
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of dimeric Securinega alkaloids (−)-flueggenines D and I†
Sangbin
Jeon
 ,
Jinwoo
Lee
,
Sangbin
Park
and
Sunkyu
Han
*
,
Jinwoo
Lee
,
Sangbin
Park
and
Sunkyu
Han
*
Department of Chemistry, Korea Advanced Institute of Science & Technology (KAIST), Daejeon 34141, South Korea. E-mail: sunkyu.han@kaist.ac.kr
First published on 7th September 2020
Abstract
We describe the total synthesis of (−)-flueggenines D and I. This features the first total synthesis of dimeric Securinega alkaloids with a C(α)–C(δ′) connectivity between two monomeric units. The key dimerization was enabled by a sequence that involves Stille reaction and conjugate reduction. The high chemofidelity of the Stille reaction enabled us to assemble two structurally complex fragments that could not be connected by other methods. Stereochemical flexibility and controllability at the δ′-junction of the dimeric intermediate render our synthetic strategy broadly applicable to the synthesis of other high-order Securinega alkaloids.
Introduction
Securinega alkaloids isolated from plants of the genera Flueggea, Securinega, Phyllanthus, Brenia and Margaritaria have fascinated the chemical community for over six decades.1 Despite numerous structural variations, a typical monomeric Securinega alkaloid consists of a tetracyclic framework (A–D rings) that is decorated with α,β–γ,δ-unsaturated ester and tertiary amine moieties (Fig. 1). This unique molecular architecture is responsible for numerous biological activities and has served as an inspiration for over 30 total syntheses.2 A breakthrough in Securinega natural products chemistry was recorded in 2006 when Yue and coworkers isolated the first dimeric Securinega alkaloids flueggenines A (2) and B from Flueggea virosa.3 Since this initial report, continued isolation campaigns of this family of secondary metabolites have resulted in discoveries of 38 dimeric and oligomeric (high-order) Securinega natural products to date.1,4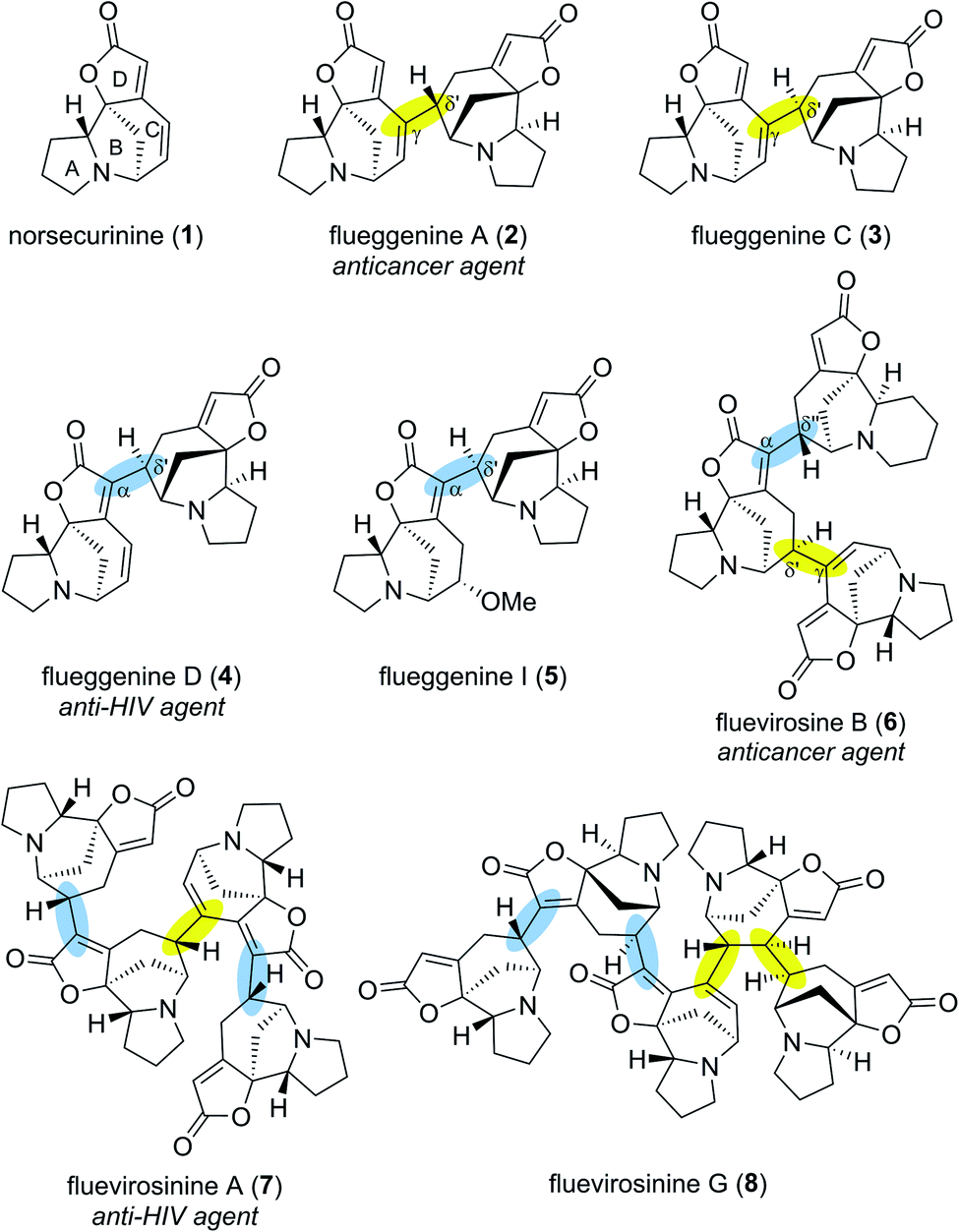 | ||
| Fig. 1 Norsecurinine (1) and representative high-order Securinega alkaloids. | ||
A closer structural examination of all known high-order Securinega alkaloids reveals that 28 of them (74%) are biosynthesized by a presumed enzymatic Rauhut–Currier (RC) reaction, a reaction that involves a carbon–carbon bond formation between two Michael acceptors.5,6 Flueggenine A (2) consists of two norsecurinine (1) units that are connected by an RC reaction-based C(γ)–C(δ′) bond (Fig. 1, highlighted in yellow).3 Flueggenine C (3) retains the atoms connectivity of flueggenine A (2) but exhibits opposite configuration at the δ′ junction implying the importance of stereochemical flexibility and controllability at this site when approaching this family of natural products by chemical synthesis.7
Another mode of connectivity within high-order Securinega alkaloids is the C(α)–C(δ′) connection (Fig. 1, highlighted in cyan) exemplified by anti-HIV agent flueggenine D (4)7 and its methoxy adduct flueggenine I (5).8 In addition to dimers, several trimeric Securinega alkaloids such as fluevirosine B (6)9 have also been isolated. It is important to note that connections between monomeric units within these trimers involve either C(γ)–C(δ′) or C(α)–C(δ′) bonds. Notably, these γ–δ′ and α–δ′ connections are the sole bonds that constitute the covalent networks of even higher-order Securinega natural products such as tetrameric fluevirosinine A (7)7 and pentameric fluevirosinine G (8).10 Hence, development of strategies that enables the C(γ)–C(δ′) and C(α)–C(δ′) bonds formation between Securinega monomers with stereochemical control is essential in synthesizing 28 known RC-based high-order Securinega alkaloids.
Despite numerous examples of monomeric Securinega alkaloids syntheses,1,11 there have been only a few examples of synthesis of dimeric Securinega natural products.12–15 Especially, synthetic access to RC reaction-based dimeric Securinega alkaloids had remained an unsolved problem until our recent total synthesis of (−)-flueggenine C (3) by an accelerated RC reaction (Scheme 1A).16 We showed that γ-hydroxy enone derivative 9 undergoes an efficient and stereoselective RC reaction to yield dimer 10. This compound could be moved forward to (−)-flueggenine C (3) with the γ–δ′ connectivity. While optimal for the synthesis of flueggenine C (3), the accelerated RC reaction could not be applied to the synthesis of its δ′-epimer, flueggenine A (2).17 Moreover, we could not expand this strategy into the formation of the C(α)–C(δ′) bond. Hence, we embarked on the development of a new dimerization strategy that overcomes these limitations and set (−)-flueggenine D (4) as our primary synthetic target.
