
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of the catalytic H-cluster of [FeFe] hydrogenase: the roles of the Fe–S maturase proteins HydE, HydF, and HydG
R. David
Britt
 *,
Guodong
Rao
and
Lizhi
Tao
*,
Guodong
Rao
and
Lizhi
Tao
Department of Chemistry, University of California, Davis, Davis, CA 95616, USA. E-mail: rdbritt@ucdavis.edu
First published on 22nd September 2020
Abstract
[FeFe] hydrogenases carry out the redox interconversion of protons and molecular hydrogen (2H+ + 2e− ⇌ H2) at a complex Fe–S active site known as the H-cluster. The H-cluster consists of a [4Fe–4S] subcluster, denoted here as [4Fe]H, linked via a cysteine sulfur to an interesting organometallic [2Fe]H subcluster thought to be the subsite where the catalysis occurs. This [2Fe]H subcluster consists of two Fe atoms, linked with a bridging CO and a bridging SCH2NHCH2S azadithiolate (adt), with additional terminal CO and CN ligands bound to each Fe. Synthesizing such a complex organometallic unit is a fascinating problem in biochemistry, complicated by the toxic nature of both the CO and CN− species and the relative fragility of the azadithiolate bridge. It has been known for a number of years that this complex biosynthesis is carried out by a set of three essential Fe–S proteins, HydE, HydF, and HydG. HydF is a GTPase, while HydE and HydG are both members of the large family of radical S-adenosylmethionine (rSAM) enzymes. In this perspective we describe the history of research and discovery concerning these three Fe–S “maturase” proteins and describe recent evidence for a sequential biosynthetic pathway beginning with the synthesis of a mononuclear organometallic [Fe(II)(CO)2CN(cysteine)] complex by the rSAM enzyme HydG and its subsequent activation by the second rSAM enzyme HydE to form a highly reactive Fe(I)(CO)2(CN)S species. In our model a pair of these Fe(I)(CO)2(CN)S units condense to form the [Fe(CO)2(CN)S]2 diamond core of the [2Fe]H cluster, requiring only the installation of the central CH2NHCH2 portion of the azadithiolate bridge, whose atoms are all sourced from the amino acid serine. This final step likely occurs with an interplay of HydE and HydF, the details of which yet remain to be elucidated.
1 Introduction to [FeFe] hydrogenase
Hydrogenase enzymes catalyze the redox interconversion of protons and electrons with molecular hydrogen, with both [FeFe] and [NiFe] variants playing important metabolic roles in their host organisms.1–4 These hydrogenase enzymes also provide important paradigms for electrocatalytic formation of H2 for renewable energy applications: for example the [FeFe] hydrogenase can generate thousands of H2 molecules per second.5,6 Direct solar generation of H2 from water has long been a goal in the “solar fuels” arena of renewable energy production. In a semiartificial approach, photosystem II and a hydrogenase enzyme can be integrated into the anode and cathode of an photoelectrochemical cell under an applied bias to generate H2 from water.7 Recently both the [NiFe] and [FeFe] hydrogenases have been fused with photosystem I for solar H2 production, with protons and electrons provided by photosystem II water splitting.8,9 This perspective focuses on the biosynthesis of the [FeFe] hydrogenase, whose catalytic core is the “H-cluster” which consists of a standard [4Fe–4S] subcluster, denoted as [4Fe]H, linked to a unique binuclear [2Fe]H subcluster via a bridging cysteine residue (Fig. 1). The [2Fe]H unit, thought to be the site where the H+ and H2 substrates bind and react,10–13 harbors the organometallic elements of the H-cluster, with the two irons exhibiting CO and CN terminal ligands, and with two bridges linking these two irons in the form of a third CO along with a unique SCH2NHCH2S azadithiolate (adt) species. | ||
| Fig. 1 The [FeFe] hydrogenase and its active site. (A) X-ray structure of Clostridium pasteurianum CpI (PDB ID: 4XDC) highlighting the H-cluster and accessory Fe–S clusters serving as electron transfer wires. (B) Structure of the catalytic H-cluster, with the subclusters [4Fe]H and [2Fe]H that are assembled by different pathways in cells. | ||
Unlike the [4MnCa] oxygen evolving complex of photosystem II, which self-assembles under illumination in a process termed photoactivation,14 the complex catalytic Fe–S clusters of enzymes such as these hydrogenases and the nitrogenases must be synthesized enzymatically.15–17 Specific challenges for the biosynthesis of the H-cluster of the [FeFe] hydrogenase include the toxic nature of the CO and CN− molecules that end up as Fe ligands and the chemical instability of the adt moiety.18 Key roles in H-cluster biosynthesis are played by a set of Fe–S “maturase” enzymes denoted HydE, HydF, and HydG, which have been under active scrutiny since the mid-2000s. This perspective focuses on the progress in our understanding of the function and reaction mechanisms of HydE, HydF, and HydG, including a description of the current state of knowledge and identification of open questions that still need to be answered. We have proposed a sequential model for the action of these three enzymes in the biosynthesis of the [FeFe] hydrogenase H-cluster, in reaction order HydG, HydE, and HydF (Fig. 2). The experimental details leading to this model are described in the following sections.
