
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminum-catalyzed tunable halodefluorination of trifluoromethyl- and difluoroalkyl-substituted olefins†
Zhong
Liu
,
Xian-Shuang
Tu
,
Le-Tao
Guo
and
Xiao-Chen
Wang
 *
*
State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin 300071, China. E-mail: xcwang@nankai.edu.cn
First published on 1st October 2020
Abstract
Herein, we report unprecedented aluminum-catalyzed halodefluorination reactions of trifluoromethyl- and difluoroalkyl-substituted olefins with bromo- or chlorotrimethylsilane. The interesting feature of these reactions is that one, two, or three fluorine atoms can be selectively replaced with bromine or chlorine atoms by modification of the reaction conditions. The generated products can undergo a variety of subsequent transformations, thus constituting a valuable stock of building blocks for installing fluorine-containing olefin motifs in other molecules.
Combined with the use of fluorine-18 for positron emission tomography, the discovery that incorporating fluorine atoms into drug molecules can improve their bioavailability, metabolic stability, and target specificity has driven the rapid development of new methods for generating C–F bonds and forming bond connections with fluorine-containing structural motifs over the past decade.1 However, synthesis of compounds bearing fluorovinyl (F–C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and gem-difluoroallyl (F2C–CC) groups remains a challenge, despite the presence of these structural motifs in numerous drugs, such as tezacitabine,2 seletracetam,3 and tafluprost4 (Scheme 1a). We envisioned that synthesis of fluorovinyls containing an allylic bromine atom (F–CC–C–Br) would facilitate the preparation of such compounds because the bromine atom would serve as a handle for a wide variety of substitution and cross-coupling reactions. The existing methods for their preparation generally rely on reactions of fluorovinyls containing an allylic hydroxyl group or gem-difluorinated vinyloxiranes with brominating reagents.5 Direct methods for their synthesis from readily accessible substrates are lacking.
C) and gem-difluoroallyl (F2C–CC) groups remains a challenge, despite the presence of these structural motifs in numerous drugs, such as tezacitabine,2 seletracetam,3 and tafluprost4 (Scheme 1a). We envisioned that synthesis of fluorovinyls containing an allylic bromine atom (F–CC–C–Br) would facilitate the preparation of such compounds because the bromine atom would serve as a handle for a wide variety of substitution and cross-coupling reactions. The existing methods for their preparation generally rely on reactions of fluorovinyls containing an allylic hydroxyl group or gem-difluorinated vinyloxiranes with brominating reagents.5 Direct methods for their synthesis from readily accessible substrates are lacking.
 | ||
| Scheme 1 Synthesis of fluorovinyls via Lewis acid activation of trifluoromethylalkenes. | ||
Elegant work from the groups of Maruoka,6 Oshima,7 Ozerov,8 Müller,9 Stephan,10 Oestreich,11 Chen,12 and Young13 on C–F bond activation reactions has proven that Lewis acid-promoted abstraction of fluoride from alkyl fluorides is a powerful tool for generating carbocations that can be trapped by nucleophiles. When trifluoromethylalkenes were studied as substrates, Ichikawa et al. reported that aryldefluorination of trifluoromethylalkenes can be accomplished with a stoichiometric amount of EtAlCl2via fluoride abstraction and subsequent Friedel–Crafts reactions between the resulting allylic carbocation and arenes (Scheme 1b).14 In addition, Braun and Kemnitz and colleagues carried out hydrodefluorination reactions of trifluoromethylalkenes with hydrosilanes catalyzed by Lewis acidic nanoscopic aluminum chlorofluoride (Scheme 1b).15 In light of these reports and our experiences in developing Lewis acid-catalyzed reactions,16 we speculated that 3,3-difluoroallyl bromides (F2CC–C–Br) could be directly prepared from trifluoromethylalkenes and a suitable bromide source via Lewis acid activation of the C–F bonds and subsequent nucleophilic attack of the bromide anion at the distal olefinic carbon of the resulting allylic carbocation, a process that has no precedent in the literature.
