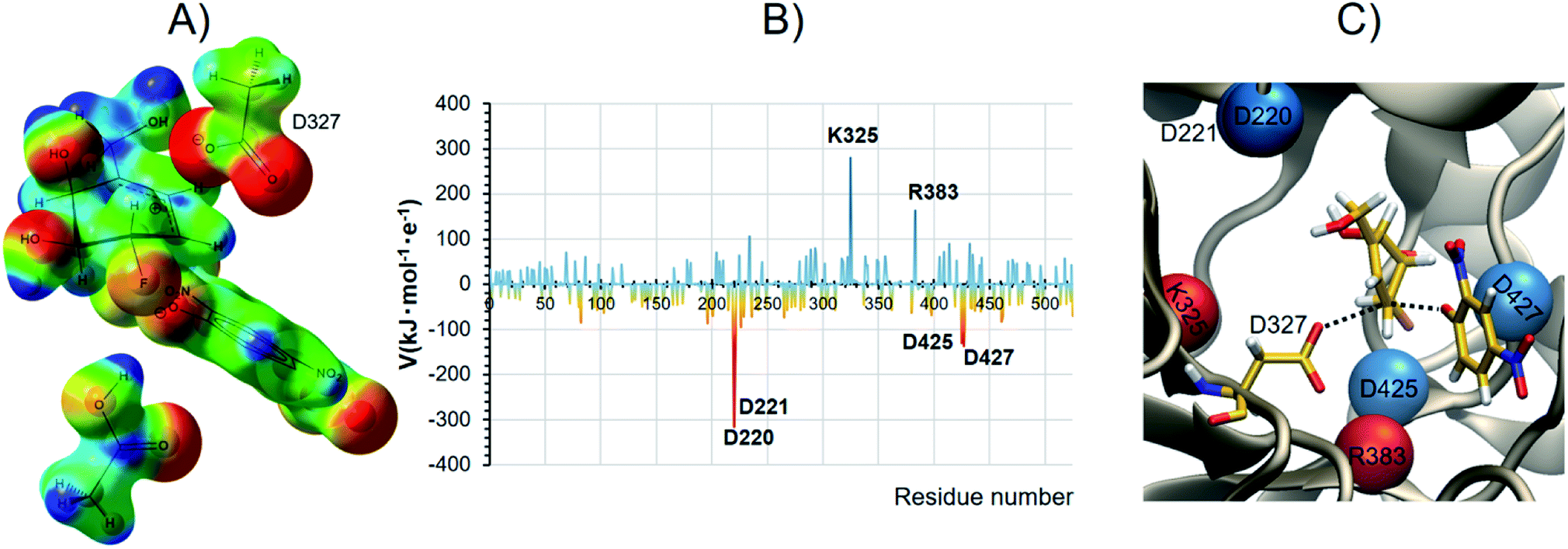 | ||
| Fig. 5 Electrostatic analysis on TSalk: Panel (A): map of electrostatic potential computed on the reacting system under the effect of the protein environment: blue positive values, red negative values. Panel (B); electrostatic potential generated by the protein, by residue, on C5 atom of the carbasugar ring. Panel (C): detail of the GH active site. Residues with a significant positive stabilizing contribution to the electrostatic potential are shown as blue spheres, while maroon spheres depict the major destabilizing residues. | ||
We also computed three TSs for the hydrolysis of the covalent intermediate CI-2 (TShyd, Fig. 3C) to give the enzyme complex of hydrolyzed inhibitor (E·5). To calculate the expected secondary α-2H KIE value, we used three ground state structures of 4 bound to TmGalA (E·4, Scheme 1), which is the ground state for the enzymatic turnover (kreact), plus the three TSs for hydrolysis of the covalent intermediate and evaluated the 3 × 3 combinations of DFT/MM optimized structures (Table 1). Again, the agreement between experimental and calculated KIE values provides validation of the theoretical TSs. It is remarkable that the standard deviations are similar to the reported experimental errors. The larger uncertainty for the QM/MM α-2H KIE values can be attributed to the nature of the isotopic substitution (increment of 100% in the mass), the flat quadratic region of the TS, and the variation in the position of the nucleophilic water.
Conclusions
In contrast to previous KIE/theoretical modeling studies using natural substrates,32,33,35 our study uses a combination of experimental KIE measurements and QM/MM calculations to demonstrate that the GH α-galactosidase from T. maritima efficiently catalyzes its own covalent labeling by allylic carbasugars through an exploded SN2 TS in which the enzyme stabilizes positive charge development at C5, a position that is far-removed from the anomeric carbon. Of critical importance to inhibitor design, these results show that GHs are capable of stabilizing positive charge at sites that are remote from those that chemists use to position weakly basic amines – specifically, the anomeric carbon, ring oxygen and glycosidic oxygen – as exemplified by the deoxynojirimycin, isofagomine and validamine families of inhibitors. We therefore propose a new avenue for the design of efficient glycoside hydrolase TSA inhibitors: the inclusion of structural elements that introduce positive charge at other ring carbon atoms, such as the tertiary C5 atom in glycosides. This prediction is supported by the improved potency that reported by Vidyasagar and Sureshan for the neutral carbasugar GH inhibitor gabosine J over the iminosugar counterpart (deoxymannojirimycin) (Fig. 1D).43Experimental
Materials and methods
All anhydrous reactions described were performed under an atmosphere of nitrogen using flame-dried glassware. Normal phase column chromatography was carried out with 230–400 mesh silica gel (Silicycle, SiliaFlash® P60). All reagents and starting materials were purchased from Sigma Aldrich, Alfa Aesar, TCI America, or Acros and were used without further purification. All solvents were purchased from Sigma-Aldrich, EMD, Anachemia, Caledon, Fisher or ACP and used without further purification unless otherwise specified. CH2Cl2 was freshly distilled over CaH2, and tetrahydrofuran (THF) was freshly distilled over Na metal/benzophenone. Cold temperatures were maintained by use of the following conditions: 0 °C, ice-water bath; −78 °C, acetone–dry ice bath; temperatures between −78 °C and 0 °C required for longer reaction times were maintained with a Neslab Cryocool Immersion Cooler (CC-100 II) in a 2-propanol bath. Detailed synthetic routes for all isotopologues, protocols for the measurement of KIEs, and all computational methods are provided in the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council (NSERC) Discovery Grants (RB 2019-06368 and AJB 2017-04910), Spanish Ministerio de Ciencia, Innovación y Universidades (Grant PGC2018-094852-B-C21 and PID2019-107098RJ-I00), Generalitat Valenciana (Grant AICO/2019/195 and SEJI/2020/007) Universitat Jaume I (UJI-B2017-31 and UJI-A2019-04), and a MSFHR Career Investigator Award (RB), K. Ś. thanks the MINECO for a Juan de la Cierva – Incorporación (ref. IJCI-2016-27503) contract. This research was enabled by computational resources provided by Servei d’Informàtica of Universitat Jaume I, West Grid (http://www.westgrid.ca), and Compute Canada (http://www.computecanada.ca).References
- B. Henrissat and A. Bairoch, Biochem. J., 1996, 316, 695–696 Search PubMed.
- B. L. Cantarel, P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard and B. Henrissat, Nucleic Acids Res., 2009, 37, D233–D238 Search PubMed.
- D. J. Vocadlo and G. J. Davies, Curr. Opin. Chem. Biol., 2008, 12, 539–555 Search PubMed.
- D. A. Comfort, K. S. Bobrov, D. R. Ivanen, K. A. Shabalin, J. M. Harris, A. A. Kulminskaya, H. Brumer and R. M. Kelly, Biochemistry, 2007, 46, 3319–3330 Search PubMed.
- M. Sinnott, in Carbohydrate Chemistry and Biochemistry: Structure and Mechanism, RSC Publishing, Cambridge, 2nd edn, 2013 Search PubMed.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 Search PubMed.
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 Search PubMed.
- A. Varki, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 3rd edn, 2017 Search PubMed.
- G. Michal, Biochemical Pathways : An Atlas of Biochemistry and Molecular Biology, Wiley, Spektrum, New York, Heidelberg, 1999 Search PubMed.
- G. Speciale, A. J. Thompson, G. J. Davies and S. J. Williams, Curr. Opin. Struct. Biol., 2014, 28, 1–13 Search PubMed.
- E. M. Sánchez-Fernández, J. M. Garcia Fernández and C. Ortiz Mellet, Chem. Commun., 2016, 52, 5497–5515 Search PubMed.
- F. M. Platt, Nat. Rev. Drug Discovery, 2018, 17, 133–150 Search PubMed.
- S. Inouye, T. Tsuruoka, T. Ito and T. Niida, Tetrahedron, 1968, 24, 2125–2144 Search PubMed.
- M. Dubernet, A. Defoin and C. Tarnus, Bioorg. Med. Chem. Lett., 2006, 16, 1172–1174 Search PubMed.
- T. M. Jespersen, W. L. Dong, M. R. Sierks, T. Skrydstrup, I. Lundt and M. Bols, Angew. Chem., Int. Ed., 1994, 33, 1778–1779 Search PubMed.
- Y. Ichikawa and Y. Igarashi, Tetrahedron Lett., 1995, 36, 4585–4586 Search PubMed.
- E. Borges de Melo, A. D. Gomes and I. Carvalho, Tetrahedron, 2006, 62, 10277–10302 Search PubMed.
- K. S. E. Tanaka, G. C. Winters, R. J. Batchelor, F. W. B. Einstein and A. J. Bennet, J. Am. Chem. Soc., 2001, 123, 998–999 Search PubMed.
- S. Horii, T. Iwasa, E. Mizuta and Y. Kamada, J. Antibiot., 1971, 24, 59–63 Search PubMed.
- H. J. Dong, T. Mahmud, I. Tornus, S. Lee and H. G. Floss, J. Am. Chem. Soc., 2001, 123, 2733–2742 Search PubMed.
- C. U. Kim, W. Lew, M. A. Williams, H. Liu, L. Zhang, S. Swaminathan, N. Bischofberger, M. S. Chen, D. B. Mendel, C. Y. Tai, W. G. Laver and R. C. Stevens, J. Am. Chem. Soc., 1997, 119, 681–690 Search PubMed.
- S. P. Clissold and C. Edwards, Drugs, 1988, 35, 214–243 Search PubMed.
- D. P. Germain, D. A. Hughes, K. Nicholls, D. G. Bichet, R. Giugliani, W. R. Wilcox, C. Feliciani, S. P. Shankar, F. Ezgu, H. Amartino, D. Bratkovic, U. Feldt-Rasmussen, K. Nedd, U. S. El Din, C. M. Lourenco, M. Banikazemi, J. Charrow, M. Dasouki, D. Finegold, P. Giraldo, O. Goker-Alpan, N. Longo, C. R. Scott, R. Torra, A. Tuffaha, A. Jovanovic, S. Waldek, S. Packman, E. Ludington, C. Viereck, J. Kirk, J. Yu, E. R. Benjamin, F. Johnson, D. J. Lockhart, N. Skuban, J. Castelli, J. Barth, C. Barlow and R. Schiffmann, N. Engl. J. Med., 2016, 375, 545–555 Search PubMed.
- C. Colombo and A. J. Bennet, Adv. Phys. Org. Chem., 2017, 51, 99–127 Search PubMed.
- G. J. Davies, A. Planas and C. Rovira, Acc. Chem. Res., 2012, 45, 308–316 Search PubMed.
- C. Colombo and A. J. Bennet, Curr. Opin. Chem. Biol., 2019, 53, 145–157 Search PubMed.
- F. S. Shidmoossavee, J. N. Watson and A. J. Bennet, J. Am. Chem. Soc., 2013, 135, 13254–13257 Search PubMed.
- R. Mosi, H. Sham, J. C. M. Uitdehaag, R. Ruiterkamp, B. W. Dijkstra and S. G. Withers, Biochemistry, 1998, 37, 17192–17198 Search PubMed.
- K. Olsen, U. Christensen, M. R. Sierks and B. Svensson, Biochemistry, 1993, 32, 9686–9693 Search PubMed.
- J. Chan, A. R. Lewis, D. Indurugalla, M. Schur, W. Wakarchuk and A. J. Bennet, J. Am. Chem. Soc., 2012, 134, 3748–3757 Search PubMed.
- H. Gu, S. M. Zhang, K. Y. Wong, B. K. Radak, T. Dissanayake, D. L. Kellerman, Q. Dai, M. Miyagi, V. E. Anderson, D. M. York, J. A. Piccirilli and M. E. Harris, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 13002–13007 Search PubMed.
- L. F. Sobala, G. Speciale, S. Zhu, L. Raich, N. Sannikova, A. J. Thompson, Z. Hakki, D. Lu, S. S. K. Abadi, A. R. Lewis, V. Rojas-Cervellera, G. Bernardo-Seisdedos, Y. M. Zhang, O. Millet, J. Jimenez-Barbero, A. J. Bennet, M. Sollogoub, C. Rovira, G. J. Davies and S. J. Williams, ACS Cent. Sci., 2020, 6, 760–770 Search PubMed.
- S. Saen-oon, S. Quaytman-Machleder, V. L. Schramm and S. D. Schwartz, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 16543–16548 Search PubMed.
- V. L. Schramm, Curr. Opin. Struct. Biol., 2005, 15, 604–613 Search PubMed.
- G. B. Evans, V. L. Schramm and P. C. Tyler, Medchemcomm, 2018, 9, 1983–1993 Search PubMed.
- T. M. Gloster, Biochem. Soc. Trans., 2012, 40, 913–928 Search PubMed.
- T. J. M. Beenakker, D. P. A. Wander, W. A. Offen, M. Artola, L. Raich, M. J. Ferraz, K. Y. Li, J. H. P. M. Houben, E. R. van Rijssel, T. Hansen, G. A. van der Marel, J. D. C. Codee, J. M. F. G. Aerts, C. Rovira, G. J. Davies and H. S. Overkleeft, J. Am. Chem. Soc., 2017, 139, 6534–6537 Search PubMed.
- W. Ren, R. Pengelly, M. Farren-Dai, S. Shamsi Kazem Abadi, V. Oehler, O. Akintola, J. Draper, M. Meanwell, S. Chakladar, K. Świderek, V. Moliner, R. Britton, T. M. Gloster and A. J. Bennet, Nat. Commun., 2018, 9, 3243 Search PubMed.
- S. Shamsi Kazem Abadi, M. Tran, A. K. Yadav, P. J. P. Adabala, S. Chakladar and A. J. Bennet, J. Am. Chem. Soc., 2017, 139, 10625–10628 Search PubMed.
- C. Adamson, R. Pengelly, S. Shamsi Kazem Abadi, S. Chakladar, J. Draper, R. Britton, T. Gloster and A. J. Bennet, Angew. Chem., Int. Ed., 2016, 55, 14978–14982 Search PubMed.
- P. M. Danby and S. G. Withers, J. Am. Chem. Soc., 2017, 139, 10629–10632 Search PubMed.
- T. K. M. Shing, Y. Chen and H. T. Wu, Synlett, 2012, 1793–1796 Search PubMed.
- A. Vidyasagar and K. M. Sureshan, Eur. J. Org. Chem., 2014, 2014, 2349–2356 Search PubMed.
- A. Kato, N. Kato, E. Kano, I. Adachi, K. Ikeda, L. Yu, T. Okamoto, Y. Banba, H. Ouchi, H. Takahata and N. Asano, J. Med. Chem., 2005, 48, 2036–2044 Search PubMed.
- P. F. Cook and W. W. Cleland, in Enzyme Kinetics and Mechanism, Garland Science, London, 2007 Search PubMed.
- A. Fersht, in Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, W.H. Freeman, New York, 1999 Search PubMed.
- J. Chan, N. Sannikova, A. Tang and A. J. Bennet, J. Am. Chem. Soc., 2014, 136, 12225–12228 Search PubMed.
- J. Chan, A. Tang and A. J. Bennet, Can. J. Chem., 2015, 93, 463–467 Search PubMed.
- L. Melander and W. H. Saunders, in Reaction Rates of Isotopic Molecules, Wiley, New York, 1980 Search PubMed.
- N. Sannikova, A. R. Lewis and A. J. Bennet, Methods Enzymol., 2017, 596, 547–571 Search PubMed.
- J. Chan, A. R. Lewis, M. Gilbert, M. F. Karwaski and A. J. Bennet, Nat. Chem. Biol., 2010, 6, 405–407 Search PubMed.
- G. Speciale, M. Farren-Dai, F. S. Shidmoossavee, S. J. Williams and A. J. Bennet, J. Am. Chem. Soc., 2016, 138, 14012–14019 Search PubMed.
- W. W. Cleland, in Isotope effects in chemistry and biology, ed. A. Kohen and H.-H. Limbach, Taylor & Francis, Boca Raton, 2006, pp. 915–930 Search PubMed.
- D. B. Northrop, Annu. Rev. Biochem., 1981, 50, 103–131 Search PubMed.
- P. J. Berti and K. S. E. Tanaka, Adv. Phys. Org. Chem., 2002, 37, 239–314 Search PubMed.
- A. J. Bennet and M. L. Sinnott, J. Am. Chem. Soc., 1986, 108, 7287–7294 Search PubMed.
- A. J. Bennet, M. L. Sinnott and W. S. S. Wijesundera, J. Chem. Soc., Perkin Trans. 2, 1985, 2, 1233–1236 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, KIE measurement protocols, computational methods, and supplementary figures and tables. See DOI: 10.1039/d0sc04401f |
| ‡ Dr W. Ren, Dr Y. Wang, Key Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 5 Yushan Road, Qingdao 266003, China and Laboratory for Marine Drugs and Bioproducts, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China. |
| This journal is © The Royal Society of Chemistry 2020 |