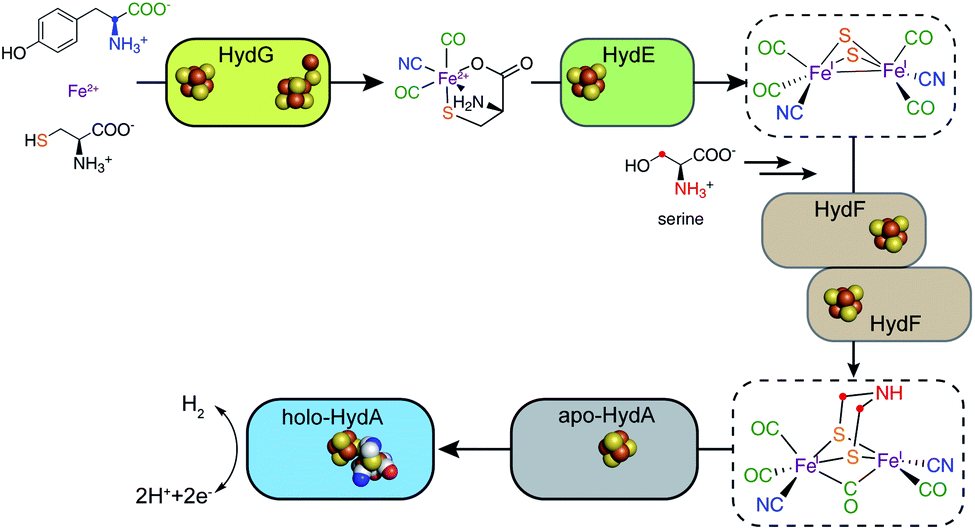 | ||
| Fig. 2 A sequential model for the roles of the HydG, HydE, and HydF maturases in the biosynthesis of the [FeFe] hydrogenase H-cluster. | ||
2 Early studies of the HydE, HydF, and HydG maturases
A breakthrough in our understanding of H-cluster synthesis was the Chlamydomonas reinhardtii genetics/molecular biology study by Posewitz et al.19 that identified genes coding for HydG and HydEF (the latter with two domains analogous to the separate HydE and HydF proteins of other organisms) as accessory genes required for formation of the H-cluster. Co-expression of these genes along with the hydrogenase HydA1 gene provided for the important new ability to produce active HydA1 hydrogenase in E. coli. Moreover it was recognized that the HydF protein contains a GTPase domain, and crucially, that HydE and HydG are members of the radical S-adenosylmethionine (rSAM) superfamily of enzymes20–22 which use a [4Fe–4S] cluster with a SAM binding site to generate the 5′-deoxyadenosyl radical (5′dAdo˙), a potent H-atom abstractor that can initiate a wide array of difficult chemical transformations. McGlynn et al.23 then demonstrated in vitro activation of [FeFe] hydrogenase, where inactive HydA (C. saccharobutylicum) expressed in E. coli was rapidly converted to active enzyme by the addition of extracts of E. coli with HydE, HydF, and HydG expressed in concert. This led to a model where the [4Fe–4S] component of the H-cluster is formed prior to activation, given that E. coli can natively synthesize and insert such clusters, e.g., via the isc operon, and that the three maturases assemble another H-cluster precursor, proposed to be the intact [2Fe]H subcluster, which when transferred to HydA provides full hydrogenase activation. To strongly reinforce this point, an X-ray structure of the HydA enzyme expressed in E. coli without coexpression of HydE, HydF, and HydF, shows only the [4Fe–4S] cluster, with an open pocket in the location of the [2Fe]H cluster in the holoenzyme.24These important studies set the stage for a number of pioneering investigations providing initial characterization of the roles played by each of these three maturase proteins, HydG, HydE, and HydF. Radical SAM enzymes such as HydE and HydG all contain a site differentiated [4Fe–4S] cluster, where one iron has an open coordination position to which SAM binds, whereas the other three irons are coordinated to the protein via cysteine ligands, canonically provided in a Cys–X3–Cys–X2–Cys sequence. The 5′dAdo˙ radical is generated by 1-electron reduction of the [4Fe–4S] cluster which leads to a homolytic cleavage between the S-atom of methionine and the C5′ of adenosine. In the most characterized subgroup of rSAM enzymes, including HydG, the resulting 5′dAdo˙ immediately abstracts an H-atom from a substrate molecule to generate the primary substrate radical and dAdoH. This requires the substrate to be bound in the active site precisely relative to the transient 5′dAdo˙ radical in order to achieve regio- and stereofidelity for this potent reaction. Along these lines, Pilet et al.25 noted the high sequence homology between HydG and ThiH,26 a rSAM enzyme that lyses L-tyrosine to generate dehydroglycine (DHG) and p-cresol. In the ThiH reaction the DHG is vectored into thiazole biosynthesis. Pilet et al. indeed confirmed that HydG SAM cleavage is stimulated by tyrosine and identified p-cresol as a product in analogy to ThiH. They went on to suggest this rSAM reactivity is used in synthesizing the azadithiolate bridge of the H-cluster. However Driesener et al.27 soon followed with a demonstration that HydG instead produces cyanide from tyrosine. Shortly after, Shepard et al.28 showed that HydG produces CO as well (as measured by CO binding to external deoxyhemoglobin), and it was therefore clear the HydG is responsible for forming both the CO and CN ligands of the [2Fe]H subcluster via a rSAM-based radical interaction with its tyrosine substrate. The HydF GTPase was also subjected to detailed investigation. HydF isolated from E. coli without subsequent iron and sulfide reconstitution showed EPR evidence for substoichiometric binding of [4Fe–4S] and [2Fe–2S] clusters.29 However HydF isolated from E. coli with HydE and HydG coexpressed showed little of the [2Fe–2S]+ EPR signal and displayed vibrational bands in the FTIR spectrum assigned to CO and CN ligation to Fe. This led to the proposal that the CO and CN formed by HydG bind to the [2Fe–2S] unit of HydF.29 But what about the role of the other rSAM enzyme HydE? With the role of HydG now assigned to CO and CN production, it was left to HydE to be the enzyme responsible for adt bridge synthesis in this early model.30–32 But there was no direct evidence for this, and a fuller characterization of the mechanism of HydE was delayed because there was no defined substrate for this rSAM enzyme, unlike HydG where there was a simple substrate, tyrosine, identified by HydG's high homology with a known rSAM tyrosine lyase. In summary, a reasonable skeletal model based on the data of the time was that the [2Fe]H subcluster is built upon an existing [2Fe–2S] cluster on HydF, with the CN and CO provided by HydG and the adt bridge provided by HydE, with the HydF assembled cluster then transferred to HydA with its extant [4Fe–4S] cluster to complete the assembly of the active H-cluster.16,28,29,31,33
3 Alternative biosynthesis approaches