Herein, we report our discovery that by using an aluminum-based Lewis acid catalyst and bromotrimethylsilane (TMSBr) or chlorotrimethylsilane (TMSCl) as a halide source, we were able to achieve the proposed C–F bond activation/substitution reaction (Scheme 1c). Furthermore, simply by adjusting the stoichiometry of the reactants and the reaction temperature, we could selectively obtain mono-, di-, or trisubstituted products. Mechanistic studies indicated the multi-substitution reaction was achieved by thermally promoted 1,3-halogen migration of the initially formed product, followed by further halodefluorination. Notably, the previously reported defluorination reactions of trifluoromethylalkenes, either Lewis acid-catalyzed14,15 or promoted via other methods,17–19 usually provide monosubstitution products; that is, our finding that we could selectively generate multiply substituted products is also unprecedented.
To test various reaction conditions, we chose α-aryl-substituted trifluoromethylalkene 1a as a model substrate (Table 1). TMSBr was selected as the bromide source because we expected the generated silyl cation to be an excellent scavenger for the displaced fluoride anion. We began by evaluating several Lewis acid catalysts and found that no reaction occurred when 1a was treated with B(C6F5)3, Zn(OTf)2, Sc(OTf)3, Al(OTf)3, or ZrCl4 (5 mol%) and 3 equiv. of TMSBr in DCE at 80 °C for 24 h (entries 1–5). However, we were encouraged to find that AlCl3 would catalyze the proposed bromodefluorination reaction, giving monobrominated product 2a and dibrominated product 3a (ref. 20) in 17% and 2% yields, respectively (entry 6). Investigation of additional aluminum-based Lewis acids showed that AlEtCl2 and Al(C6F5)3(tol)0.5 (ref. 21) had higher activities: AlEtCl2 gave 2a and 3a in 5% and 38% yields, respectively (entry 7), and Al(C6F5)3(tol)0.5 gave 16% and 32% yields, respectively (entry 8). Because Al(C6F5)3(tol)0.5 is a solid and therefore easier to store and handle than AlEtCl2 (a liquid), we chose Al(C6F5)3(tol)0.5 for further investigation. Changing the solvent to toluene inhibited the formation of 3a, but failed to improve the yield of 2a (entry 9). Coordinative solvents (acetonitrile and dioxane) shut down the reaction entirely (entries 10 and 11). When the reaction temperature was increased to 120 °C, 2a and 3a were obtained in 13% and 68% yields, respectively (entry 13). Gratifyingly, when 4 equiv. of TMSBr relative to 1a was used, 3a was generated as the sole reaction product in 90% yield (Z/E = 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45, entry 14). Next, we tried using TMSBr as the limiting reagent to determine whether we could obtain the monobrominated product (2a) as the major product. Indeed, when 3 equiv. of 1a was treated with 1 equiv. of TMSBr at 80 °C, 2a was obtained as the sole product, although the yield was only 30% (entry 15). Further screening of reaction conditions revealed that using 9.0 mol% of Al(C6F5)3(tol)0.5 and running the reaction at 60 °C for 48 h (entry 16) gave the highest yield of 2a (76%; the yield of 3a was 8%).
:45, entry 14). Next, we tried using TMSBr as the limiting reagent to determine whether we could obtain the monobrominated product (2a) as the major product. Indeed, when 3 equiv. of 1a was treated with 1 equiv. of TMSBr at 80 °C, 2a was obtained as the sole product, although the yield was only 30% (entry 15). Further screening of reaction conditions revealed that using 9.0 mol% of Al(C6F5)3(tol)0.5 and running the reaction at 60 °C for 48 h (entry 16) gave the highest yield of 2a (76%; the yield of 3a was 8%).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lewis acid | 1a/TMSBr | T (°C) | Solvent | Yieldb2a (%) | Yieldb3a (%) |
|
a Unless otherwise specified, reactions were performed with 0.1 mmol of 1a and 5 mol% of a Lewis acid in 1 mL of solvent for 24 h under N2.
b Yields were determined by 1H NMR using CH2Br2 as the internal standard; the 2a/3a ratios were determined by 19F NMR; n.d. = not detected.
c 4.5 mol% Al(C6F5)3(tol)0.5 was used as catalyst.
d The Z/E ratio was 55:45.
e The reaction was carried out with 9.0 mol% of Al(C6F5)3(tol)0.5 for 48 h.
|
||||||
| 1 | B(C6F5)3 | 1:3 |
80 | DCE | n.d. | n.d. |
| 2 | Zn(OTf)2 | 1:3 |
80 | DCE | n.d. | n.d. |
| 3 | Sc(OTf)3 | 1:3 |
80 | DCE | n.d. | n.d. |
| 4 | Al(OTf)3 | 1:3 |
80 | DCE | n.d. | n.d. |
| 5 | ZrCl4 | 1:3 |
80 | DCE | Trace | n.d. |
| 6 | AlCl3 | 1:3 |
80 | DCE | 17 | 2 |
| 7 | AlEtCl2 | 1:3 |
80 | DCE | 5 | 38 |
| 8c | Al(C6F5)3(tol)0.5 | 1:3 |
80 | DCE | 16 | 32 |
| 9c | Al(C6F5)3(tol)0.5 | 1:3 |
80 | Toluene | 16 | n.d. |
| 10c | Al(C6F5)3(tol)0.5 | 1:3 |
80 | CH3CN | n.d. | n.d. |
| 11c | Al(C6F5)3(tol)0.5 | 1:3 |
80 | Dioxane | n.d. | n.d. |
| 12c | Al(C6F5)3(tol)0.5 | 1:3 |
100 | DCE | 25 | 48 |
| 13c | Al(C6F5)3(tol)0.5 | 1:3 |
120 | DCE | 13 | 68 |
| 14c | Al(C6F5)3(tol)0.5 | 1:4 |
120 | DCE | n.d. | 90d |
| 15c | Al(C6F5)3(tol)0.5 | 3:1 |
80 | DCE | 30 | n.d. |
| 16e | Al(C6F5)3(tol)0.5 | 3:1 |
60 | DCE | 76 | 8 |

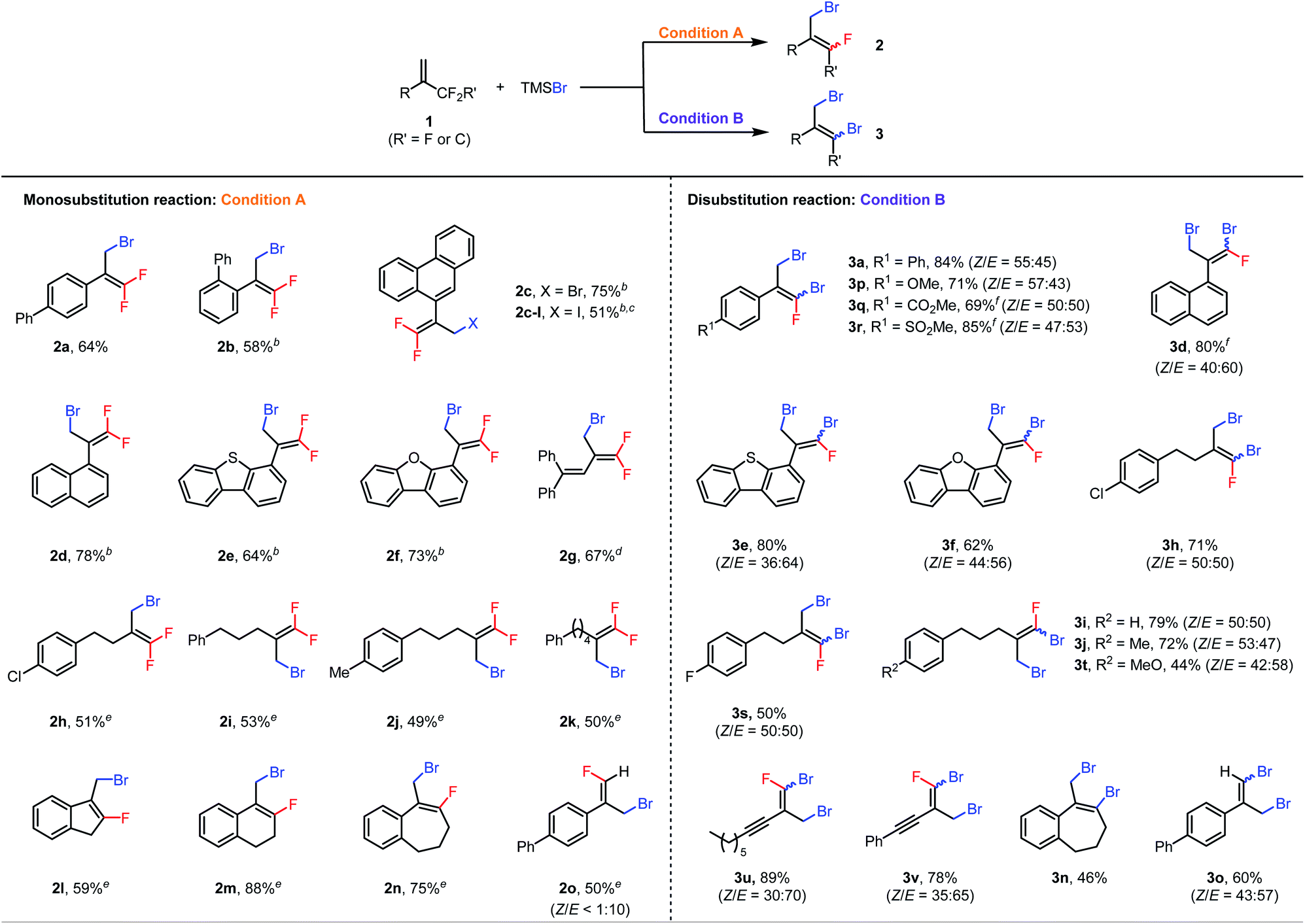
With the optimal conditions in hand, we first explored the scope of the monosubstitution reaction by testing various trifluoromethyl- and difluoroalkyl-substituted olefins 1 (Table 2, left column). From 1a, monobrominated product 2a could be isolated in pure form in 64% yield by means of preparative HPLC. When the α-phenyl ring bore an ortho-phenyl substituent, the reaction still afforded 2b in 58% yield despite the increased steric bulk around the reaction site. When the α substituent was changed to a 9-phenanthryl group (1c), monobrominated product 2c was isolated in 75% yield. We also tested other halogenating reagents with 1c: TMSI gave iodinated product 2c-I in 51% yield, whereas TMSCl was poorly reactive, giving a <10% yield of product. Furthermore, substrates with 1-naphthyl (2d), 4-dibenzothiophenyl (2e), and 4-dibenzofuranyl (2f) moieties at the α position were all suitable. Interestingly, even the reaction of conjugated diene 1g was feasible, giving brominated diene 2g in 67% isolated yield. In addition, a series of α-alkyl-substituted trifluoromethylalkenes gave the desired