
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coligand role in the NHC nickel catalyzed C–F bond activation: investigations on the insertion of bis(NHC) nickel into the C–F bond of hexafluorobenzene†
Maximilian W.
Kuntze-Fechner
a,
Hendrik
Verplancke
 b,
Lukas
Tendera
a,
Martin
Diefenbach
b,
Ivo
Krummenacher
ac,
Holger
Braunschweig
ac,
Todd B.
Marder
ac,
Max C.
Holthausen
*b and
Udo
Radius
*a
b,
Lukas
Tendera
a,
Martin
Diefenbach
b,
Ivo
Krummenacher
ac,
Holger
Braunschweig
ac,
Todd B.
Marder
ac,
Max C.
Holthausen
*b and
Udo
Radius
*a
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: u.radius@uni-wuerzburg.de
bInstitute for Inorganic and Analytical Chemistry, Goethe-Universität Frankfurt, Max-von-Laue-Strasse 7, 60438 Frankfurt, Germany. E-mail: max.holthausen@chemie.uni-frankfurt.de
cInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 6th October 2020
Abstract
The reaction of [Ni(Mes2Im)2] (1) (Mes2Im = 1,3-dimesityl-imidazolin-2-ylidene) with polyfluorinated arenes as well as mechanistic investigations concerning the insertion of 1 and [Ni(iPr2Im)2] (1ipr) (iPr2Im = 1,3-diisopropyl-imidazolin-2-ylidene) into the C–F bond of C6F6 is reported. The reaction of 1 with different fluoroaromatics leads to formation of the nickel fluoroaryl fluoride complexes trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = 4-CF3-C6F42, C6F53, 2,3,5,6-C6F4N 4, 2,3,5,6-C6F4H 5, 2,3,5-C6F3H26, 3,5-C6F2H37) in fair to good yields with the exception of the formation of the pentafluorophenyl complex 3 (less than 20%). Radical species and other diamagnetic side products were detected for the reaction of 1 with C6F6, in line with a radical pathway for the C–F bond activation step using 1. The difluoride complex trans-[Ni(Mes2Im)2(F)2] (9), the bis(aryl) complex trans-[Ni(Mes2Im)2(C6F5)2] (15), the structurally characterized nickel(I) complex trans-[NiI(Mes2Im)2(C6F5)] (11) and the metal radical trans-[NiI(Mes2Im)2(F)] (12) were identified. Complex 11, and related [NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) and [NiI(Mes2Im)2(2,3,5-C6F3H2)] (14), were synthesized independently by reaction of trans-[Ni(Mes2Im)2(F)(ArF)] with PhSiH3. Simple electron transfer from 1 to C6F6 was excluded, as the redox potentials of the reaction partners do not match and [Ni(Mes2Im)2]+, which was prepared independently, was not detected. DFT calculations were performed on the insertion of [Ni(iPr2Im)2] (1ipr) and [Ni(Mes2Im)2] (1) into the C–F bond of C6F6. For 1ipr, concerted and NHC-assisted pathways were identified as having the lowest kinetic barriers, whereas for 1, a radical mechanism with fluoride abstraction and an NHC-assisted pathway are both associated with almost the same kinetic barrier.
Introduction
Fluorinated organic compounds have exceptional properties that are being exploited in many applications including materials, pharmaceuticals and agrochemicals. The development of methods to introduce fluorinated aromatic building blocks selectively into organic molecules is thus of fundamental interest in many areas of chemical research.1 One strategy for such transformations is the selective activation and subsequent functionalization of C–F bonds of readily available fluoroorganic compounds such as fluoroaromatics. The challenge here is the selective cleavage of very stable C–F bonds.2 We have recently established a protocol for the transformation of commercially available fluoroaromatics via a selective C–F defluoroborylation process to obtain polyfluorinated arylboronic esters,3 which may be further used in late stage functionalization, for example in Suzuki–Miyaura cross-coupling reactions.4 Defluoroborylation of polyfluoroaromatics can be achieved by a thermal [Ni(Mes2Im)2]-catalyzed (Mes2Im = 1,3-dimesityl-imidazolin-2-ylidene) transformation of polyfluoroarenes into fluoroaryl boronic acid pinacol esters via C–F bond activation and transmetalation with bis(pinacolato)diboron (B2pin2) as the boron source (see Scheme 1).3a Various arenes with different degrees of fluorination were converted into their corresponding boronate esters in this way. One particularly interesting finding of our study was that activation of the C–F bond by the nickel(0) complex is fast at ambient temperature. This step yields the oxidative addition product trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = fluoroaryl), which represents the resting state in the catalytic cycle. The subsequent defluoroborylation step with B2pin2 is the rate determining step and requires elevated temperatures. A boryl complex trans-[Ni(Mes2Im)2(Bpin)(ArF)], a likely intermediate, was never observed and stoichiometric reactions of trans-[Ni(Mes2Im)2(F)(ArF)] with B2pin2 led directly to the formation of ArF–Bpin. This finding implied that reductive elimination is very fast and that [Ni(Mes2Im)n(Bpin)(ArF)], once formed, will eliminate ArF–Bpin immediately (Scheme 1).5a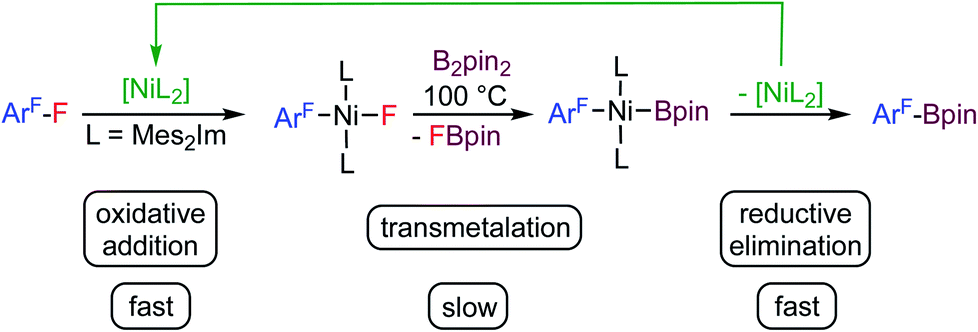 | ||
| Scheme 1 Thermal borylation of fluoroarenes with B2pin2 mediated by [Ni(Mes2Im)2] via the oxidative addition product trans-[Ni(Mes2Im)2(F)(ArF)] as the resting state of the catalysis. | ||
As an alternative to the thermally-induced C–F bond activation and subsequent borylation of fluoroarenes, we have recently developed a process that employs visible-light photocatalysis, which has emerged as a powerful tool in organic synthesis.6 Our highly selective and general photocatalytic C–F borylation protocol3b employs a rhodium biphenyl complex7 as triplet sensitizer combined with the nickel catalyst [Ni(Mes2Im)2] (1) for the C–F bond activation step and the defluoroborylation process. This Rh/Ni tandem catalyst system operates with visible light (400 nm) and achieves the highly selective borylation of a wide range of polyfluoroarenes with B2pin2 at room temperature in excellent yields. Both procedures, the thermal and photochemical defluoroborylation, work well for partially fluorinated aromatics but fail, or afford only low yields, for perfluoroaromatics such as hexafluorobenzene or octafluorotoluene.
Utilizing the dinuclear complex [Ni2(iPr2Im)4(μ-(η2:η2)-COD)] (iPr2Im = 1,3-diisopropyl-imidazolin-2-ylidene) or the ethylene complex [Ni(iPr2Im)2(η2-C2H4)]8 as sources of [Ni(iPr2Im)2] (1iPr), we previously found that both readily undergo C–F bond insertion with a wide variety of per- and polyfluoroaromatics on a time scale suitable for catalysis (exemplarily shown for C6F6 in Scheme 2).4c,k,9 Mechanistic investigations9a of the insertion process were performed using the ethylene complex [Ni(iPr2Im)2(η2-C2H4)] as nickel precursor. Ethylene exchange at the [Ni(iPr2Im)2(η2-C2H4)] complex with hexafluorobenzene and octafluoronaphthalene occurs at low temperatures (−80 °C and −30 °C, respectively; Scheme 2). Subsequent insertion reactions occur at higher temperatures (0 °C and 20 °C, respectively) to form the trans-[Ni(iPr2Im)2(F)(C6F5)] and trans-[Ni(iPr2Im)2(F)(C10F7)] fluoroaryl fluoride complexes.9 We studied the C–F bond activation kinetics and, based on the decay rates of the octafluoronaphthalene complex [Ni(iPr2Im)2(η2-C10F8)] determined by variable-temperature NMR spectroscopy, we derived an activation enthalpy of ΔH‡ = 27.7 ± 1.9 kcal mol−1 (ΔS‡ = 8.8 ± 6.0 cal K−1 mol−1).
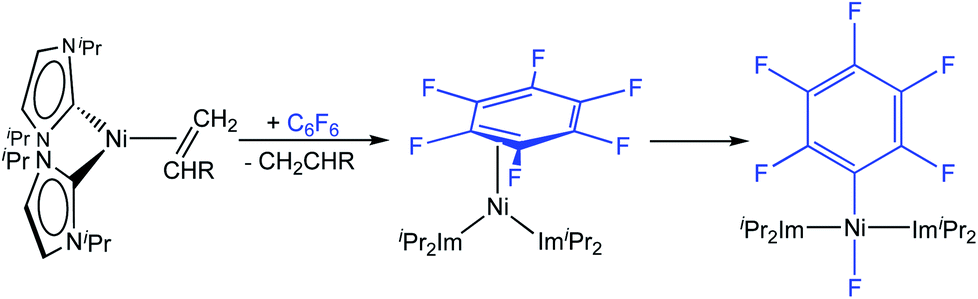 | ||
| Scheme 2 Stoichiometric C–F bond activation of C6F6 using sources of [Ni(iPr2Im)2] 1iPr. | ||
We report herein on the reactivity of 1 with polyfluorinated arenes. We compare the results with those of earlier studies on C–F bond activation processes using nickel complexes with sterically less demanding NHCs, employing iPr instead of Mes substituents, i.e., using [Ni(iPr2Im)2] (1ipr) as the nickel source. We demonstrate that the complex of the small NHC ligand iPr2Im favors a concerted oxidative addition proceeding through an η2(C,C) intermediate in reactions with fluoroarenes to yield trans-[NiII(NHC)2(F)(ArF)] complexes, whereas the complex of the larger Mes2Im ligand leads to fluorine atom abstraction to yield [NiI(NHC)2(F)] and a phenyl radical. For both mechanisms, competitive NHC-assisted pathways are found which account for the formation of diamagnetic products by a C–F bond activation step across the Ni–CNHC bond. These NHC-assisted pathways play an important role for complexes of both sterically demanding and less bulky NHC ligands, and should thus be of general importance and widely applicable for the reactivity of NHC-stabilized transition metal complexes.
Results and discussion
C–F bond activation of fluoroaromatics
To gain insight into the C–F bond activation process using [Ni(Mes2Im)2] (1), we first investigated stoichiometric reactions of perfluorotoluene, perfluorobenzene, perfluoropyridine and the partially fluorinated arenes pentafluorobenzene, 1,2,3,5-tetrafluorobenzene and 1,3,5-trifluorobenzene with 1 (see Scheme 3). We monitored the reactions by 1H and 19F{1H} NMR spectroscopy and observed a significant effect of the degree of fluorination on both reaction rate and yield. Reactions of 1 with hexafluorobenzene and octafluorotoluene proceed within seconds at room temperature, whereas the reactions with tetra- and pentafluorobenzene take minutes to complete. With 1,3,5-trifluorobenzene, full conversion of 1 takes weeks at room temperature (see ESI, Fig. S1†), but can be accelerated at 80 °C in thf to reach completion after 5 days.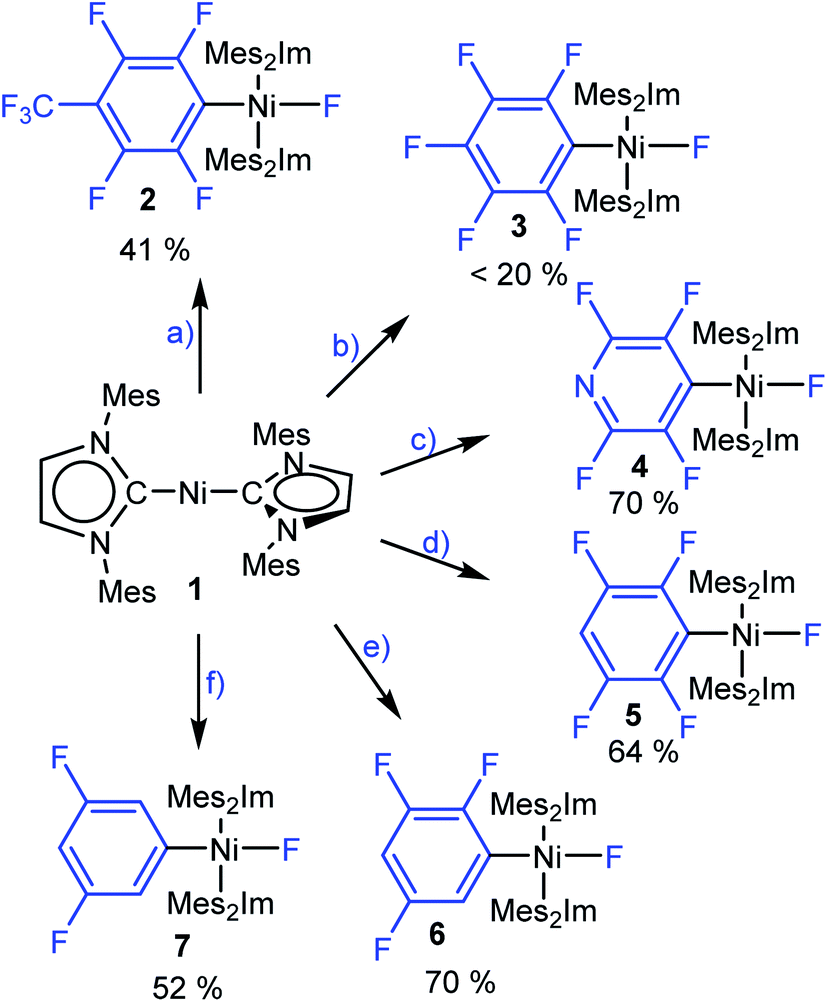 | ||
| Scheme 3 The reactions of [Ni(Mes2Im)2] (1) with (a) octafluorotoluene, (b) hexafluorobenzene, (c) perfluoropyridine, (d) pentafluorobenzene, (e) 1,2,3,5-tetrafluorobenzene and (f) 1,3,5-trifluorobenzene to give the complexes trans-[Ni(Mes2Im)2(F)(4-CF3-C6F4)] (2), trans-[Ni(Mes2Im)2(F)(C6F5)] (3), trans-[Ni(Mes2Im)2(F)(2,3,5,6-C5F4N)] (4), trans-[Ni(Mes2Im)2(F)(2,3,5,6-C6F4H)] (5), trans-[Ni(Mes2Im)2(F)(2,3,5-C6F3H2)] (6) and trans-[Ni(Mes2Im)2(F)(3,5-C6F2H3)] (7), respectively. Isolated yields are given. | ||
These reactions can be performed in thf, toluene or hexane at room temperature and lead, in each case, to the insertion of the nickel complex into the C–F bond of the fluoroarene to form the nickel fluoroaryl fluoride complexes trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = 4-CF3-C6F42, C6F53, 2,3,5,6-C5F4N 4, 2,3,5,6-C6F4H 5, 2,3,5-C6F3H26, 3,5-C6F2H37) in fair to good isolated yields. Notably, however, the reaction with C6F6 yields less than 20% of the pentafluorophenyl complex 3. Higher temperature, different solvents (thf, toluene, hexane) or added [NMe4]F does not seem to affect the yield of the insertion product 3. Complexes 2–7 were characterized by elemental analysis, 1H, 19F{1H} and 13C{1H} NMR spectroscopy (see ESI†). In the 19F{1H} NMR spectra of these complexes, the resonances of the nickel-bound fluoride ligand were observed in the typical range between −361.9 and −333.1 ppm. Within the series presented (see ESI, Table S1†), the NMR shift of this resonance depends on the degree of fluorination of the fluoroaryl ligands, i.e., an increase of the degree of fluorination of the aryl ligand leads to an upfield shift of the Ni–F resonance.
