
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLate stage C–H functionalization via chalcogen and pnictogen salts
Christopher B.
Kelly
 * and
Rosaura
Padilla-Salinas
* and
Rosaura
Padilla-Salinas
Discovery Process Research, Janssen Research & Development LLC, 1400 McKean Road, Spring House, Pennsylvania 19477, USA. E-mail: ckelly5@its.jnj.com
First published on 7th September 2020
Abstract
Late-stage functionalization (LSF) of heteroarenes can dramatically accelerate SAR studies by enabling the installation of functional groups that would otherwise complicate a synthetic sequence. Although heteroaryl halides and boronic esters have well-established chemistries for LSF, alternatives that enable site-selective C–H functionalization are highly attractive. Recently, three unrelated cationic groups (phosphonium, pyridinium, and thianthrenium), which can replace C–H bonds late stage, have been identified as precursors to various functional groups. This review will discuss the synthesis and application of these three salts with an emphasis on their use for LSF and application to medicinal chemistry.
1. Introduction
Reaction efficiency is an important consideration in many industrial sectors and is paramount within the chemical community.1 The most efficient processes focus on preparing materials from commodity chemicals using non-hazardous reagents while simultaneously reducing waste. One practical means of achieving improved synthetic efficiency is through C–H functionalization. C–H functionalization processes allow materials like petroleum products to become viable starting points for the construction of complex molecules that treat disease, monomers for the polymers that comprise everyday items, or crop protectants to ensure adequate food supplies.2In addition to dramatically increasing molecular complexity of simple molecules in short order, in the context of the pharmaceutical industry, C–H functionalization offers opportunities for the late-stage functionalization (LSF) of pre-clinical candidates. LSF allows for improvements of targets in many ways such potency/solubility or mitigation of off-target interactions/metabolism.3 Further, LSF via C–H functionalization can simplify the overall synthetic sequence to an active pharmaceutical ingredient (API) or enable structure–activity relationship (SAR) studies that would be otherwise intractable using traditional approaches. Some notable examples of this are given in Scheme 1.4
 | ||
| Scheme 1 Strategies towards C–H functionalization and pharmaceutical applications. | ||
Although the merits of C–H functionalization are clear, the challenges associated with this synthetic paradigm are plentiful. Selectivity in C–H functionalization and controlled reactivity are chief concerns when developing such processes. Many of the recent developments in C–H functionalization stem from the realm of transition metal catalysis5 or photoredox chemistry (Scheme 1).6 In the case of the former (which has seen broad adoption in both medicinal and process chemistry), some type of directing group is typically employed to tackle the challenge of targeting a specific C–H bond and to facilitate the now canonical concerted metalation–deprotonation (CMD) component of many of these reactions.7a Alternatively, a two-step approach to installing certain groups may be used (e.g. iridium-catalyzed borylation followed by classical cross-coupling processes) that circumvents the sometimes restrictive nature of CMD processes by proceeding through an oxidative addition-based mechanism for C–H functionalization.7b Regardless of the operative mechanism, transition metal-mediated C–H functionalization reactions have seen a flurry of attention in recent years, leading to the creation of institutes like the NSF's Center for Selective C–H Functionalization (CCHF).5c Although a powerful synthetic tool, one caveat of transition metal-mediated C–H functionalization is the need for chemists to tailor their synthetic approach around the C–H functionalization event which may require directing groups that are difficult to remove.
In the case of radical processes, selectivity is generally a function of radical structure and/or the radical acceptor. The venerable Minisci reaction8 is the archetype for the benefits and pitfalls of radical processes; the process is highly LSF amenable but can lead to undesired reactivity (regiochemical mixtures, over-alkylation etc.). These facets of radical reactions can be largely attributable to the sometimes-indiscriminate nature of odd-electron intermediates. The advent of Ni/photoredox dual catalysis9 has alleviated some of the challenges of radical-mediated arene functionalization but comes at the expense of programmed reactivity (i.e. pre-functionalization of one of the components of the reaction). Not surprisingly, this often takes the form of halogenated or pseudo-halogenated species with most cases being heteroaryl or aryl bromides. Further, from an industrial standpoint, photoredox processes can present challenges beyond medicinal chemistry applications and advances in photoflow technology and photochemical reactor design are key to making these reactions more feasible on scale.10,11
Recently, a series of seemingly unrelated reports have revealed a paradigm by which net arene C–H functionalization can be achieved (and in some cases without the need for any transition metal catalysts) (Scheme 2). The crux of these approaches is the installation of a chalcogen or pnictogen cationic motif onto the arene in place of a C–H bond. The regiochemical selectivity of functionalization is dictated by the innate electronics of the arene and, in some cases, by the reaction conditions used in the activation process. These chalcogen or pnictogen salts enable an array of disconnections to be realized, ranging from net C–H trifluoromethylation to net C–H amination to net C–H deuteration. This Minireview will examine the C–H functionalization processes used to prepare these salts and their unique reactivities, with special attention given to potential pharmaceutical applications such as LSF.
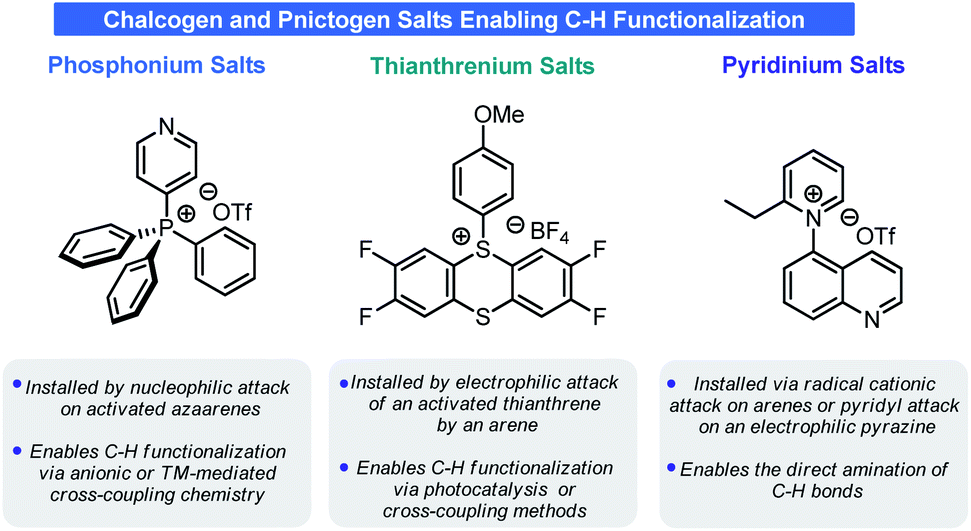 | ||
| Scheme 2 Examples of chalcogen and pnictogen salts examined in this Minireview. | ||
2. Pnictogen
Pyridinium salts
Pyridinium salts are a structurally diverse and well-established class of nitrogen-based salts that feature a six-membered heteroaromatic. Not surprisingly, pyridinium salts have been used as acylating agents, surfactants, phase transfer catalysts, and even biocides/bioactive agents. Recently, so called “Katritzky salts” and other N-functionalized pyridinium salts have experienced a surge in popularity, largely driven by their redox and photophysical properties.12 Zincke was the first to describe the synthesis of N-alkyl pyridinium salts in 1904.13 Zincke's route involves an amine exchange process that first converts pyridine into N-2(2,4-dinitrophenyl)pyridinium chloride, also known as the Zincke salt, followed by SNAr with the appropriate alkyl or aromatic amine. In the 1980s, Katritzky discovered that N-alkyl pyridinium salts could also be synthesized from the condensation of pyrylium salts with primary and secondary amines via SNAr (Scheme 3A).14,15 This approach was notable because several nucleophilic functional groups could be reacted to forge C–N, C–O, C–C, C–P, S–X, and C–S bonds. In subsequent work, he demonstrated alkyl pyridinium salts reacted with nitroalkane and ethylmalonate anions by a radical nucleophilic substitution mechanism. Although this indicated pyridinium ions were redox-active, their redox properties were largely overlooked given that, until recently, radical chemistry had the connotation of being uncontrolled and impractical. Thus, classical reactivity modes of pyridinium salts that were recognized were: (i) the enhanced susceptibility to nucleophilic addition and displacement at the 2, 4, and 6-positions (ii) ring opening after displacement depending on the reaction conditions and (iii) deprotonation and subsequent reactivity of the alkyl group of alkyl pyridiniums.12 It should be noted that aryl 2,4,6-substituted pyridinium salts are unreactive in SNAr at the ipso carbon due to steric congestion from the 2- and 6-substituents (Scheme 3B).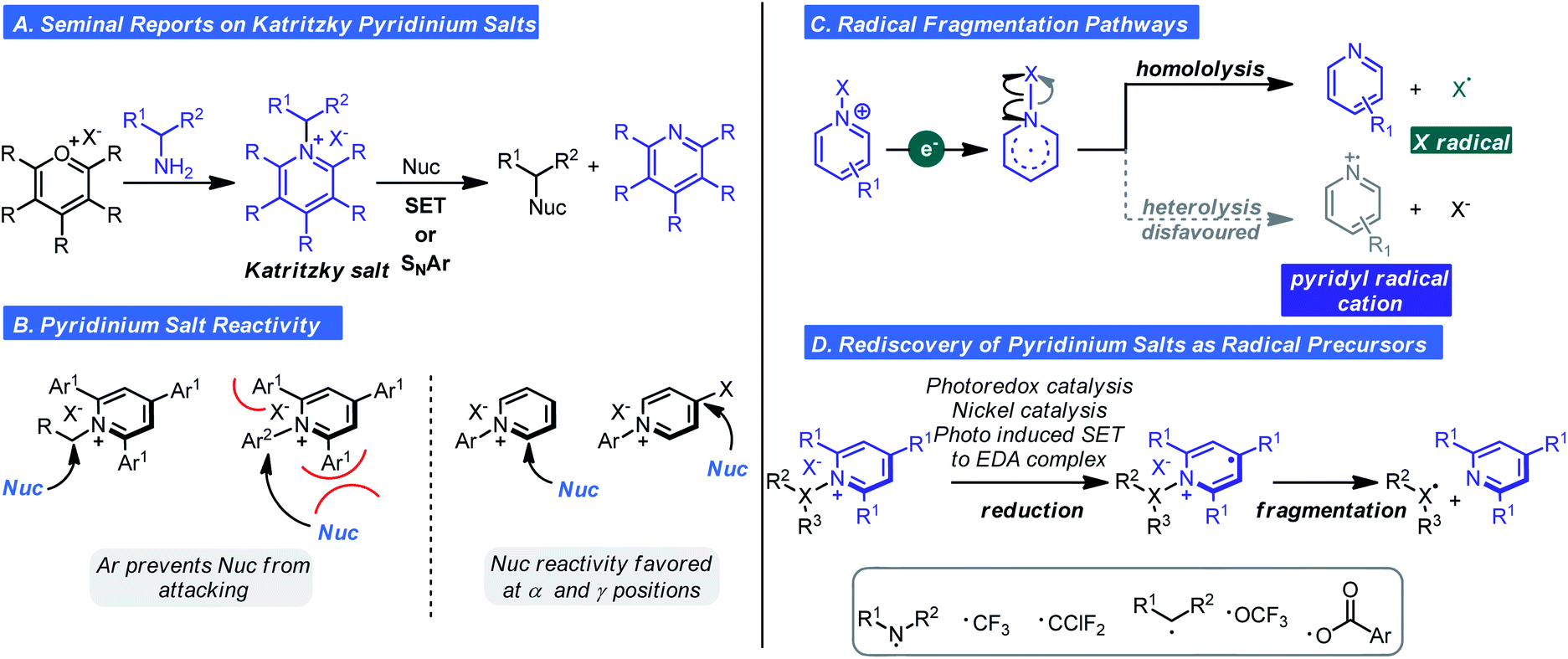 | ||