products (2h–2k) in moderate yields. Difluoroalkyl-substituted alkenes were also reactive; specifically, benzene-fused methylenecycloalkanes 1l–1n gave the corresponding products (2l–2n) in 59–88% yields. Finally, acyclic substrate 1o afforded (E)-2o as the predominant isomer (E/Z > 10:1) in 50% yield.
| a Condition A: reactions were performed with 0.6 mmol of 1, 0.2 mmol of TMSBr, and 9.0 mol% of Al(C6F5)3(tol)0.5 in 1.5 mL of DCE at 60 °C for 48 h; condition B: reactions were performed with 0.2 mmol of 1, 0.8 mmol of TMSBr, and 4.5 mol% of Al(C6F5)3(tol)0.5 in 1.5 mL of DCE at 120 °C for 24 h; isolated yields are reported. b The reaction was performed at 80 °C. c TMSI was used instead of TMSBr. d The reaction was carried out with 13.5 mol% of Al(C6F5)3(tol)0.5. e 4 equiv. of 1 was used. f The reaction was performed with 5 equiv. of TMSBr. |
|---|
|
Next the scope of the disubstitution reaction was investigated (Table 2, right column). Trifluoromethyl-substituted alkenes bearing electron-donating or electron-withdrawing groups on the α-aryl ring were reactive, affording the corresponding products (3a and 3p–3r) in 69–85% yields with Z/E ratios of approximately 1:1. 1-Naphthyl (3d), 4-dibenzothiophenyl (3e), 4-dibenzofuranyl (3f), and aliphatic (3h–3j, 3s, and 3t) substituents at the α position were well tolerated. Interestingly, even alkynyl-substituted trifluoromethylalkenes afforded the desired disubstituted products (3u and 3v) in good yields. In addition, difluoroalkyl-substituted alkenes 1n and 1o gave completely defluorinated products 3n and 3o in 46% and 60% yields, respectively. Notably, under these conditions, the monobrominated products either did not form or formed in only trace amounts, as indicated by GC-MS or NMR spectroscopy. Moreover, the E and Z isomers of dibrominated products were found interconvertible under the reaction conditions (for details, see the ESI†) so the Z/E ratios of products might be the result of the thermodynamic equilibrium.
It is also worth mentioning that some substrates shown in Table 2 were not compatible either with the monosubstitution reaction or with the disubstitution reaction. For example, substrates bearing coordinative functional groups, such as methoxy, carbonyl, sulfonyl and alkyne (1p, 1q, 1r, 1t, 1u, and 1v), gave very low yields (<20%) for monosubstitution, perhaps because the relatively low reaction temperature (60 °C) was not sufficient to break the coordination of these functional groups to the Lewis acid catalyst. Furthermore, Al(C6F5)3(tol)0.5 is probably a precatalyst because Al(C6F5)3(tol)0.5 rapidly decomposes in DCE to give a mixture of unidentified aluminum species21b that are active for the halodefluorination reaction (for details, see the ESI†).
We performed several control experiments to explore the reaction mechanism. When substrate 1a was treated with mesitylene in the presence of 1 equiv. of Al(C6F5)3(tol)0.5, Friedel–Crafts allylation of the aromatic ring generated product 4 in 96% yield (Scheme 2a).22 This result demonstrates that the aluminum Lewis acid could abstract fluoride from the trifluoromethylalkene to generate an allylic carbocation. Furthermore, when 2a was subjected to the conditions used for the disubstitution reaction, 3a was isolated in 65% yield (Scheme 2b), indicating that the dibrominated products were generated via monobrominated intermediates. However, subjecting nonbrominated 5 to the same conditions did not result in substitution of the vinylic fluorine atom by the bromine atom (6, Scheme 2c), which excludes the vinylic nucleophilic substitution (SNV) mechanism23 for the conversion from 2a to 3a. We thus suspected that the allylic bromine atom in 2a was involved in this conversion. Indeed, when 2a was heated at 120 °C in toluene for 12 h, 1,3-migration of the bromine atom gave bromodifluoromethylalkene 7 in 83% NMR yield (Scheme 2d).24 And, treatment of 7 with TMSBr in the presence of the catalyst at 120 °C gave 3a in 77% yield (Scheme 2e). Taken together, these results indicate that dibrominated products were generated via isomerization of the monobrominated product to form bromodifluoromethylalkenes, which then underwent a second bromodefluorination reaction. In addition, silylium Et3Si[B(C6F5)4]25 was found incapable of catalyzing the bromodefluorination reaction (Scheme 2f). This result suggests that the Lewis acidic aluminum is probably a catalyst, rather than an initiator, and TMS+ from TMSBr abstracts the fluoride from the aluminum–fluoride adduct to regenerate the active catalyst.
 | ||
| Scheme 2 Control experiments. | ||
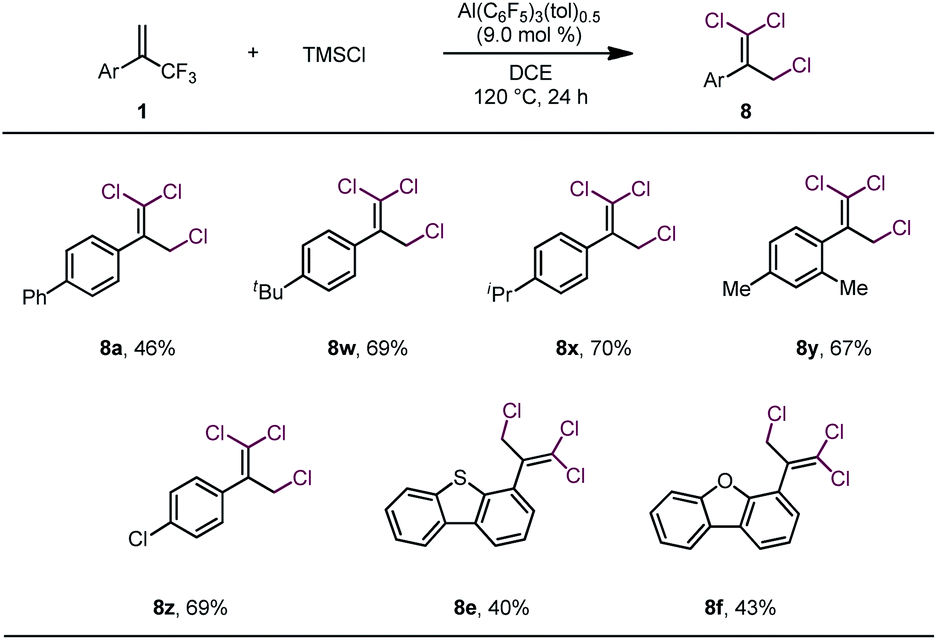
These results led us to wonder whether all three fluorine atoms of a trifluoromethylalkene could be replaced with bromine atoms via a 1,3-bromo migration reaction of the dibrominated product to give a dibromofluoromethylalkene, which would then undergo bromodefluorination. After screening various reaction conditions, we discovered that tribrominated products could be obtained by using a large excess (e.g., 10 equiv.) of TMSBr and extending the reaction time; however, in all cases, substantial amounts of the dibrominated products were always produced as well (see Table S1 in the ESI†), which made separation of the product difficult. However, we were delighted to find that when TMSCl was used in large excess (7 equiv.) and the reaction temperature was 120 °C, trichlorinated compounds were the major or only products (Table 3). However, these conditions were suitable only for substrates bearing α-aryl substituents. The moderate to low yields of these reactions were due mainly to decomposition of the starting materials rather than to the formation of mono- or dichlorinated byproducts.
| a Unless otherwise specified, reactions were performed with 0.2 mmol of 1, 1.4 mmol of TMSCl, and 9.0 mol% of Al(C6F5)3(tol)0.5 in 1.5 mL of DCE at 120 °C for 24 h; isolated yields are reported. |
|---|
|
As mentioned above, bromine atoms are among the most useful substituents for introducing other functional groups. To explore the utility of the above-described reactions, we carried out some transformations of the products (Scheme 3). For example, treatment of monobrominated product 2d with estrone under basic conditions delivered phenoxy-substituted product 9 in 65% yield via an SN2′ reaction. Additionally, azide and an indole were also suitable nucleophiles for SN2′ reactions, giving 10 and 11 in 50% and 79% isolated yields, respectively. Furthermore, a Suzuki coupling reaction of 2d with an arylboronic acid delivered coupling product 12 in 61% yield, and treatment of 2d with hexaldehyde gave alcohol 13 (63% yield) via an indium-mediated gem-difluoroallylation reaction.5b Reaction of dibrominated product 3a with an allyl Grignard reagent selectively replaced the allylic bromide to give compound 14. Subsequent electrophilic fluorination of 14 with Selectfluor in the presence of MeOH afforded α-CF2Br-substituted ether 15 in 41% yield. In addition, 14 could undergo a Pd-catalyzed intramolecular Heck reaction to generate fluoro-substituted cyclopentene 16 in 54% yield. Notably, both (Z)- and (E)-14 underwent these last two transformations to give a single product, thus eliminating the need to separate the isomers.
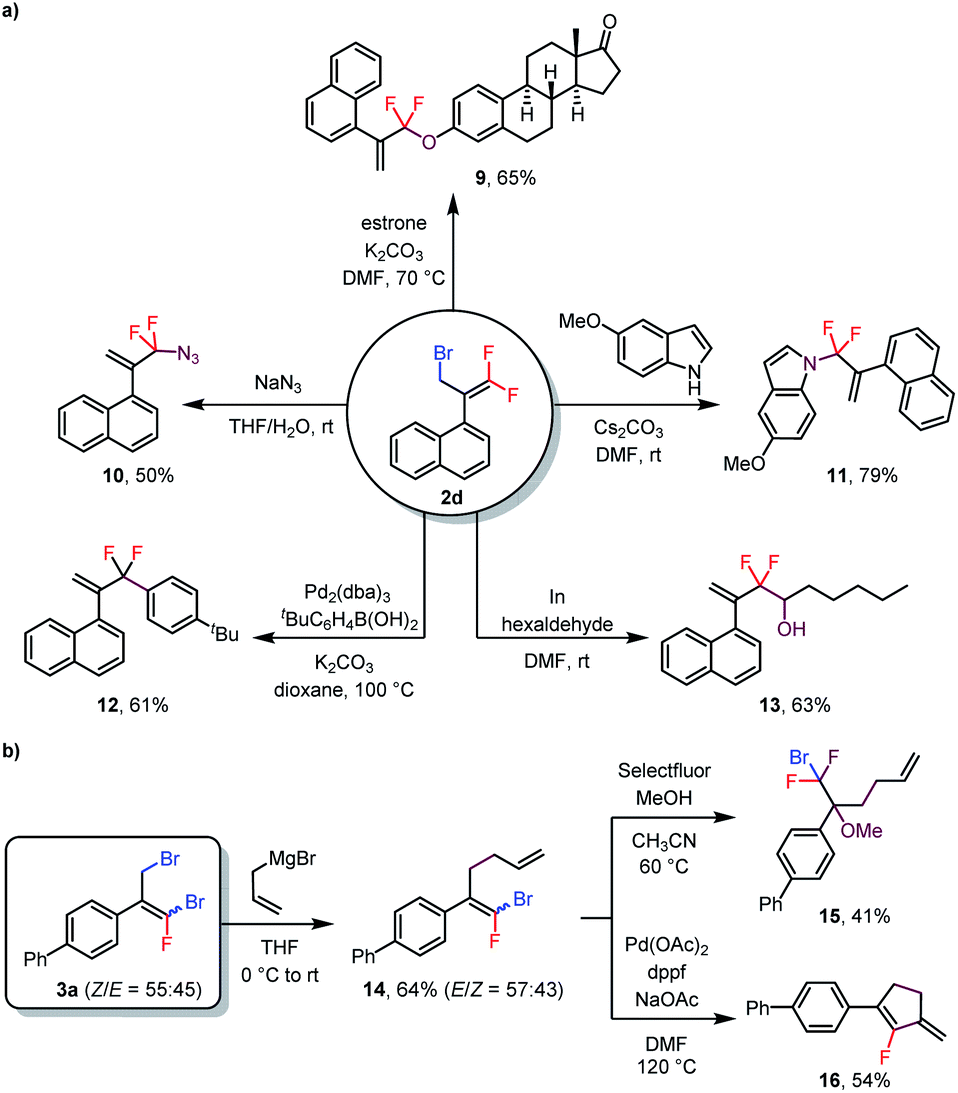 | ||
| Scheme 3 Transformations of products 2d and 3a. | ||
Conclusions
In summary, we have developed a protocol for aluminum-catalyzed halodefluorination reactions of trifluoromethyl- and difluoroalkylalkenes, which provide convenient access to fluorovinyls bearing an allylic halogen atom. Furthermore, di- and trisubstituted products could also be selectively synthesized by 1,3-migration of a halogen atom in the initially formed products and subsequent halodefluorination. The fluorine-containing products are useful building blocks for a variety of transformations. We are currently investigating the use of other nucleophiles for these aluminum-promoted defluorination reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21871147, 91956106, and 21602114) and the Fundamental Research Funds for Central Universities (2122018165). X.-C. W. thanks the Tencent Foundation for support via the Xplorer Prize.Notes and references
- For reviews, see: (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS; (c) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS; (d) Q. Liu, C. Ni and J. Hu, Natl. Sci. Rev., 2017, 4, 303 CrossRef CAS.
- P. Taverna, K. Rendahl, D. Jekic-McMullen, Y. Shao, K. Aardalen, F. Salangsang, L. Doyle, E. Moler and B. Hibner, Biochem. Pharmacol., 2007, 73, 44 CrossRef CAS.
- B. Bennett, A. Matagne, P. Michel, M. Leonard, M. Cornet, M.-A. Meeus and N. Toublanc, Neurotherapeutics, 2007, 4, 117 CrossRef CAS.
- D. Pozarowska, Clin. Ophthalmol., 2010, 4, 1229 CrossRef.
- (a) F. Tellier and R. Sauvêtre, J. Fluorine Chem., 1996, 76, 79 CrossRef CAS; (b) P. R. Mears and E. J. Thomas, Tetrahedron Lett., 2015, 56, 3980 CrossRef CAS; (c) H. Ueki and T. Kitazume, J. Org. Chem., 2005, 70, 9354 CrossRef CAS.
- T. Ooi, D. Uraguchi, N. Kagoshima and K. Maruoka, Tetrahedron Lett., 1997, 38, 5679 CrossRef CAS.
- K. Hirano, K. Fujita, H. Yorimitsu, H. Shinokubo and K. Oshima, Tetrahedron Lett., 2004, 45, 2555 CrossRef CAS.
- (a) V. J. Scott, R. Çelenligil-Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852 CrossRef CAS; (b) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS; (c) W. Gu, M. R. Haneline, C. Douvris and O. V. Ozerov, J. Am. Chem. Soc., 2009, 131, 11203 CrossRef CAS; (d) C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946 CrossRef CAS.
- R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676 CrossRef CAS.
- (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS; (b) J. Zhu, M. Pérez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417 CrossRef CAS; (c) J. Zhu, M. Pérez and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 8448 CrossRef CAS; (d) C. B. Caputo and D. W. Stephan, Organometallics, 2012, 31, 27 CrossRef CAS.
- (a) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248 CrossRef CAS; (b) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578 CrossRef CAS.
- J. Chen and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2015, 54, 6842 CrossRef CAS.
- (a) K. K. K. Goh, A. Sinha, C. Fraser and R. D. Young, RSC Adv., 2016, 6, 42708 RSC; (b) D. Mandal, R. Gupta and R. D. Young, J. Am. Chem. Soc., 2018, 140, 10682 CrossRef CAS; (c) A. K. Jaiswal, P. K. Prasad and R. D. Young, Chem.–Eur. J., 2019, 25, 6290 CrossRef CAS; (d) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572 CrossRef CAS.
- K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890 CrossRef CAS.
- G. Meißner, K. Kretschmar, T. Braun and E. Kemnitz, Angew. Chem., Int. Ed., 2017, 56, 16338 CrossRef.
- (a) Z.-Y. Zhang, Z.-Y. Liu, R.-T. Guo, Y.-Q. Zhao, X. Li and X.-C. Wang, Angew. Chem., Int. Ed., 2017, 56, 4028 CrossRef CAS; (b) X.-S. Tu, N.-N. Zeng, R.-Y. Li, Y.-Q. Zhao, D.-Z. Xie, Q. Peng and X.-C. Wang, Angew. Chem., Int. Ed., 2018, 57, 15096 CrossRef CAS; (c) Z.-Y. Liu, M. Zhang and X.-C. Wang, J. Am. Chem. Soc., 2020, 142, 581 CrossRef CAS.
- For selected reports on SN2′ reactions, see: (a) M. Bergeron, T. Johnson and J.-F. Paquin, Angew. Chem., Int. Ed., 2011, 50, 11112 CrossRef CAS; (b) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059 CrossRef CAS; (c) L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701 RSC.
- For selected reports of methods involving photoredox catalysis, see: (a) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908 CrossRef CAS; (b) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073 CrossRef CAS; (c) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723 CrossRef CAS.
- For selected reports of method involving transition-metal catalysis, see: (a) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947 CrossRef CAS; (b) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Chem, 2018, 4, 2201 CrossRef CAS; (c) S.-S. Yan, D.-S. Wu, J.-H. Ye, L. Gong, X. Zeng, C.-K. Ran, Y.-Y. Gui, J. Li and D.-G. Yu, ACS Catal., 2019, 9, 6987 CrossRef CAS; (d) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809 RSC; (e) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245 CrossRef CAS; (f) Z. Lin, Y. Lan and C. Wang, ACS Catal., 2019, 9, 775 CrossRef CAS; (g) Z. Lin, Y. Lan and C. Wang, Org. Lett., 2019, 21, 8316 CrossRef CAS.
- CCDC 2003727 ((Z)-3a) and 2003728 (8a) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- Al(C6F5)3(tol)0.5 is heat- and shock-sensitive, so extra caution and use of protective equipment (e.g. a blast shield) is necessary when handling this material. For the preparation and reactivities of Al(C6F5)3(tol)0.5, see: (a) S. Feng, G. R. Roof and E. Y.-X. Chen, Organometallics, 2002, 21, 832 CrossRef CAS; (b) D. Chakraborty and E. Y.-X. Chen, Inorg. Chem. Commun., 2002, 5, 698 CrossRef CAS; (c) J. Chen and E. Y.-X. Chen, Dalton Trans., 2016, 45, 6105 RSC.
- This reaction does not work catalytically.
- For selected reports of SNV reactions of gem-difluoroalkenes, see: (a) J. Ichikawa, Y. Wada, H. Miyazaki, T. Mori and H. Kuroki, Org. Lett., 2003, 5, 1455 CrossRef CAS; (b) Y. Xiong, X. Zhang, T. Huang and S. Cao, J. Org. Chem., 2014, 79, 6395 CrossRef CAS; (c) T. Fujita, M. Takazawa, K. Sugiyama, N. Suzuki and J. Ichikawa, Org. Lett., 2017, 19, 588 CrossRef CAS.
- This migration reaction is known, but was never utilized for multidefluorination reaction. See ref. 5b.
- J. B. Lambert and S. Zhang, J. Chem. Soc., Chem. Commun., 1993, 383 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2003727 and 2003728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03883k |
| This journal is © The Royal Society of Chemistry 2020 |