Berggren et al.34 provided a fresh breakthrough in the synthesis of the H-cluster, showing that a synthetic precursor of the [2Fe]H complex, [Fe2(adt)(CN)2(CO)4]2−, can be integrated with the [4Fe–4S] cluster form of HydA1 to form a highly active H-cluster. Active hydrogenase only resulted with an azadithiolate bridge in the synthetic di-iron cluster, not with complexes substituting its bridgehead NH by O or CH2, clearly demonstrating that the light atom at the center of this dithiolate bridge is nitrogen. This study included the HydF maturase (but not HydE or HydG) in the reaction mix, but it was quickly shown that the semisynthesis can be carried out without even HydF,35 thus clearly reinforcing the picture that the three maturases are required for building the natural [2Fe]H subcluster since they can all be deleted if the appropriate synthetic analog is instead provided. Importantly, this approach also allows for the incorporation of isotopically labeled and chemically modified forms of the [2Fe]H subcluster.The James Swartz laboratory at Stanford had been working on cell free synthetic approaches to [FeFe] hydrogenase production, and Kuchenreuther et al.36 carried out the in vitro maturation of [FeFe] hydrogenase using individually expressed and purified HydE, HydF, and HydG. This allowed them to vary the concentration of each maturase and to measure how each affects the kinetics of hydrogenase activation. This study introduced an alternative mechanistic proposal wherein HydG's role is to generate an Fe complex with bound CO and CN ligands, rather than free CO and CN. An advantage of this cell free synthesis approach is that isotope labels and chemical analogs can be added efficiently as substrates and cofactors in the in vitro reactions, allowing isotope and element sensitive spectroscopies such as pulse EPR and EXAFS to track their incorporation into an isotope or element edited H-cluster.17,37–41
4 Characterization of reaction intermediates
In this earlier phase of studying HydE, HydF, and HydG, the work described above characterized the maturases themselves, the products of their reactions, and modeled the interplay of the three together in building the H-cluster. More recently there have been advancements not only in further characterizing the resting states of these enzyme and their ultimate products, but also in trapping and characterizing reaction intermediates. These studies have been enabled by combining of the use of specific isotope or chemical labels incorporated in substrates and cofactors with isotope or element sensitive spectroscopic techniques. The ability to selectively incorporate isotopes or alternate atoms into the H-cluster, either through the chemical semisynthesis method or the in vitro maturation method, have reinforced interpretations of spectroscopic results obtained on kinetically resolved intermediates in the reactions with one or more maturases. In addition, the characterization of early maturase products provides a guide for new semi-synthesis methods enabling detailed investigations of specific enzyme mechanisms and the precise sequence in which the enzymes act in generating the H-cluster. We begin this section with a focus on the radical SAM enzyme HydG, both because it is now the best characterized maturase, and because that understanding reveals that the biosynthesis of the [2Fe]H cluster is initiated by this very interesting bifunctional Fe–S enzyme.4.1 HydG and the radical mechanism of tyrosine lysis
Kuchenreuther et al.42 targeted the chemistry of HydG driven by 5′-deoxyadenosyl radical generation using EPR spectroscopy of HydG reacting with isotopically labeled tyrosine, with samples frozen rapidly after reaction initiation in order to quench and trap reaction intermediates at different timepoints. The use of specific magnetic nuclear labelled substrates was crucial to definitively assign any detected radical in such experiments and to analyze its electronic structure through site-specific hyperfine (electron – nuclear spin magnetic coupling) interactions. In this case the EPR spectrum of the trapped radical was assigned to a 4-oxidobenzyl (or hydroxybenzyl) radical (Fig. 3A) produced following Cα–Cβ bond lysis of some initial tyrosine radical produced by the 5′dAdo˙-driven H-atom abstraction. A quantum chemistry study26 of the homologous ThiH tyrosine lysis reaction had proposed that the initial 5′dAdo˙ abstracts the phenolic H of tyrosine to form a neutral tyrosine radical such as observed in photosystem II and ribonucleotide reductase,43,44 which would then be cleaved at the Cα–Cβ bond via a homolytic cleavage, forming a transient glycyl radical. Instead the experimental EPR data pointed to a heterolytic cleavage resulting in the 4-oxidobenzyl radical plus dehydroglycine (DHG), with DHG being the ultimate source of the CO and CN ligands in the [2Fe]H subcluster. Other recent data have led to a reevaluation of the specific H-atom abstraction that leads to this tyrosine cleavage. The X-ray structure of a related rSAM tryptophan lyase NosL included a tryptophan oriented to favor 5′dAdo˙ H-atom abstraction from this amino acid's amino group, and the authors suggested that a similar amino H-atom abstraction drives the tyrosine fragmentation in ThiH and HydG.45 Sayler et al.46 studied the HydG reaction with a non-native substrate, 4-hydroxy phenyl propanoic acid (HPPA) (Fig. 3B), where a simple C(2)H2 replaces the CαH–NH2 of tyrosine. A strong new HPPA-derived radical EPR signal was observed, with majority spin density on the C2 carbon: when the HPPA C2 carbon was specifically 13C-labeled, a strong 13C-hyperfine splitting was observed. Moreover, mass spectrometry of dAdoH with the HPPA reactions run in buffer with natural abundance water versus2H-enriched water showed that the abstracted H-atom is not exchangeable with HPPA as a substrate, eliminating the H of the HPPA phenol OH group as the abstracted hydrogen. Thus it is clear it is the amino group of the tyrosine that is the target of the H-atom abstraction by 5′dAdo˙, generating a transient, and yet to be observed, nitrogen centered tyrosine radical that rapidly fragments to form the DHG and 4-oxidobenzyl radical products of the rSAM component of HydG. Interestingly, when the more oxidized cis-p-coumaric acid was employed in place of HPPA, Sayler et al.46 were able to trap the primary 5′dAdo˙ which is accumulated because it cannot abstract the hydrogen atom from the sp2-carbon.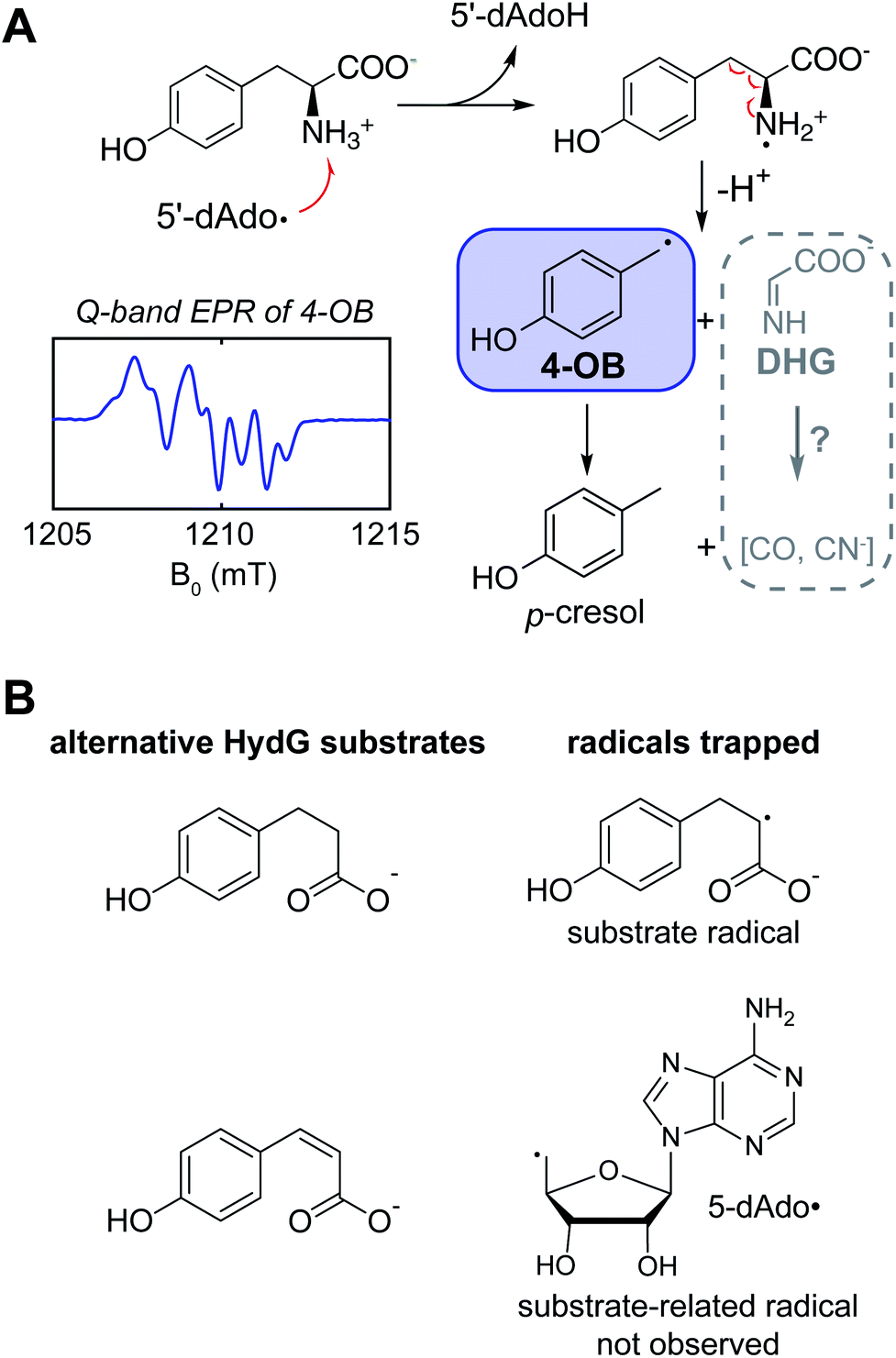 | ||
| Fig. 3 The rSAM chemistry performed by HydG. (A) Reaction mechanism of tyrosine cleavage into the diatomic CO and CN–ligands. (B) Alternative substrates tested with HydG, 4-hydroxy phenyl propanoic acid and cis-p-coumaric acid, and the corresponding radicals trapped in their reactions. | ||
We next turn to the other “auxiliary” Fe–S center of the enzyme and the evolution of our understanding of its structure and its role in the assembly of the [2Fe]H subcluster.