| Scheme 3 Historical context and new applications of pyridinium salt reactivity. | ||
The advent of photoredox chemistry16 inspired chemists to reconsider the utility of pyridinium salts in radical-mediated transformations. In 2015, Studer reported a method for the direct amidation of indoles, and pyrroles with pyridinium salts under photoredox conditions.17 The pyridinium salts described by Studer were those prepared from hydrazines to generate N-amino pyridinium ions. Reduction of this species by the excited state of a Ru photocatalysis results in generation of an N-centered radical by way of N–N bond homolysis. Aminyl radicals are electrophilic and will engage electron-rich π systems, thus explaining the observed reactivity. This report spawned a concerted effort to examine the reactivity of pyridinium ions in odd-electron reaction manifolds. A related important finding was reported shortly afterward, wherein Watson and co-workers found alkyl radicals could be generated from pyridinium ions by SET/homolytic C–N fragmentation and used in Ni-catalyzed cross-coupling of arylboronic acids (Scheme 3C).18 Homolytic fragmentation by these pathways (photoredox or non-photoredox) has become the focal point of research for pyridinium ion chemistry in the past five years and has enabled the generation of carbon-, nitrogen-, and oxygen-based radicals (Scheme 3D).19
An alternative pathway for the fragmentation of pyridinium ions was largely ignored in these studies: heterolysis thus generating a pyridinium radical cation. This was likely due to the types of groups appended to the cationic nitrogen and methods of activation employed favouring the homolysis pathway (Scheme 3C). However, as observed with Studer's aminyl radicals or radical cations are extremely electrophilic and will react with arenes by way of C–H functionalization. With these considerations in mind, Togni,20,21 Carreira,21 and Ritter22 concurrently and independently reported that pyridyl radical cations could be accessed from select N-substituted pyridinium reagents. The first observation of heterolysis was by Togni whom observed this phenomenon when developing a trifluoromethylation process involving pyridinium ions.20 It was noted that a side product was C–H functionalization to give a new N-aryl pyridinium salt. The observation inspired the hypothesis that heterolysis could become the major pathway depending on pyridinium structure. Specifically, it was posited that pyridinium substitution pattern and the nature of the N–Y bond would dictate the balance between heterolysis and homolysis (Scheme 3C). As such, Carreira and Togni found that if the Y group were a triflate (1 in Scheme 4), heterolysis would be the predominant pathway. Using this reagent, they found they could readily affect the pyridination of arenes under photoredox conditions using a Ru photocatalyst (Scheme 4A).21 Mechanistically, SET reduction of the pyridinium reagent occurs by way of oxidative quenching of the excited state of the photocatalyst (Scheme 4C). Expulsion of the stable triflate anion is the driving force for the heterolytic cleavage of the N–X bond. The pyridyl radical (II) undergoes addition to the arene generating intermediate (III), which undergoes oxidation and deprotonation affording the pyridinium salt (IV). Control experiments confirmed light and the Ru-photocatalyst are essential and evidence for the existence of the pyridyl radical cation was obtained by photoinduced EPR studies.
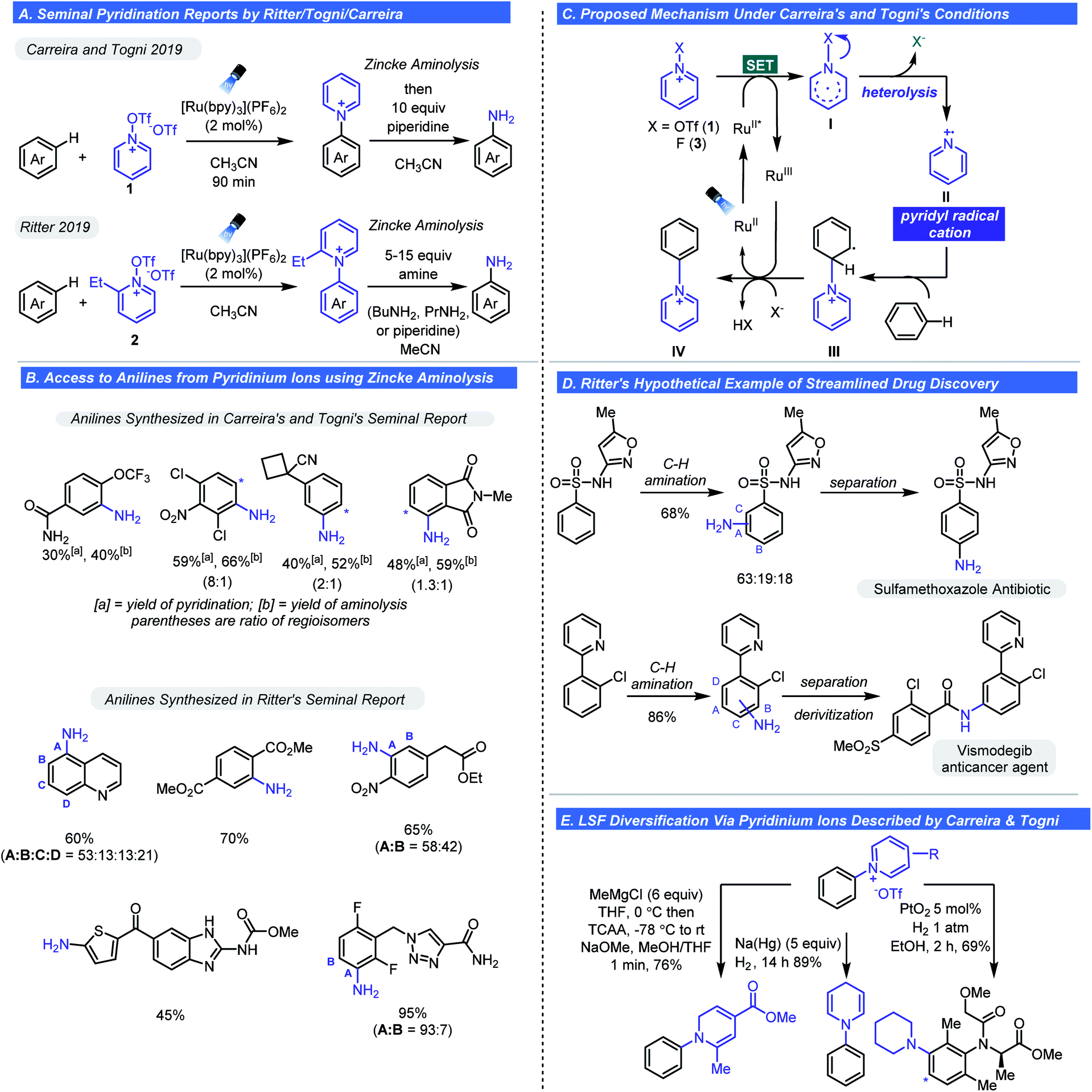 | ||
| Scheme 4 Seminal reports on late-stage C–H amination enabled by the pyridyl radical cation. | ||
A wide array of complex electron-rich and electron-poor arenes were pyridinated to give their N-aryl pyridinium triflate salts. Notably, electron-poor arenes that were unreactive with previous approaches could be successfully pyridinated in moderate yields. The reaction conditions tolerate nitrile, ester, nitro, and amide functional groups. Lastly, heteroarenes such as pyridine and pyrazine could be tolerated whereas pyrimidines afforded low yields. Although these salts are academically interesting, the synthetic utility of this transformation was expounded by the in situ conversion of pyridinium salts into anilines via Zincke aminolysis with piperidine (Schemes 4A and C). Likewise, direct reduction of the pyridinium adducts enables facile access to piperidyl substituted arenes. One slight drawback is a consequence of the promiscuity of the pyridyl radical cation. The pyridination reaction gives regiochemical mixtures with certain arenes but is, on a whole, not especially selective. The origins for positional selectivity were not clear; however, it was noted that steric hindrance has little impact on the regioselectivity. Although potentially problematic for scale-up, this could be useful in an LSF setting (i.e. SAR for medicinal chemistry efforts) as these isomers could likely be separated by preparative LC methods.
At the same time, the Ritter group disclosed their direct C–H amination method using a slightly modified pyridinium triflate, 2 (Scheme 4A).22 The reagent can be easily synthesized from triflation of 2-ethylpyridine N-oxide or it can be prepared in situ. Electronically and structurally complex arenes were successfully pyridinated in moderate yields and selectivity (Scheme 4B). Ritter chose to focus mostly on complex substrates to emphasize LSF compatibility. The broad substrate scope is attributed to an early transition state for the fast-irreversible addition of the pyridinium radical cation ion to the arene (Scheme 4C).
Hammett and Brown–Stock analysis showed the pyridinium radical cation exhibits the lowest electronic selectivity profile with respect to regiochemical outcome compared to other cationic nitrogen and neutral radicals used for substitution reactions.
The reactivity of these new aminating species can have a profound impact on aniline synthesis in drug discovery and other industries. The introduction of the ArNH2 group into complex drug-like structures continues to be an arduous task that relies on nitration of the corresponding aromatic precursor followed by reduction, Buchwald–Hartwig amination, and SNAr chemistry. These approaches require additional synthetic manipulations to install protecting groups or groups required for cross-coupling (i.e. halide and -OTf) or specific electronic properties on the arene (SNAr and EAS nitration). Although amenable to LSF, the inherent restrictions or required pre-programing can limit what chemical space can be sampled. The C–H pyridination/aminolysis sequence is poised to change how medicinal chemists can synthesize complex anilines and use N-functionalized pyridinium triflates as redox-active functional group transfer reagents to access diverse chemical matter. Using a hypothetical scenario, Ritter elegantly showed how a medicinal chemist can apply the amination chemistry for LSF of complex drug molecules (Scheme 4D). The synthesis of two marketed drugs via C–H pyridination–aminolysis sequence was achieved in moderate yields. Carreira also investigated the application of pyridinium salts as synthetic handles for late-stage diversification (Scheme 4E). N-Phenyl pyridiniums were selectively functionalized using diverse sets of conditions to afford N-arylated piperidines, 1,2- and 1,4-dihydropyridines, and dienes in moderate to high yields. This approach is ideal for SAR because structurally diverse compounds can be rapidly accessed from a single pyridinium intermediate without having to design a different route for each analogue.