Crystals of 3, 4, and 5 suitable for X-ray diffraction were obtained from saturated solutions of these compounds either in pentane or hexane at −30 °C (Fig. 1, Table 1; see also ESI Fig. S34–S36 and Table S2†). The crystal structure of 6 was published previously.3a All complexes of the type trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = C6F53, 2,3,5,6-C5F4N 4, 2,3,5,6-C6F4H 5, 2,3,5-C6F3H26) adopt a square planar structure with a trans arrangement of the NHC ligands. An increasing degree of fluorination of the fluoroaryl ligand leads to a slight shortening of the Ni–F bond lengths (Ni–F: 6: 1.874(2) Å, 5: 1.856(2) Å, 4: 1.859(2) Å, 3: 1.844(2) Å), while the distances of the nickel center to the fluoroaryl ligand become gradually longer (Ni–C3: 6: 1.854(5) Å, 5: 1.896(3) Å, 4: 1.883(3) Å, 3: 1.944(5) Å). We assume that both the upfield shift of the Ni–F 19F NMR resonance and the shortening of the Ni–F bond lengths with increasing degree of aryl fluorination are indications of stronger Ni–F bonding.
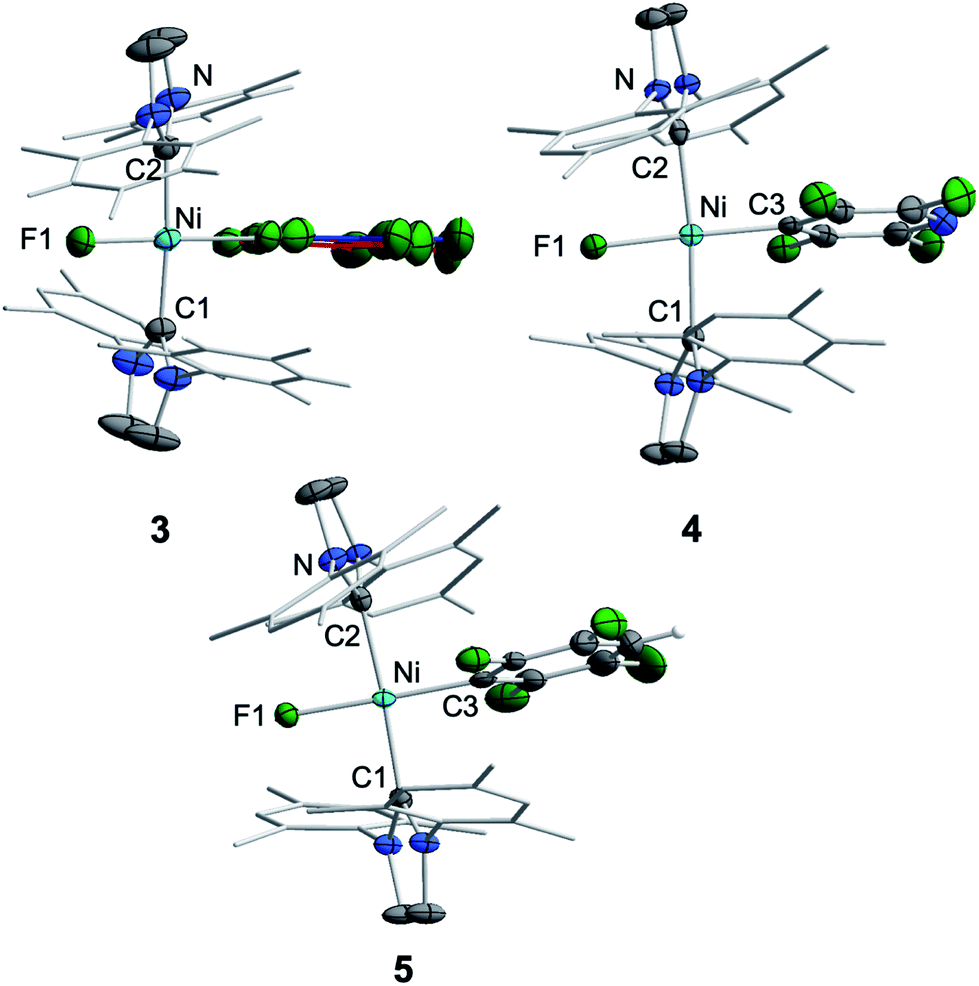 | ||
| Fig. 1 Molecular structures of trans-[Ni(Mes2Im)2(F)(C6F5)] (3) (top left), trans-[Ni(Mes2Im)2(F)(2,3,5,6-C5F4N)] (4) (top right) and trans-[Ni(Mes2Im)2(F)(2,3,5,6-C6F4H)] (5) (bottom) in the solid state (ellipsoids drawn at the 50% probability level). Hydrogens atoms, with exception of the proton at the fluoroaromatic of 5, are omitted for clarity. | ||
| d Ni–C1/C2 | d Ni–C3(ArF) | d Ni–F | ∠C1–Ni–C2 | ∠NHC(C1)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NHC(C2) :NHC(C2) |
|
|---|---|---|---|---|---|
| [Ni(Mes2Im)2] 1 | 1.827(6) | — | — | 176.4 | 53.0 |
| 1.830(6) | |||||
| 3 | 1.923(3) | C3: 1.882(7) | 1.844(2) | 175.6(1) | 37.07(2) |
| 1.922(3) | C3′: 1.944(5) | ||||
| 4 | 1.923(3) | 1.883(3) | 1.859(2) | 174.3(1) | 36.01(2) |
| 1.920(3) | |||||
| 5 | 1.921(2) | 1.896(3) | 1.856(2) | 176.4(1) | 33.81(1) |
| 1.924(2) | |||||
| 6 | 1.912(3) | 1.854(5) | 1.874(2) | 176.7(1) | 31.65(2) |
| 1.912(3) | |||||
| 8 | 1.894(3) | — | — | 174.5(1) | 57.99(1) |
| 1.894(3) | |||||
| 9 | 1.903(3) | — | F1: 1.845(2) | 178.5(1) | 53.34(1) |
| 1.902(3) | F2: 1.823(2) | ||||
| 11 | 1.923(2) | 1.984(3) | — | 159.8(8) | 82.37(1) |
| 1.923(2) | |||||
| 13 | 1.930(2) | 1.987(3) | — | 157.3(8) | 82.11(1) |
| 1.930(2) | |||||
| 14 | 1.918(1) | C3: 1.869(1) | — | 159.5(5) | 82.46(8) |
| 1.917(1) | C3′: 2.046(1) | ||||
| [NiI(6-Mes)2][Br] | 1.939(3) | — | — | 179.3(1) | 57.99(1) |
| 1.941(3) | |||||
| [NiI(PiPr3)2(C6F5)] | P1: 2.243(5) | 1.973(2) | — | P1–Ni–P2 | — |
| P2: 2.233(5) | 145.2(2) |
As the low yield of trans-[Ni(Mes2Im)2(F)(C6F5)] (3) is in sharp contrast with the results we obtained previously for the reaction of [Ni2(iPr2Im)4(μ-(η2:η2)-COD)] or [Ni(iPr2Im)2(η2-C2H4)] with C6F6,9 we decided to take a closer look at the corresponding reaction using [Ni(Mes2Im)2] (1). Performing the stoichiometric reaction of 1 with C6F6 in an NMR tube in C6D6 led to an immediate color change from dark-violet, the color of concentrated complex 1, to orange after addition of C6F6 at room temperature. A quantitative conversion of 1 was achieved after 5 min as monitored by 1H NMR spectroscopy (see ESI, Fig. S2†). However, the spectroscopic yield determined by 19F{1H} NMR spectroscopy after 5 min at room temperature, vs. a Ph–F containing capillary as internal standard, revealed the formation of 3 in approximately 17% yield and, in addition, the formation of small amounts of fluoride-containing side products (see ESI, Fig. S3†). Even after 72 h at room temperature, no increase in the spectroscopic yield of 3 was observed. In further control experiments, neither the use of an excess of 1 (2.85 equiv.) nor C6F6 (2.5 equiv.) increased the yield of 3 substantially. These experiments demonstrate that the low isolated yield of 3 is not a problem of the isolation process for this complex, but rather an intrinsic problem associated with its formation and the C–F bond activation step. Low temperature NMR experiments (−50 °C to +20 °C) revealed that a nickel fluoride resonance at −358 ppm appeared for this reaction in the 19F{1H} NMR spectrum already at −50 °C (see ESI, Fig. S4†), but also that, at these temperatures, all resonances are significantly broadened in the 1H NMR spectrum of the reaction mixture (see ESI, Fig. S5†). Although we previously observed some line broadening for the N-alkyl groups of the related complex trans-[Ni(iPr2Im)2(F)(C6F5)],9a which arose due to hindered rotation of the NHC ligand about the Ni–C axis, all resonances observed for the reaction of 1 with C6F6 are involved in the broadening. This led to the assumption that radical species are involved in the process. Subsequent EPR experiments were performed at −203 °C for the reaction of 1 with C6F6 which confirmed the presence of metal-centered radicals in the mixture.
For EPR spectroscopic investigations, 1 and C6F6 were combined in an EPR tube with thf at −78 °C and the sample was frozen immediately in liquid nitrogen. The EPR tube containing the frozen reaction mixture was transferred to the cooled EPR cavity at −203 °C and a spectrum was recorded.10 The resulting EPR spectrum displays a superposition of resonances of three different products, of which I and II represent the two dominant species (Fig. 2, I: 40%, II: 50%, III: 10%).
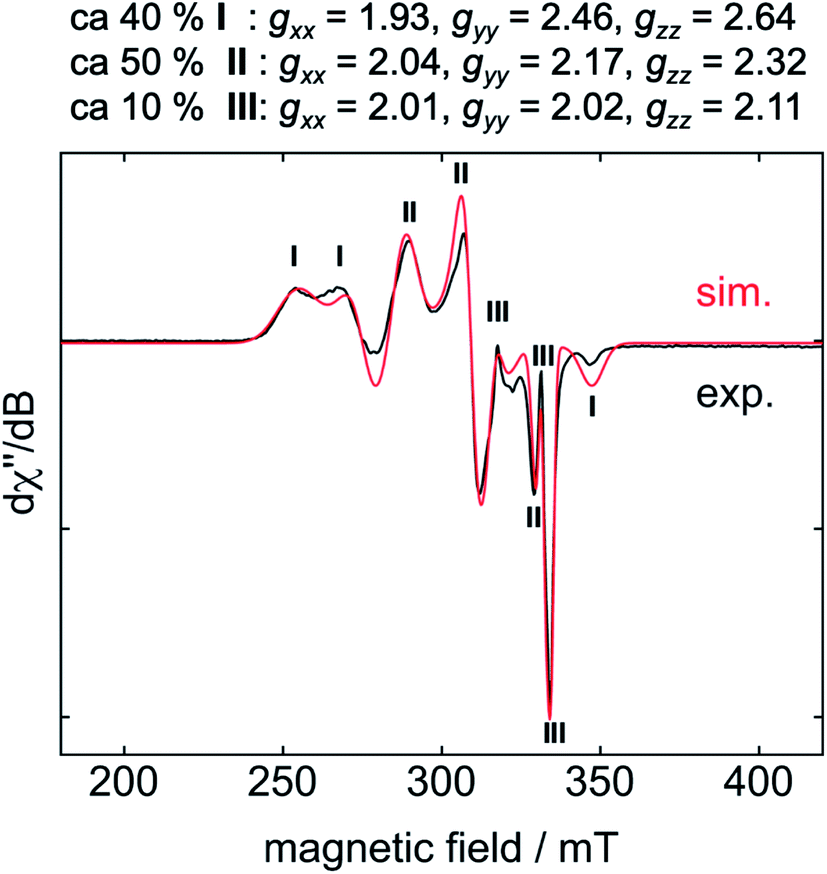 | ||
| Fig. 2 EPR spectrum (−203 °C) of the reaction mixture of 1 with C6F6 after 5 s at −78 °C in thf. | ||
Cyclic voltammetry results exclude a simple electron transfer from 1 to C6F6 as the origin of radical generation in the reaction mixture (see ESI, Fig. S6†), as 1 shows a reversible oxidation/reduction associated with a redox potential of −2.03 V for the redox-couple Ni0/NiI, and an irreversible oxidation at 0.14 V for the redox-couple NiI/NiII. Although the reduction of C6F6 at −2.87 V is irreversible, we exclude simple one electron transfer because of the large separation of 0.84 V.
For further scrutiny, complex 1 was oxidized by adding ferrocenium tetrafluoroborate in thf at room temperature to a suspension of 1 in thf. A few min after addition of the ferrocenium salt the metal-centered radical [Ni(Mes2Im)2][BF4] (8) precipitated as an off-white solid (83% isolated yield, Scheme 4), which is only sparingly soluble in common organic solvents. The NiI complex 8 was characterized by 11B{1H} and 19F{1H} NMR spectroscopy in acetonitrile (decomposition occurs after some time) and IR spectroscopy, elemental analysis and high-resolution mass spectroscopy. The 11B{1H} and 19F{1H} NMR spectra revealed an intact counter anion [BF4]− (see ESI, Fig. S64†). Crystals of 8 suitable for X-ray diffraction (Fig. 3, Table 1; see also ESI, Table S2 and Fig. S37†) were obtained by slow evaporation of a saturated solution of 8 in a 1:1 toluene/ethanol mixture under an argon atmosphere at room temperature. The X-ray crystal structure reveals a nearly linear alignment of the NHC ligands with slightly elongated Ni–C distances compared to those of the starting material 1.
 | ||
| Scheme 4 Synthesis of [Ni(Mes2Im)2][BF4] (8). | ||
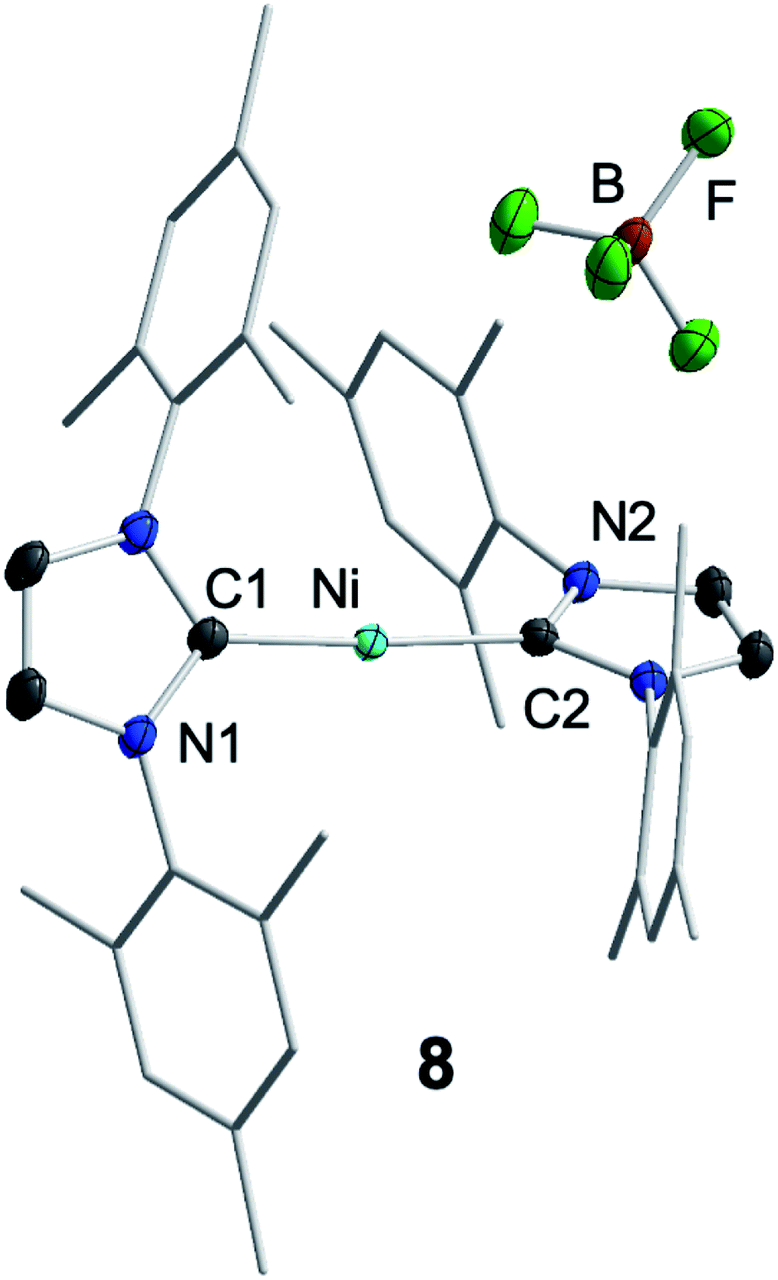 | ||
| Fig. 3 Molecular structure of [Ni(Mes2Im)2][BF4] (8) in the solid state (ellipsoids drawn at the 50% probability level). Hydrogens atoms are omitted for clarity. | ||
The results of the EPR spectroscopic investigations10 performed on solid-state samples of [Ni(Mes2Im)2][BF4] (8) are shown in Fig. 4. The general insolubility of 8 precluded determination of its magnetic moment by the Evans method. The spectrum reveals two sets of signals, i.e., 8a (gxx = 2.02, gyy = 2.47, gzz = 2.62; 70%) and 8b (gxx = 1.98, gyy = 2.06, gzz = 2.13; 30%), both in line with nickel-centered radicals. Note that in previous studies of two other homoleptic two-coordinate cationic d9-nickel(I) complexes, [Ni(6-Mes)2][Br] (6-Mes = 1,3-bis(2,4,6-trimethylphenyl)-3,4,5,6-tetrahydropyrimidin-2-ylidene) and [Ni(PtBu3)2][Al(OC(CF3)3)4], no EPR signals were observed.11 To obtain further insight, we performed density functional theory (DFT) calculations on the molecular geometries, electronic structures and EPR parameters (g tensors) of a variety of potential candidates for 8 (PBE0-D/pcSseg-2, see the ESI for details†).12
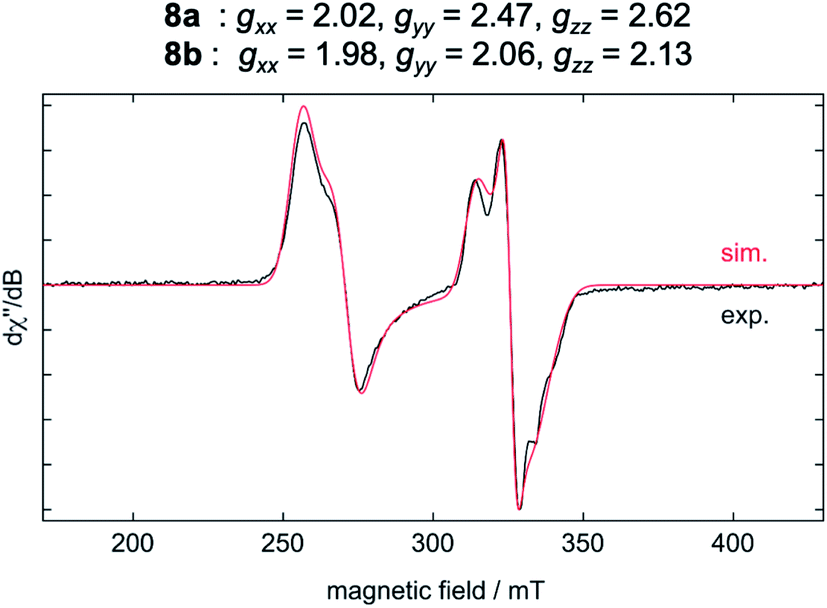 | ||
| Fig. 4 EPR spectrum of 8 in the solid state at −203 °C with NBu4Br. | ||
The DFT-optimized, D2-symmetric geometry of the [Ni(Mes2Im)2]+ radical cation of 8 agrees very well with the X-ray structure (Fig. 5; e.g., dNi–C = 1.89 Å, exp: 1.894(3) Å). In the 2A electronic ground state, the spin density is localized at the metal center, with the unpaired electron residing in an s/dz2-type orbital (Fig. 5a).
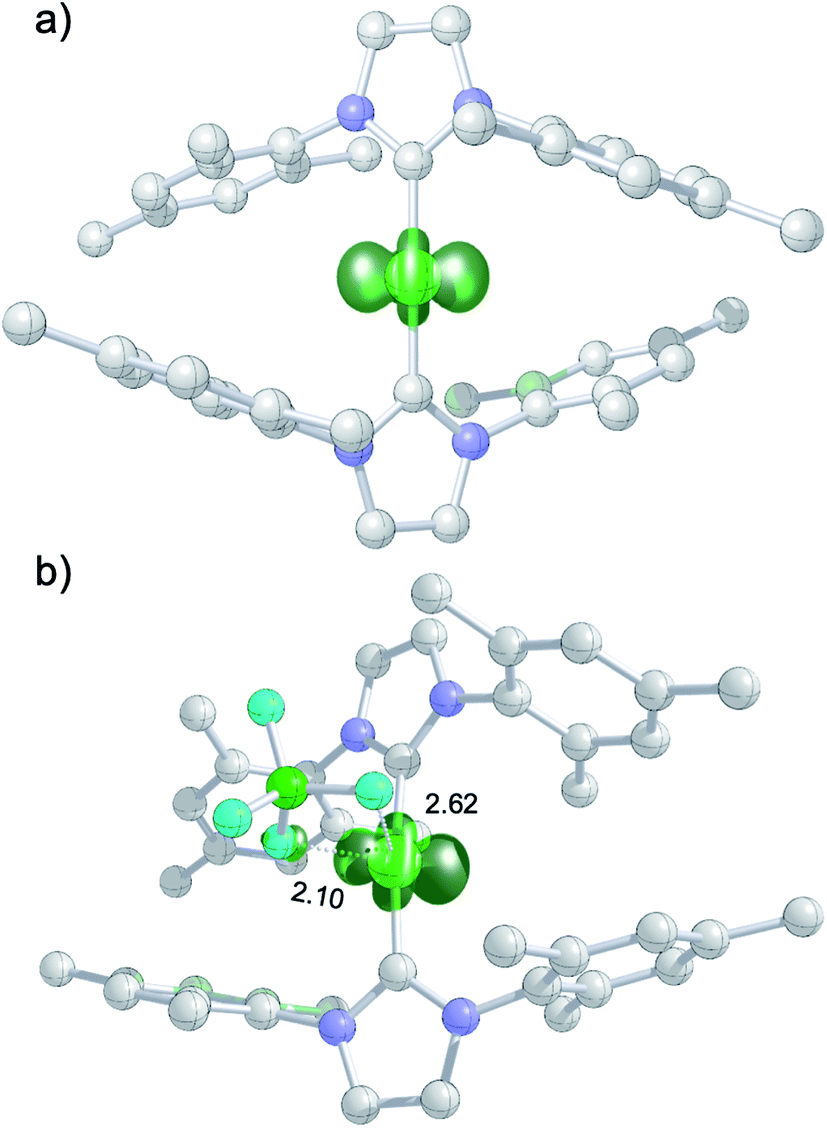 | ||
| Fig. 5 (a) Spin density plot for [Ni(Mes2Im)2]+; (b) molecular structure of 8DFT1 showing Ni–FBF4− contacts (isovalue ± 0.0075 a0−3; lengths of Ni–F contacts in Å; hydrogen atoms not shown). | ||
The calculated g values for the radical cation [Ni(Mes2Im)2]+ (gxx = 2.01, gyy = 2.65, gzz = 2.98), computed under gas-phase conditions, strongly differ from the experimental data with a maximum deviation of 0.36 (8a) and 0.85 (8b; see Table 2 and ESI, Table S3†). However, computations in the presence of the counter ion result in further structural motifs with impact on the computed g tensors (see ESI, Fig. S7†). A Ni–F contact with the counter ion in 8DFT1 (Table 2, entry 4, Fig. 5b) results in g tensor components closely corresponding to those of 8a (maximum deviation: 0.03), while no species matching the EPR parameters of 8b were identified in our computational exploration. However, none of the EPR signatures detected for the electrochemically-formed complex 8 appeared during the reaction of 1 with C6F6 (Fig. 2) and, in light of our CV results, it is unlikely that the [Ni(Mes2Im)2]+ cation is involved here.
| Compound | g tensor components | |||
|---|---|---|---|---|
| g xx | g yy | g zz | ||
| a DFT-optimized structure with Ni–FBF4− contacts. | ||||
| 8a | Exp. (solid state) | 2.02 | 2.47 | 2.62 |
| 8b | Exp. (solid state) | 1.98 | 2.06 | 2.13 |
| [Ni(Mes2Im)2]+(gas phase) | DFT | 2.01 | 2.65 | 2.98 |
| 8DFT1 | DFT | 2.03 | 2.50 | 2.59 |
We then focused on identifying the byproducts of the reaction of 1 with C6F6. Stoichiometric reaction of 1 with C6F6 in thf overnight at room temperature led to a very small amount of a dark-green precipitate which was removed by filtration. After removal of all volatiles from the filtrate, the residue was washed with a large amount of hexane to extract the C–F bond activation product. The yellow residue, which remained after washing, was identified as the difluoride complex trans-[Ni(Mes2Im)2(F)2] (9) by elemental analysis, X-ray diffraction and 1H, 19F{1H} and 13C{1H} NMR spectroscopy (see ESI†). Most significantly, the fluoride resonance, detected as a singlet at −560 ppm in the 19F{1H} NMR spectrum, is shifted ca. 200 ppm to higher field compared to those of the mono-fluoride complexes 2–7 (−333 ppm to −362 ppm, vide supra). A similar high-field shifted fluoride resonance was also observed for the phosphine-stabilized platinum complex [Pt(PiPr3)2(F)2] (−455.9 ppm) compared to [Pt(PPh3)2(F)(C6H5)] (−107.6 ppm).13 Crystals of 9 suitable for X-ray diffraction (Fig. 6, Table 1; see also ESI, Table S2 and Fig. S38†) were obtained after storing a saturated solution of the complex at room temperature in C6D6. Crystallographic analysis revealed a square planar coordination environment about the NiII center with a trans-arrangement of NHC and fluoride ligands.
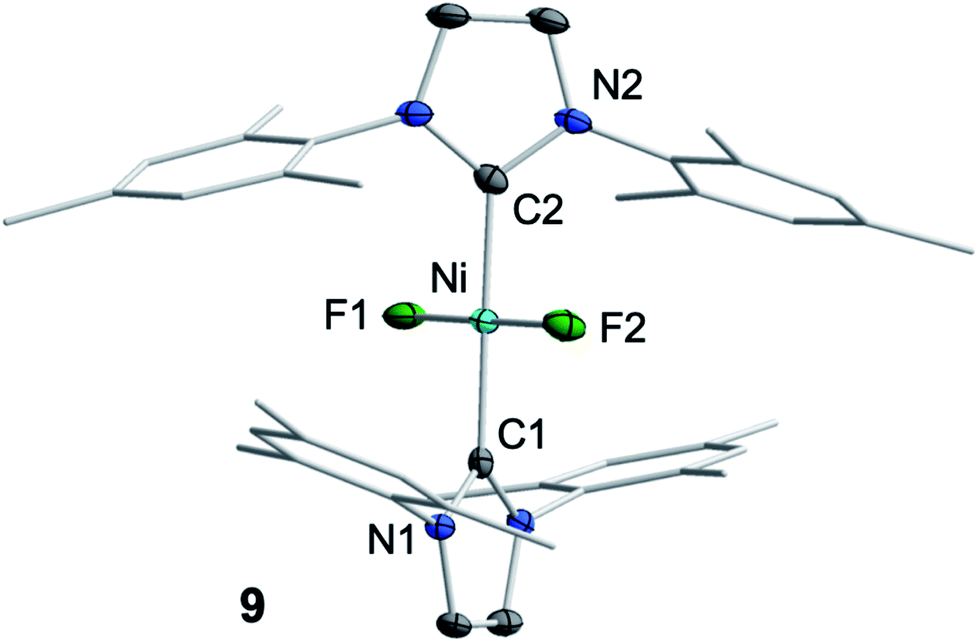 | ||
| Fig. 6 Molecular structure of trans-[Ni(Mes2Im)2(F)2] (9) in the solid state (ellipsoids drawn at the 50% probability level). Hydrogens atoms are omitted for clarity. | ||
An independent sample of complex 9 was synthesized in 38% yield by fluorination of [Ni(Mes2Im)2(I)2] (10) using an excess (2.5 equiv.) of silver(I) fluoride in CH2Cl2 at 0 °C (Scheme 5). Complex 10 was synthesized by reaction of 1 with I2, isolated in 80% yield and characterized by elemental analysis, and 1H and 13C{1H} NMR spectroscopy (see ESI†). Interestingly, the resonance of the carbene carbon atoms is almost unaffected by substitution of the fluoride by the more electropositive iodide ligand, and was detected at 176.5 ppm (cf. [Ni(Mes2Im)2(F)2] (9): 174.6 ppm).
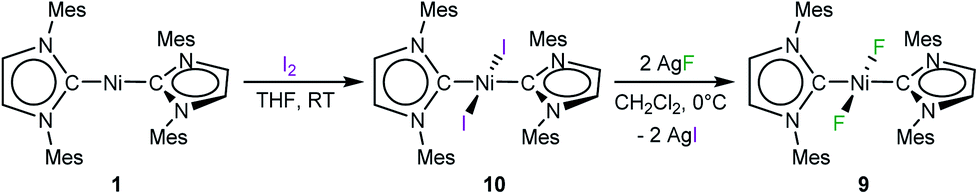 | ||
| Scheme 5 Synthesis of [Ni(Mes2Im)2(I)2] (10) and [Ni(Mes2Im)2(F)2] (9). | ||
Thus, [Ni(Mes2Im)2(F)2] (9) was clearly identified as one of the side products of the reaction of 1 with C6F6. This complex is formed in low yield (17%) but in an amount similar to that of the insertion product trans-[Ni(Mes2Im)2(F)(C6F5)] (3). The amounts of complexes 9 and 3 total ca. 40% when the reaction of 1 with C6F6 is performed at room temperature, and thus the majority of the products formed in this reaction is still unaccounted for.
Storing the concentrated hexane mother liquor of the extract from the isolation of 9 (vide supra) for 3 days at −30 °C led to crystallization of the remaining C–F bond insertion product trans-[Ni(Mes2Im)2(F)(C6F5)] (3) and a novel nickel(I) complex trans-[NiI(Mes2Im)2(C6F5)] (11) as yellow (3) and orange (11) crystals, respectively, which were manually separated in a glovebox (see ESI, Fig. S8†). The paramagnetic compound 11 was characterized by elemental analysis, EPR spectroscopy and X-ray diffraction. Determination of the room-temperature magnetic moment of 11 in solution (Evans method) gave a μeff value of 1.80 μB, which is consistent with the presence of one unpaired electron. The molecular structure (Fig. 7, top, Table 1; see also ESI, Table S2 and Fig. S39†) and the EPR spectrum (Fig. 7, bottom) of 11 confirm that this complex is a three-coordinate nickel(I) radical. Simulation of the EPR spectrum of 11 gave a g tensor of gxx = 2.04, gyy = 2.16 and gzz = 2.31, which was also observed in the EPR spectrum of the crude reaction mixture of 1 and C6F6 (Fig. 2). With the experimentally obtained g tensors and the molecular structure of the radical species 11 in hand, we carried out computational studies of the electronic properties of complex 11 and a likely radical counterpart from the reaction of 1 and C6F6, [NiI(Mes2Im)2(F)] (12) (Fig. 8). Both complexes 11 and 12 would be the result of a one-electron oxidative addition reaction of two equiv. of 1 with one equiv. C6F6 (Scheme 6).
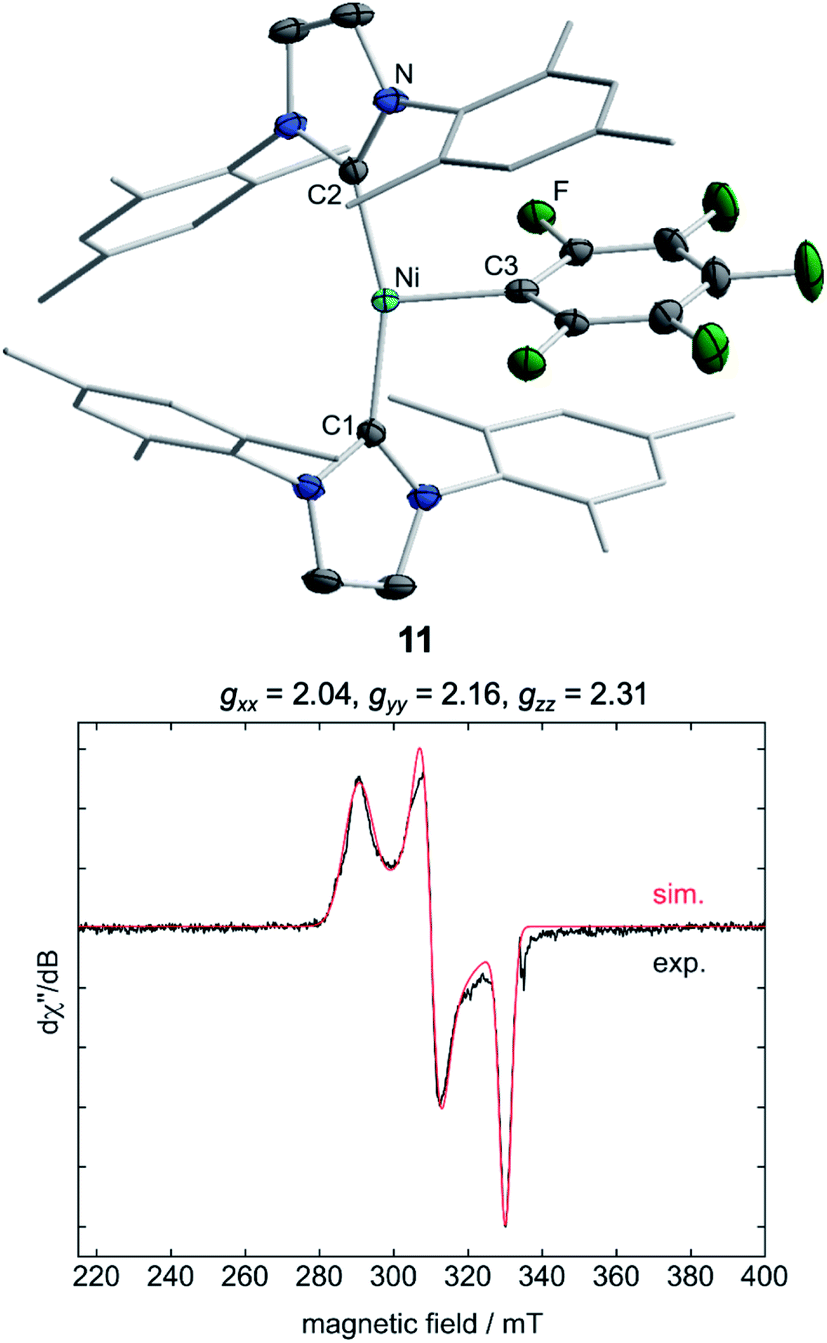 | ||
| Fig. 7 Molecular structure of trans-[NiI(Mes2Im)2(C6F5)] (11) (top) in the solid state (ellipsoids drawn at the 50% probability level) and EPR spectrum at −203 °C of the isolated compound 11 (bottom). Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 8 Spin density plots for trans-[NiI(Mes2Im)2(C6F5)] (11) (top) and trans-[NiI(Mes2Im)2(F)] (12) (bottom) (isovalue 0.0075 a0−3; hydrogen atoms are omitted for clarity). | ||
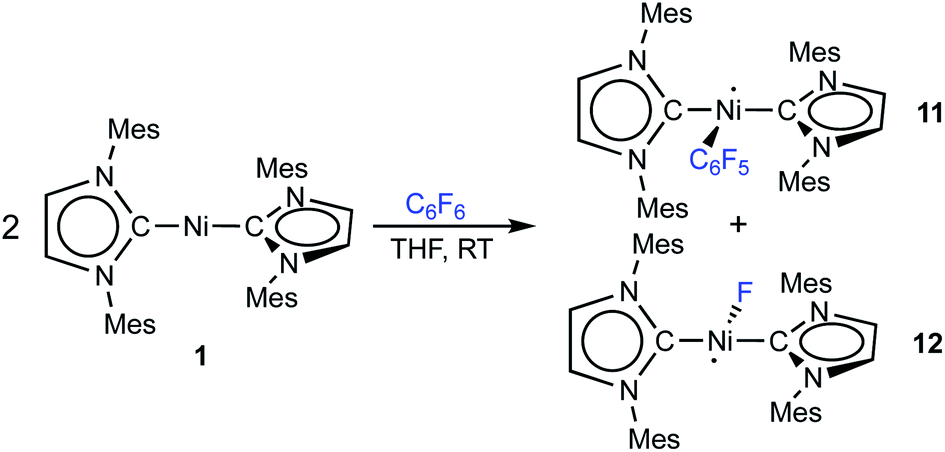 | ||
| Scheme 6 One-electron oxidative addition of C6F6 to [Ni(Mes2Im)2] (1) to yield the metal radicals trans-[NiI(Mes2Im)2(C6F5)] (11) and trans-[NiI(Mes2Im)2(F)] (12). | ||
Molecular geometries, electronic structures and EPR parameters (g tensors) were thus calculated for the metal radicals trans-[NiI(Mes2Im)2(C6F5)] (11) and trans-[NiI(Mes2Im)2(F)] (12) (Fig. 8) in order to connect the experimentally observed EPR spectra from the reaction mixture of 1 and C6F6 (Fig. 2), the EPR spectra of the isolated compound 11, and the corresponding isotropic g tensor components with the assigned structure of 11 (Fig. 7).
According to DFT calculations, complexes 11 and 12 are C2-symmetric doublet ground state species. The spin density is located at the metal center and the unpaired electron resides in an s/dz2-type orbital, yielding 2A electronic ground states (Fig. 8). Calculated and experimental g tensor components are in good agreement for species 11, with a maximum difference of 0.03 in gzz. With the largest deviation being 0.08 for 12, the agreement is still reasonable (Table 3).
| Compound | DFT/Expa | g tensor componentsb | ||
|---|---|---|---|---|
| g xx | g yy | g zz | ||
| a The experimental g-tensor components are reorganized in ascending order from gxx to gzz. b EPR parameter have been calculated using DFT. The calculated values are rounded to match the number of digits of the experimental values. c Exp. (isol.): see Fig. 7 (bottom). d Exp. (react. mix.): see Fig. 2I and II (I corresponds to compound 12; II corresponds to compound 11). | ||||
| 11 | Exp. (isol.)c | 2.04 | 2.16 | 2.31 |
| Exp. (react. mix.)d | 2.04 | 2.17 | 2.32 | |
| DFT | 2.06 | 2.17 | 2.29 | |
| 12 | Exp. (react. mix.)d | 1.93 | 2.46 | 2.64 |
| DFT | 2.01 | 2.42 | 2.57 | |
To provide further evidence for the existence of trans-[NiI(Mes2Im)2(C6F5)] (11) and trans-[NiI(Mes2Im)2(F)] (12), we attempted to synthesize these complexes independently. The reaction of [Ni(Mes2Im)2][BF4] (8) with CsF led to a mixture of two complexes, which we were not able to separate. One of them was identified via19F{1H} NMR spectroscopy as trans-[Ni(Mes2Im)2(F)2] (9) (19F{1H} NMR resonance at −560 ppm), and the resulting mixture reveals an EPR resonance with g tensors (gxx = 2.05, gyy = 2.42, gzz = 2.61) which are close to the g-tensors calculated for trans-[NiI(Mes2Im)2(F)] (12). We are thus confident that the second metal radical obtained in the reaction mixture is the monofluoride complex trans-[NiI(Mes2Im)2(F)] (12).
The complex trans-[NiI(Mes2Im)2(C6F5)] (11) as well as related trans-[NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) and trans-[NiI(Mes2Im)2(2,3,5-C6F3H2)] (14) can be synthesized from the reaction of trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = C6F53, 2,3,5,6-C6F4H 5, 2,3,5-C6F3H26) with PhSiH3 (Scheme 7, see also ESI Fig. S9 and S10†).14
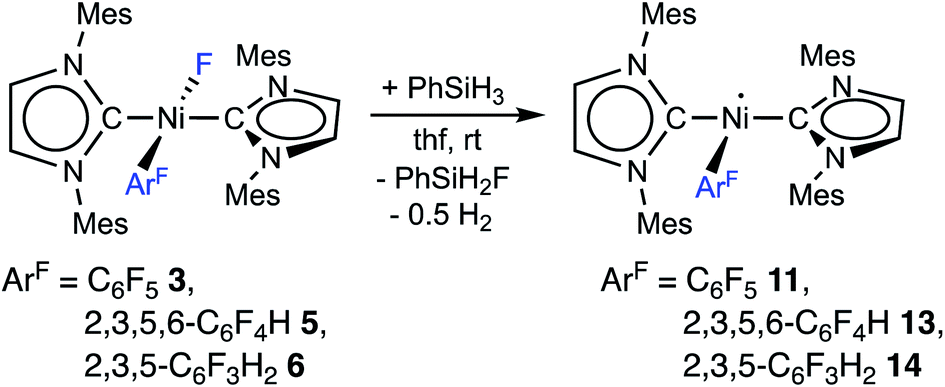 | ||
| Scheme 7 Synthesis of the metal radicals [NiI(Mes2Im)2(C6F5)] (11), [NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) and [NiI(Mes2Im)2(2,3,5-C6F3H3)] (14). | ||
The metal radicals were characterized by elemental analysis, IR and EPR spectroscopy as well as single-crystal X-ray diffraction. All compounds are stable in the solid state as well as in solution for several days. If the reactions are performed in an NMR tube and followed by 1H and 19F{1H} NMR spectroscopy (see ESI; Fig. S9 and S10†), the resonances for the Mes2Im, pentafluorophenyl and fluoride ligands vanish, indicating the formation of a paramagnetic species. For complexes of the type trans-[Ni(NHC)2(H)(ArF)], we expect hydride resonances in the region of ca. −13 ppm in the 1H NMR spectrum,9b,d and a strong absorption in the IR spectrum in the region between 1600 and 2200 cm−115 (we expect the Ni–H stretch to be at ca. 1850 cm−1 based on DFT calculations). However, such signals were absent for 11, 13 and 14. Thus, although complexes of the type trans-[NiI(Mes2Im)2(ArF)] cannot easily be distinguished from the corresponding hydride complexes trans-[NiI(Mes2Im)2(H)(ArF)] by X-ray diffraction (see below), we are confident that 11, 13 and 14 are the metal radicals. Crystals of trans-[NiI(Mes2Im)2(C6F5)] (11), trans-[NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) and trans-[NiI(Mes2Im)2(2,3,5-C6F3H2)] (14) suitable for X-ray diffraction (Fig. 9, Table 1; see also ESI Table S2 and Fig. S39–S41†) were obtained by storing saturated solutions of these compounds either in pentane or hexane at −30 °C. Complexes 11–13 adopt a distorted T-shaped structure, in which the NHC ligands occupy mutually trans positions. Due to the absence of the fluoride ligand, 11, 13 and 14 exhibit shortened Ni–C distances to the fluoroaryl ligand and reduced C1–Ni–C2 angles compared to nickel(II) complexes 3, 4, 5 and 6, which is also a further indication of the absence of a metal hydride. The data is in line with the data observed for [NiI(PiPr3)2(C6F5)] reported by Johnson and co-workers previously (Table 1, see also ESI Table S2†).16 EPR spectra of compounds 11, 13 and 14 were recorded in frozen thf solutions and reveal similar g tensors for the complexes, which are in good agreement with the calculated parameters (see ESI, Fig. S11–S13 and Table S4†).
 | ||
| Fig. 9 Molecular structures of trans-[NiI(Mes2Im)2(C6F5)] (11) (top left), trans-[NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) (top right) and trans-[NiI(Mes2Im)2(2,3,5-C6F3H2)] (14) (bottom) in the solid state (ellipsoids drawn at the 50% probability level). Hydrogens atoms (with exception of the protons at fluoroarene rings) are omitted for clarity. Because of disorder of the fluoroaryl ligand of 14, the ligand is represented by a ball and stick model in two different colors for clarity. | ||
Thus, the reaction of 1 with C6F6 affords the insertion product trans-[Ni(Mes2Im)2(F)(C6F5)] (3) in approximately 20% isolated yield, the difluoride complex trans-[Ni(Mes2Im)2(F)2] (9) in approximately 17% isolated yield, the three-coordinate nickel(I) metal radicals trans-[NiI(Mes2Im)2(C6F5)] (11) (isolated yield: 10%), trans-[NiI(Mes2Im)2(F)] (12) (not isolated), and a small amount of a decomposition product, i.e., a dark green precipitate which was not characterized. Trans-[NiI(Mes2Im)2(F)] (12) was not isolated; it was only observed in the EPR spectra after 5 s at −78 °C, and the signals vanish after about 10 s during the course of the reaction. Further investigation of the hexane mother liquor of the reaction of 1 and C6F6 revealed that the bis(aryl) nickel(II) complex [Ni(Mes2Im)2(C6F5)2] (15) remains in solution and was identified in the reaction mixture by 19F{1H} NMR spectroscopy. The radical species 11 and 12 were identified by EPR spectroscopy in a frozen thf solution at −78 °C (Fig. 2). The diamagnetic products trans-[Ni(Mes2Im)2(F)(C6F5)] (3), trans-[Ni(Mes2Im)2(F)2] (9), and trans-[Ni(Mes2Im)2(C6F5)2] (15) were identified by NMR spectroscopy (see Fig. S14 of the ESI†).
To expand our study to less fluorinated systems, we reacted 1 with pentafluorobenzene. After 48 h at room temperature, the 19F{1H} and 19F NMR spectra recorded in C6D6 reveal the formation of the C–F bond activation product trans-[Ni(Mes2Im)2(F)(C6F4H)] (5), the nickel difluoride complex [Ni(Mes2Im)2(F)2] (9), and the corresponding bis(aryl) nickel(II) complex [Ni(Mes2Im)2(C6F4H)2] (see ESI, Fig. S15†). Furthermore, an EPR spectrum of the frozen reaction mixture of 1 with pentafluorobenzene in thf recorded after 5 s at −78 °C (see ESI, Fig. S16†) revealed resonances for three different products, one of which is in accordance with trans-[NiI(Mes2Im)2(F)] (12) and another has the same g tensor as observed for isolated [NiI(Mes2Im)2(C6F4H)] (13). Thus, the reaction of 1 with C6F5H also follows a radical reaction mechanism akin to the reaction of 1 with C6F6 below.
Mechanistic investigations
Experimental investigations and DFT studies reported previously9a for the reaction of [Ni2(iPr2Im)4(μ-(η2:η2)-COD)] and [Ni(iPr2Im)2(η2-C2H4)], used as source of [Ni(iPr2Im)2] (1ipr), with C6F6 suggested a concerted mechanism for the insertion of 1ipr into the C–F bond, and no indications for radical reactivity were obtained. As presented above, however, paramagnetic complexes clearly emerge in the reaction of 1 and C6F6. To obtain further insight, we performed a quantum-chemical investigation (COSMO(THF)-PBE0-D/def2-TZVP, for details see ESI†)17 on the reaction pathways of C6F6 with [Ni(Mes2Im)2] (1) and with the sterically less encumbered [Ni(iPr2Im)2] (1ipr).C–F bond activation in the latter reaction commences with the formation of a rather stable 16-electron η2 adduct between 1ipr and C6F6 (I1, Scheme 8; see ESI, Fig. S17†). The DFT-optimized geometry of I1 is in good agreement with the structure of the closely related complex [Ni(iPr2Im)2(η2-C10F8)].9a Three distinct reaction pathways are then possible. First, direct oxidative addition of the C–F bond to the nickel atom proceeds through TS1 to yield the trans product 3ipr with an effective activation barrier of Δ‡G = 23 kcal mol−1 relative to I1 (see ESI, Fig. S18†). Alternative formation of the corresponding cis-[Ni(iPr2Im)2(F)(C6F5)] (I2) and subsequent isomerization is kinetically disfavored (Δ‡Geff = 27 kcal mol−1, see ESI Fig. S19 and S20†), as is dissociation of an NHC ligand (ΔG298 = 28 kcal mol−1, see Fig. S32†).
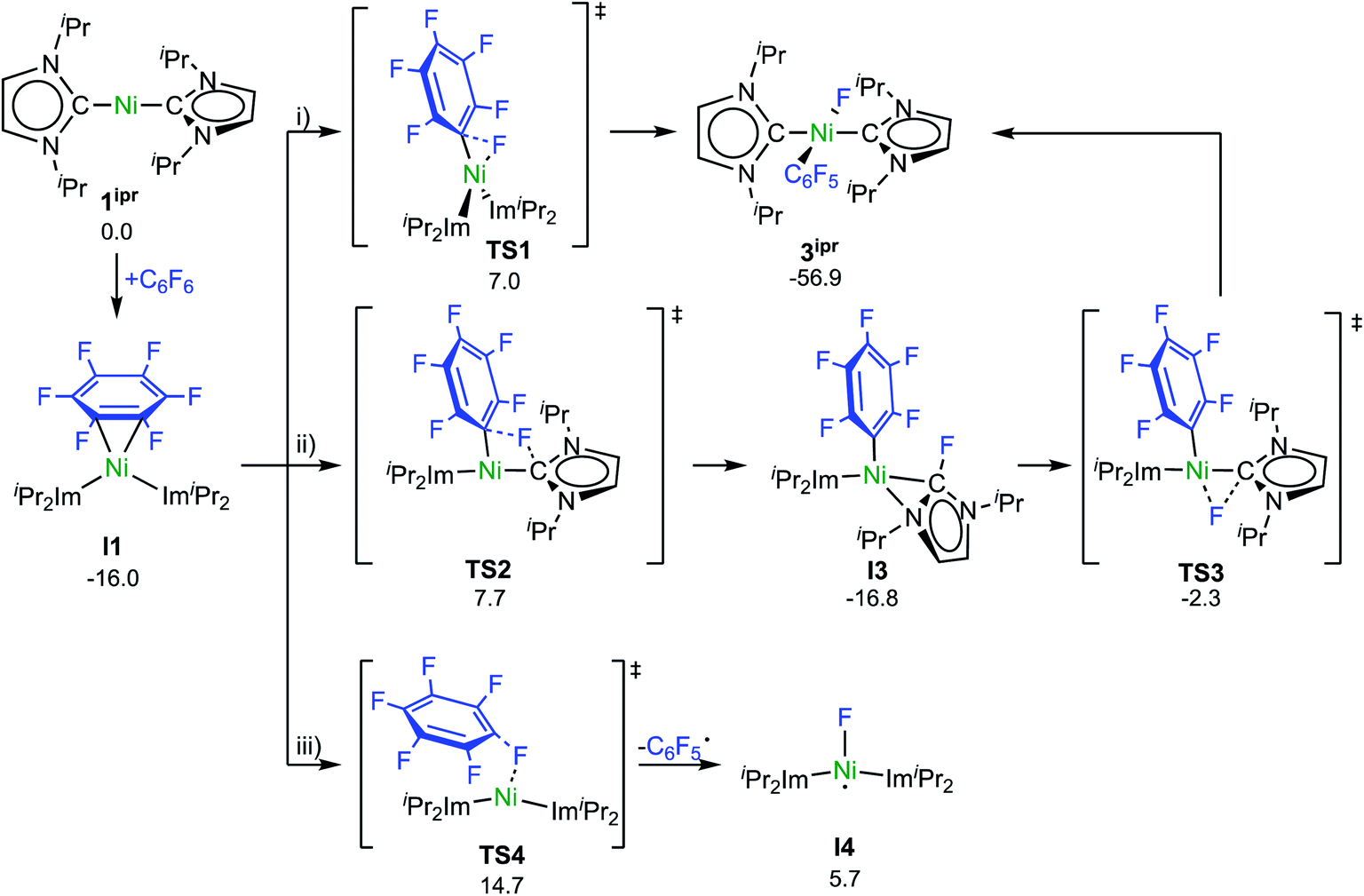 | ||
| Scheme 8 Calculated pathways for the C–F bond activation of C6F6 with 1ipr (ΔG298 in kcal mol−1). | ||
Second, NHC ligand cooperativity (see ESI; Fig. S21 and S23†) opens a kinetically competitive pathway to the trans-product 3ipr, that is, addition of the C–F bond across the Ni–CNHC bond through TS2 to yield intermediate I3, in which coordination of the fluorinated NHC–F ligand to the nickel atom involves a bridging C–N bonding interaction. In TS2, the Caryl–F bond of 1.93 Å is strongly elongated compared to C6F6 (Caryl–F bond: 1.32 Å) and TS1 (Caryl–F bond: 1.77 Å), while NHC–F bond formation is hardly visible (C/F distance: 2.40 Å). From I3, fluoride migration onto the nickel ion (TS3, with a low barrier of Δ‡G = 15 kcal mol−1) leads to 3ipr with an overall barrier of Δ‡Geff = 24 kcal mol−1. Third, homolytic C–F bond cleavage involves an effective barrier of Δ‡Geff = 31 kcal mol−1 (TS4) and, hence, radical abstraction is kinetically disfavored here (see ESI, Fig. S22†).
C–F bond activation with the sterically more congested Mes–NHC complex 1 shows marked differences. Formation of the η2-C6F6 adduct I5 (see ESI, Fig. S24†) is now endergonic by 12 kcal mol−1, and consecutive oxidative C–F bond addition viaTS5 (Δ‡Geff = 21 kcal mol−1, see ESI; Fig. S25†) leads to the cis-product I6. We attribute the endergonicity of the η2-C6F6 adduct formation (I5, ΔΔG = 28 kcal mol−1 compared to the exergonic formation of I1) mainly to the increased steric demand of the mesityl groups. A trajectory to the trans-product is precluded by the steric demand of the mesityl substituents. NHC dissociation to yield [Ni(Mes2Im)(η6-C6F6)] and subsequent insertion into the C–F bond is associated with a large barrier (Δ‡Geff = 34 kcal mol−1, see ESI; Fig. S32 and S33†) and is irrelevant here. Note that an alternative adduct formation stabilized by π-stacking interactions between C6F6 and one of the NHC mesityl substituents,18 such as I7 (see ESI; Fig. S26†), is also endergonic and less favorable than I5. Furthermore, a “concerted” NHC-assisted process as in the iPr system does not exist. We found a multi-step sequence for the mesityl system instead (Scheme 9 and ESI; Fig. S27†), commencing with heterolytic C–F bond cleavage in I5, which exhibits a partially reduced C6F6 fragment (qNPA(C6F6) = −0.69). The fluoride anion expelled from the nickel coordination sphere is loosely held within the cleft formed by the mesityl substituents in I8. A similar stabilizing association of a fluoride anion by the methyl groups of mesityl substituents has been reported by Macgregor et al. for the C–F bond activation step in hydrodefluorination reactions.19 Formation of the trans-product 3 from here involves binding to the carbene carbon atom and subsequent F-shift onto the Ni center. The overall path involves a low effective barrier of 16 kcal mol−1 (TS6).
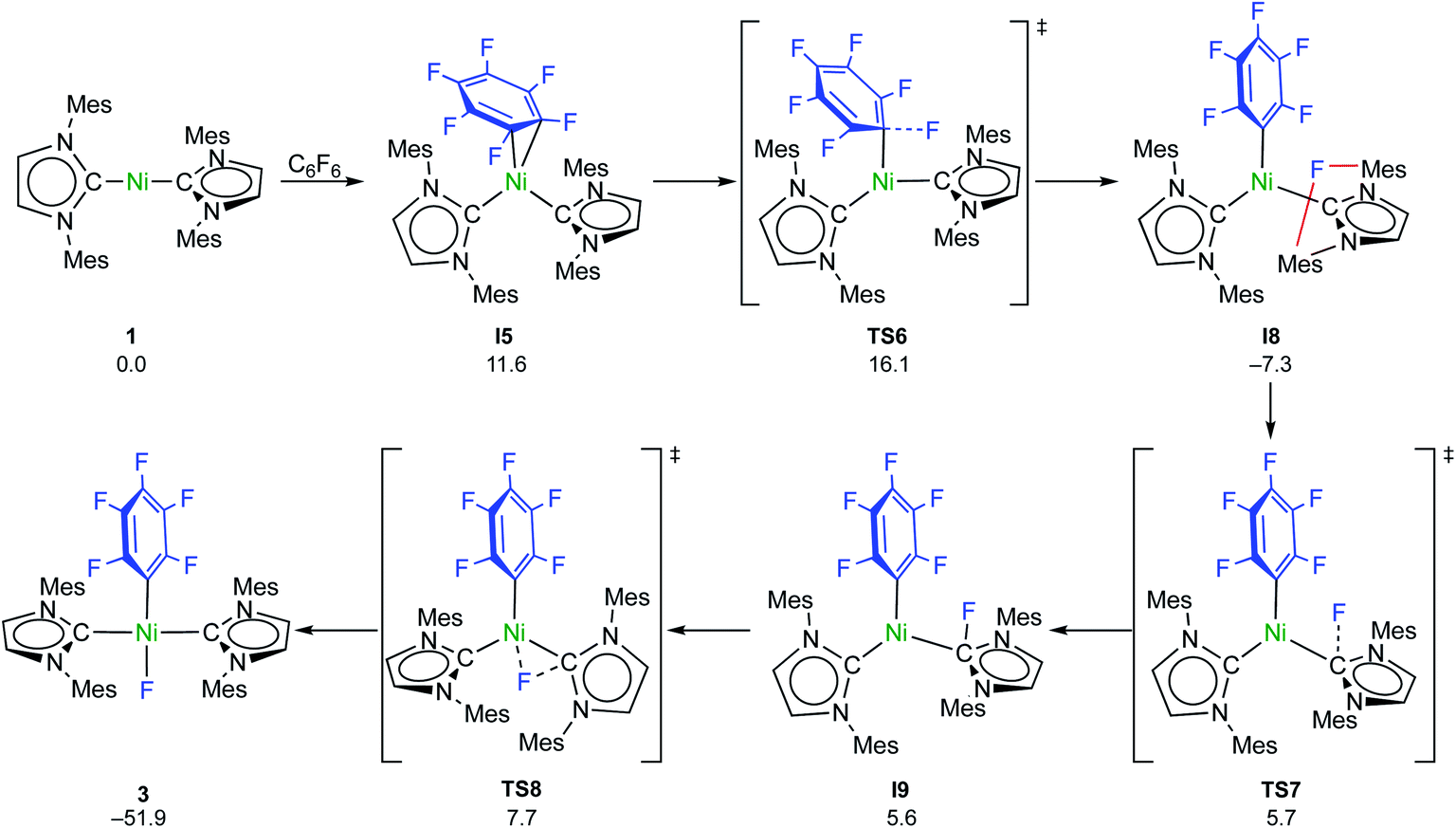 | ||
| Scheme 9 Calculated pathways for the heterolytic C–F bond cleavage of C6F6 by 1 and further reaction steps (ΔG298 in kcal mol−1). | ||
Fluorine radical abstraction to yield 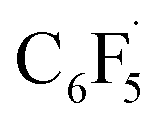 and radical complex 12viaTS9 is slightly endergonic and exhibits a barrier of 16 kcal mol−1 (Scheme 10 and ESI, Fig. S26 and S28†). Recombination of
and radical complex 12viaTS9 is slightly endergonic and exhibits a barrier of 16 kcal mol−1 (Scheme 10 and ESI, Fig. S26 and S28†). Recombination of 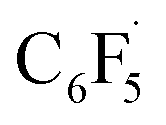 and 12 to 3 then provides a large thermodynamic driving force. Alternative addition of
and 12 to 3 then provides a large thermodynamic driving force. Alternative addition of 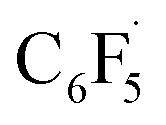 to the initial complex 1 to yield radical species 11 is also a highly exergonic process (−69.5 kcal mol−1), as well as addition of a second equivalent of
to the initial complex 1 to yield radical species 11 is also a highly exergonic process (−69.5 kcal mol−1), as well as addition of a second equivalent of 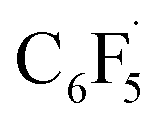 to yield 15 (−108.0 kcal mol−1). Endergonic formation of difluoride complex 9 from 12 and another equiv. of C6F6, can be compensated by consumption of
to yield 15 (−108.0 kcal mol−1). Endergonic formation of difluoride complex 9 from 12 and another equiv. of C6F6, can be compensated by consumption of 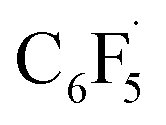 ; however, a second fluorine abstraction step is prevented by the high kinetic barrier of 37 kcal mol−1viaTS10 (see ESI; Fig. S29†). The mechanism for the formation of 9 remains obscure to us thus far. We compute the ligand exchange reaction 3 + 3 → 9 + 15 to be exceedingly endergonic (25.7 kcal mol−1), and also the disproportionation reactions of radicals 11 and 12 yielding 1 + 15 (31.0 kcal mol−1) or 1 + 9, (14.9 kcal mol−1), are unlikely to contribute to the formation of 9 (see ESI, Fig. S30†). A dinuclear complex [{Ni(Mes2Im)2}2(μ-(η2:η2)-C6F6)], which would be an intermediate for an one-electron oxidative addition, is too high in energy to be considered (35 kcal mol−1, see ESI; Fig. S31†). Hence both, the radical pathway and the NHC-assisted multistep pathway represent kinetically competitive C–F bond activation steps in the reaction with [Ni(Mes2Im)2] (1).
; however, a second fluorine abstraction step is prevented by the high kinetic barrier of 37 kcal mol−1viaTS10 (see ESI; Fig. S29†). The mechanism for the formation of 9 remains obscure to us thus far. We compute the ligand exchange reaction 3 + 3 → 9 + 15 to be exceedingly endergonic (25.7 kcal mol−1), and also the disproportionation reactions of radicals 11 and 12 yielding 1 + 15 (31.0 kcal mol−1) or 1 + 9, (14.9 kcal mol−1), are unlikely to contribute to the formation of 9 (see ESI, Fig. S30†). A dinuclear complex [{Ni(Mes2Im)2}2(μ-(η2:η2)-C6F6)], which would be an intermediate for an one-electron oxidative addition, is too high in energy to be considered (35 kcal mol−1, see ESI; Fig. S31†). Hence both, the radical pathway and the NHC-assisted multistep pathway represent kinetically competitive C–F bond activation steps in the reaction with [Ni(Mes2Im)2] (1).
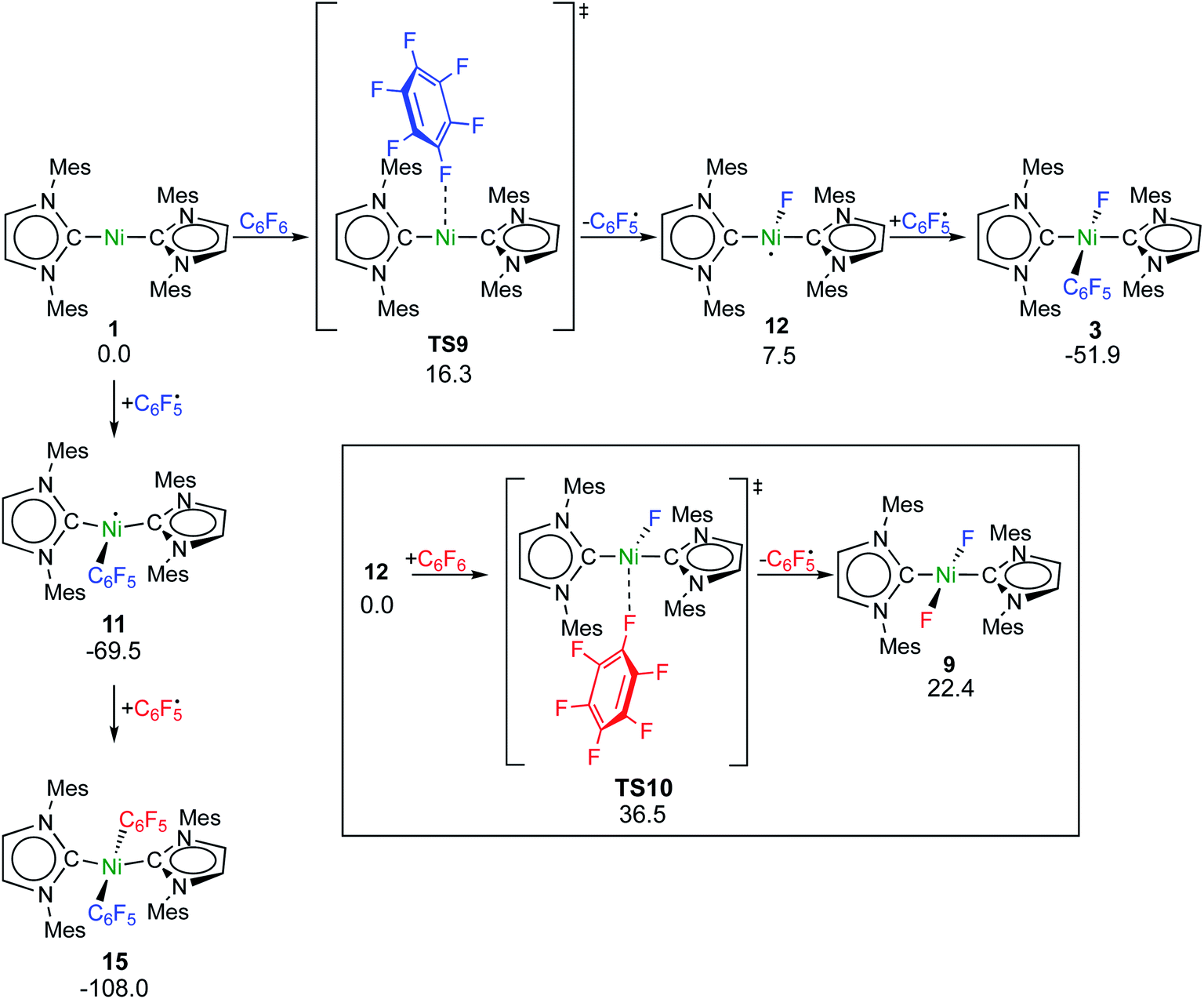 | ||
| Scheme 10 Calculated pathways for the homolytic C–F bond cleavage of C6F6 by 1 and further radical reaction steps (ΔG298 in kcal mol−1; energies of TS10 and 9 are given relative to 12 + C6F6). | ||
Discussion
It is now well established that nickel(0) complexes with phosphine, carbene, and even some nitrogen ligands undergo C–F oxidative addition with perfluoroarenes to yield complexes trans-[Ni(L)2(F)(C6F5)].1,9,20 Although the lack of clean kinetics for many of the C–F oxidative additions indicate complex mechanistic scenarios, there were strong indications that the conversion of C6F6 to the aryl fluoride complex follows the same type of mechanism as observed for typical C–H activation reactions of benzene. It has been demonstrated, for nickel NHC and phosphine complexes, that the first stage of C–F oxidative addition is the η2-coordination of the fluoroarene.1g,9a,21,22 The introduction of fluorine substituents on the arene results in a lower lying LUMO, which renders the fluorinated arene a better electron acceptor compared to H-arenes and makes the reaction of electron-poor C6F6 with an electron-rich, suitable nickel precursor more exothermic. The fluoroarene of [Ni(L)2(η2-C6F6)] is ene–diene distorted, and the arene fluoride substituents are bent out of the plane, as observed for I1 and I5. Subsequent C–F oxidative addition is strongly exothermic for trans-[Ni(iPr2Im)2(F)(C6F5)] (ΔG298 = −57 kcal mol−1) and trans-[Ni(Mes2Im)2(F)(C6F5)] (ΔG298 = −52 kcal mol−1). Computational studies reported previously9a,23 of the reaction pathways have supported the idea of concerted mechanisms involving a σ-complex as a three-center transition state between the C6F6 carbon and fluorine atoms and the transition metal atom. The transition state structures typically show limited elongation of the C–F bond and interaction of the electron-rich transition metal ion with the C–F σ* orbital leads to C–F bond breaking and formation of the M–C and M–F bond. We have demonstrated now for [Ni2(iPr2Im)4(μ-(η2:η2)-COD)] and the related [Ni(iPr2Im)2] (1ipr) synthon complexes that C–F bond activation of C6F6 occurs via both a concerted and an NHC-assisted pathway, as both are associated with very similar kinetic barriers of Δ‡Geff = 23 kcal mol−1 for the concerted and of Δ‡Geff = 24 kcal mol−1 for the NHC-assisted pathway. This situation will probably change if other substrates with other leaving groups, such as partially fluorinated arenes, fluoropyridines or other aryl halides, are involved in the reaction with the nickel complex; however, our calculations demonstrate that both reaction paths are feasible, at least for fluoroarenes.The direction of the concerted oxidative addition in TS1 to give the trans product is rather unusual.24 For the oxidative addition of A–B to d10-ML2 the important orbital interactions of the transition state are those between the filled σ(A–B) orbital and the empty dσ-type orbital of the metal, leading to electron donation from A–B to the metal center, and a second interaction between the filled dπ-orbital of the metal and the σ*(A–B), leading to electron transfer from the metal to the ligand. Strong back-donation will lead to fission of the A–B bond. This back-donation is strongest if A–B lies within the bent-d10-ML2 plane and the σ*(A–B) orbital can interact with the dx2–y2 orbital (actually a d–p hybrid orbital), which is pointing at the two ligands L.24
However, it was also shown previously that concerted oxidative addition reactions may take place through a nonplanar transition state structure even for non-polar substrates with dihedral angle between ML2 and M(A–B) planes larger than 70°.25 It was demonstrated that this nonplanar transition state is connected to the planar product on the singlet surface and suggested that steric rather than electronic factors are responsible for the nonplanar transition state structure. Martin et al.,25c for example, calculated at the B3LYP/LanL2DZ-level of theory a nonplanar transition state for the oxidative addition of C6H5–I to [Pd(dmpe)] (dmpe = bis{dimethylphosphino}ethane), in which the P–Pd–P and C–Pd–I planes are almost perpendicular to one another. Another example was provided by Jones et al.25d for the oxidative addition of the C–CN σ-bond of organonitriles to the low-valent nickel complex [Ni(dmpe)]. The C–C–N plane of the transition state (calculated at the B3LYP/6-31G(d,p)-level of theory), which leads to C–CN bond cleavage, is rotated by 38° relative to the P–Ni–P plane.
The η2(C,C)-bonded complex [Ni(iPr2Im)2(η2-C6F6)] (I1) is also the crucial reaction intermediate for the NHC-assisted pathway. The key step here is the addition of the C–F bond across the Ni–CNHC bond and, thus, the unoccupied NHC pπ-orbital plays a central role for this pathway as intramolecular fluoride acceptor. Fluoride transfer from the arene to the NHC leads to a η2-fluoro-imidazolyl intermediate (I3; Scheme 8) which rearranges with a second fluoride transfer step from the NHC to the nickel atom to give trans-[Ni(iPr2Im)2(F)(C6F5)] (3ipr).
A phosphine-assisted process has been proposed before for the C–F bond activation of pentafluoropyridine with [Ni(PR3)2], based on the experimental observation of an unusual selectivity for the insertion into the 2-position of C5NF5 and on DFT calculations.26 However, another study performed on the reaction of pentafluoropyridine with [Ni(PEt3)2] suggested that pathways other than a concerted oxidative addition or a phosphine-assisted pathway account for the unusual selectivity.27 The detailed experimental analysis of the reactivity of a [Ni(PEt3)2] precursor with perfluoropyridine demonstrated the formation of a mononuclear adduct [Ni(PEt3)2(η2-C5F5N)], of dinuclear adducts [{Ni(PEt3)2}2)(μ-(η2:η2)-C5F5N)], some of which exhibit C–F bond activation, and a nickel(I) radical species [Ni(PEt3)2(2-C5F4N)]. Other heteroatom-assisted C–F bond activation processes have also been proposed for other metals mainly including boryl or silyl moieties.28
Despite precedent in the oxidative addition of other aryl carbon–halide bonds to nickel,29,30 there is only little experimental evidence for the involvement of radicals in C–F bond activation processes. It is known that some polyfluoro pyridines react with [Ni(PR3)2] to yield EPR-active complexes as likely intermediates,16,27 and some studies on C–F bond activation have shown unusual products with highly-fluorinated arenes that may be indicative of radical pathways.16,22,31 However, the clear identification of radical intermediates has not been possible so far and alternate mechanisms cannot be ruled out. Although DFT calculations were performed to examine the traditional concerted oxidative addition and phosphine-assisted pathways for C–F bond activation, radical pathways involving Ni(I) intermediates were rarely considered computationally.
Thus, the reaction of 1 with different fluoroarenes leads to nickel insertion into the C–F bond to give the nickel fluoroaryl fluoride complexes trans-[Ni(Mes2Im)2(F)(ArF)], but EPR spectroscopy also provided evidence that at least three paramagnetic species are intermediates or products of the reaction of C6F6 with 1. We provide evidence that simple electron transfer from [Ni(Mes2Im)2] (1) to C6F6, often considered as the first step in radical oxidative additions at nickel,29 is unlikely to occur. The redox potentials are not in line with intermolecular electron transfer to yield [Ni(Mes2Im)2]+ and C6F6− and the EPR resonance of [Ni(Mes2Im)2]+, which has been established for the authentic complex [Ni(Mes2Im)2][BF4] (8), was not detected in the reaction mixture. Furthermore, many diamagnetic and radical products of the reaction of [Ni(Mes2Im)2] (1) to C6F6 were identified, namely the insertion product trans-[Ni(Mes2Im)2(F)(C6F5)] (3), the difluoride complex trans-[Ni(Mes2Im)2(F)2] (9), the bis(aryl) complex trans-[NiII(Mes2Im)2(C6F5)2] (15), the nickel(I) complex trans-[NiI(Mes2Im)2(C6F5)] (11), and the metal-centered radical trans-[NiI(Mes2Im)2(F)] (12). DFT calculations performed on the reaction of [Ni(Mes2Im)2] (1) with C6F6 explain the occurrence of the radical species observed. Both an NHC-assisted and a radical process are kinetically equally favored routes for this reaction. Fluorine radical abstraction from C6F6 by 1 is associated with a barrier of only 16 kcal mol−1 and subsequent radical recombination steps provide the thermodynamic driving force required.
Matsubara et al. and Louie et al. reported the clean isolation of T-shaped three-coordinate radical species [NiI(NHC)2(X)] (X = Cl, Br, I; NHC = Mes2Im, Dipp2Im) from the reaction of [Ni(NHC)2] with aryl halides.30a,b,d We have demonstrated earlier that [Ni2(iPr2Im)4(μ-(η2:η2)-COD)], a source of [Ni(iPr2Im)2] (1ipr), reacts cleanly with aryl chlorides to yield the nickel(II) complexes trans-[Ni(NHC)2(Cl)(Ar)].32 Our calculations show now that a trajectory to the trans-product by a concerted oxidative addition is precluded for [Ni(Mes2Im)2] (1) (and most probably also for [Ni(Dipp2Im)2]) by the steric demand of the mesityl substituents. As a consequence, other pathways such as electron transfer and radical abstraction must occur which are responsible for a limited or altered reactivity of complex [Ni(Mes2Im)2] (1) and analogues containing even more bulky N-aryl substituents compared to complexes of sterically less demanding NHCs. However, fluoride abstraction occurs for the reaction of 1 and C6F6 even at −78 °C to yield trans-[NiI(Mes2Im)2(C6F5)] (11) and trans-[NiI(Mes2Im)2(F)] (12). The latter is, in contrast to the complexes of the heavier homologues, very reactive and has defied thus far isolation. In turn, the complexes trans-[NiI(Mes2Im)2(C6F5)] (11), [NiI(Mes2Im)2(2,3,5,6-C6F4H)] (12) and [NiI(Mes2Im)2(2,3,5-C6F3H2)] (13) seem to be much more stable than [NiI(NHC)2(C6H5)] and have been synthesized and characterized. The increased stability of [NiI(Mes2Im)2(2,3,5,6-C6F4H)] (12) can be explained by the increased Ni–CAr bond strength of the fluoroaryl ligand with respect to C6H5.33
Nelson and Maseras34 reported computational investigations of the reaction of [Ni(NHC)2] complexes with aryl halides Ph–X (X = Cl, Br, I) and demonstrated that steric effects determine the mechanism. Small NHC ligands (NHC = Me2ImMe) favor concerted oxidative addition via a η2(C,C) π-coordinated intermediate leading to trans-[NiII(NHC)2(X)(Ar)] complexes whereas larger NHC ligands (e.g. NHC = Mes2Im) lead to halide abstraction to form [NiI(X)(NHC)2] and a phenyl radical. We confirm here, by means of experiment and theory, that [Ni(NHC)2] complexes of sterically less demanding NHCs favor the reaction with fluoroarenes via a concerted oxidative addition proceeding through an η2(C,C) intermediate, and that for the bulkier NHC Mes2Im, C–F bond activation is achieved more easily by fluorine atom abstraction. However, for both mechanisms, we found an NHC-assisted pathway which is competitive, that accounts for the formation of diamagnetic products by a C–F bond activation step across the Ni–CNHC bond. NHC-assisted pathways play an important role for complexes of both sterically demanding and less bulky NHC ligand. We believe that this dual reaction pathway concept, including NHC-assisted reaction pathways, should be of general importance and widely applicable for the reactivity of NHC transition metal complexes.
Conclusions
We present herein a detailed account of the C–F bond activation of polyfluoroaromatics, especially of C6F6 using the nickel(0) complex [Ni(Mes2Im)2] (1). The reaction of 1 with different fluoroarenes leads to insertion of nickel into the C–F bond of the fluoroarene to give the nickel(II) complexes trans-[Ni(Mes2Im)2(F)(ArF)] (ArF = 4-CF3-C6F42, C6F53, 2,3,5,6-C6F4N 4, 2,3,5,6-C6F4H 5, 2,3,5-C6F3H26, 3,5-C6F2H37) in good to fair yields with the exception of the formation of the pentafluorophenyl complex trans-[Ni(Mes2Im)2(F)(C6F5)] (3) (less than 20%). Whereas the C–F bond activation process of C6F6 using [Ni(iPr2Im)2] (1ipr) follows a concerted or NHC-assisted mechanism to give the insertion product via η2-coordinated intermediates, metal radical species were detected for the reaction of 1 with C6F6. EPR spectroscopy provided evidence that at least three paramagnetic products are intermediates or products of this reaction. The experiments reveal that simple electron transfer from [Ni(Mes2Im)2] (1) to C6F6 is unlikely to occur as (i) the redox potentials do not match for an electron transfer between [Ni(Mes2Im)2] (1) and C6F6 to give [Ni(Mes2Im)2]+ and C6F6−, and (ii) the EPR resonance for [Ni(Mes2Im)2]+, as established for the stable, isolated complex [Ni(Mes2Im)2][BF4] (8), was not detected in the reaction mixture. Several other byproducts were identified aside from the insertion product 3, namely the difluoride complex trans-[Ni(Mes2Im)2(F)2] (9), the bis(aryl) complex trans-[NiII(Mes2Im)2(C6F5)2] (15), the structurally-characterized nickel(I) complex trans-[NiI(Mes2Im)2(C6F5)] (11) and the metal radical trans-[NiI(Mes2Im)2(F)] (12). Complex 11 and related complexes [NiI(Mes2Im)2(2,3,5,6-C6F4H)] (13) and [NiI(Mes2Im)2(2,3,5-C6F3H2)] (14) were synthesized and characterized independently from the reaction of trans-[Ni(Mes2Im)2(F)(ArF)] with PhSiH3.DFT calculations were performed on the insertion of [Ni(iPr2Im)2] (1ipr) and [Ni(Mes2Im)2] (1) into the C–F bond of C6F6, which explain the formation of radical species for the reaction with [Ni(Mes2Im)2] (1). For [Ni(iPr2Im)2] (1ipr), the crucial reaction intermediate is an η2(C,C)-bonded complex [Ni(iPr2Im)2(η2-C6F6)], from which two favorable pathways with almost identical barriers, i.e., a concerted oxidative addition pathway and a NHC-assisted pathway, lead to the formation of trans-[Ni(iPr2Im)2(F)(C6F5)]. For [Ni(Mes2Im)2] (1), an NHC-assisted and a radical pathway were identified with similar kinetic barriers. Fluorine atom abstraction from C6F6 at [Ni(Mes2Im)2] (1) occurs via end-on attack of C6F6, while the key intermediate for the NHC-assisted pathway is the η2(C,C) intermediate [Ni(Mes2Im)2(η2-C6F6)]. The NHC-assisted pathway can be interpreted as heterolytic C–F bond cleavage to yield ionic intermediates trans-[Ni(Mes2Im)2(C6F5)]+F−, in which the fluoride anion is stabilized within the sphere of the trans-[Ni(Mes2Im)2(C6F5)]+ cation. Several fluoride transfer steps, i.e., migration to the NHC, NHC rotation, and fluoride transfer to the metal cation lead to the formation of trans-[Ni(Mes2Im)2(F)(C6F5)] (3).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
U. R., H. B. and T. B. M. thank the Julius-Maximilians-Universität Würzburg and U. R. the Deutsche Forschungsgemeinschaft (Ra720/12) for support. Quantum-chemical calculations were performed at the Center for Scientific Computing (CSC) Frankfurt on the Goethe-HLR computer cluster.References
- (a) N. M. Doherty and N. W. Hoffmann, Chem. Rev., 1991, 91, 553–573 CrossRef CAS; (b) W. R. Dolbier, J. Fluorine Chem., 2005, 126, 157–163 CrossRef CAS; (c) K. Uneyama, Organofluorine Chemistry, Wiley-Blackwell, Oxford, U.K., 2006 CrossRef; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (e) A. D. Sun and J. A. Love, Dalton Trans., 2010, 39, 10362–10374 RSC; (f) P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS; (g) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS; (h) S. A. Johnson, J. A. Hatnean and M. E. Doster, Prog. Inorg. Chem., 2011, 57, 255–352 Search PubMed; (i) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328–3348 CrossRef; (j) L. Keyes and J. A. Love, Aromatic C-F Activation: Converting Fluoroarenes to Useful Building Blocks, in C-H and C-X Bond Functionalization: Transition Metal Mediation, ed. X. Ribas, RSC, Cambridge, U.K., 2013, pp. 159–192 Search PubMed; (k) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS; (l) J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413–7428 CrossRef CAS; (m) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Acena, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS; (n) D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC; (o) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS.
- (a) K. Daasbjerg, J. Chem. Soc., Perkin Trans. 2, 1994, 1275–1277 RSC; (b) Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, FL, 2007 CrossRef; (c) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS; (d) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS.
- (a) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS; (b) Y.-M. Tian, X.-N. Guo, M. W. Kuntze-Fechner, I. Krummenacher, H. Braunschweig, U. Radius, A. Steffen and T. B. Marder, J. Am. Chem. Soc., 2018, 140, 17612–17623 CrossRef CAS.
- (a) J. W. J. Kennedy and D. G. Hall, Boronic Acids, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) Boronic acids: preparation, applications in organic synthesis and medicine, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed; (c) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS; (d) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS; (e) I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS; (f) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS; (g) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC; (h) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (i) S. K. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562–4565 CrossRef CAS; (j) S. K. Bose, A. Deissenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843–11847 CrossRef CAS; (k) J. Zhou, J. H. J. Berthel, M. W. Kuntze-Fechner, A. Friedrich, T. B. Marder and U. Radius, J. Org. Chem., 2016, 81, 5789–5794 CrossRef CAS; (l) Y. P. Budiman, A. Friedrich, U. Radius and T. B. Marder, ChemCatChem, 2019, 11, 5387–5396 CrossRef CAS; (m) Z. Liu, Y. P. Budiman, Y. Tian, A. Friedrich, M. Huang, S. A. Westcott, U. Radius and T. B. Marder, Chem.–Eur. J., 2020 DOI:10.1002/chem.202002888.
- (a) M. S. Cheung, F. K. Sheong, T. B. Marder and Z. Lin, Chem.–Eur. J., 2015, 21, 7480–7488 CrossRef CAS; (b) L. Kuehn, M. Stang, S. Würtemberger-Pietsch, A. Friedrich, H. Schneider, U. Radius and T. B. Marder, Faraday Discuss., 2019, 220, 350–363 RSC.
- (a) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS; (b) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985–2988 CrossRef CAS; (c) A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408–8411 CrossRef CAS; (d) K. Chen, S. Zhang, P. He and P. Li, Chem. Sci., 2016, 7, 3676–3680 RSC; (e) K. Chen, M. S. Cheung, Z. Lin and P. Li, Org. Chem. Front., 2016, 3, 875–879 RSC; (f) A. M. Mfuh, B. D. Schneider, W. Cruces and O. V. Larionov, Nat. Protoc., 2017, 12, 604–610 CrossRef CAS; (g) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS.
- C. Sieck, M. G. Tay, M. H. Thibault, R. M. Edkins, K. Costuas, J. F. Halet, A. S. Batsanov, M. Haehnel, K. Edkins, A. Lorbach, A. Steffen and T. B. Marder, Chem.–Eur. J., 2016, 22, 10523–10532 CrossRef CAS.
- (a) T. Schaub and U. Radius, Chem.–Eur. J., 2005, 11, 5024–5030 CrossRef CAS; (b) T. Schaub, M. Backes and U. Radius, Organometallics, 2006, 25, 4196–4206 CrossRef CAS; (c) P. Fischer, T. Linder and U. Radius, Z. Anorg. Allg. Chem., 2012, 638, 1491–1496 CrossRef CAS; (d) A. P. Prakasham and P. Ghosh, Inorg. Chim. Acta, 2015, 431, 61–100 CrossRef CAS; (e) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS.
- (a) T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius and A. Mix, J. Am. Chem. Soc., 2008, 130, 9304–9317 CrossRef CAS; (b) T. Schaub, M. Backes and U. Radius, Eur. J. Inorg. Chem., 2008, 2680–2690 CrossRef CAS; (c) T. Schaub, P. Fischer, T. Meins and U. Radius, Eur. J. Inorg. Chem., 2011, 3122–3126 CrossRef CAS; (d) P. Fischer, K. Götz, A. Eichhorn and U. Radius, Organometallics, 2012, 31, 1374–1383 CrossRef CAS; (e) U. S. D. Paul and U. Radius, Chem.–Eur. J., 2017, 23, 3993–4009 CrossRef CAS; (f) M. W. Kuntze-Fechner, C. Kerpen, D. Schmidt, M. Häring and U. Radius, Eur. J. Inorg. Chem., 2019, 1767–1775 CrossRef CAS.
- W. R. Hagen, Dalton Trans., 2006, 4415–4434 RSC.
- (a) R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. Lopez, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640–13643 CrossRef CAS; (b) M. M. Schwab, D. Himmel, S. Kacprzak, V. Radtke, D. Kratzert, P. Weis, M. Wernet, A. Peter, Z. Yassine, D. Schmitz, E. W. Scheidt, W. Scherer, S. Weber, W. Feuerstein, F. Breher, A. Higelin and I. Krossing, Chem.–Eur. J., 2018, 24, 918–927 CrossRef CAS.
- Molecular structures of the radical species discussed in this section were obtained at the RI-PBE-D/def2-SVP/W06 level of DFT. The PBE0-D functional and the pcSseg-2 basis set, which is specifically optimized for the calculation of magnetic properties, were employed to obtain EPR parameters (PBE0-D/pcSseg-2).
- (a) P. Nilsson, F. Plamper and O. F. Wendt, Organometallics, 2003, 22, 5235–5242 CrossRef CAS; (b) A. Yahav, I. Goldberg and A. Vigalok, Inorg. Chem., 2005, 44, 1547–1553 CrossRef CAS.
- (a) R. K. Marat and A. F. Janzen, Can. J. Chem., 1977, 55, 3845–3849 CrossRef CAS; (b) R. Damrauer, R. A. Simon and B. Kanner, Organometallics, 1988, 7, 1161–1164 CrossRef CAS; (c) A. Kunai, T. Sakurai, E. Toyoda and M. Ishikawa, Organometallics, 1996, 15, 2478–2482 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, CA, 2010, ch. 3.8 Search PubMed.
- J. A. Hatnean, M. Shoshani and S. A. Johnson, Inorg. Chim. Acta, 2014, 422, 86–94 CrossRef CAS.
- Optimized molecular structures and thermal/vibrational contributions to Gibbs free energies were obtained at the PBE0-D/def2-SVP level of DFT. The def2-TZVP basis set and the COSMO solvation model were employed to obtain improved relative energies (COSMO(THF)-PBE0-D/def2-TZVP).
- (a) A. Hori, Arene-Perfluoroarene Interactions in Coordination Architectures, in The Importance of Pi-Interactions in Crystal Engineering: Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. Zuckerman-Schpector, John Wiley & Sons, Chichester, UK, 2012, pp. 163–185 Search PubMed; (b) T. Dahl, Acta Chem. Scand., 1988, 42, 1–7 CrossRef; (c) J. C. Collings, K. P. Roscoe, R. L. Thomas, A. S. Batsanov, L. M. Stimson, J. A. K. Howard and T. B. Marder, New J. Chem., 2001, 25, 1410–1417 RSC; (d) J. C. Collings, K. P. Roscoe, E. G. Robins, A. S. Batsanov, L.-M. Stimson, J. A. K. Howard, S. J. Clark and T. B. Marder, New J. Chem., 2002, 26, 1740–1746 RSC; (e) Y. P. Budiman, A. Friedrich, U. Radius and T. B. Marder, ChemCatChem, 2019, 11, 5387–5396 CrossRef CAS.
- S. A. Macgregor, D. McKay, J. A. Panetier and M. K. Whittlesey, Dalton Trans., 2013, 42, 7386–7395 RSC.
- M. E. Doster and S. A. Johnson, Angew. Chem., Int. Ed., 2009, 48, 2185–2187 CrossRef CAS.
- T. Braun, L. Cronin, C. L. Higgitt, J. E. McGrady, R. N. Perutz and M. Reinhold, New J. Chem., 2001, 25, 19–21 RSC.
- S. A. Johnson, E. T. Taylor and S. J. Cruise, Organometallics, 2009, 28, 3842–3855 CrossRef CAS.
- M. Reinhold, J. E. McGrady and R. N. Perutz, J. Am. Chem. Soc., 2004, 126, 5268–5276 CrossRef CAS.
- (a) R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley-VCH, New York, 6th edn, 2014, ch. 6 Search PubMed; (b) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, CA, 2010, ch. 6–7 Search PubMed; (c) T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley & Sons, Hoboken, N. J., 2nd edn, 2013, ch. 19.6 CrossRef; (d) S. Niu and M. B. Hall, Chem. Rev., 2000, 100, 353–405 CrossRef CAS; (e) K. C. Lam, T. B. Marder and Z. Lin, Organometallics, 2007, 26, 758–760 CrossRef CAS; (f) L. P. Wolters and F. M. Bickelhaupt, Struct. Bonding, 2016, 167, 139–162 CrossRef CAS.
- (a) S. Sakaki, N. Mizoe, Y. Musashi, B. Biswas and M. Sugimoto, J. Phys. Chem. A, 1998, 102, 8027–8036 CrossRef CAS; (b) W. Guan, F. B. Sayyed, G. Zeng and S. Sakaki, Inorg. Chem., 2014, 53, 6444–6457 CrossRef CAS; (c) A. Sundermann, O. Uzan and J. M. L. Martin, Chem.–Eur. J., 2001, 7, 1703–1711 CrossRef CAS; (d) T. A. Atesin, T. Li, S. Lachaize, W. W. Brennessel, J. J. Garcia and W. D. Jones, J. Am. Chem. Soc., 2007, 129, 7562–7569 CrossRef CAS.
- (a) L. Cronin, C. L. Higgitt, R. Karch and R. N. Perutz, Organometallics, 1997, 16, 4920–4928 CrossRef CAS; (b) A. Nova, S. Erhardt, N. A. Jasim, R. N. Perutz, S. A. Macgregor, J. E. McGrady and A. C. Whitwood, J. Am. Chem. Soc., 2008, 130, 15499–15511 CrossRef CAS; (c) A. Nova, M. Reinhold, R. N. Perutz, S. A. Macgregor and J. E. McGrady, Organometallics, 2010, 29, 1824–1831 CrossRef CAS; (d) E. Jiao, F. Xia and H. Zhu, Comput. Theor. Chem., 2011, 965, 92–100 CrossRef CAS.
- J. A. Hatnean and S. A. Johnson, Organometallics, 2012, 31, 1361–1373 CrossRef CAS.
- See for example: (a) M. Aizenberger and D. Milstein, Science, 1994, 265, 359–361 CrossRef; (b) R. J. Lindup, T. B. Marder, R. N. Perutz and A. C. Whitwood, Chem. Commun., 2007, 3664–3666 RSC; (c) T. Braun, F. Wehmeier and K. Altenhöhner, Angew. Chem., Int. Ed., 2007, 46, 5321–5324 CrossRef CAS; (d) M. Teltewskoi, J. A. Panetier, S. A. MacGregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS.
- T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS.
- (a) S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932–1934 RSC; (b) K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS; (c) A. Manzoor, P. Wienefeld, M. C. Baird and P. H. M. Budzelaar, Organometallics, 2017, 36, 3508–3519 CrossRef CAS; (d) C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- S. A. Johnson, N. M. Mroz, R. Valdizon and S. Murray, Organometallics, 2011, 30, 441–457 CrossRef CAS.
- (a) T. Zell, M. Feierabend, B. Halfter and U. Radius, J. Organomet. Chem., 2011, 696, 1380–1387 CrossRef CAS; (b) T. Zell, P. Fischer, D. Schmidt and U. Radius, Organometallics, 2012, 31, 5065–5073 CrossRef CAS; (c) L. Kuehn, D. G. Jammal, K. Lubitz, T. B. Marder and U. Radius, Chem.–Eur. J., 2019, 25, 9514–9521 CrossRef CAS.
- (a) M. E. Evans, C. L. Burke, S. Yaibuathes, E. Clot, O. Eisenstein and W. D. Jones, J. Am. Chem. Soc., 2009, 131, 13464–13473 CrossRef CAS; (b) E. Clot, M. Besora, F. Maseras, C. Mégret, O. Eisenstein, B. Oelckers and R. N. Perutz, Chem. Commun., 2003, 490–491 RSC; (c) E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817–7827 CrossRef CAS.
- D. J. Nelson and F. Maseras, Chem. Commun., 2018, 54, 10646–10649 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2017693–2017700. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04237d |
| This journal is © The Royal Society of Chemistry 2020 |