4.2 HydG and the formation of an organometallic [Fe(II)(CO)2CN(cysteine)] “synthon” precursor to the binuclear subcluster
In addition to the rSAM [4Fe–4S] cluster, HydG harbors an additional auxiliary Fe–S cluster (Fig. 4) that carries out a second key reaction, using the CO and CN ligands formed by two sequential tyrosine lysis reactions to form a organometallic Fe(II) species with cyanide, carbon monoxide, and cysteine ligands.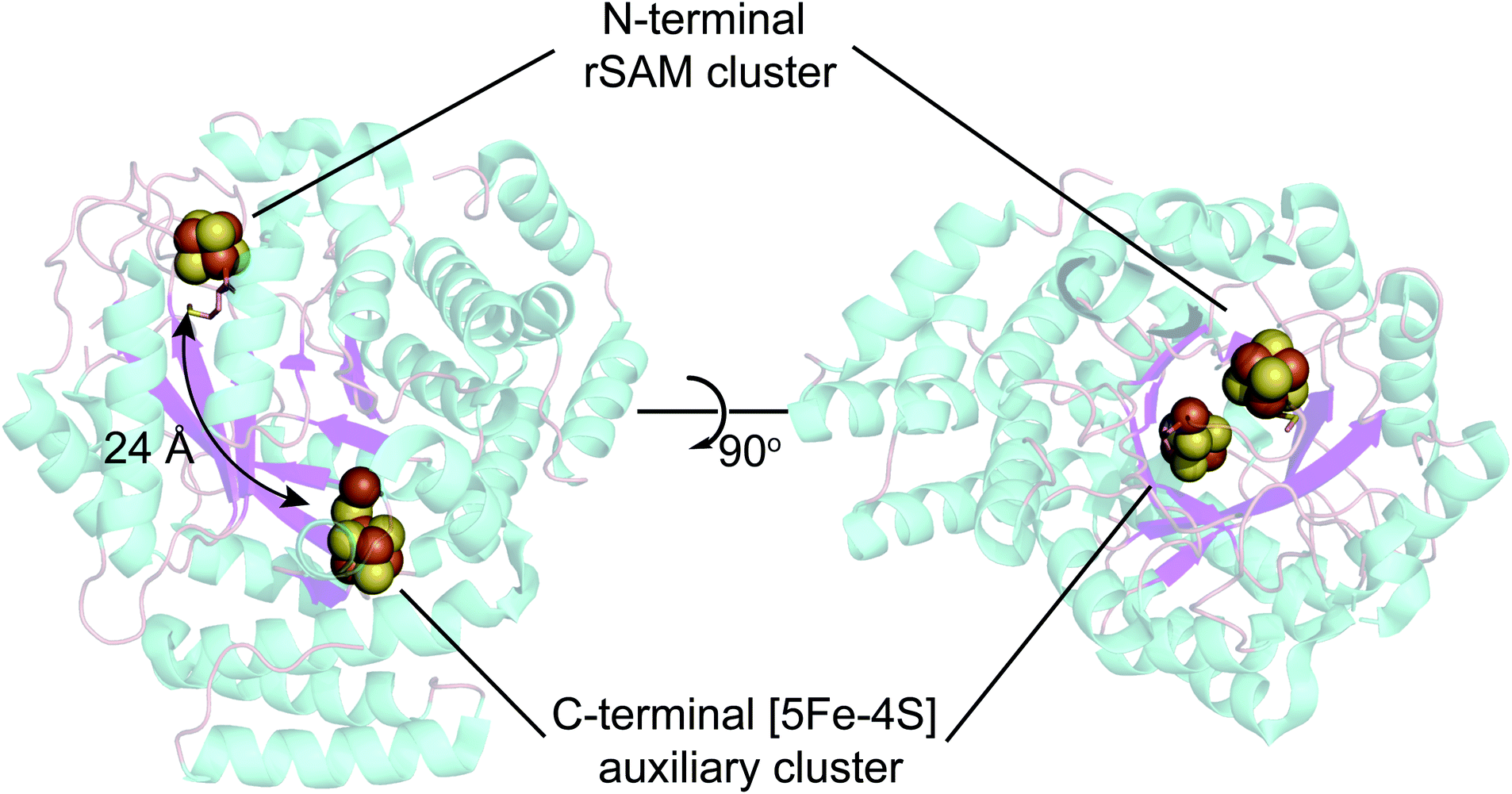 | ||
| Fig. 4 X-ray structure of Shewanella oneidensis HydG (PDB ID: 4WCX). The two Fe–S clusters in HydG are located at each end of the TIM-barrel channel (magenta). | ||
The existence of this HydG auxiliary Fe–S cluster was first predicted by sequence analysis, with a CysX2CysX22Cys sequence near the C-terminus in addition to a parallel sequence near the N-terminus that binds the [4Fe–4S]-SAM cluster. The first HydG EPR study47 did not find clear evidence of the second cluster. Driesener et al.48 characterized C. acetobutylicum HydG, including knockout mutants alternatively eliminating the rSAM Fe–S cluster or the auxiliary Fe–S cluster, as expressed in E. coli and purified via a chelating sepharose column charged with NiSO4 and eluted via an imidazole gradient (to 250 mM). Fe–S centers were then reconstituted with FeCl3 and Na2S. EPR spectroscopy clearly demonstrated that both Fe–S clusters, as reduced by deazariboflavin photoreduction, were of the [4Fe4S]+ type. It was also observed that the mutant in which the auxiliary cluster is deleted was able to carry out SAM driven tyrosine cleavage to p-cresol, but did not produce CO (also see Nicolet et al.49). The HydG Fe–S cluster EPR signals reported by Kuchenreuther et al.42 of Strep-tag isolated Shewanella oneidensis (So) HydG anaerobically expressed in E. coli were different, in that a broad, low-field signal with maximum intensities at g ≈ 9.5 and g ≈ 5 was observed. This was assigned to a high-spin (HS) S = 5/2 Fe–S cluster form of the auxiliary cluster.37 Suess et al.50 also observed this S = 5/2 signal in non-reconstituted strep-tag isolated S. oneidensis HydG and using alternative cluster knockout mutants assigned its origin to the auxiliary Fe–S cluster. This raises a couple of clear questions:
(1) What is the origin of the unusual HS Fe–S EPR signal?
(2) Why are there two different HydG EPR results from the different labs? Is the discrepancy organism or preparation dependent?
As to (1), Kuchenreuther et al.42 noted that such HS signals have not been associated with [4Fe–4S]+ clusters, and suggested that the signal could arise from a linear [3Fe–4S]+ form (ref. 51) of the auxiliary cluster which could bind a fourth iron along with the tyrosine substrate. We now know this to be incorrect, and the correct assignment for the HS EPR cluster signal was to prove quite interesting, providing a key foundation to the understanding of the role of HydG in H-cluster assembly. An issue at the time was the lack of a structure for HydG, along with the ongoing spectroscopic assignment of the auxiliary cluster to a [4Fe–4S] form. Nicolet et al.52 published the first HydG structure in 2015. This showed a (β/α)8 TIM barrel extending from the [4Fe–4S] SAM binding cluster to the CysX2CysX22Cys region where the auxiliary cluster was modeled (as a [4Fe–4S] cluster) to bind, although this cluster itself was missing in the structure. However it was clear from the structure that the auxiliary cluster binding domain is too far from the rSAM cluster where the 4-oxidobenzyl radical must be formed via tyrosine lysis to support the model suggested by Kuchenreuther et al.42 with the tyrosine substrate bound directly to the auxiliary cluster. On the continuing spectroscopy front, a combination of time resolved FTIR and57 Fe ENDOR led Kuchenreuther et al.37 to propose that the CO and CN produced by the rSAM chemistry of HydG bind to an Fe of the auxiliary cluster to form an Fe(CO)2CN “synthon” precursor en route to the synthesis of the [2Fe]H cluster. The 2015 Thermoanaerobacter italicus (Ti) HydG structure by Dinis et al.53 provided a big breakthrough in showing that the auxiliary cluster is not simply a traditional [4Fe–4S], but instead includes a fifth Fe atom linked to the site differentiated Fe (the one lacking a cysteine residue ligand) of a [4Fe–4S] cluster via a sulfide bridge (Fig. 4). In this structure assignment the other ligands to the fifth Fe are a conserved histidine (265) trans to the sulfide, an unassigned nonproteinaceous amino acid, and two waters. The reported occupancy of the fifth Fe site was relatively low, with 0.73 in one monomer of the structure and none in the other. Nevertheless the presence of a high spin S = 2 Fe(II) linked to a [4Fe–4S] cluster opened a window to the assignment of the S = 5/2 HS signal, with S = 5/2 to S = 1/2 interconversion verified by EPR experiments. This structure also provided a likely site for the formation of the proposed synthon, as the fifth Fe is located at the other end of the 24 Å barrel from the rSAM cluster, and therefore positioned to bind CO and CN created by the rSAM tyrosine lysis.
The next step in the evolution of the HydG structure was provided by Suess et al.,50 who used CW and pulse EPR and Mössbauer spectroscopies to show that in the strep tag isolated So HydG, the fifth “dangler” Fe is chelated by L-cysteine, with the cysteine providing the bridging sulfur and further ligation by its amino nitrogen and a carboxylate oxygen. Therefore in the strep tag isolated So HydG, the cysteine functionally replaces the bridging sulfide and the unassigned amino acid of the prior Ti HydG structure. The Suess et al. experiments also provided a clear answer to (2), why different laboratories reported different EPR signals. The five-Fe auxiliary cluster form provides the S = 5/2 HS EPR signal. Chelation and removal of the fifth dangler Fe converts the auxiliary cluster to an S = 1/2 [4Fe–4S]+ form. Reconstitution with Fe2+ and cysteine restore the HS S = 5/2 signal, but reconstitution with Fe2+ and inorganic sulfide but without cysteine, ala Driesener et al.,48 results in only the S = 1/2 [4Fe–4S] EPR signal, as the cysteine is needed to strongly stabilize the dangler Fe bound form of the cluster. The cysteine stabilized dangler Fe is also needed to generate the FTIR signals54 that had been crucial in demonstrating the HydG formation of an organometallic synthon.37
Suess et al.50 also proposed a mechanism for the reaction catalyzed by the auxiliary cluster of HydG, with details of the intermediates characterized by spectroscopy aided by specific isotope labeling (Fig. 5).37,50,55 The resting state of the enzyme is formed with Fe2+ binding in the site provided by the cysteine bound to the [4Fe–4S] cluster along with the conserved histidine ligand. The cleavage of the first tyrosine leads to the formation of the first pair of CO and CN ligands to the dangler Fe (complex A). The cleavage of the second tyrosine results in the second CO binding to the dangler Fe (complex B), but the CN attacks the unique Fe of the [4Fe–4S] cluster, forming a [4Fe–4S]-CN complex, and releasing the proposed product, a Fe(II)Cys(CO)2CN organometallic precursor to the [2Fe]H component of the H-cluster.
 | ||
| Fig. 5 The catalytic cycle of HydG to generate the [Fe(CO)2(CN)(cysteinate)] synthon, along with the reaction intermediates and their corresponding spectroscopic features. The pulse EPR methods such as hyperfine sublevel correlation (HYSCORE) and electron nuclear double resonance (ENDOR) are designed to probe weaker hyperfine interactions than may be evidenced in the conventional CW EPR.56 | ||
Though much of the HydG reaction chemistry seems well understood at this point, a few key questions remain:
(a) How is the DHG intermediate converted stoichiometrically to CO and CN−? Is the auxiliary 5Fe cluster directly involved in this specific DHG reaction?
(b) Is there further on-pathway radical chemistry involving the transient 4-oxidobenzyl radical, or is this radical simply quenched to form the p-cresol product?
 | ||
| Fig. 6 The molecular sources of the adt ligands. (A) The sulfur atoms in adt are sourced from cysteine in the synthon, and can be replaced with selenium by using the Se–Cys analogue of the synthon in the in vitro maturation reaction. The presence of Se vs. S atoms in the H-cluster is revealed by EXAFS spectroscopy. (B) The carbon and nitrogen atoms in adt are sourced from serine C3 carbon and amino nitrogen. 13C and 15N labels can be introduced into adt by using selectively labeled serine and detected by ENDOR spectroscopy. | ||
It is noteworthy that the two sulfurs of the adt bridge come from cysteine of the HydG product synthon. Suess et al.50 showed that cysteine chelates the dangler Fe in the resting state of the HydG enzyme, before any Fe–CO and Fe–CN bonds are formed. It is now clear that this Fe–S bond is maintained all the way into the H-cluster, even when the rest of the cysteine moiety is stripped away via S–C3 cleavage. Remaining questions include:
(a) What is the enzymology that drives the cysteine cleavage?