Based on earlier work, Ritter subsequently developed a one-pot, two-step, direct C–H pyridonation of (hetero-)arenes using chloropyridinium radical cations.23 This approach enables an unprecedented net C–H functionalization of an arene with the nitrogen atom of a pyridone under mild conditions. Pyridones are challenging substrates because of their low nucleophilicity and ambidentate nature but often appear in therapeutic molecules and in SAR studies. To overcome these challenges, Ritter cleverly repurposed the pyridyl radical cation reagents as masked pyridone surrogates (A and B, Scheme 5A). The reactivity of the scaffold was fine-tuned through installation of a chloro substituent at the 2- or 4-position. In addition to priming the scaffold for hydrolysis to either a 2- or 4-pyridione, the electron-withdrawing nature of chlorine lowers the reduction potential. Ultimately this promotes fragmentation to the pyridyl radical cation.
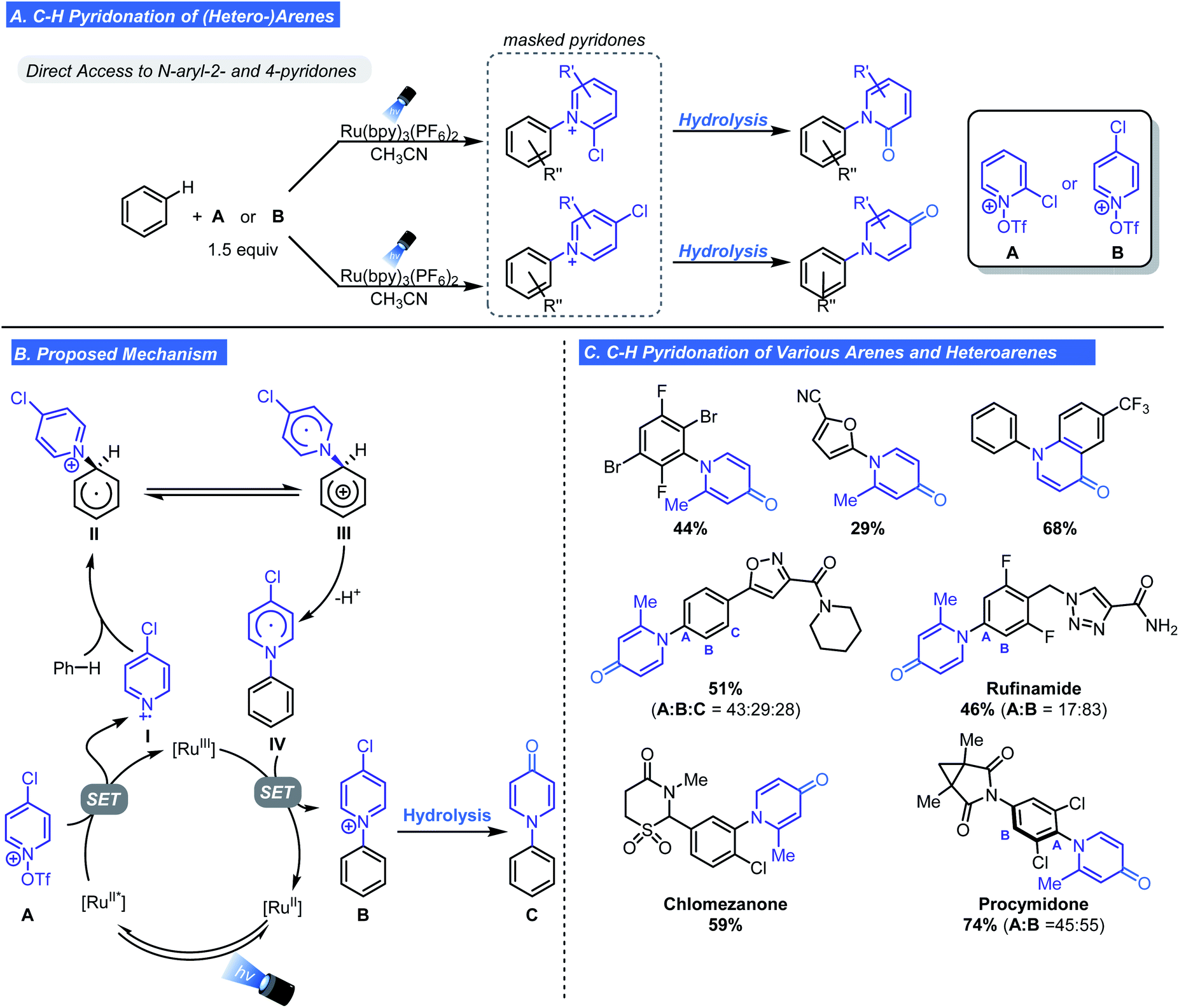 | ||
| Scheme 5 Ritter's C–H pyridonation of arenes. | ||
Scheme 5B outlines the proposed mechanism which is near identical to the one proposed in the Ritter/Togni/Carreira seminal reports. A key detail is the equilibrium between II and III which permits re-aromatization of the aryl ring via deprotonation. Subsequent single electron oxidation generates the aryl pyridinium salt from IV. To generate the desired product, a second hydrolysis step is necessary to give N-arylpyridone C. The utilization of tert-butylhydroperoxide during the hydrolysis step is crucial for minimizing competing decomposition pathways such as nucleophilic attack at the 2-position forming the free aniline. This method proved effective for the introduction of 4-pyridone motif onto an array of substituted arenes including highly electron-deficient and halogen-substituted arenes (Scheme 5C). The authors identified basic functional groups as problematic, rendering N-heterocycles incompatible. Additionally, C5-selectivity was observed for five membered heteroarenes, and bulky ortho substituents prevent C–H pyridonation. LSF of drug molecules was demonstrated with the successful pyridonation of rufinamide, procymidone, and chlormezanone (Scheme 5C). A notable feature that makes this method suitable for SAR exploration is the ability to access a variety of N-aryl pyridone regioisomers.
As one might note in the scope of the respective pyridyl radical cation reports, the compatibility with basic heterocycles is limited. Thus, late stage C–H amination of pyridines or pyrazines using pyridinium ions poises a problem. However, a recent report by Fier and co-workers has revealed a way to take advantage of the pyridinium ions as intermediates for pyridine C–H functionalization. They recently developed a new multifunctional reagent that converts pyridines to Boc-protected 2-aminopyridines (Scheme 6A).24 Using the dicyanopyrazine (A), activation of Lewis basic pyridines by way of an SNAr reaction is possible (Scheme 6B). Reactive intermediate B is primed for C–N bond formation at the 2-position of the attacking pyridine, giving C. Collapse of this intermediate by deprotonation/N–O bond cleavage is aided by N,O-bis-(trimethylsilyl)acetamide (BSA) and gives imine D. It should be noted that BSA was critical as an appropriate HCl scavenger and expanded the tolerance of this C–H functionalization process to include protic and nucleophilic functionalities. Thermal isomerization of D gives E and subsequent Zn-mediated reduction with AcOH removes the dicyanopyrazine, affording the desired C–H aminated product (Scheme 6B). Experimental support for the proposed mechanism includes observation of the dihydropyridine species C and compound E by NMR spectroscopy. Isomerization of D to E was confirmed by LC/MS analysis of reaction at rt and 80 °C and structural assignment of Evia 1- and 2D NMR spectroscopy. The new multifunctional reagent can be prepared in bulk quantities (>200 g) from commercially available A and is a bench stable crystalline solid.
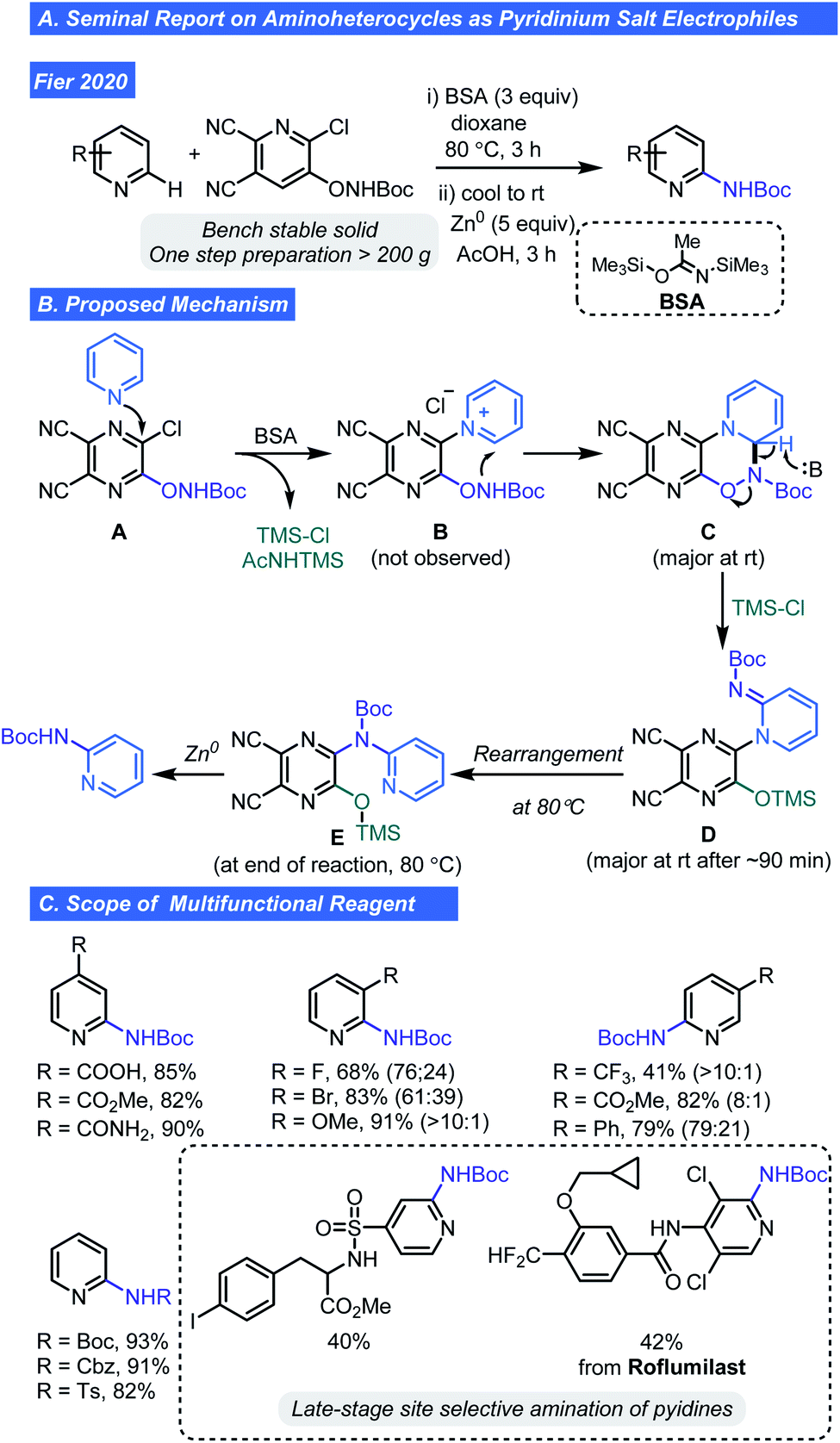 | ||
| Scheme 6 Seminal report site selective amination of pyridines. | ||
The authors were able to show the mild reaction conditions are robust and compatible with alcohols, carboxylic acids, N–H sulfonamides, amides, NH heterocycles, thioethers, aldehydes, and phenols (Scheme 6C). Selective formation of 2,3-disubstituted pyridines was observed for unsymmetrical 3-substituted pyridines bearing a –F, –Br, or –OMe groups and 2,5-selectivity for hetero(aryl), methoxycarbonyl, and trifluoromethyl substituted pyridines. Numbers in parentheses in Scheme 6 indicate the distribution of 2-positional regioisomers. The authors speculate that inductive and resonance effects including the relief of eclipsing interactions during rehybridization, are factors contributing to increased reactivity at C2. Low yields (<10%) were obtained with 2-substituted pyridines and diazines thus denoting a limitation of this approach. Successful late-stage amination of two drug like molecules containing an N–H bearing sulfonamide and amide functional group (e.g. roflumilast) clearly demonstrates the potential impact this chemistry can have on LSF of pyridines in drug discovery.
Taken together, these pyridinium-based methods, while powerful, seem to be restricted to C–H amination. However, the ability to generate pyridinium ions (Ritter/Togni/Carreira reports) or aminopyridines (Fier) provides a hypothetical avenue for further diversification using chemistry recently reported by Cornella. Cornella has shown that unsubstituted N-aryl pyridinium tertrafluoroborates (preparable by the Katritzky pyrylium salts exchange with aminoarenes or potentially by the methods reviewed here) can be engaged in SNAr chemistry. Thus, a wide range of disconnections become possible (Scheme 7).25 Indeed, Cornella showed that substitution of pyridinium ions by an array of nucleophiles was quite feasible and tolerated high levels of complexity on both the nucleophile and pyridinium salt (Scheme 7A). Key to the success of this transformation was the fact that 2- and 6-positions of the pyridinium ion were unoccupied which translates to lower steric hindrance and thus allows for SNAr reactivity. Further, the development of bench stable pyrylium tetrafluoroborate (Pyr-BF4) reagent was also critical.15 A pyrylium salt with a BF4 counter-ion was favoured because it has a high decomposition temperature and is stable/non-explosive unlike pyrylium perchlorate. With the developments in this section, a process involving C–H pyridination followed by SNAr can be envisioned to generate an array of complex molecules (Scheme 7B).
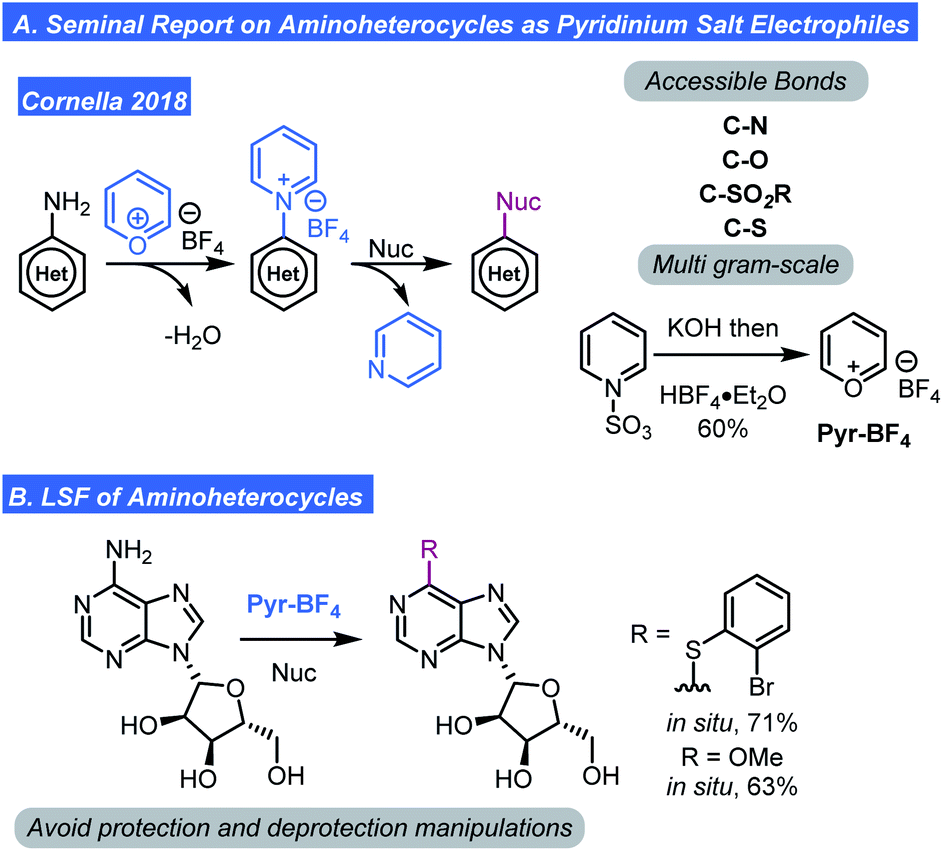 | ||
| Scheme 7 Selective functionalization of aminoheterocycles. | ||
Phosphonium salts
Most commonly associated with Wittig chemistry, phosphonium salts have also become competent intermediates for C–H functionalization. In 1987, Anders reported that treatment of pyridine or pyrazine with Tf2O and triphenylphosphine in the presence of Et3N results in the generation of heterocyclic triphenylphosphonium salts by way of a dearomatized intermediate (Scheme 8).26 Although other methods exist for generating such salts,27 this approach is notable as it relies on simple commodity materials and proceeds in a regioselective manner. Specifically, phosphination occurred at the 4-position of pyridines and the 2-position of pyrazines. However, the developments by Anders went largely unnoticed until McNally and co-workers saw this as an opportunity for selective C–H functionalization of basic nitrogen heterocycles. Building on Anders' initial report, the McNally group has thoroughly explored this unique C–H functionalization reaction. Beginning in 2016, it was demonstrated that the Anders–McNally phosphination process could be used in far more complex systems than the original 1987 report.28 An array of drug molecules (e.g. Chantix, Claritin etc.), natural products (e.g. nicotine, cinchonidine etc.), and complex building blocks (Scheme 8) readily underwent phosphination to give their corresponding triphenylphosphonium triflate salts (herein called Anders–McNally salts). Despite being limited to pyridines, pyrazines, and pyrimidines, the structural diversity was quite high and tolerated electron-rich and electron-poor variants of these basic heterocycles.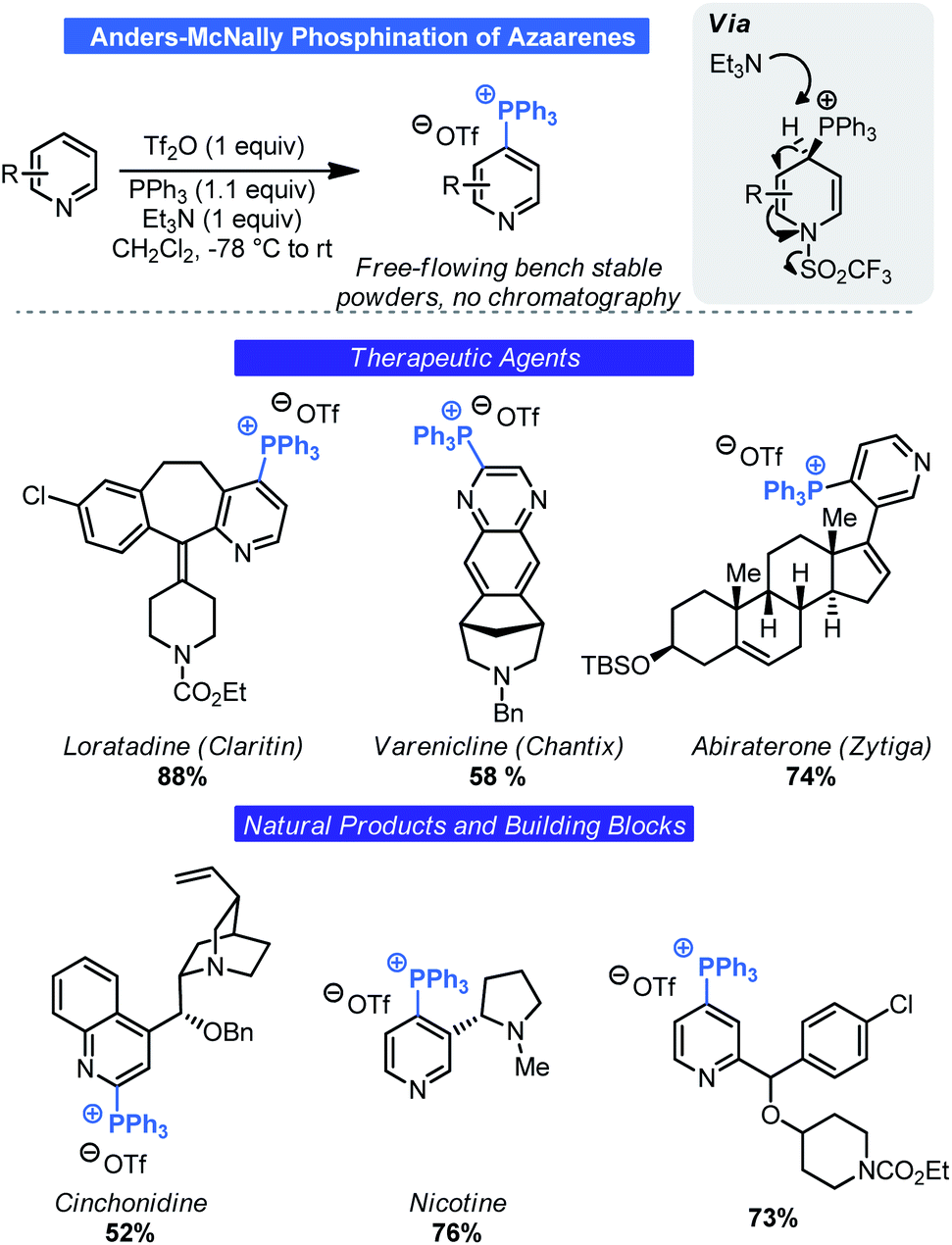 | ||
| Scheme 8 Phosphination of azaarenes described by McNally and Anders. | ||
Although impressive in its own right, one potential criticism of this initial report was that small-molecule therapeutic agents will often contain multiple azaarene motifs (e.g. Ibrance, Tasigna, Gleevec).29 Recognizing this as a potential complication for LSF, McNally investigated approaches for selective phosphination in poly azaarene systems.30 Using a mechanistic approach (Scheme 9), three parameters were identified that could control selectivity in phosphination namely: acyl blocking, base-switching, and kinetic trapping. A fourth, order of addition, was also considered. Each parameter was governed by a different facet of reactivity (azaarene Lewis basicity, steric congestion around the site of deprotonation or triflation, and base structure) and thus initially it was assumed that no singular approach could be uniformly applied. Although McNally devised four distinct approaches that could be used to impart selectivity (Scheme 9), ultimately it was found that “base switching” (involving double-dearomatization) and inverse addition (addition of PPh3 first then Tf2O) were the most efficacious. Both rely on kinetic control to facilitate selectivity. In the case of the former, judicious choice of base allows for selective re-aromatization over extrusion of PPh3 upon workup by way of a site selective deprotonation event. This is especially advantageous in instances where only subtle differences exist between azaarenes (e.g. bipyridyl systems). Large differences in electronics could take advantage of the acylation-blocking strategy. Acylation switching is viable for substrates with major steric differences around the pyridine nitrogen (e.g. 2-substituted versus non-2-unsubstituted pyridines). Likewise, inverse addition enables kinetic trapping of the dearomatized intermediate that forms fastest. One potential improvement from the lens of process chemistry would be to devise a means to conduct the reaction without the need for highly cryogenic temperatures.
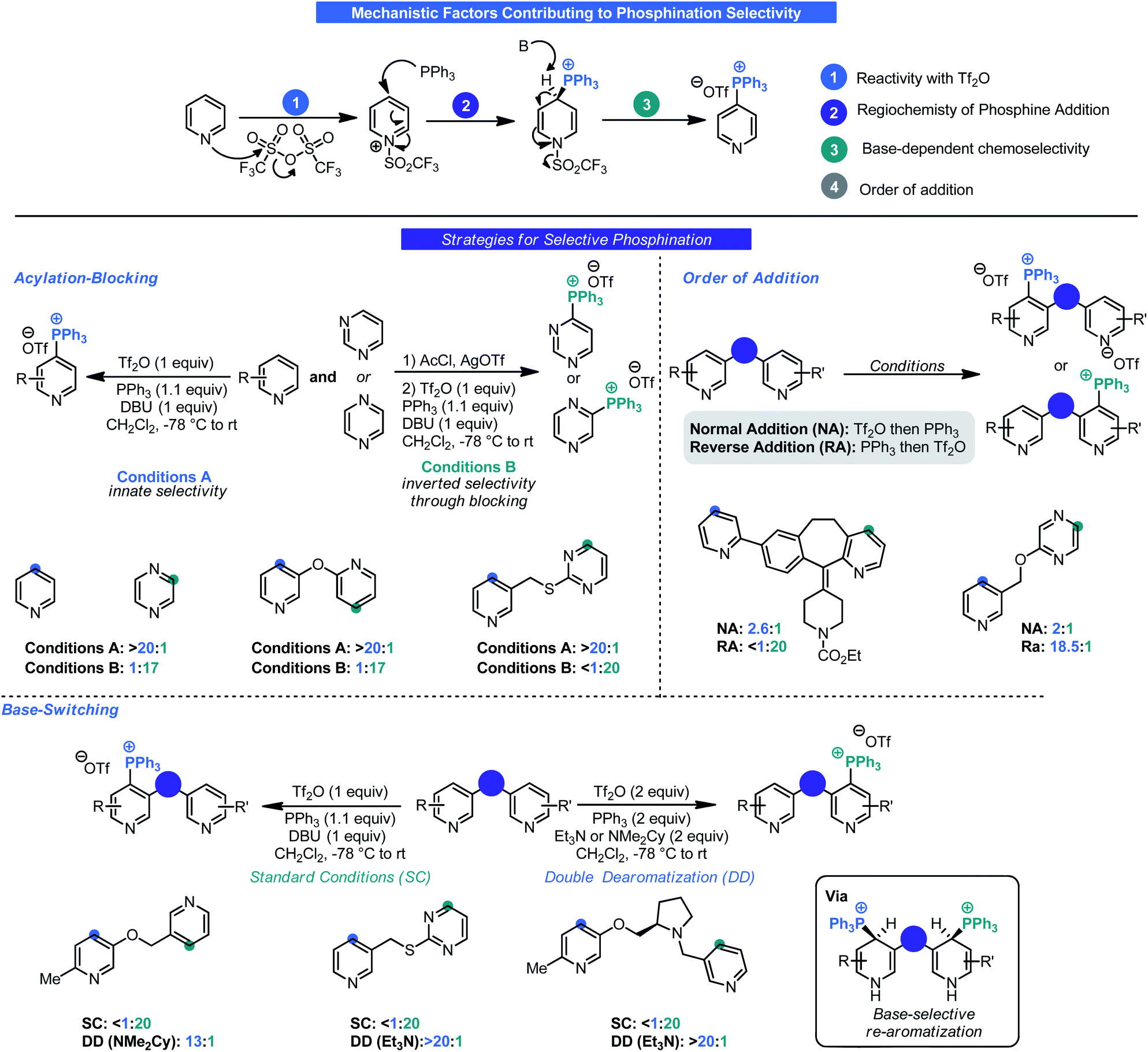 | ||
| Scheme 9 Various approaches to selective C–H phosphination. | ||
Without direct applications of these salts, the phosphination process, although academically intriguing, would likely have remained a novelty. McNally and co-workers have revealed the true power of this approach by examining the general reactivity of Anders–McNally salts for forging an array of bonds. A summary of a series of comprehensive studies is given in Scheme 10. Thioetherification,31a etherification,31b amination31c deuteration,31d halogenation,31e arylation,31f and alkylation31g are all feasible using these salts. Although no standard set of conditions exists for all these transformations, each has been optimized for the task at hand and are ideal for LSF. It is clear from these transformations that the potential to access new chemical space at a specified position from a common intermediate is unmatched, especially given that 4-position C–H functionalization processes of pyridines are rare. This is ideal for SAR campaigns where variation at a specific position to improve binding or to prevent metabolic degradation may be key.
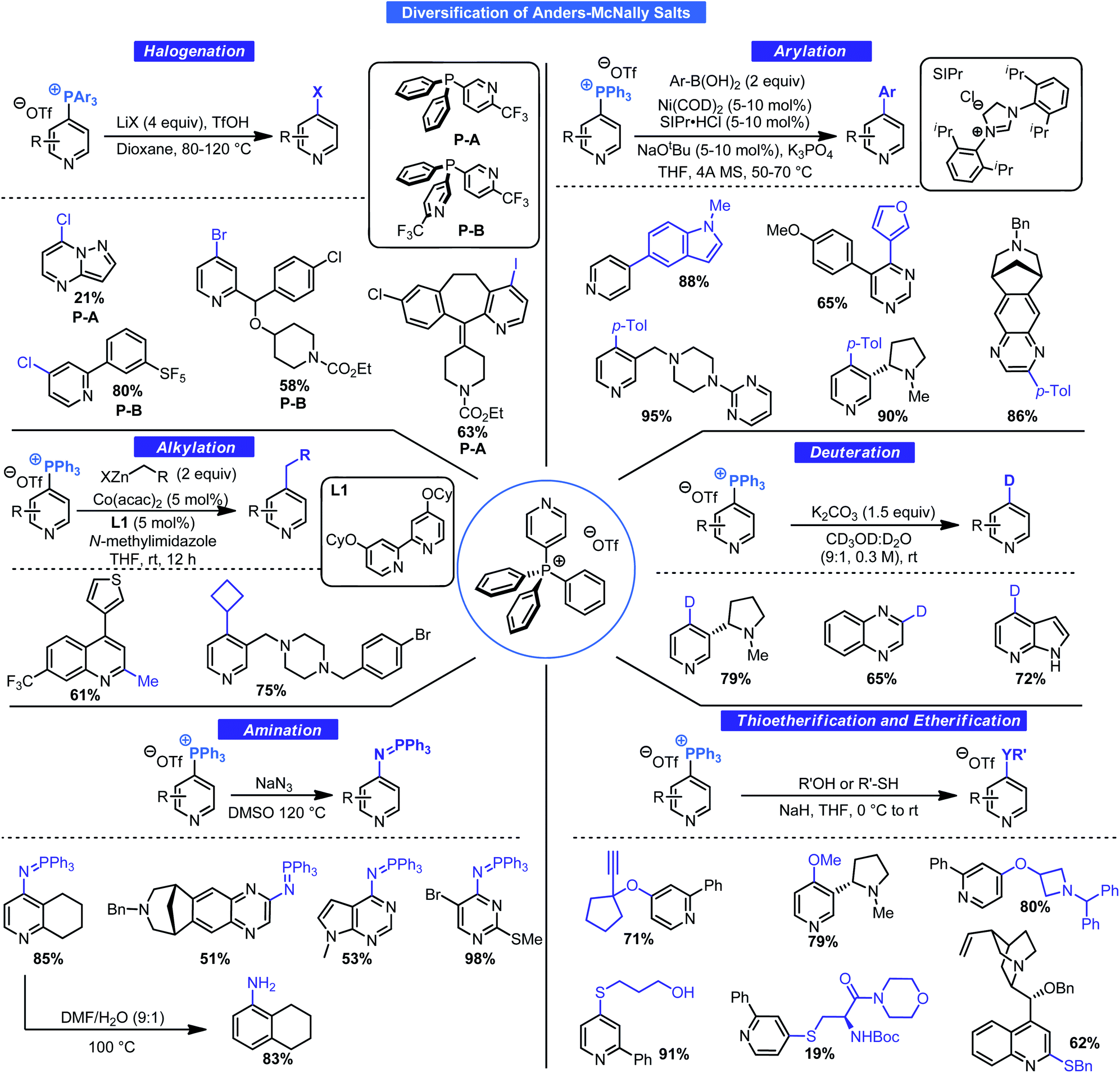 | ||
| Scheme 10 Interconversion of Anders–McNally salts into various functional groups. | ||
Mechanistically, it was noted that these functionalization reactions likely do not proceed through a classical SNAr-type mechanism but rather through attack at phosphorous followed by “contractive coupling” (CC), a process reminiscent of reductive elimination (RE) in transition metal catalysis. With this in mind, the possibility of mimicking classical RE to forge a Csp2–Csp2 bond by way of CC was examined by McNally.32 One key challenge in this process was devising a means to retain the “C–H functionalization” aspect of phosphination. To do so, a custom phosphine was prepared from diphenyl phosphine and methyl acrylate. The necessary phosphonium salt was then assembled by way of a two-step double C–H phosphination. Treatment of this phosphonium salt with base did facilitate CC but also gave a mixture of protodephosphination products. Treatment with triflic acid in a polar solvent, however, furnished the desired CC outcome. A Hammett-type analysis demonstrated that the rate of contractive coupling was directly related to the structure of the aryl groups on phosphorus; with more electron-rich arenes retarding the rate and more electron-withdrawing substituents accelerating CC. McNally and co-workers demonstrated that this reaction could not only be used with small azaarene species but even in an LSF fashion on drug molecules (Scheme 11). One caveat of this reaction is the sequence in which the two C–H functionalization events are performed is critical for reaction success. When using 3-subsituted pyridines, it is imperative that these be coupled onto phosphorous during the second C–H functionalization event. Because of a combination of steric and electronic factors, triflation favors the 3-subsituted azaarenes phosphine and can lead to deactivation (and hence prevent attack on another arene by phosphorous). Thus, the 2-substituted pyridine (or non-3-substituted pyridine) must be phosphinated first followed by C–H phosphination of the 3-substituted pyridine. It was later found that chloropyridines underwent phosphination in an SNAr-type fashion and could be combined with the approach here to prepare an array of bis-azines.33
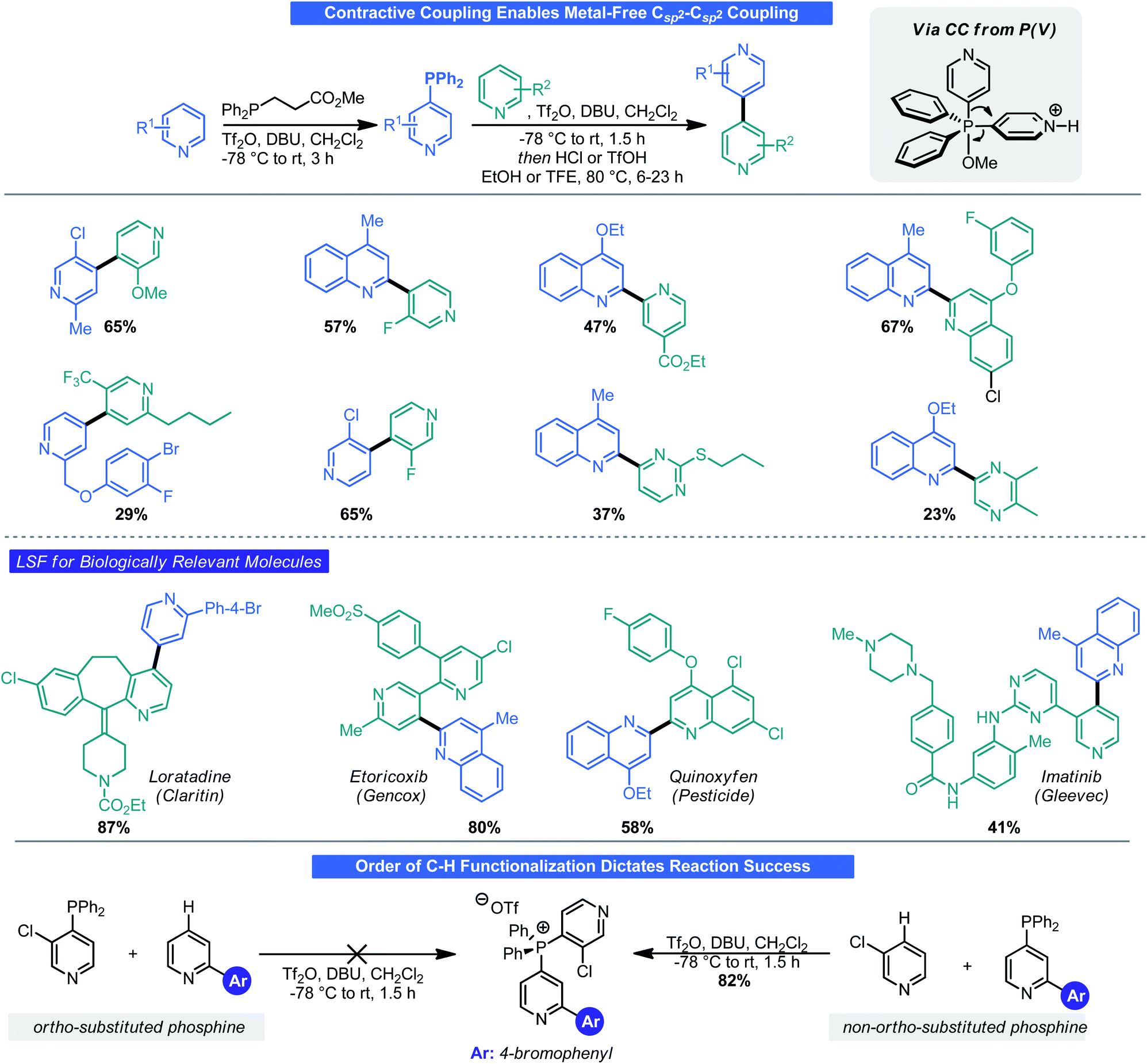 | ||
| Scheme 11 Contractive coupling via P(V) generates poly azaarenes. Yields given are of contractive coupling step. Yields for C–H functionalization steps ranged from ∼50–90%. | ||
Given recent attention by the chemical community towards odd-electron processes34 and the tendency for electron-deficient pyridines to engage in single-electron transfer (SET) events,35 it comes as no surprise that the phosphonium salts discussed here are redox-active. In attempting a borylation process, McNally and co-workers serendipitously discovered a radical–radical coupling process that forges the same types of bonds as the aforementioned CC work.36 Although initially employing photoredox catalysis, it was later discovered that this process proceeded without the aid of visible light. Indeed, the combination of B2Pin2 and 4-cyanopyridine results in a redox-active species which can transfer an electron via an inner sphere mechanism to Anders–McNally salts (Scheme 12). The now reduced salt can engage a 4-cyanopyridine radical in a radical–radical coupling event. Overall, this surmounts to a net Minisci process between two pyridines which is generally kinetically prohibited because of radical/π electronic mismatch. McNally and co-workers demonstrated this process was amenable to a wide array of phosphonium salts and 4-cyanopyridines. The reaction tolerates numerous functional groups including olefins, various saturated/unsaturated heterocycles, protic groups, and drug-like molecules.
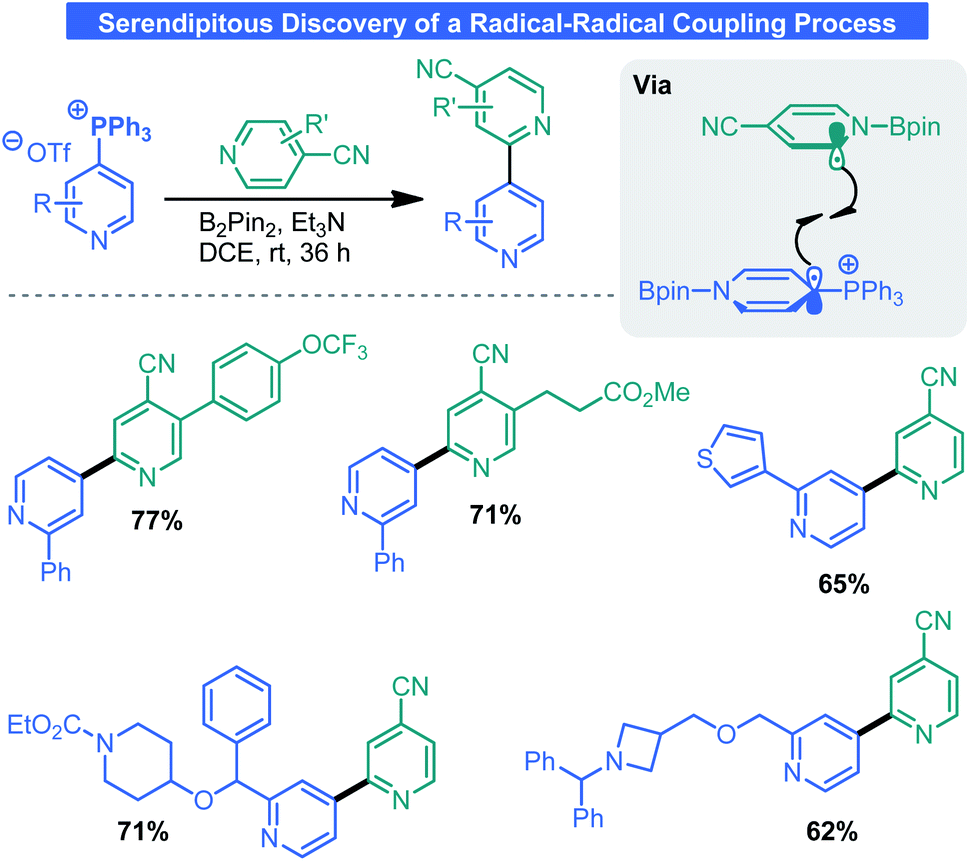 | ||
| Scheme 12 Radical–radical coupling enables 2-4′-bipyridyl synthesis. | ||
3. Chalcogen
Thianthrenium salts
Unlike the pnictogen salts, chalcogen salts are, in general, much less common, owing to octet restrictions with one of its members (oxygen). Thus, sulfonium salts are much more common than oxonium salts (with the aforementioned pyrylium salts being the notable exception). Indeed, sulfonium salts have seen use in a range of reactions either as critical intermediates (e.g. the Swern oxidation) or as bench stable reagents (e.g. Corey–Chaykovsky reagents).37 More recently these cationic sulfur species have found use in cyclopropanation,38 electrophilic trifluoromethylation,39 and even as electrophiles in cross-coupling processes.40 However, in most cases, these species are prepared by treating the corresponding sulfide with a strongly electrophilic alkylating reagent, thus diminishing their abilities to participate in LSF-type processes.In 1971, Shine reported that thianthrenium perchlorate underwent electrophilic aromatic substitution with electron-rich arenes such as anisole, to generate the corresponding aryl sulfonium salts (Scheme 13A).41 This unusual reactivity stems from the radical cationic nature of thianthrenium perchlorate (TP), which was first synthesized and described in 1962 by Lucken.42 This same type of reactivity could be replicated with AlCl3 and thiantherene sulfoxide.41 These discoveries went largely unnoticed by the chemical community because of the inherent instability of TP (detonates upon drying). Still, this was notable as it is one of the few methods where aryl sulfonium ions can be generated by C–H functionalization.
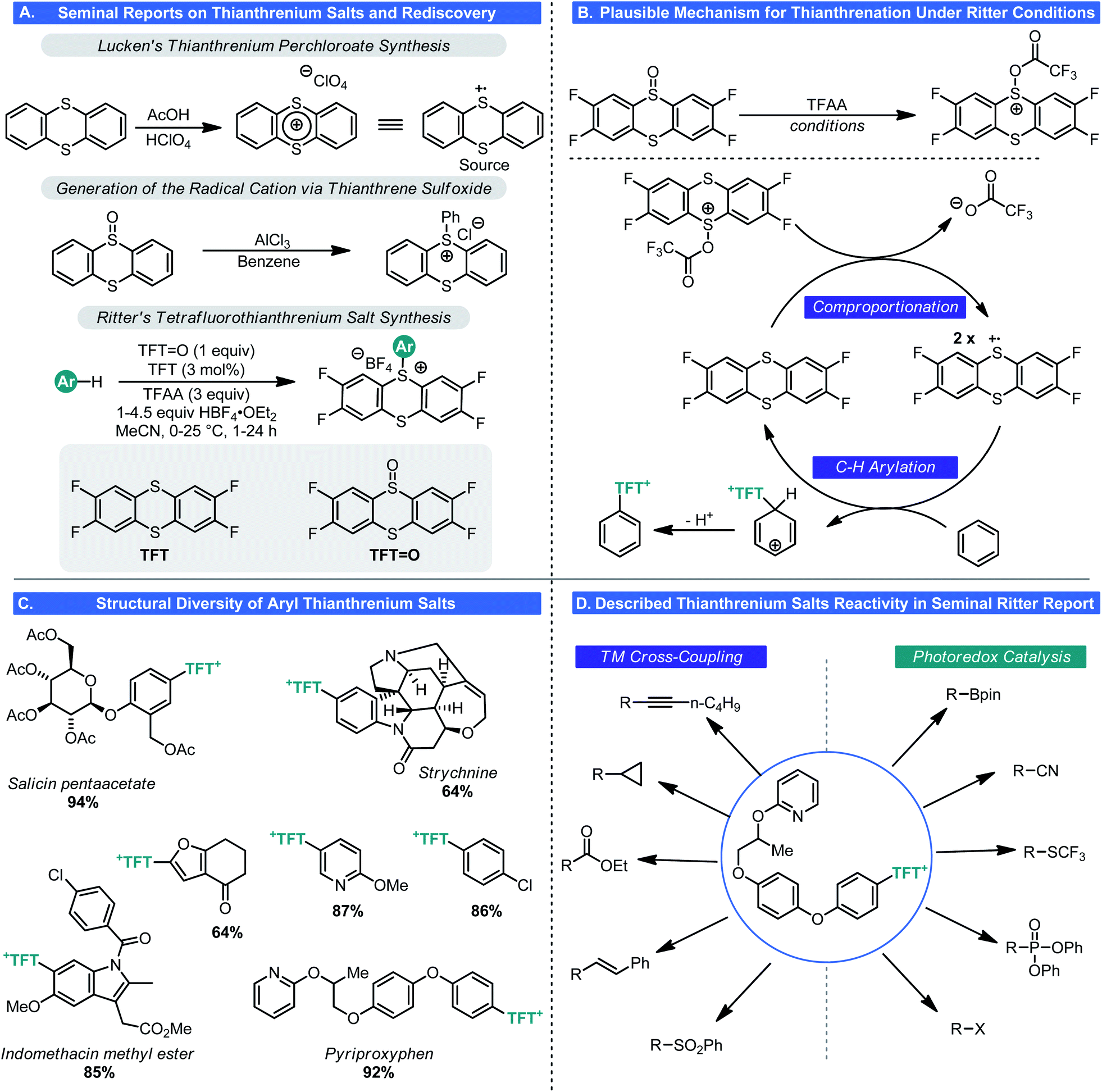 | ||
| Scheme 13 Discovery, development, and applications of TFT+ salts. | ||
As part of a program to devise new C–H functionalization process, Ritter and co-workers revisited these salts and their synthesis in 2019.43 In the modern era of chemistry, it is generally inadvisable to work with perchlorate-based reagents and hence Ritter examined alternative means to generate thianthrenium salts (Scheme 13A). A route to a tetrafluoro analogue was realized wherein 1,2-difluorobenzene can be treated with disulfur dichloride and AlCl3 to give the corresponding tetrafluorothianthrene (TFT). Oxidation of this disulfur species to the monosulfoxide using iron(III) nitrate gave tetrafluorothianthrene-S-oxide (TFT![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). Rather than formally generating the tetrafluorothianthrenium salt, a process whereby the thianthrenium could be generated from these two species was envisioned. Electrophilic activation of TFTO by the combined action of HBF4 and TFAA results in a comproportionation reaction giving the tetrafluorothianthrenium radical cation, which has a violet appearance in solution (Scheme 13B). Electrochemical studies confirmed that this was likely the pathway by which the radical cation was being generated. From there, the inherent reactivity of this odd-electron species results in facile C–H bond functionalization to give S-aryl tetrafluorothianthrenium tetrafluoroborate (TFT+) salts. Similar to the Anders–McNally salts, a wide array of structurally diverse and complex compounds are amenable to C–H functionalization (Scheme 13C). These include natural products and pharmacons as well as small building blocks. In general, the types of arene most compatible with this process are those one would expect to fare best in EAS process: electron-rich aromatics or heteroaromatics though this is not exclusive. This nicely complements the reactivity of the Anders–McNally phosphination process as most azaarenes fail to undergo thianthrenation unless they bear an electron-donating group. Those that do allow for C–H functionalization at sites not observed when attempting C–H phosphination. Olefins also underwent thianthrenation, though a thianthrene-S-oxide (TO)/thianthrene system was used, giving thianthrenium tetrafluoroborate (TT+) salts instead.44 Very recently, Proctor has reported a similar approach to arene functionalization using dibenzothiophene S-oxide (DBTSO), demonstrating the generality for this approach to C–H functionalization.45
O). Rather than formally generating the tetrafluorothianthrenium salt, a process whereby the thianthrenium could be generated from these two species was envisioned. Electrophilic activation of TFTO by the combined action of HBF4 and TFAA results in a comproportionation reaction giving the tetrafluorothianthrenium radical cation, which has a violet appearance in solution (Scheme 13B). Electrochemical studies confirmed that this was likely the pathway by which the radical cation was being generated. From there, the inherent reactivity of this odd-electron species results in facile C–H bond functionalization to give S-aryl tetrafluorothianthrenium tetrafluoroborate (TFT+) salts. Similar to the Anders–McNally salts, a wide array of structurally diverse and complex compounds are amenable to C–H functionalization (Scheme 13C). These include natural products and pharmacons as well as small building blocks. In general, the types of arene most compatible with this process are those one would expect to fare best in EAS process: electron-rich aromatics or heteroaromatics though this is not exclusive. This nicely complements the reactivity of the Anders–McNally phosphination process as most azaarenes fail to undergo thianthrenation unless they bear an electron-donating group. Those that do allow for C–H functionalization at sites not observed when attempting C–H phosphination. Olefins also underwent thianthrenation, though a thianthrene-S-oxide (TO)/thianthrene system was used, giving thianthrenium tetrafluoroborate (TT+) salts instead.44 Very recently, Proctor has reported a similar approach to arene functionalization using dibenzothiophene S-oxide (DBTSO), demonstrating the generality for this approach to C–H functionalization.45
Again, similar to the aforementioned phosphonium salts, this transformation would be of little use if these TFT+s were inert species. Ritter showed that this was indeed not the case. In the seminal report in this area,43 Ritter demonstrated that aryl TFT+s could be engaged in virtually every canonical cross-coupling process (Suzuki, Negishi, Heck, Sonogashira, and even carbonylation chemistry) as well as photoredox processes (halogenation, borylation, trifluoromethylthiolation, cyanation, phosphorylation, and Minisci chemistry) (Scheme 13D). Later, similar chemistry with alkenyl TFT+s was reported.44
Subsequent reports in this area dealt with diversifying the chemistry of these species further (Scheme 14). Amination (Scheme 14D),46a fluorination (Scheme 14C),46b trifluoromethylation (Scheme 14C),46c etherification (Scheme 14B),46d and thioetherification (Scheme 14B)46d processes were all realized. These processes were made possible by photoredox catalysis. Under photochemical conditions, SET reduction of the aryl–TFT+ salt by the photocatalyst results in aryl radical generation. This reactive odd-electron intermediate can be engaged by a metal catalyst (typically copper) resulting in C–Y bond formation after reductive elimination. Unlike in typical transition-metal/photoredox chemistry, only the photoredox cycle is a closed cycle; stoichiometric quantities of the transition metal species are needed to facilitate catalysis (Scheme 14). The only exception to this rule is amination as both photoredox and non-photoredox processes were developed. The structure of the amine dictated which of these two approaches is required. Recently, Schoenebeck showed that these sulfonium salts can be used to affect the germylation of arenes using her Pd(I) dimer catalyst.47
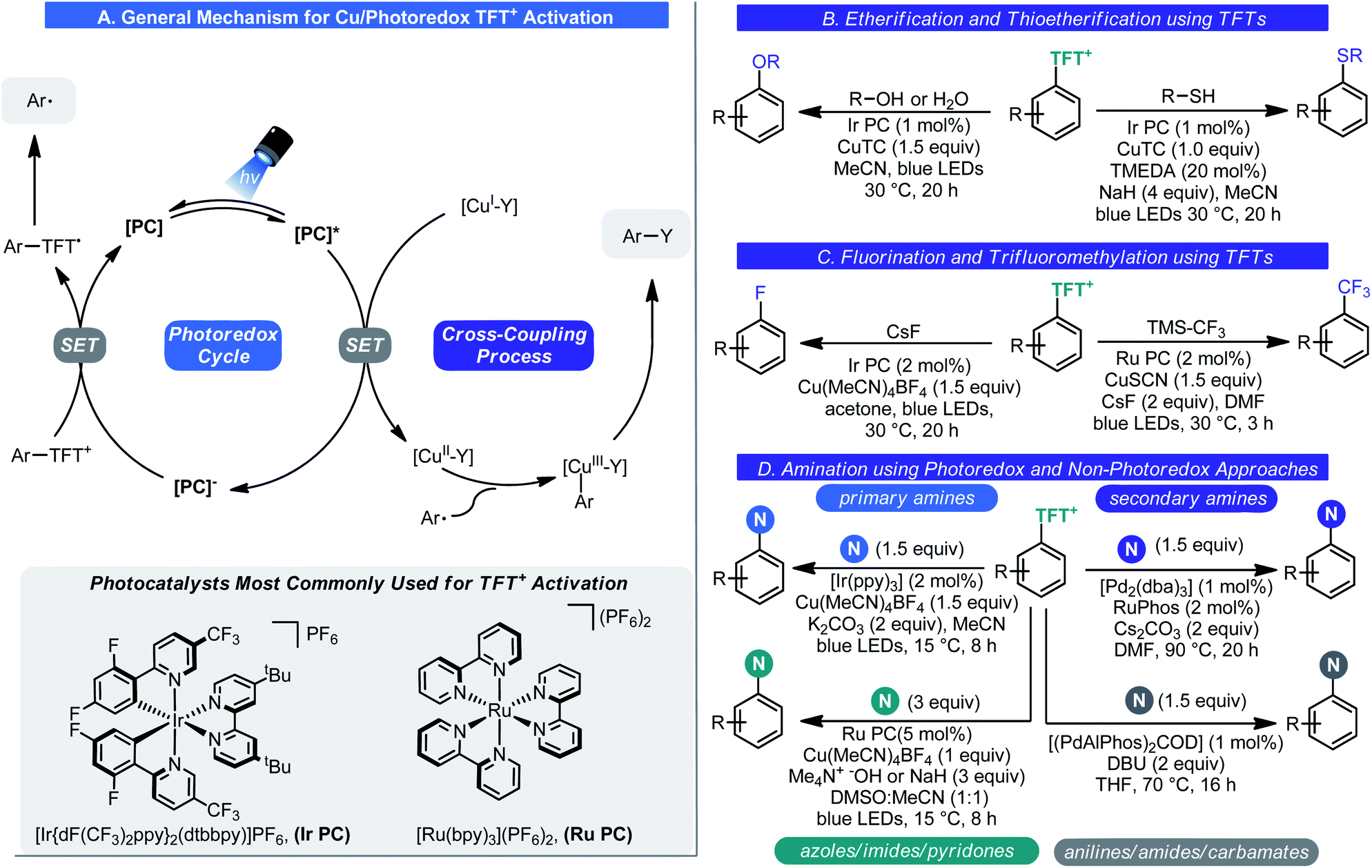 | ||
| Scheme 14 Cu/photoredox chemistry enables diversification of TFT+ salts. | ||
4. Conclusions and outlook
The chalcogen and pnictogen salts examined in this Minireview are clearly powerful reagents for the rapid diversification of C–H bonds. These bench stable powders can be assembled under mild conditions from commodity chemicals with precise regiochemical fidelity. The repertoire of reactions available between the three salts is unmatched for virtually any other aryl reagent class. More specifically, phosphonium and thianthrenium salts reviewed here rival classical diazonium salts in terms of reactivity while retaining desirable attributes for medicinal chemistry applications (ease of preparation, amenable to LSF in complex systems, and dump and stir-type reactivity). Broad adoption of the methods described here by the medicinal chemistry and agrochemical communities is expected. Further, the chemistry of these “hidden gem” salts is still in its infancy and thus opportunities for reaction discovery in both academic and industrial research remain. Ultimately, attention in this area of research and using the philosophy of two-stage directed C–H functionalization is expected to continue and push the boundaries of organic synthesis. Like the salts discussed here, future developments may lie in the curiosities of the past.Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC.
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (b) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- (a) J. Bien, A. Davulcu, A. J. DelMonte, K. J. Fraunhoffer, Z. Gao, C. Hang, Y. Hsiao, W. Hu, K. Katipally, A. Littke, A. Pedro, Y. Qiu, M. Sandoval, R. Schild, M. Soltani, A. Tedesco, D. Vanyo, P. Vemishetti and R. E. Waltermire, Org. Process Res. Dev., 2018, 22, 1393 CrossRef CAS; (b) M. A. Fitzgerald, O. Soltani, C. Wei, D. Skliar, B. Zheng, J. Li, J. Albrecht, M. Schmidt, M. Mahoney, R. J. Fox, K. Tran, K. Zhu and M. D. Eastgate, J. Org. Chem., 2015, 80, 6001 CrossRef CAS; (c) S. R. Wisniewski, J. M. Stevens, M. Yu, K. J. Fraunhoffer, E. O. Romero and S. A. Savage, J. Org. Chem., 2019, 84, 4704 CrossRef CAS; (d) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS.
- (a) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925 RSC; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS; (c) http://www.nsf-cchf.com/ .
- L. Revathi, L. Ravindar, W.-Y. Fang, K. P. Rakes and H.-L. Qin, Adv. Synth. Catal., 2018, 360, 4652 CrossRef CAS.
- (a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (b) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS.
- (a) R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666 CrossRef CAS; (b) M. A. J. Duncton, MedChemComm, 2011, 2, 1135 RSC.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS , for reviews, see: ; (c) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429 CrossRef CAS; (d) Y.-Y. Gui, L. Sun, Z.-P. Lu and D.-G. Yu, Org. Chem. Front., 2016, 3, 522 RSC; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS; (f) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152 CrossRef CAS; (g) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS.
- (a) B. Y. Park, M. T. Pirnot and S. L. Buchwald, J. Org. Chem., 2020, 85, 3234 CrossRef CAS; (b) Z. G. Brill, C. B. Ritts, U. F. Mansoor and N. Sciammetta, Org. Lett., 2020, 22, 410 CrossRef CAS; (c) F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085 CrossRef CAS.
- (a) Z. J. Garlets, J. D. Nguyen and C. R. J. Stephenson, Isr. J. Chem., 2014, 54, 351 CrossRef CAS; (b) Y. Su, N. J. W. Straathof, V. Hessel and T. Noel, Chem.–Eur. J., 2014, 20, 10562 CrossRef CAS; (c) J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2012, 51, 4144 CrossRef CAS.
- S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453 RSC.
- (a) T. Zincke, Liebigs Ann. Chem., 1904, 330, 361 CrossRef; (b) T. Zincke, G. Heuser and W. Möller, Liebigs Ann. Chem., 1904, 333, 296 CrossRef; (c) T. Zincke and W. Wurker, Liebigs Ann. Chem., 1905, 338, 107 CrossRef.
- (a) A. R. Katritzky and S. S. Thind, J. Chem. Soc., Perkin Trans. 1, 1980, 1895 RSC; (b) J. B. Bapat, R. J. Blade, A. J. Boulton, J. Epsztajn, A. R. Katritzky, J. Lewis, P. Molina-Buendia, P. Lin and C. A. Ramsden, Tetrahedron Lett., 1976, 17, 2691 CrossRef; (c) A. R. Katritizky, W. K. Yeung and R. C. Patel, J. Chem. Soc., Perkin Trans. 1, 1982, 2365 RSC.
- Y. Pang, D. Moser and J. Cornella, Synthesis, 2020, 52, 489 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS.
- T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254 CrossRef CAS.
- C. H. Basch, J. Liao, J. Xu, J. J. Piane and M. P. Watson, J. Am. Chem. Soc., 2017, 139, 5313 CrossRef CAS.
- (a) S. L. Rössler, B. J. Jelier, E. Magnier, G. Dagousset, E. M. Carreira and A. Togni, Angew. Chem., Int. Ed., 2020, 59, 9264 CrossRef; (b) J. Wang, M. E. Hoerrner, M. P. Watson and D. J. Weix, Angew. Chem., Int. Ed., 2020, 59, 13484 CrossRef CAS.
- B. J. Jelier, P. F. Tripet, E. Pietrasiak, I. Franzoni, G. Jeschke and A. Togni, Angew. Chem., Int. Ed., 2018, 57, 13784 CrossRef CAS.
- S. L. Rössler, B. J. Jelier, P. F. Tripet, A. Shemet, G. Jeschke, A. Togni and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 526 Search PubMed.
- W. S. Ham, J. Hillenbrand, J. Jacq, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 532 CrossRef CAS.
- J. Hillenbrand, W. S. Ham and T. Ritter, Org. Lett., 2019, 21, 5363 CrossRef CAS.
- P. S. Fier, S. Kim and R. D. Cohen, J. Am. Chem. Soc., 2020, 142, 8614 CrossRef CAS.
- D. Moser, Y. Duan, F. Wang, Y. Ma, M. J. O'Neil and J. Cornella, Angew. Chem., Int. Ed., 2018, 57, 11035 CrossRef CAS.
- E. Anders and F. Markus, Tetrahedron Lett., 1987, 28, 2675 CrossRef CAS.
- (a) F. M. J. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 3037 CAS; (b) D. Marcoux and A. B. Charette, J. Org. Chem., 2008, 73, 590 CrossRef CAS; (c) D. Marcoux and A. B. Charette, Adv. Synth. Catal., 2008, 350, 2967 CrossRef CAS; (d) L. Horner, G. Mummenthey, H. Moser and P. Beck, Chem. Ber., 1966, 99, 2782 CrossRef; (e) E. Redmund, A. R. Leroux, J. Bayardon and S. Juge, Org. Lett., 2010, 12, 1568 CrossRef; (f) A. F. Fearnley, J. An, M. Jackson, P. Lindovska and R. M. Denton, Chem. Commun., 2016, 52, 4987 RSC.
- M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806 CrossRef CAS.
- M. Baumann and I. R Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265 CrossRef.
- R. D. Dolewski, P. J. Fricke and A. McNally, J. Am. Chem. Soc., 2018, 140, 8020 CrossRef CAS.
- (a) R. G. Anderson, B. M. Jett and A. McNally, Tetrahedron, 2018, 74, 3129 CrossRef CAS; (b) R. G. Anderson, B. M. Jett and A. McNally, Angew. Chem., Int. Ed., 2018, 57, 12514 CrossRef CAS; (c) C. Patel, M. Mohnike, M. C. Hilton and A. McNally, Org. Lett., 2018, 20, 2607 CrossRef CAS; (d) J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990 CrossRef CAS; (e) J. N. Levy, J. V. Alegre-Requena, R. Liu, R. S. Paton and A. McNally, J. Am. Chem. Soc., 2020, 142, 11295 CrossRef CAS; (f) X. Zhang and A. McNally, Angew. Chem., Int. Ed., 2017, 56, 9833 CrossRef CAS; (g) X. Zhang and A. McNally, ACS Catal., 2019, 9, 4862 CrossRef CAS.
- M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Science, 2018, 362, 799 CrossRef CAS.
- B. T. Boyle, M. C. Hilton and A. McNally, J. Am. Chem. Soc., 2019, 141, 15441 CrossRef CAS.
- S. Z. Zard, Org. Lett., 2017, 19, 1257 CrossRef CAS.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711 CAS.
- J. L. Koniarczyk, J. W. Greenwood, J. V. Alegre-Requena, R. S. Paton and A. McNally, Angew. Chem., Int. Ed., 2019, 58, 14882 CrossRef CAS.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701 CrossRef CAS.
- P. Cyr, J. Flynn-Robitaille, P. Boissarie and A. Marinier, Org. Lett., 2019, 21, 2265 CrossRef CAS.
- S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806 CrossRef CAS.
- (a) C. Savarin, J. Srogl and L. S. Liebeskind, Org. Lett., 2000, 2, 3229 CrossRef CAS; (b) S. Zhang, D. Marshall and L. S. Liebeskind, J. Org. Chem., 1999, 64, 2796 CrossRef CAS; (c) J. Srogl, G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1997, 119, 12376 CrossRef CAS.
- (a) J. J. Silber and H. J. Shine, J. Org. Chem., 1971, 36, 2923 CrossRef; (b) K. Kim, V. J. Hull and H. J. Shine, J. Org. Chem., 1974, 39, 2534 CrossRef CAS.
- E. A. C. Lucken, J. Chem. Soc., 1962, 4963 RSC.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS.
- J. Chen, J. Li, M. B. Plutschack, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 5616 CrossRef CAS.
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163 CrossRef CAS.
-
(a) P. S. Engl, A. P. Haring, F. Berger, G. Berger, A. Perez-Bitrian and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346 CrossRef CAS;
(b) J. Li, J. Chen
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) , R. Sang, W. S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56 CrossRef CAS;
(c) F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615 CrossRef CAS;
(d) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161 CrossRef CAS.
, R. Sang, W. S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56 CrossRef CAS;
(c) F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615 CrossRef CAS;
(d) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161 CrossRef CAS. - A. Selmani, A. G. Gevondian and F. Schoenebeck, Org. Lett., 2020, 22, 4802 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |