
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioorthogonal release of anticancer drugs via gold-triggered 2-alkynylbenzamide cyclization†
Kenward
Vong
 *ab,
Tomoya
Yamamoto
a,
Tsung-che
Chang
a and
Katsunori
Tanaka
*abcd
*ab,
Tomoya
Yamamoto
a,
Tsung-che
Chang
a and
Katsunori
Tanaka
*abcd
aBiofunctional Synthetic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan. E-mail: kotzenori@riken.jp; kenward.vong@riken.jp
bGlycoTargeting Research Laboratory, RIKEN Baton Zone Program, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan
cBiofunctional Chemistry Laboratory, A. Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Street, Kazan, 420008, Russia
dDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1 O-okayama, Meguro-ku, Tokyo 152-8552, Japan
First published on 2nd September 2020
Abstract
Metal-based uncaging of biomolecules has become an emerging approach for in vivo applications, which is largely due to the advantageous bioorthogonality of abiotic transition metals. Adding to the library of metal-cleavable protecting groups, this work introduces the 2-alkynylbenzamide (Ayba) moiety for the gold-triggered release of secondary amines under mild and physiological conditions. Studies were further performed to highlight some intrinsic benefits of the Ayba protecting group, which are (1) its amenable nature to derivatization for manipulating prodrug properties, and (2) its orthogonality with other commonly used transition metals like palladium and ruthenium. With a focus on highlighting its application for anticancer drug therapies, this study successfully shows that gold-triggered conversion of Ayba-protected prodrugs into bioactive anticancer drugs (i.e. doxorubicin, endoxifen) can proceed effectively in cell-based assays.
Introduction
Dissociative bioorthogonal reactions are defined as reactions that can activate a substrate (prodrug) in vivo to release a bioactive payload (drug) in the presence of an abiotic trigger (Fig. 1).1,2 Methodologies can become further viable if the cleavable (masking) moiety renders the prodrug significantly inactive compared to the bioactive agent.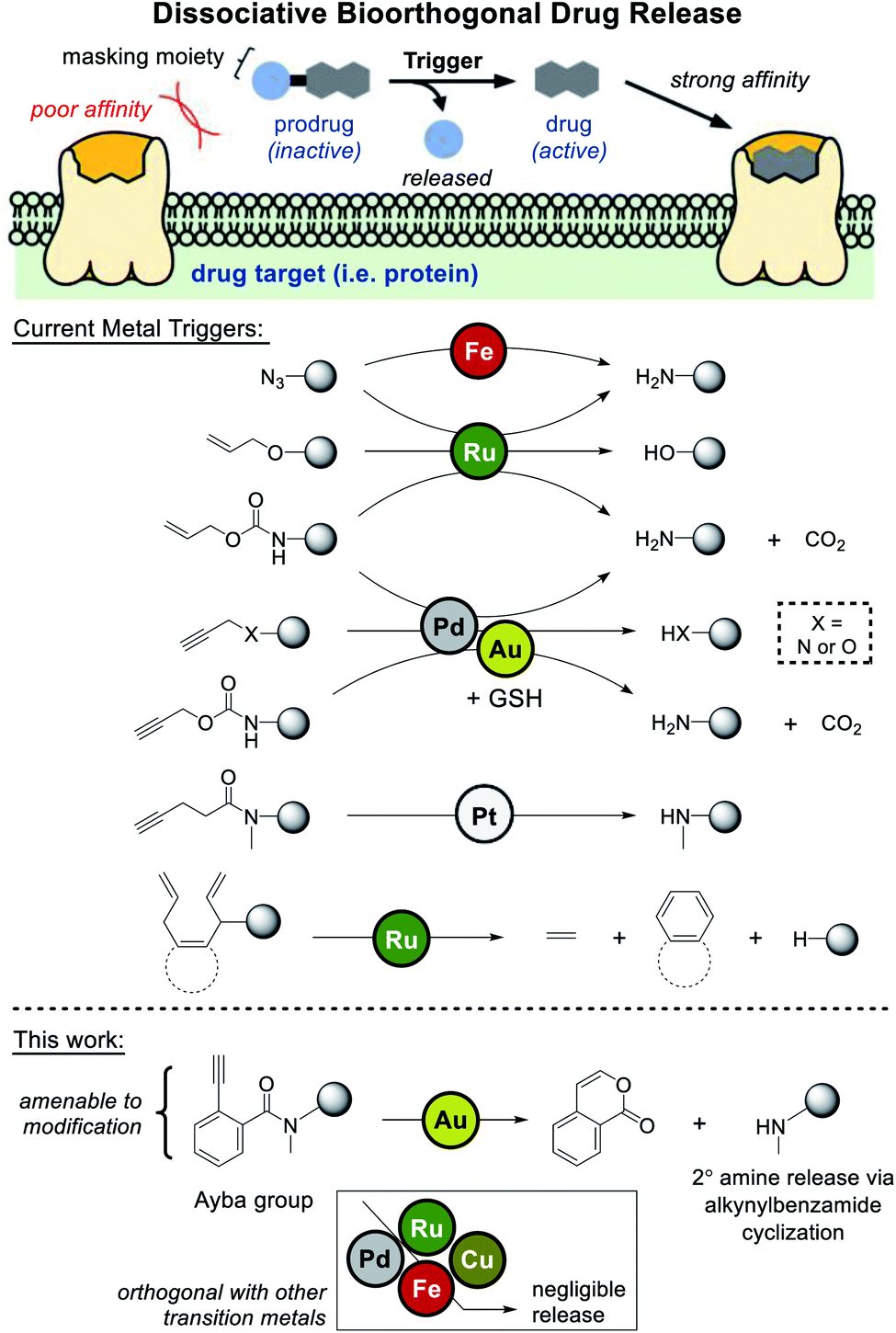 | ||
| Fig. 1 Metal-based triggers for dissociative bioorthogonal drug release have largely employed Fe-, Ru-, and Pd-based reactions. This work presents the release of secondary amine containing drugs via gold-triggered 2-alkynylbenzamide cyclization. | ||
In an emerging field of study, researchers have extensively investigated metal-based uncaging of drugs/fluorophores applicable in cell- or animal-based models.3 Mainly employing metals like palladium and ruthenium, a number of examples have leaned heavily on some iteration of amine/alcohol release through masking groups that include azide,4–7 allene,8 allyl,9,10 propargyl,11–22 propargyloxycarbonyl (Proc),19–29 and allyloxycarbonyl (Alloc).10–12,28–40
Looking at current literature, a few areas of need can be clearly identified. First, given the overreliance of palladium- and ruthenium-based uncaging, it would be beneficial to develop drug release reactions based on other transition metals. One relevant example that addresses this issue is the work done by Unciti-Broceta and coworkers, whom discovered that gold catalysts could also be used to facilitate depropargylation via glutathione assistance.41 However it should be noted that since palladium also facilitates uncaging, these metals cannot be orthogonally used in tandem. Recently, Bernardes and coworkers have also uncovered a method of pentynoyl tertiary amide/N-propargyl decaging via platinum complexes. Using an elegant combination therapy based on cisplatin, prodrug activation was shown to proceed in zebrafish models.42
A further area of need is for masking groups to be amenable to derivatization. For drugs where the protected amine/alcohol is critical for target receptor binding, most of current strategies would work well. However, for drugs where key binding interactions are distributed throughout the molecule, masking groups need to have a greater influence over binding. In one recent study that can likely address this issue, Ward and coworkers developed a “close-to-release” reaction that functions via alcohol uncaging triggered by ring-closing metathesis.43 In this case, the naphthalene precursor masking groups should be robustly amenable to derivatization.
Considering these points, the primary aim of this study was to develop an amine-releasing reaction that could meet all the following criteria: (1) be compatible under aqueous/physiological conditions, (2) be orthogonally catalyzed by gold without interference from other transition metals (i.e. palladium and ruthenium), and (3) possess a masking group that is amenable to derivatization. To do this, 2-alkynylbenzamides (Ayba) protecting groups were thus developed.
Results
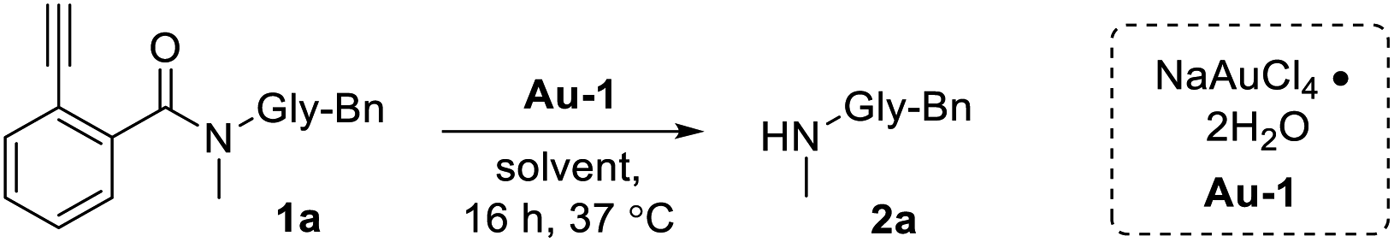
Looking at the literature, 2-alkynylbenzoates first presented themselves as potential gold-activatable masking groups. Originally conceived as a means for glycosylation,44–46 later adaptations developed Au(I)/Au(III) sensing probes that functioned by releasing alcohol-containing fluorophores.47,48 Unfortunately, one major shortcoming of this reaction is the susceptibility of esters to hydrolysis.49 A search of available literature revealed no successful attempts had been made to replace the ester with an amide, likely due to its added chemical stability. This was confirmed in our own hands as the release of Ayba-protected primary amines was not found to proceed. However, we were encouraged to find that the Au-triggered release of Ayba-protected secondary amines could proceed under mild and aqueous conditions.As a model reaction, Ayba-deprotection of 1a to the N-methyl amine product 2a was first monitored under various conditions (Table 1). As expected, increasing levels of Au-1 under aqueous conditions led to the release of 2a (7 to 72% yield, entries 2–5). In addition, reactivity was shown to be time and temperature dependent (Table S2 in the ESI†). Further investigations were then done to explore changes to solvent, where reactions carried out in water-miscible solvents generally gave excellent yields (i.e. 75–99% for THF, dioxane, methanol), while water-immiscible solvents were poor (2–8% for hexanes, chloroform). These observations can be attributed to the importance of hydrolysis as the last step for amine release. Additionally, tests were done in solutions containing rat serum (15%, entry 16) and DMEM culture media (6%, entry 17) to mimic biological settings. In these cases, the lowered observed yields are likely due to metal quenching from complex biological mixtures.
|
|
|||
|---|---|---|---|
| Entry | Mol% | Solvent | Yield of 2aa (%) |
| a Yields determined by HPLC (peak retention times compared to product standards, followed by MS analysis for confirmation, and then calculation of yields based on product standard curves). b Isolated yields obtained by column chromatography purification. All reactions were standardized to 10 μmol of 1a in 1 ml of solvent (10 mM). | |||
| 1 | — | 10% DMF in PBS buffer | 0 |
| 2 | 10 | 10% DMF in PBS buffer | 7 |
| 3 | 25 | 10% DMF in PBS buffer | 18 |
| 4 | 50 | 10% DMF in PBS buffer | 52 |
| 5 | 75 | 10% DMF in PBS buffer | 72 |
| 6 | 50 | 50% DMF in PBS buffer | 7 |
| 7 | 50 | 50% THF in PBS buffer | 17 |
| 8 | 50 | 50% THF in H2O | 24 |
| 9 | 50 | THF | 99 |
| 10 | 50 | Dioxane | 91 (85)b |
| 11 | 50 | Methanol | 75 |
| 12 | 50 | CHCl3 | 8 |
| 13 | 50 | Hexanes | 2 |
| 14 | 50 | 10% DMSO in PBS buffer | 57 |
| 15 | 50 | 10% DMF in MES buffer | 21 |
| 16 | 50 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:8 rat serum/DMF/PBS :1:8 rat serum/DMF/PBS |
15 |
| 17 | 50 | 10% DMF in DMEM media | 6 |
Summarized in Table S3 in the ESI,† a number of Ayba group derivatives were next prepared to explore the factors that contribute to amine release. Important observations of note include the fact that N-substitution is critical for facilitating amine release. This is most apparent with the Ayba-protection of primary amines, which showed no detectable levels of Au-triggered release. Another significant observation was that despite numerous derivatizations made to the Ayba moiety, only a few instances saw decreased yields of release. As such, the amenable nature of the Ayba-masking group may potentially be advantageous for designing better non-toxic prodrugs.
In the next step of this study, the effects of different gold metal complexes were investigated. In Table 2, prodrugs 3a,m were tested for the release of the secondary-amine containing drug known as endoxifen. First, the reactivity of Au(III) complexes were investigated using Au-2 (prepared as described50,51) and Au-3. Although good yields were obtained using pure water (Table S4 in the ESI†), observed reactivities experienced a noticeable drop when switched to buffer (i.e. PBS buffer pH 7.4). Likely, this effect is brought about by the instability of Au(III) complexes. As a result, the more stable Au(I) complex Au-4 was next tested, giving release yields of roughly 60% in PBS buffer for prodrugs 3a,m. Additionally, since amine release is dependent on hydrolysis, it was theorized that increased reactivity is likely correlated with basicity. As a verification, 3a was also tested under similar conditions with Au-4 in phosphate buffer pH 8 (77%) and BBS buffer pH 8.5 (80%), both of which gave higher yields.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of endoxifen releasea (%) | |
| Substrate 3a | Substrate 3m | ||
| a Yields determined by HPLC (peak retention times compared to product standards, followed by MS analysis for confirmation, and then calculation of yields based on product standard curves). All reactions were standardized to 30 nmol of 3a,m and 30 nmol of catalyst in 50 μl of solvent (600 μM). n.d. = not detectable. | |||
| 1 | — | n.d. | n.d. |
| 2 | Au-2 | 6 | 3 |
| 3 | Au-3 | 19 | 22 |
| 4 | Au-4 | 60 | 60 |
| 5 | Pd-1 | 10 | 7 |
| 6 | Pd-2 | 9 | 5 |
| 7 | Pd-3 | 5 | 6 |
| 8 | Ru-1 | n.d. | n.d. |
| 9 | Ru-2 | 1 | 1 |
| 10 | Cu-1 | n.d. | n.d. |
| 11 | Cu-2 | n.d. | n.d. |
| 12 | Fe-1 | n.d. | n.d. |
| 13 | Fe-2 | n.d. | n.d. |
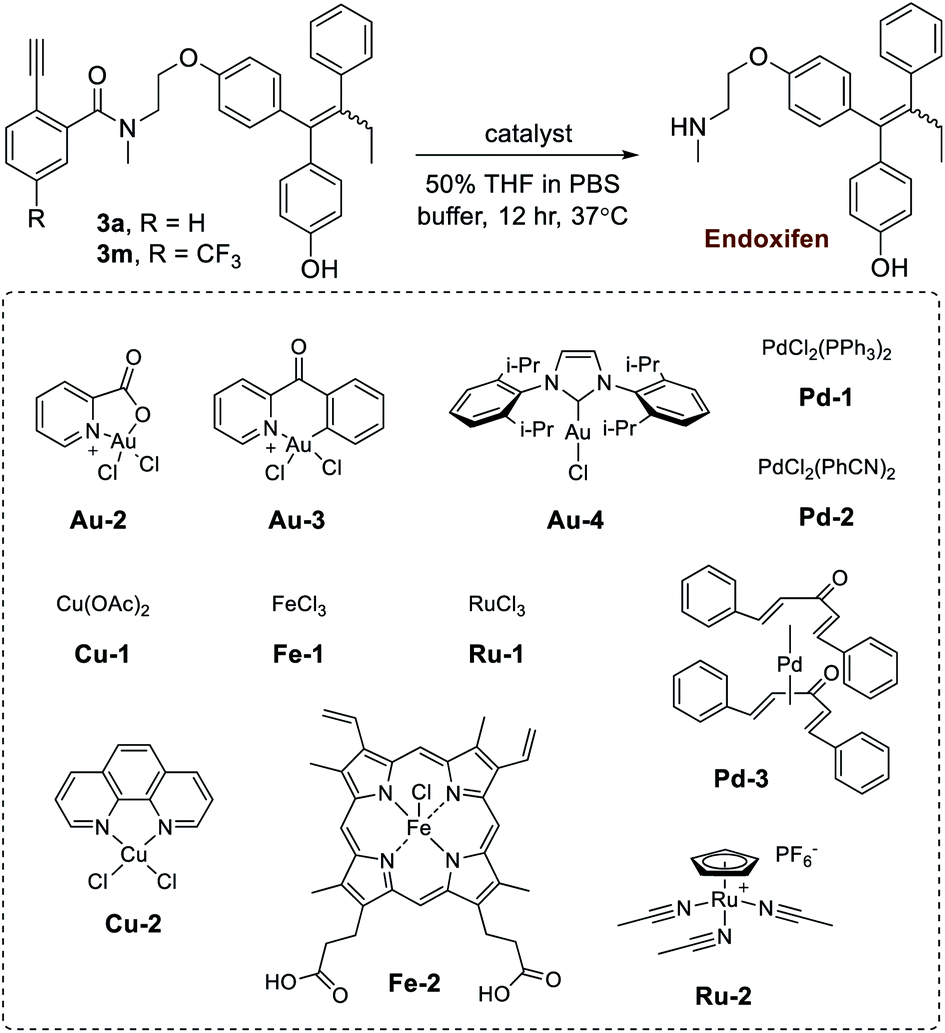
Given the potential benefits of mutually-orthogonal, tandem metal applications,9 a further test was conducted to monitor gold-based Ayba-decaging in the presence of other commonly used abiotic transition metals (i.e. palladium and ruthenium). In addition, biological metals such as copper and iron were also tested. Shown in Table 2, drug release from prodrugs 3a,m was generally found to be negligible in the given cases (entries 5–13).
Based on the preliminary data, the capacity of Ayba-protected prodrugs to be used in parallel with other metal-triggered drug release systems was next explored. Alloc-doxorubicin 4 (ruthenium decaged) and Proc-doxorubicin 5 (palladium decaged) were chosen as model alternative prodrugs. For the mixtures of 3a/4 (Fig. 2A), observations show that doxorubicin release largely occurs only in the presence of Ru-2, while endoxifen release takes place with Au-4 addition. A similar trend in reactivity was also observed for the mixtures of 3a/5 (Fig. 2B).
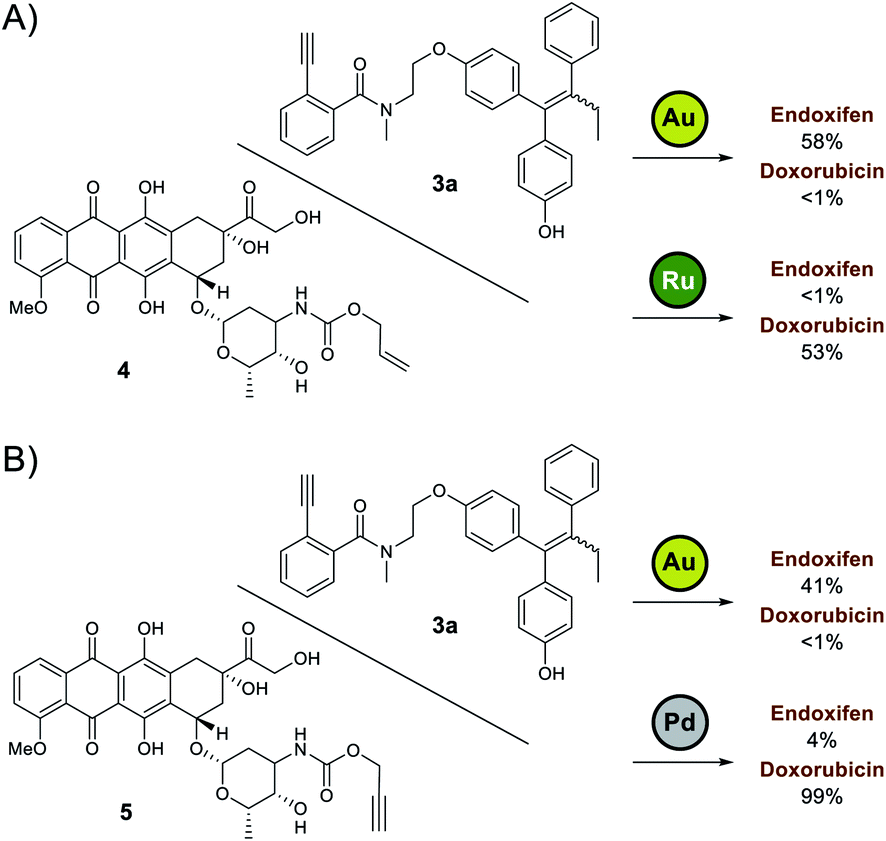 | ||
| Fig. 2 Orthogonal drug release study. (A) To a mixture of Ayba-endoxifen 3a and Alloc-doxorubicin 4, addition of Au-4 led to preferential release of endoxifen while Ru-2 led to the release of doxorubicin. (B) To a mixture of Ayba-endoxifen 3a and Proc-doxorubicin 5, addition of Au-4 led to preferential release of endoxifen while Pd-3 led to the release of doxorubicin. | ||
Overall, the proposed mechanism for gold-triggered alkynylbenzamide cyclization/amine release is depicted in Fig. 3. In this process, gold-activation of the alkynyl group elicits nucleophilic attack from the proximal carbonyl oxygen. Based on the type of ligated Au complex,52 6-endocyclization is favored to generate the oxonium intermediate that is susceptible to base-dependent hydrolysis. As a result, the secondary amine-containing molecule can then be released in conjunction with a corresponding isocoumarin derivative. To reason why gold-triggered release is favored for secondary amines over primary amines, it can be theorized that the alpha effects of the N-substituted methyl group enhances the nucleophilicity of the carbonyl oxygen. This rationale would be consistent with a similar report that showed the presence of a Weinreb amide can increase rates of N-alkoxy-2-alkynylbenzamide cyclization via copper catalysis.53 To address the poor catalytic activity, it may be hypothesized that hydrolysis is a limiting factor under physiological conditions. Also, Au(I) disproportionation could play a factor in the decomposition of the gold complexes.54
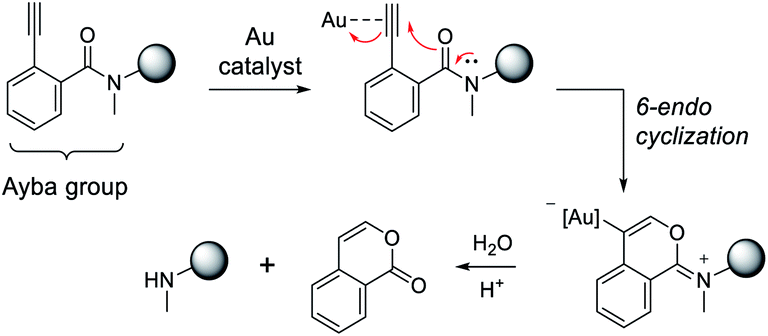 | ||
| Fig. 3 Proposed mechanism for Ayba deprotection via Au-catalysed cyclization. Subsequent hydrolysis then leads to the release of a secondary amine. | ||
In the next part of this study, the focus shifted to investigating the viability of Ayba-based prodrugs as anticancer therapeutics. Designed to directly release endoxifen, the cytotoxic profiles of prodrugs 3a,m were first tested against ER-positive MCF7 breast cancer cells (Table S5 and Fig. S16 in the ESI†). However, only a low difference in EC50 values between prodrugs 3a,m (55–57 μM) and endoxifen (25 μM) was observed. Given the moderate cytotoxicity of the parent drug endoxifen in these trials, the desire thus shifted to search for an alternative and more robust anticancer drug.
To bypass the constraints of secondary-amine containing drug usage, the final portion of this study focused on adapting the use of p-methylamino-benzyloxycarbonyl (PMBC) spacers, which are also capable of undergoing spontaneous 1,6-elimination to release an attached amine.55 As depicted in Fig. 4A, compounds 6a,m were synthesized and tested for the release of the anticancer agent doxorubicin. Under aqueous in vitro conditions, doxorubicin release was found to range from 63–86% for Au-4 and 53–79% for Au-5.
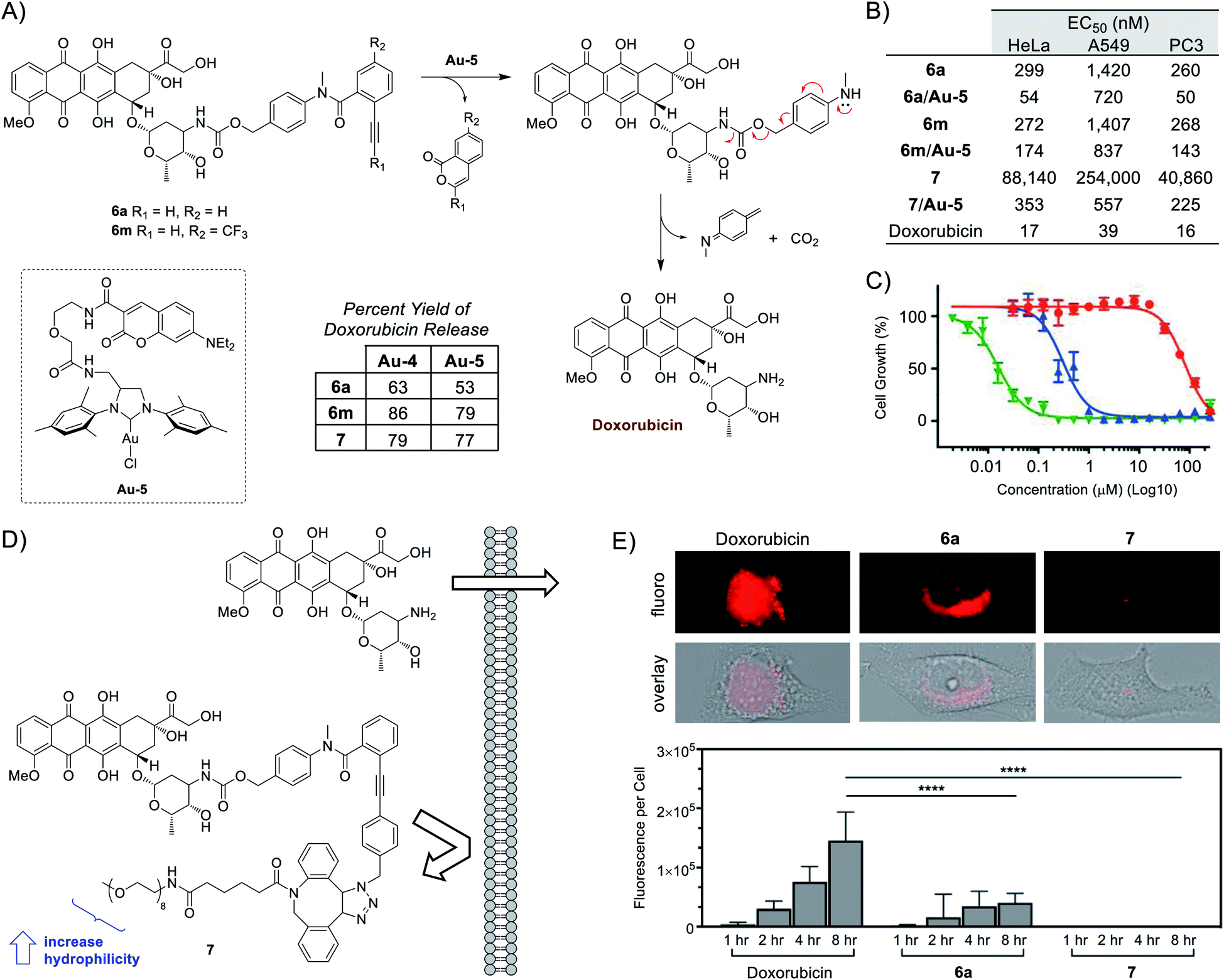 | ||
| Fig. 4 Ayba-based doxorubicin prodrugs. (A) Mechanism of indirect release (via PMBC spacer) of primary amine-containing doxorubicin from 6a,m. Reactions were standardized to 30 nmol of prodrug and 30 nmol of catalyst in 50 μl of THF/PBS buffer pH 7.4 (600 μM). (B) Summary of cytotoxic activity (EC50 values) against various cancer cell lines. (C) Example growth curves for HeLa cancer cells incubated with either prodrug 7 (red), mixture 7/Au-5 (blue), or doxorubicin (green). (D) Depiction of the strategy to prevent passive diffusion of prodrug 7 by increasing bulk and hydrophilicity. (E) Imaging studies to investigate the cellular penetration of doxorubicin, 6a, and 7 (10 μM) into HeLa cancer cells over time. | ||
To carry out subsequent cell-based experiments, a preference was made to utilized Au-5 due to its improved solubility in 1% DMSO compared to Au-4. Additionally, control experiments (Fig. S15 in the ESI†) determined that a final concentration of 10 μM Au-5 would be non-toxic under all cell assay conditions. As shown in Fig. 4B and S17–S19 in the ESI,† addition of Au-5 to Ayba-protected prodrugs were all found to help increase their cytotoxic activity. For example, gold-activation of prodrug 6a led to a drop in EC50 values for several cancer cells lines; 299 to 52 nM for HeLa, 1420 to 720 nM for A549, and 260 to 50 nM for PC3.
From the collected data, the intrinsic cytotoxicity of prodrugs 6a,m (260–1420 nM) compared to the active drug (16–39 nM) is at most about a 16–36 fold difference. To explain this modest difference, it should be noted that anthracyclines (i.e. doxorubicin) are generally known to enter cells through passive diffusion,56 followed by nuclear transport through proteasome binding and subsequent DNA intercalation.57,58 Given this mechanism, it was theorized that the hydrophobic nature of the unsubstituted Ayba masking group is unlikely to have a substantial effect in preventing cellular uptake of doxorubicin-based prodrugs.
With the opportunity for derivatization, the freedom exists to manipulate the molecular properties of Ayba-protected prodrugs on a situational basis. Therefore, as depicted in Fig. 4D, compound 7 was prepared with the reasoning that cellular uptake could be prevented via controlling size (increase bulk) and polarity (increase hydrophilicity), both of which are factors known to govern passive diffusion.
In order to study cellular uptake, imaging studies were performed with HeLa cells incubated with either doxorubicin, 6a, or 7 (Fig. 4E). As anticipated, the fastest and highest levels of uptake occurred with doxorubicin. In this case, fluorescence is strongly distributed throughout the cytoplasm and nucleus. For the unsubstituted Ayba-prodrug 6a, fluorescence was instead detectable only within the cytoplasm while being faint/absent in the nucleus. This observation could be interpreted as nuclear transport (via proteasome binding) being affected by the presence of the Ayba group. Finally, imaging studies conducted with the PEG-containing prodrug 7 was shown to be poorly uptaken by HeLa cells under all timepoints. Given the significant impairment of cellular uptake, it thus becomes apparent why the EC50 values of prodrug 7 (41–254 μM) are so high compared to doxorubicin, which effectively translates to about a 2554–6512 fold difference. Ultimately, this translated to more effective biological effects of the 7/Au-5 mixture, where gold-activation of prodrug 7 led to significant drops in EC50 values for the tested cancer cells lines; 88 to 0.4 μM for HeLa, 254 to 0.6 μM for A549, and 41 to 0.2 μM for PC3.
Conclusions
In conclusion, this study demonstrates the design and development of the 2-alkynylbenzamide (Ayba) group for gold-dependent release of secondary amines under mild and physiological conditions. Although not shown to proceed catalytically, one of the principal benefits of the Ayba protecting group is that decaging can be triggered via gold complexes that proceed orthogonally in the presence of transition metals like palladium and ruthenium. Furthermore, this study showed that the amenable nature of the Ayba group can allow for the manipulation of prodrugs properties, such as size and lipophilicity. Overall, we believe this work will be a useful addition to the growing library of metal-triggered dissociative bioorthogonal reactions, which can serve as a versatile means to uncage primary/secondary amines for biological applications or for developing future drug therapies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Kind acknowledgement is given to GlyTech Inc. for supporting this research. This work was financially supported by the JSPS KAKENHI Grant Numbers JP16H03287, JP18K19154, JP15H05843 (to K. T.), JP18K14347 (to K. V.), and JP19K15708 (to T. C. C). Funding was also provided by the AMED Grant JP15KM0908001 (to K. T.), as well as additional support from a RIKEN Incentive Research Project grant (to K. V.), and the Russian Government Program for Competitive Growth (to Kazan Federal University).Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- J. Tu, M. Xu and R. M. Franzini, ChemBioChem, 2019, 20, 1615–1627 CrossRef CAS.
- J. G. Rebelein and T. R. Ward, Curr. Opin. Biotechnol., 2018, 53, 106–114 CrossRef CAS.
- P. K. Sasmal, S. Carregal-Romero, A. A. Han, C. N. Streu, Z. Lin, K. Namikawa, S. L. Elliott, R. W. Köster, W. J. Parak and E. Meggers, ChemBioChem, 2012, 13, 1116–1120 CrossRef CAS.
- K. K. Sadhu, E. Lindberg and N. Winssinger, Chem. Commun., 2015, 51, 16664–16666 RSC.
- K. K. Sadhu, T. Eierhoff, W. Römer and N. Winssinger, J. Am. Chem. Soc., 2012, 134, 20013–20016 CrossRef CAS.
- K. K. Sadhu and N. Winssinger, Chem.–Eur. J., 2013, 19, 8182–8189 CrossRef CAS.
- J. Wang, S. Zheng, Y. Liu, Z. Zhang, Z. Lin, J. Li, G. Zhang, X. Wang, J. Li and P. R. Chen, J. Am. Chem. Soc., 2016, 138, 15118–15121 CrossRef CAS.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1913 CrossRef.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef.
- J. T. Weiss, C. Fraser, B. Rubio-Ruiz, S. H. Myers, R. Crispin, J. C. Dawson, V. G. Brunton, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, Front. Chem., 2014, 2, 59 Search PubMed.
- M. Sancho-Albero, B. Rubio-Ruiz, A. M. Pérez-López, V. Sebastián, P. Martín-Duque, M. Arruebo, J. Santamaría and A. Unciti-Broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS.
- T. L. Bray, M. Salji, A. Brombin, A. M. Pérez-López, B. Rubio-Ruiz, L. C. A. Galbraith, E. E. Patton, H. Y. Leung and A. Unciti-Broceta, Chem. Sci., 2018, 9, 7354–7361 RSC.
- C. Adam, A. M. Pérez-López, L. Hamilton, B. Rubio-Ruiz, T. L. Bray, D. Sieger, P. M. Brennan and A. Unciti-Broceta, Chem.–Eur. J., 2018, 24, 16783–16790 CrossRef CAS.
- B. Rubio-Ruiz, J. T. Weiss and A. Unciti-Broceta, J. Med. Chem., 2016, 59, 9974–9980 CrossRef CAS.
- P. Destito, A. Sousa-Castillo, J. R. Couceiro, F. López, M. A. Correa-Duarte and J. L. Mascareñas, Chem. Sci., 2019, 10, 2598–2603 RSC.
- C. Torres-Sánchez, A. M. Pérez-López, M. N. Alqahtani, A. Unciti-Broceta and B. Rubio-Ruiz, New J. Chem., 2019, 43, 1449–1458 RSC.
- J. T. Weiss, N. O. Carragher and A. Unciti-Broceta, Sci. Rep., 2015, 5, 9329 CrossRef CAS.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef.
- J. Clavadetscher, E. Indrigo, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Angew. Chem., Int. Ed., 2017, 56, 6864–6868 CrossRef CAS.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Megia-Fernandez, A. Lilienkampf and M. Bradley, Chem. Commun., 2017, 53, 6712–6715 RSC.
- E. Indrigo, J. Clavadetscher, S. V. Chankeshwara, A. Lilienkampf and M. Bradley, Chem. Commun., 2016, 52, 14212–14214 RSC.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185–4189 RSC.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364–5368 CrossRef CAS.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207–1218 CrossRef CAS.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645–5648 CrossRef CAS.
- P. K. Sasmal, S. Carregal-Romero, W. J. Parak and E. Meggers, Organometallics, 2012, 31, 5968–5970 CrossRef CAS.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef.
- T. Völker and E. Meggers, ChemBioChem, 2017, 18, 1083–1086 CrossRef.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814–12826 CrossRef CAS.
- R. Das, R. F. Landis, G. Y. Tonga, R. Cao-Milán, D. C. Luther and V. M. Rotello, ACS Nano, 2019, 13, 229–235 CrossRef CAS.
- A. Gupta, R. Das, G. Yesilbag Tonga, T. Mizuhara and V. M. Rotello, ACS Nano, 2018, 12, 89–94 CrossRef CAS.
- M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901–1907 RSC.
- A. M. Perez-Lopez, B. Rubio-Ruiz, V. Sebastian, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaria and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548–12552 CrossRef CAS.
- B. L. Oliveira, B. J. Stenton, V. B. Unnikrishnan, C. R. de Almeida, J. Conde, M. Negrão, F. S. S. Schneider, C. Cordeiro, M. G. Ferreira, G. F. Caramori, J. B. Domingos, R. Fior and G. J. L. Bernardes, J. Am. Chem. Soc., 2020, 142, 10869–10880 CrossRef CAS.
- V. Sabatino, J. G. Rebelein and T. R. Ward, J. Am. Chem. Soc., 2019, 141, 17048–17052 CrossRef CAS.
- Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS.
- Y. Li, X. Yang, Y. Liu, C. Zhu, Y. Yang and B. Yu, Chem.–Eur. J., 2010, 16, 1871–1882 CrossRef CAS.
- Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS.
- H. Seo, M. E. Jun, O. A. Egorova, K. H. Lee, K. T. Kim and K. H. Ahn, Org. Lett., 2012, 14, 5062–5065 CrossRef CAS.
- N. T. Patil, V. S. Shinde, M. S. Thakare, P. Hemant Kumar, P. R. Bangal, A. K. Barui and C. R. Patra, Chem. Commun., 2012, 48, 11229–11231 RSC.
- H. Seo, M. E. Jun, K. Ranganathan, K. H. Lee, K. T. Kim, W. Lim, Y. M. Rhee and K. H. Ahn, Org. Lett., 2014, 16, 1374–1377 CrossRef CAS.
- Y. Lin, K. Vong, K. Matsuoka and K. Tanaka, Chem.–Eur. J., 2018, 24, 10595–10600 CrossRef CAS.
- K. Tsubokura, K. K. H. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579–3584 CrossRef CAS.
- Y. Yang, Y. Liu, R. Zhu, C. Liu and D. Zhang, J. Org. Chem., 2019, 84, 9705–9713 CrossRef CAS.
- M. Jithunsa, M. Ueda and O. Miyata, Org. Lett., 2011, 13, 518–521 CrossRef CAS.
- C. H. Gammons, Y. Yu and A. E. Williams-Jones, Geochim. Cosmochim. Acta, 1997, 61, 1971–1983 CrossRef CAS.
- A. N. Dinaut and S. D. Taylor, Chem. Commun., 2001, 15, 1386–1387 RSC.
- T. Skovsgaard and N. I. Nissen, Pharmacol. Ther., 1982, 18, 293–311 CrossRef CAS.
- K. Kiyomiya, S. Matsuo and M. Kurebe, Cancer Res., 2001, 61, 2467–2471 CAS.
- K. Kiyomiya, J. Satoh, H. Horie, M. Kurebe, H. Nakagawa and S. Matsuo, Int. J. Oncol., 2002, 21, 1081–1085 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04329j |
| This journal is © The Royal Society of Chemistry 2020 |