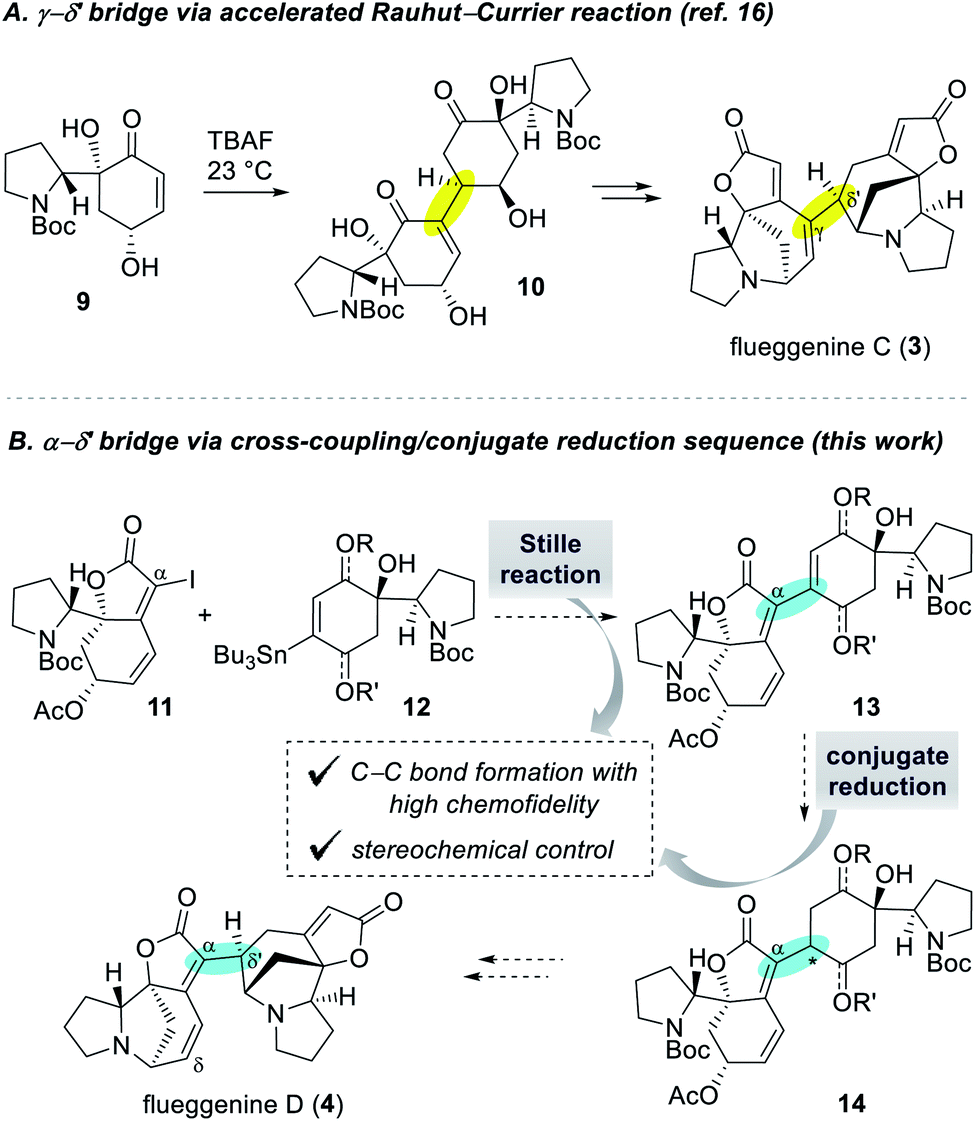 | ||
| Scheme 1 Dimerization strategies for the synthesis of high-order Securinega alkaloids. | ||
Extensive preliminary studies aimed at the C(α)–C(δ′) bond formation involving metal catalyzed conjugate additions, (reductive) Heck reactions, and radical-based approaches turned unfruitful. At this juncture, we envisioned to utilize Stille cross-coupling reaction for the key C(α)–C(δ′) bond formation due to its proven high chemofidelity18 in complex molecular settings (Scheme 1B).19 We planned to couple iodide 11 and organostannane 12via a Stille reaction to obtain dimeric precursor 13. Conjugate reduction of enone derivative 13 would yield compound 14 with controllable configuration at the connection junction. Precursor 14 with the desired C(α)–C(δ′) bond connectivity and stereochemistry was projected to be transformed to (−)-flueggenine D (4) and its C(δ)-methoxy adduct (−)-flueggenine I (5).
Results and discussion
Our studies commenced with the synthesis of the electrophilic coupling partner for Stille cross-coupling reaction (Scheme 2A). Our previously accessed enone 15 (8 steps from N-Boc-D-proline)16 was allowed to react with lithium phenylacetylide generated in situ from phenoxy dichloroethene 16 to produce propargyl alcohol intermediate 17.20 Treatment of 17 with NIS followed by stirring of the reaction mixture with silica gel provided α-iodobutenolide 18 in 54% yield over 3 steps. A plausible reaction mechanism for the transformation of 17 to 18 might involve an iodo Meyer–Schuster rearrangement21 but we cannot rule out a direct iodine-induced 5-endo-dig cyclization.22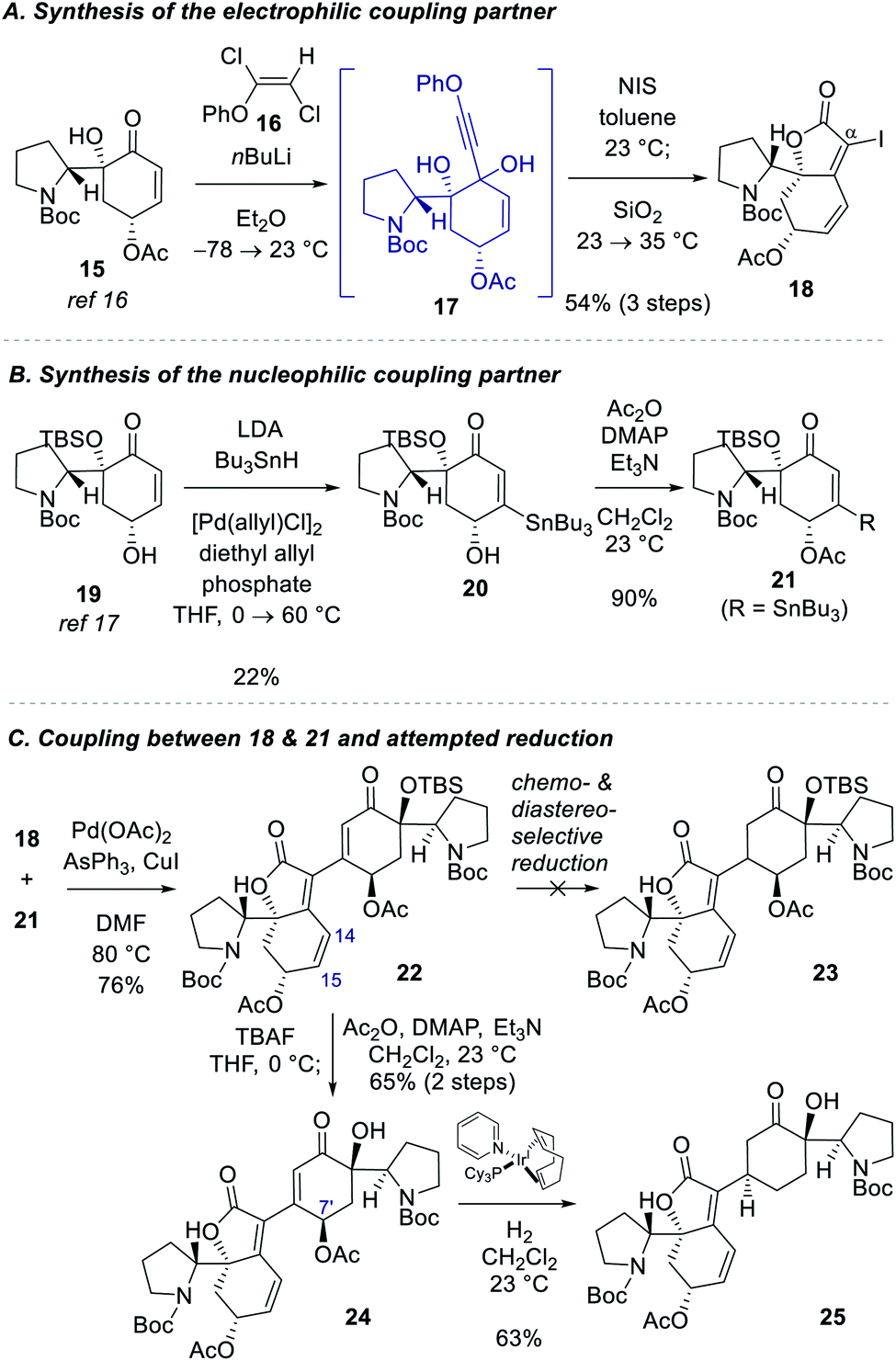 | ||
| Scheme 2 Initial synthetic approach toward (−)-flueggenine D (4). | ||
For the synthesis of the nucleophilic coupling partner of the Stille reaction, we invoked a Pd-catalyzed oxidative β-stannylation of enone that was recently reported by the Newhouse group (Scheme 2B).23 We found that our previously reported enone 1917 delivered stannane 20 in 22% yield under Newhouse's conditions. The hydroxyl group of 20 was subsequently acylated to yield stannane 21 in 90% yield. With both coupling partners 18 and 21 available, we attempted the Stille cross-coupling reaction between them. In the presence of Pd(OAc)2, triphenylarsine, and CuI, iodide 18 and stannane 21 were coupled to yield enone 22 in 76% yield (Scheme 2C).
However, the chemoselective conjugate reduction of 22 to 23 turned problematic. We could not obtain any desired product under various conjugate reduction or Pd-catalyzed hydrogenation conditions. We only observed the reduction at the C14–C15 double bond of the unsaturated ester moiety in the left fragment under these conditions. Attempted hydroxyl-directed hydrogenation of cyclohexenone 24 derived from 22 in 65% yield over 2 steps using Crabtree's catalyst24 resulted in allylic deacetoxylation at the C7′ position followed by a stereoselective hydrogenation of the enone group to provide saturated ketone 25 in 63% yield. Modest yield of the β-stannylation of enone 19 and unsuccessful conjugate reduction of the β-substituted cyclohexenones 22 and 24 necessitated us to modify the synthetic approach.
We envisioned that problems encountered in our initial synthetic route (Scheme 2B and C) can be solved by introducing a ketone group at the C7 position of bicyclic compound 15 (Scheme 3). Firstly, it would make the introduction of the stannyl group at C15 (flueggenine D numbering) easier because there are more reported methods for the α-functionalization of enone than its β-functionalization. Secondly, the substrate for a conjugate reduction post-Stille reaction would be a “β-unsubstituted” cyclohexenone derivative which has more successful precedences compared to the β-substituted cyclohexenone derivative. Thirdly, after the dimerization and conjugate reduction, the ketone group juxtaposed to the C15′ methine can provide stereochemical flexibility at this junction due to possible deprotonation.
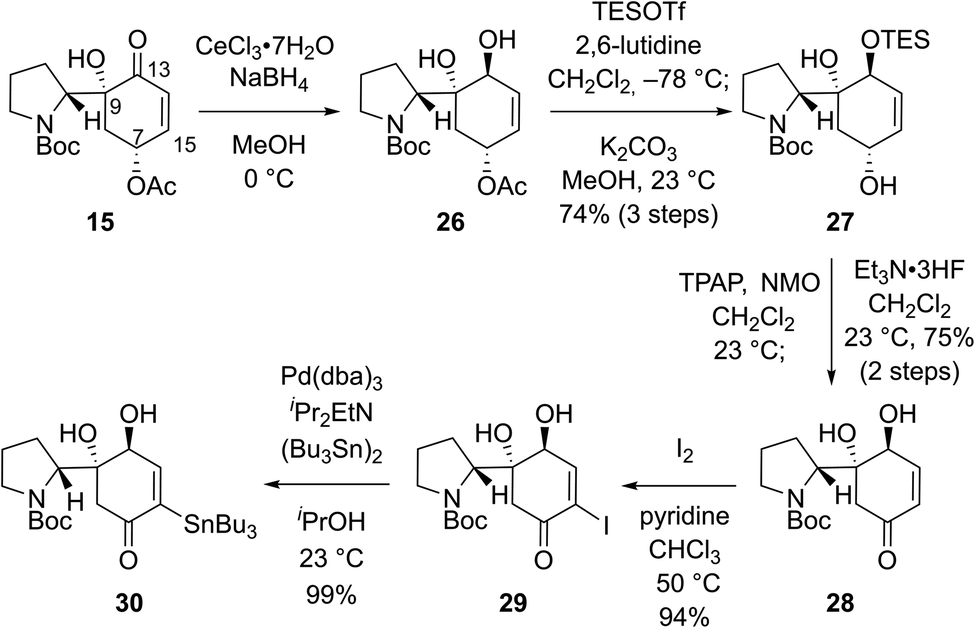 | ||
| Scheme 3 Re-design of the organostannane coupling partner. | ||
Based on these rationales, ketone 15 was diastereoselectively reduced to diol 26 under Luche's conditions (Scheme 3). Notably, deacetylation and oxidation at C7 of 15 caused immediate dehydration and quinone derivative formation. Silylation of the allylic alcohol moiety in 26 followed by methanolysis of the acetyl group yielded allylic alcohol 27 in 74% yield over 3 steps. Ley oxidation of allylic alcohol 27 and subsequent desilylation produced enone 28 in 75% yield over 2 steps. As expected, α-functionalization of enone 28 was facile. α-Iodination of enone 28 (94% yield) and consequent Pd-catalyzed stannylation using bis(tributyltin) (99% yield) afforded stannane 30 in a reliable manner.
With robust synthetic accesses to both 18 and 30, we allowed them to react in the presence of catalytic Pd(PPh3)4 (20 mol%) and CuI additive. Coupled product 31 was obtained in 99% yield (Scheme 4) under these conditions highlighting the robustness of Stille reaction in a complex molecular setting. We next set out for the conjugate reduction of the enone moiety in 31. Delightfully, treatment of enone 31 with 0.3 equiv. of Stryker's reagent and 5.0 equiv. of phenylsilane afforded conjugatively reduced ketone 34 in 67% yield along with <5% of its C15′ epimer.25 The lower energy of chair-conformation transition state with minimal torsional strains resulting from the protonation of silyl enol ether intermediate 33 from the top face might have contributed to the observed diastereoselectivity. Intramolecular proton delivery from the C13′ hydroxyl group during the protonation of silyl enol ether intermediate 33 might have also contributed to the observed high diastereoselectivity. Silylation of the secondary alcohol moiety in 34 required for the later chemodifferentiation was achieved in 67% yield.
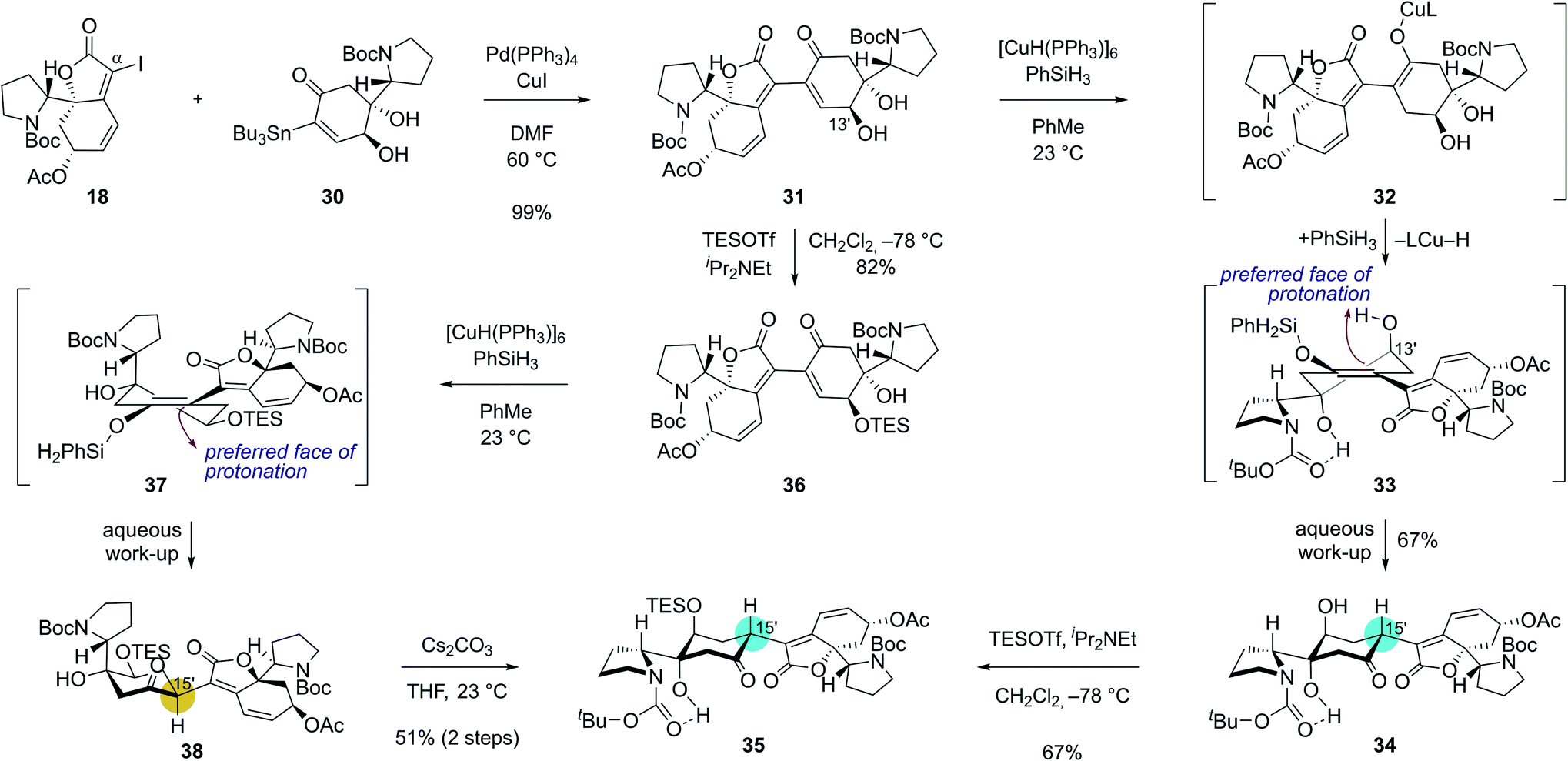 | ||
| Scheme 4 Key dimerization sequence with complete stereochemical flexibility and controllability at the C15′ connection junction. | ||
Interestingly, the stereochemical outcome of the Stryker's reagent-catalyzed conjugate reduction could be reversed when silyl ether 36 that is derived from alcohol 31 upon silylation at C13′ hydroxyl group was utilized as a substrate (Scheme 4). When 36 was treated with Stryker's reagent and phenylsilane for the conjugate reduction, saturated ketone 38 with an S-configuration at C15′ was obtained as a single diastereomer. The high diastereoselectivity during the protonation of the silyl enol ether intermediate 37 is reasoned to stem from its preferential protonation from the bottom face that leads to a chair conformation transition state with minimized torsional strains. Importantly, ketone 38 underwent complete epimerization at the C15′ position upon treatment with cesium carbonate in THF to yield the thermodynamic product 35 (51% yield over 2 steps). These observations highlight the stereochemical flexibility and controllability of our synthetic strategy at the pivotal δ' connection junction.
Reduction of the C7′ ketone group of 35 under Luche's conditions followed by acetylation of the resulting secondary alcohol produced compound 39 in 67% yield over 2 steps (Scheme 5). The observed equatorial attack of the hydride during the reduction step can be rationalized by a hindered axial Bürgi–Dunitz trajectory by the C9′ hydroxyl group. Desilylation of 39 and subsequent Dess–Martin periodinane-mediated oxidation of the resulting alcohol afforded ketone 40 in 62% yield over 2 steps. For the construction of the butenolide moiety, hydroxyketone 40 was treated with Bestmann ylide at 100 °C in toluene.26 Subsequent methanolysis produced diol 41 in 59% yield over 2 steps. Parallel mesylations of both alcohol groups in 41, Boc deprotections, and subsequent N-alkylations under basic conditions resulted in the first synthetic sample of (−)-flueggenine D (4) in 51% yield over 3 steps. NMR, HRMS, and specific rotation data of our synthetic sample were in agreement with the reported data from the isolation paper.7 Finally, (−)-flueggenine D (4) could be converted to (−)-flueggenine I (5) upon its treatment with sodium methoxide in methanol solution in 87% yield enabling its first total synthesis as well. The complete stereoselectivity during this 1,6-conjugate addition reaction is notable.27
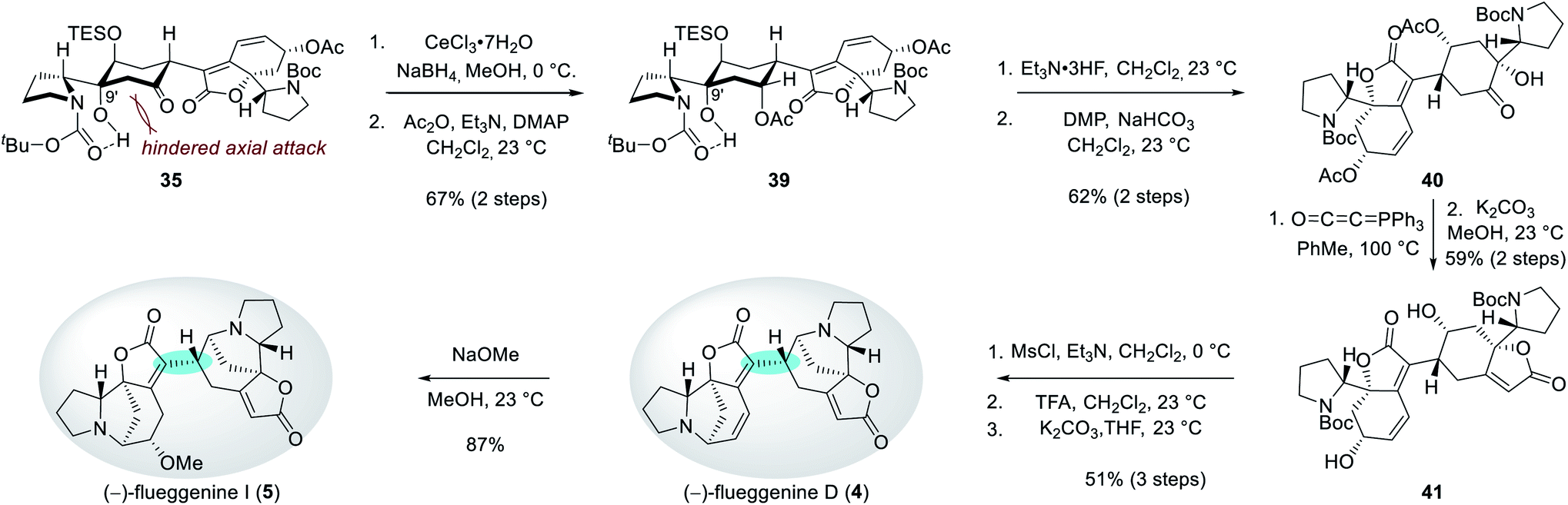 | ||
| Scheme 5 Total synthesis of (−)-flueggenines D (4) and I (5). | ||
Conclusions
In conclusion, we have completed the first total synthesis of dimeric Securinega alkaloids (−)-flueggenines D (4) and I (5) via a Stille reaction/conjugate reduction-based dimerization strategy. This constitutes the first synthetic entry to dimeric Securinega alkaloids with the C(α)–C(δ′) connectivity between monomeric units. The high chemofidelity of the Stille reaction enabled us to link two structurally complex coupling partners that could not be connected by other methods. Our synthetic strategy features stereochemical flexibility and controllability at the α–δ′ connection junction that are indispensable in accessing other RC reaction-based high-order Securinega alkaloids. With chemical technologies to make both α–δ′ (this study) and γ–δ′16 connections, we can now envision to synthesize trimer or even higher-order Securinega natural products that contain both connection modes. Those will be the subjects of our forthcoming reports.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Gyumin Kang for helpful discussions. This work was supported by Samsung Science and Technology Foundation under Project Number SSTF-BA1701-13.Notes and references
- (a) E. Chirkin, W. Atkatlian, F.-H. Porée, in The Alkaloids, ed, H.-J. Knölker, Academic Press, London, 2015, vol. 74, pp. 1–120 Search PubMed; (b) S. M. Weinreb, Nat. Prod. Rep., 2009, 26, 758–775 Search PubMed.
- R. Wehlauch and K. Gademann, Asian J. Org. Chem., 2017, 6, 1146–1159 Search PubMed.
- L.-S. Gan, C.-Q. Fan, S.-P. Yang, Y. Wu, L.-P. Lin, J. Ding and J.-M. Yue, Org. Lett., 2006, 8, 2285–2288 Search PubMed.
- Z.-L. Wu, X.-J. Huang, M.-T. Xu, X. Ma, L. Li, L. Shi, W.-J. Wang, R.-W. Jiang, W.-C. Ye and Y. Wang, Org. Lett., 2018, 20, 7703–7707 Search PubMed and references therein.
- P. Xie and Y. Huang, Eur. J. Org. Chem., 2013, 2013, 6213–6226 Search PubMed.
- C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069–4084 Search PubMed.
- H. Zhang, W. Wei and J.-M. Yue, Tetrahedron, 2013, 69, 3942–3946 Search PubMed.
- H. Zhang, C.-R. Zhang, Y.-S. Han, M. A. Wainberg and J.-M. Yue, RSC Adv., 2015, 5, 107045–107053 Search PubMed.
- H. Zhang, C.-R. Zhang, K.-K. Zhu, A.-H. Gao, C. Luo, J. Li and J.-M. Yue, Org. Lett., 2013, 15, 120–123 Search PubMed.
- H. Zhang, Y.-S. Han, M. A. Wainberg and J.-M. Yue, Tetrahedron, 2015, 71, 3671–3679 Search PubMed.
- For recent synthesis of monomeric Securinega alkaloids, see: (a) E. Chirkin, C. Bouzidi and F.-H. Porée, Synthesis, 2019, 51, 2001–2006 Search PubMed; (b) K. Antien, A. Lacambra, F. P. Cossío, S. Massip, D. Deffieux, L. Pouységu, P. A. Peixoto and S. Quideau, Chem.–Eur. J., 2019, 25, 11574–11580 Search PubMed; (c) S. Lee, G. Kang, G. Chung, D. Kim, H.-Y. Lee and S. Han, Angew. Chem., Int. Ed., 2020, 59, 6894–6901 Search PubMed.
- S. Jeon, J. Park and S. Han, Synlett, 2017, 28, 2353–2359 Search PubMed.
- B.-X. Zhao, Y. Wang, C. Li, G.-C. Wang, X.-J. Huang, C.-L. Fan, Q.-M. Li, H.-J. Zhu, W.-M. Chen and W.-C. Ye, Tetrahedron Lett., 2013, 54, 4708–4711 Search PubMed.
- H. Wei, C. Qiao, G. Liu, Z. Yang and C.-C. Li, Angew. Chem., Int. Ed., 2013, 52, 620–624 Search PubMed.
- N. Ma, Y. Yao, B.-X. Zhao, Y. Wang, W.-C. Ye and S. Jiang, Chem. Commun., 2014, 50, 9284–9287 Search PubMed.
- S. Jeon and S. Han, J. Am. Chem. Soc., 2017, 139, 6302–6305 Search PubMed.
- J. Park, S. Jeon, G. Kang, J. Lee, M.-H. Baik and S. Han, J. Org. Chem., 2019, 84, 1398–1406 Search PubMed.
- S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 Search PubMed.
- M. M. Heravi and L. Mohammadkhani, J. Organomet. Chem., 2018, 869, 106–200 Search PubMed.
- M. Egi, Y. Ota, Y. Nishimura, K. Shimizu, K. Azechi and S. Akai, Org. Lett., 2013, 15, 4150–4153 Search PubMed.
- S. Puri, N. Thirupathi and M. S. Reddy, Org. Lett., 2014, 16, 5246–5249 Search PubMed.
- M. S. Reddy, N. Thirupathi, M. H. Babu and S. Puri, J. Org. Chem., 2013, 78, 5878–5888 Search PubMed.
- A. W. Schuppe, D. Huang, Y. Chen and T. R. Newhouse, J. Am. Chem. Soc., 2018, 140, 2062–2066 Search PubMed.
- R. H. Crabtree and M. W. Davis, J. Org. Chem., 1986, 51, 2655–2661 Search PubMed.
- For a review on CuH-catalyzed reactions, see: C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 Search PubMed.
- R. Wehlauch, S. M. Grendelmeier, H. Miyatake-Ondozabal, A. H. Sandtorv, M. Scherer and K. Gademann, Org. Lett., 2017, 19, 548–551 Search PubMed.
- S. G. Klochkov, S. V. Afanas'eva and V. V. Grigor'ev, Chem. Nat. Compd., 2008, 44, 197–202 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive experimental procedures, spectroscopic data, spectrometric data, and copies of 1H, 13C NMR spectra of all new compounds. See DOI: 10.1039/d0sc03057k |
| This journal is © The Royal Society of Chemistry 2020 |