(b) How is serine enzymatically processed, directing its C3 (×2) and N to be installed in the adt bridge?
The reaction mediated by HydE requires only the [4Fe–4S] SAM binding cluster, as not all HydE gene sequences show the conserved cysteines needed to bind a second Fe–S cluster.58 An early EPR spectroscopy study of Tm HydE expressed in E. coli favored the existence of a second [4Fe–4S] cluster,47 although this was not observed in the first X-ray structures of Tm HydE, which revealed alternatively either no second cluster or a [2Fe–2S] cluster bound at the enzyme surface 20 Å from the SAM cluster, coordinated by three cysteines and a water molecule.58 As far as the previously unassigned substrate for this rSAM enzyme, Betz et al.32 interpreted SAM turnover studies to favor a possible thiol-containing substrate, though no specific reactivity was defined. Rohac et al.59 uncovered an interesting result in a series of crystal structures of Tm HydE incubated with 1,3-thiazolidines, showing that the 5′dAdo˙ reacts with these to form a C5′–S1 bond, with this new adduct bound into a large cavity observed near the SAM binding site.
Tao et al.57 tested a sequential synthesis model, where first HydG generates its product synthon, which then serves as the substrate for the HydE rSAM enzyme. The first intermediate characterized by EPR, resulting from the 5′dAdo˙ attack on the cysteine sulfur of the [Fe(II)(CO)2CN(cysteine)] provided by the syn-B carrier, forms in about 10 s (Fig. 7). Analysis of cw and pulse EPR spectra shows this to be an adenosylated Fe(I) species with the CO, CN, and cysteine ligands still present, but with the cysteine now covalently attached to the adenosine via a new S–C5′ linkage. This reaction has clear analogies to the HydE reactions with the 1,3-thiazolidines,59 but here with a substrate known to be a direct precursor to the [2Fe]H subcluster.40 It is particular interesting that instead of the classic rSAM enzyme H-atom abstraction, HydE uses its 5′dAdo˙ to attack an electron rich sulfur, in turn forming a biologically rare mononuclear Fe(I) complex. This transient species undergoes a further reaction on the ≈10 m timescale, with a cleavage of the cysteine S–C3 bond, just as reported in the prior HydG-minus activation study.40 This demonstrates that HydE is the enzyme that carries out this crucial conversion of the HydG [Fe(II)(CO)2CN(cysteine)] product to a new [Fe(I)(CO)2(CN)S] center. This activated organometallic unit could in turn condense pairwise60 to a [Fe(I)2(CO)4(CN)2S2] dimer that only needs the addition of the serine-derived CH2NHCH2 component of the adt bridge to complete the [2Fe]H subcluster. Remaining questions include:
 | ||
| Fig. 7 Role of HydE. (A) X-ray structure of Thermotoga maritima HydE (PDB ID: 5FEZ) showing the essential [4Fe–4S] SAM binding cluster, with the auxiliary Fe–S cluster knocked out. (B) Reactions catalyzed by HydE with its authentic substrate, the HydG product synthon, as provided by the syn-B synthetic carrier. (C) CW EPR and ENDOR spectroscopic features of two mononuclear low-spin Fe(I) intermediates in HydE catalytic reaction. | ||
(a) Do two [Fe(I)(CO)2(CN)S] monomers dimerize within HydE itself, specifically in the large pocket adjacent to the SAM cluster, or are two such monomers transferred to HydF for assembly of the [Fe(I)2(CO)4(CN)2S2] core of the [2Fe]H cluster?
(b) What enzyme(s) and mechanistic sequences activate two serines to build the CH2NHCH2 component of the bridge and link it to the bridging cysteine-derived sulfurs? Does this reaction occur on HydE or HydF? Is the reaction directly coupled to the cleavage of the S-ribose linkage?
A recent X-ray structure of HydF from Thermosipho melanesiensis (Tm HydF) (Fig. 8) reveals a [4Fe–4S] cluster with one glutamate and three cysteine ligands.62 The glutamate is exchangeable and was modeled as the site of formation of a cyanide linkage to the synthetic [Fe2(adt)(CN)2(CO)4]2− along the lines indicated earlier by EPR spectroscopy.34 However no X-ray structure was reported with the [4Fe–4S] cluster bound by either the [Fe2(adt)(CN)2(CO)4]2− or an analog with a propanedithiolate replacing the adt bridge.62 Additionally, this [4Fe–4S] cluster was the only Fe–S center revealed in this HydF structure. Specifically no additional [2Fe–2S] cluster was found as suggested by prior spectroscopic studies,29,63 and Caserta et al.62 suggest the prior evidence for such was obtained in HydF samples with incompletely loaded and degraded Fe–S clusters.
 | ||
| Fig. 8 Role of HydF. (A) X-ray structure of Thermosipho melanesiensis HydF (PDB ID: 5KH0) in a dimeric form. In the left-hand chain, the three domains in HydF are shown in magenta (GTPase domain), orange (dimerization surface) and cyan (Fe–S cluster binding domain), respectively. (B) Space-filling model of HydF showing the cleft between the dimerization domain (orange) and the Fe–S cluster binding domain (cyan) that is proposed to be the cavity to bind the [2Fe]H cluster. Shown in red is the glutamate residue (E305) that is thought to bind the non-cysteinyl-coordinated apical Fe in the 4Fe–4S cluster in apo-HydF. (C) The proposed binding mode between the HydF 4Fe–4S cluster and the [2Fe]H cluster. | ||
Also, while the three cysteinyl ligands are strictly conserved for the binding of the [4Fe–4S] cluster, the fourth ligand varies in HydF proteins from different organisms. Instead of the glutamate in the TmeHydF X-ray structure, pulse EPR spectroscopic studies (especially HYSCORE) of HydF from Clostridium acetobutylicum64 and Thermotoga neapolitana65 suggest a water/hydroxide and a histidinyl residue as the fourth ligand of the [4Fe–4S] cluster, respectively.
Experiments are now revealing details of how this synthetic cluster, as installed in HydF, is transferred to HydA.66,67 Of course such studies do not directly indicate how nature introduces the binuclear cluster when it is not preassembled by talented synthetic chemists. Is some binuclear precursor first formed by HydE in the cavity adjacent to the HydE's SAM cluster, to then be transferred to HydF en route to HydA1 along parallel lines as observed for the synthetic binuclear cluster? Or are a pair of HydE-activated mononuclear [Fe(I)(CO)2(CN)S] species transferred to a binding site in HydF where the [2Fe]H cluster is formed with the completion of the adt bridge and the reconfiguration of one of the terminal COs into the final bridging CO? This is one of the final big puzzles of H-cluster biosynthesis.
5 Conclusions
The HydG, HydE, and HydF Fe–S proteins carry out fascinating chemistry en route to building the organometallic binuclear [2Fe]H cluster. Advancement in our knowledge of their specific roles have come from recent X-ray structures as well as studies combining isotope and element labeling with spectroscopic probes of the enzymes, their reaction intermediates, and their products (Fig. 2). There is now strong evidence for a sequential model that begins with the radical SAM enzyme HydG, which over its full reaction cycle lyses two tyrosines, harvesting two CO and two CN− molecules. The two COs and one CN− bind to a “dangler” Fe(II) that is chelated by a nonproteinaceous cysteine whose sulfur bridges to a [4Fe–4S] cluster, forming a unique 5 Fe auxiliary cluster at the other end of a 24 Å TIM barrel from the SAM binding [4Fe–4S] cluster that initiates the radical chemistry that drives the tyrosine fragmentation. The second CN− produced in the cycle releases the HydG product, a [Fe(II)(CO)2CN(cysteine)] organometallic complex, by attacking the site differentiated Fe of the [4Fe–4S] cluster where the chelating cysteine was previously bound. This [Fe(II)(CO)2CN(cysteine)] product of HydG serves as the long sought for substrate of the second radical SAM enzyme HydE, which activates it for incipient dimerization by forming an adenosylated Fe(I)(CO)2CN(cysteine) intermediate within which the cysteine S–3C bond is subsequently cleaved, forming a highly reactive Fe(I)(CO)2(CN)S species that can pairwise provide all the [2Fe]H atoms other than the CH2NHCH2 central component of the azadithiolate bridge, which is sourced from serine, specifically the 3C and amino N of this amino acid. The last major issues involve the sequence of dimerization of two reactive Fe(I)(CO)2(CN)S units, the mechanism by which the CH2NHCH2 unit is built and attached including the required serine fragmentation, the de-adenylation of the HydE intermediate, and the exact interplay of HydE and HydF in this set of reactions. In the end the resultant [2Fe]H cluster, presumably bound in HydF, is transferred to the HydA and linked to the extant [4Fe–4S] cluster to complete the H-cluster. It is likely that these final open issues will soon be resolved using the same isotope/element labeling and spectroscopy techniques that proved so essential in the earlier stages of the H-cluster biosynthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the National Institutes of Health (1R35GM126961 to R. D. B.).Notes and references
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 Search PubMed.
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 Search PubMed.
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1350–1369 Search PubMed.
- K. Schuchmann, N. P. Chowdhury and V. Müller, Front. Microbiol., 2018, 9, 2911 Search PubMed.
- C. Madden, M. D. Vaughn, I. Diez-Perez, K. A. Brown, P. W. King, D. Gust, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2012, 134, 1577–1582 Search PubMed.
- K. Pandey, S. T. A. Islam, T. Happe and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3843–3848 Search PubMed.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 Search PubMed.
- A. Kanygin, Y. Milrad, C. Thummala, K. Reifschneider, P. Baker, P. Marco, I. Yacoby and K. E. Redding, Energy Environ. Sci., 2020, 13, 2903–2914 Search PubMed.
- J. Appel, V. Hueren, M. Boehm and K. Gutekunst, Nat. Energy, 2020, 5, 458–467 Search PubMed.
- D. W. Mulder, Y. S. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 Search PubMed.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. X. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 16894–16902 Search PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 Search PubMed.
- H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 Search PubMed.
- N. Tamura and G. Cheniae, Biochim. Biophys. Acta, 1987, 890, 179–194 Search PubMed.
- E. M. Shepard, E. S. Boyd, J. B. Broderick and J. W. Peters, Curr. Opin. Chem. Biol., 2011, 15, 319–327 Search PubMed.
- B. R. Duffus, T. L. Hamilton, E. M. Shepard, E. S. Boyd, J. W. Peters and J. B. Broderick, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 1254–1263 Search PubMed.
- R. D. Britt, G. Rao and L. Tao, Nat. Rev. Chem., 2020, 9, 2911 Search PubMed.
- R. Angamuthu, C. S. Chen, T. R. Cochrane, D. L. Gray, D. Schilter, O. A. Ulloa and T. B. Rauchfuss, Inorg. Chem., 2015, 54, 5717–5724 Search PubMed.
- M. C. Posewitz, P. W. King, S. L. Smolinski, L. P. Zhang, M. Seibert and M. L. Ghirardi, J. Biol. Chem., 2004, 279, 25711–25720 Search PubMed.
- P. A. Frey and O. T. Magnusson, Chem. Rev., 2003, 103, 2129–2148 Search PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 Search PubMed.
- J. Bridwell-Rabb, T. A. Grell and C. L. Drennan, Annu. Rev. Biochem., 2018, 87, 555–584 Search PubMed.
- S. E. McGlynn, S. S. Ruebush, A. Naumov, L. E. Nagy, A. Dubini, P. W. King, J. B. Broderick, M. C. Posewitz and J. W. Peters, JBIC, J. Biol. Inorg. Chem., 2007, 12, 443–447 Search PubMed.
- D. W. Mulder, E. S. Boyd, R. Sarma, R. K. Lange, J. A. Endrizzi, J. B. Broderick and J. W. Peters, Nature, 2010, 465, 248–251 Search PubMed.
- E. Pilet, Y. Nicolet, C. Mathevon, T. Douki, J. C. Fontecilla-Camps and M. Fontecave, FEBS Lett., 2009, 583, 506–511 Search PubMed.
- M. Kriek, F. Martins, M. R. Challand, A. Croft and P. L. Roach, Angew. Chem., Int. Ed., 2007, 46, 9223–9226 Search PubMed.
- R. C. Driesener, M. R. Challand, S. E. McGlynn, E. M. Shepard, E. S. Boyd, J. B. Broderick, J. W. Peters and P. L. Roach, Angew. Chem., Int. Ed., 2010, 49, 1687–1690 Search PubMed.
- E. M. Shepard, B. R. Duffus, S. J. George, S. E. McGlynn, M. R. Challand, K. D. Swanson, P. L. Roach, S. P. Cramer, J. W. Peters and J. B. Broderick, J. Am. Chem. Soc., 2010, 132, 9247–9249 Search PubMed.
- E. M. Shepard, S. E. McGlynn, A. L. Bueling, C. S. Grady-Smith, S. J. George, M. A. Winslow, S. P. Cramer, J. W. Peters and J. B. Broderick, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10448–10453 Search PubMed.
- Y. Nicolet and J. C. Fontecilla-Camps, J. Biol. Chem., 2012, 287, 13532–13540 Search PubMed.
- E. M. Shepard, F. Mus, J. N. Betz, A. S. Byer, B. R. Duffus, J. W. Peters and J. B. Broderick, Biochemistry, 2014, 53, 4090–4104 Search PubMed.
- J. N. Betz, N. W. Boswell, C. J. Fugate, G. L. Holliday, E. Akiva, A. G. Scott, P. C. Babbitt, J. W. Peters, E. M. Shepard and J. B. Broderick, Biochemistry, 2015, 54, 1807–1818 Search PubMed.
- A. S. Byer, E. M. Shepard, M. W. Ratzloff, J. N. Betz, P. W. King, W. E. Broderick and J. B. Broderick, JBIC, J. Biol. Inorg. Chem., 2019, 24, 783–792 Search PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–70 Search PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Nothl, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 Search PubMed.
- J. M. Kuchenreuther, R. D. Britt and J. R. Swartz, PLoS One, 2012, 9, 1–9 Search PubMed.
- J. M. Kuchenreuther, W. K. Myers, D. L. M. Suess, T. A. Stich, V. Pelmenschikov, S. A. Shiigi, S. P. Cramer, J. R. Swartz, R. D. Britt and S. J. George, Science, 2014, 343, 424–427 Search PubMed.
- W. K. Myers, T. A. Stich, D. L. M. Suess, J. M. Kuchenreuther, J. R. Swartz and R. D. Britt, J. Am. Chem. Soc., 2014, 136, 12237–12240 Search PubMed.
- G. D. Rao and R. D. Britt, Inorg. Chem., 2018, 57, 10935–10944 Search PubMed.
- G. Rao, S. A. Pattenaude, K. Alwan, N. J. Blackburn, R. D. Britt and T. B. Rauchfuss, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20850–20855 Search PubMed.
- G. D. Rao, L. Z. Tao and R. D. Britt, Chem. Sci., 2020, 11, 1241–1247 Search PubMed.
- J. M. Kuchenreuther, W. K. Myers, T. A. Stich, S. J. George, Y. NejatyJahromy, J. R. Swartz and R. D. Britt, Science, 2013, 342, 472–475 Search PubMed.
- B. A. Diner and R. D. Britt, in Photosystem II: The light-driven water:plastoquinone oxidoreductase, ed. T. J. Wydrzynski, K. Satoh and J. A. Freeman, Springer Netherlands, Dordrecht, 2005, pp. 207–233 Search PubMed.
- C. J. Bender, M. Sahlin, G. T. Babcock, B. A. Barry, T. K. Chandrashekar, S. P. Salowe, J. Stubbe, B. Lindstrom, L. Petersson, A. Ehrenberg and B. M. Sjoberg, J. Am. Chem. Soc., 1989, 111, 8076–8083 Search PubMed.
- Y. Nicolet, L. Zeppieri, P. Amara and J. C. Fontecilla-Camps, Angew. Chem., Int. Ed., 2014, 53, 11840–11844 Search PubMed.
- R. I. Sayler, T. A. Stich, S. Joshi, N. Cooper, J. T. Shaw, T. P. Begley, D. J. Tantillo and R. D. Britt, ACS Cent. Sci., 2019, 5, 1777–1785 Search PubMed.
- J. K. Rubach, X. Brazzolotto, J. Gaillard and M. Fontecave, FEBS Lett., 2005, 579, 5055–5060 Search PubMed.
- R. C. Driesener, B. R. Duffus, E. M. Shepard, I. R. Bruzas, K. S. Duschene, N. J. R. Coleman, A. P. G. Marrison, E. Salvadori, C. W. M. Kay, J. W. Peters, J. B. Broderick and P. L. Roach, Biochemistry, 2013, 52, 8696–8707 Search PubMed.
- Y. Nicolet, L. Martin, C. Tron and J. C. Fontecilla-Camps, FEBS Lett., 2010, 584, 4197–4202 Search PubMed.
- D. L. M. Suess, I. Burstel, L. De La Paz, J. M. Kuchenreuther, C. C. Pham, S. P. Cramer, J. R. Swartz and R. D. Britt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11455–11460 Search PubMed.
- M. C. Kennedy, T. A. Kent, M. Emptage, H. Merkle, H. Beinert and E. Munck, J. Biol. Chem., 1984, 259, 4463–4471 Search PubMed.
- Y. Nicolet, A. Pagnier, L. Zeppieri, L. Martin, P. Amara and J. C. Fontecilla-Camps, ChemBioChem, 2015, 16, 397–402 Search PubMed.
- P. Dinis, D. L. M. Suess, S. J. Fox, J. E. Harmer, R. C. Driesener, L. De La Paz, J. R. Swartz, J. W. Essex, R. D. Britt and P. L. Roach, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1362–1367 Search PubMed.
- D. L. M. Suess, C. C. Pham, I. Burstel, J. R. Swartz, S. P. Cramer and R. D. Britt, J. Am. Chem. Soc., 2016, 138, 1146–1149 Search PubMed.
- G. D. Rao, L. Z. Tao, D. L. M. Suess and R. D. Britt, Nat. Chem., 2018, 10, 555–560 Search PubMed.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, 2001 Search PubMed.
- L. Tao, S. A. Pattenaude, S. Joshi, T. P. Begley, T. B. Rauchfuss and R. D. Britt, J. Am. Chem. Soc., 2020, 142, 10841–10848 Search PubMed.
- Y. Nicolet, J. K. Rubach, M. C. Posewitz, P. Amara, C. Mathevon, M. Atta, M. Fontecave and J. C. Fontecilla-Camps, J. Biol. Chem., 2008, 283, 18861–18872 Search PubMed.
- R. Rohac, P. Amara, A. Benjdia, L. Martin, P. Ruffie, A. Favier, O. Berteau, J. M. Mouesca, J. C. Fontecilla-Camps and Y. Nicolet, Nat. Chem., 2016, 8, 491–500 Search PubMed.
- W. Hieber and J. Gruber, Zeitschrift für anorganische und allgemeine Chemie, 1958, 296, 91–103 Search PubMed.
- F. Vallese, P. Berto, M. Ruzzene, L. Cendron, S. Sarno, E. De Rosa, G. M. Giacometti and P. Costantini, J. Biol. Chem., 2012, 287, 36544–36555 Search PubMed.
- G. Caserta, L. Pecqueur, A. Adamska-Venkatesh, C. Papini, S. Roy, V. Artero, M. Atta, E. Reijerse, W. Lubitz and M. Fontecave, Nat. Chem. Biol., 2017, 13, 779–784 Search PubMed.
- E. M. Shepard, A. S. Byer, J. N. Betz, J. W. Peters and J. B. Broderick, Biochemistry, 2016, 55, 3514–3527 Search PubMed.
- P. Berto, M. Di Valentin, L. Cendron, F. Vallese, M. Albertini, E. Salvadori, G. M. Giacometti, D. Carbonera and P. Costantini, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 2149–2157 Search PubMed.
- M. Albertini, P. Berto, F. Vallese, M. Di Valentin, P. Costantini and D. Carbonera, J. Phys. Chem. B, 2015, 119, 13680–13689 Search PubMed.
- B. Németh, H. Land, A. Magnuson, A. Hofer and G. Berggren, J. Biol. Chem., 2020, 295, 11891–11901 Search PubMed.
- B. Németh, C. Esmieu, H. J. Redman and G. Berggren, Dalton Trans., 2019, 48, 5978–5986 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |